

Abstract
As of 2004, >73 million people were prescribed antiinflammatory medication. Despite the extensive number of current products, many people still suffer from their diseases or the pharmacological properties (side effects) of the medications. Therefore, developing therapeutic strategies to treat inflammation remains an important endeavor. Here, we demonstrate that the soluble epoxide hydrolase (sEH) is a key pharmacologic target for treating acute systemic inflammation. Lipopolysaccharide-induced mortality, systemic hypotension, and histologically evaluated tissue injury were substantially diminished by administration of urea-based, small-molecule inhibitors of sEH to C57BL/6 mice. Moreover, sEH inhibitors decreased plasma levels of proinflammatory cytokines and nitric oxide metabolites while promoting the formation of lipoxins, thus supporting inflammatory resolution. These data suggest that sEH inhibitors have therapeutic efficacy in the treatment and management of acute inflammatory diseases.
Keywords: cyclooxygenase, lipoxin A4, lipoxygenase, proinflammatory mediators, epoxygenase
The oxidative metabolism of polyunsaturated fatty acids produces potent inflammatory mediators (1). The bulk of research has focused on the arachidonic acid derivatives processed by cyclooxygenase (COX) (prostaglandins) and lipoxygenases (LOX) (leukotrienes), as well as cytokines and oxygen/nitrogen radicals. To this end, many pharmaceuticals have been produced to alleviate inflammatory conditions in rheumatoid arthritis, psoriasis, osteoarthritis, and asthma. These drugs include nonsteroidal antiinflammatory drugs (acetylsalicylic acid), specific COX-2 inhibitors (Rofecoxib), and 5-LOX inhibitors (Zileuton).
One critical pathway, still relatively unexplored, is mediated by cytochrome P450 enzymes, transforming arachidonic and linoleic acids to various biologically active compounds, including epoxyeicosatrienoic acids (EETs) or hydroxyeicosatrienoic acids (HETEs) (2, 3) and epoxyoctadecenoic acids (EpOMEs), respectively. EETs are endothelium-derived hyperpolarizing factor candidates that mediate vascular relaxation responses (4) and possess antiinflammatory properties (5–8). EETs and EpOMEs are further metabolized by soluble epoxide hydrolase (sEH) to their corresponding diols, dihydroxyeicosatrienoic acids (DHETs; also known as DiHETs) and dihydroxyoctadecenoic acids (DiHOMEs) (Fig. 5, which is published as supporting information on the PNAS web site) (9).
Based on the antiinflammatory action of EETs, we surmised that increasing cellular EETs by inhibition of sEH would decrease the inflammatory effects of acute endotoxin [i.e., lipopolysaccharide (LPS)] exposure. Endotoxin exposure is a common model of septicemia, a disease with mortality rates of 40–70% (1). LPS is the primary Gram-negative bacteria surface antigen responsible for eliciting immunologic responses. These responses include leukocyte activation, cytokine production, enhanced proinflammatory gene expression, increased reactive oxygen/nitrogen species production, and enhanced biosynthesis of oxidized lipids.
To investigate the role of sEH in acute systemic inflammation, we assessed the effects of sEH inhibitors on mortality and blood pressure in male C57BL/6 mice challenged with 10 mg/kg LPS. Mortality was eliminated and blood pressure was preserved. To enhance our understanding of these results, a second study evaluated the underlying changes in various inflammatory markers during the first 24 h after LPS exposure. Finally, different dosing regimens were evaluated to determine a minimum effective dose.
Materials and Methods
Animals and Chemicals. Male, 7- to 8-week-old C57BL/6 mice (Charles River Laboratories) weighing between 22 and 28 g were used in all experiments. All appropriate vehicle and saline controls were tested in the experiments. The LPS administered was Escherichia coli serotype 0111:B4 from Sigma–Aldrich. All reagents administered to the mice were tested to ensure that they were endotoxin free. Blood was collected by cardiac puncture with an EDTA-rinsed syringe. Each sample was immediately spun, the plasma was separated, and a combination of triphenylphosphine and butylated hydroxytoluene (0.2% wt/wt) was added. All samples were stored at –80°C until analysis. Both sEH inhibitors, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid butyl ester (AUDA-BE) and 1-adamantan-3-(5-(2-(2-ethylethoxy)ethoxy)pentyl)urea (compound 950), were synthesized in house (10, 11).
Lethality. Mice were housed five per cage in a controlled environment and were fed mouse chow ad libitum. To establish that the sEH inhibitor AUDA-BE did not cause lethality, the animals were treated with the sEH inhibitor (20 mg/kg) s.c. for 2 days or the corresponding volume of olive oil or saline during the first week of experimentation. From previous pharmacokinetic experiments we have shown that AUDA-BE is cleared from the animals within 48 h. During the second week of experimentation, the mice were again injected with sEH inhibitor (20 mg/kg) s.c. or the corresponding volume of olive oil or saline; 24 h later, the mice were given a single injection i.p. of LPS (10 mg/kg) in freshly prepared endotoxin-free PBS. Immediately after the LPS exposure, another dose (20 mg/kg) of the AUDA-BE was administered s.c. Mice were examined every 2 hours for lethality. Food and water consumption, urinary output, and weight were monitored throughout the experiment.
AUDA-BE Temporal Study. Mice were administered 20 mg/kg AUDA-BE s.c. 24 h before LPS (10 mg/kg) i.p. exposure. Immediately after the LPS exposure, another dose of 20 mg/kg AUDA-BE was administered. The animals were then overdosed with pentobarbital at various time points (0, 6, 12, and 24 h), and blood and tissue (liver, spleen, and kidney) were collected.
AUDA-BE Dose Response. Mice were administered various doses of AUDA-BE (0, 5, 10, or 20 mg/kg) s.c. 24 h before LPS (10 mg/kg) i.p. exposure. Immediately after the LPS exposure, another dose of the AUDA-BE (0, 5, 10, or 20 mg/kg) was administered. The dosing was designed so that every combination was administered to four mice. Olive oil (vehicle) was administered at the corresponding volume for the 0 mg/kg dose. Twenty-four hours after LPS administration, the mice were killed. Part of the livers, spleens, and kidneys were fixed by formaldehyde (6%) in PBS for pathological examination. Another section of the livers was collected for Western analysis.
Biochemical Analysis. Oxylipin concentrations were measured by HPLC coupled with mass spectrometry (12). For statistical analysis, if the peak was below the limit of detection, the value was assigned at the limit of detection. Quantitative measurements of total blood plasma nitrite (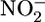 ) and nitrate (
) and nitrate (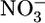 ) were performed as an index of global NO production 24 h after each LPS injection by using the methods described in refs. 13 and 14. TNF-α, IL-6, and monocyte chemoattractant protein 5 (MCP-5) sandwich ELISAs were performed according to the manufacturer's instructions by employing commercially available DuoSet kits (R & D Systems).
) were performed as an index of global NO production 24 h after each LPS injection by using the methods described in refs. 13 and 14. TNF-α, IL-6, and monocyte chemoattractant protein 5 (MCP-5) sandwich ELISAs were performed according to the manufacturer's instructions by employing commercially available DuoSet kits (R & D Systems).
Immunoblot Analysis. Western immunoblot analysis was performed with proteins isolated from liver for inducible nitric oxide synthase (iNOS), COX-2, and sEH. The isolated proteins were separated by electrophoresis by 10% SDS/PAGE and then transferred onto polyvinylidene fluoride membranes (Immobilon P, Millipore). iNOS and COX-2 were detected with polyclonal antibodies from Santa Cruz Biotechnology. sEH was detected with a rabbit polyclonal antibody against affinity-purified recombinant murine sEH prepared in-house. The membranes were incubated with rabbit anti-mouse horseradish peroxidase-linked IgG or donkey anti-mouse horseradish peroxidase-linked IgG whole antibody (Amersham Biosciences) at 1:5,000 or 1:10,000 dilution, respectively, and washed. Then secondary antibodies were visualized by a SuperSignal West Femto Substrate chemiluminescence detection system (Pierce) and detected by autoradiography. The immunodetectable bands were quantified by densitometry with 1d image analysis 3.5.4 (Kodak).
Blood Pressure. A Visitech BP-2000 (Visitech Systems, Apex, NC) blood pressure analysis system was used to determine systolic blood pressure in mice. There were two sessions 25 h apart. The first session was 1 h before LPS injection, and the second session was 24 h after LPS exposure. The computer was programmed to run 10 routine cuff inflations and deflations before recording measurements. Thereafter, the computer was programmed to record 10 measurements per set, with 2 sets, so that a total of 20 measurements were used to determine the systolic blood pressure of each mouse. For individual mouse measurements to be included, we required that the computer successfully identify a systolic blood pressure in at least eight of the 10 measurements within the set. The limit of detection was set at 40 mmHg (1 mmHg = 133 Pa) (15).
Results and Discussion
Mortality Study. The mortality study used a sEH inhibitor, (AUDA-BE) (Fig. 1). This inhibitor has an in vitro IC50 of 50 nM and 100 nM with a colorimetric substrate and recombinant mouse and human sEH, respectively (10). AUDA-BE is metabolized in vivo to the corresponding acid, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid, an equally potent sEH inhibitor. The highest soluble dose of AUDA-BE (20 mg/kg in olive oil) was administered by s.c. injection 24 h before LPS injection and again immediately after LPS exposure. This treatment resulted in a AUDA-BE peak blood concentration of 282 ± 25 nM at 12 h after injection. Without therapeutic intervention, all LPS-exposed mice developed severe hypotension (<40 mmHg systolic blood pressure) and expired within 4 days (Fig. 1). In contrast, treatment with AUDA-BE prevented mortality, restored 79 ± 7% of preLPS systolic blood pressure by 24 h (Fig. 1), and mice resumed normal food and water consumption, grooming, and urine excretion by 48 h (data not shown). Therefore, pharmacologic inhibition of sEH protected mice from the adverse effects of LPS, but how?
Fig. 1.

The sEH inhibitor AUDA-BE prevented LPS-induced mortality and preserved systolic blood pressure in mice. (A) Results from the mortality study show the percentage of animals surviving a 10 mg/kg LPS challenge exposed to 20 mg/kg AUDA-BE (gray triangle), saline (black diamond), or vehicle (olive oil; white square). The differences in mortality between AUDA-BE treatment and untreated mice is significant at all points after 36 h (P < 0.05), but there is not enough power to say whether there is a significant difference in saline- vs. olive oil-treated animals with LPS alone. (B) Systolic blood pressure was measured 1 h before (white bars) and 24 h after (gray bars) LPS challenge ± AUDA-BE. In the absence of AUDA-BE, blood pressure measurements were below the detection limit (40 mmHg). In the presence of AUDA-BE, blood pressure was ≈80% of normal. Data represent the average ± SD of 10 measurements on each of five mice. (C) The chemical structures of the sEH inhibitors described in this manuscript.
AUDA-BE Temporal Study. We began to address this question by examining histological changes at 24 h and temporal shifts in inflammatory mediators including NO, cytokines, chemokines, and oxylipins (a collection of oxygenated fatty acid metabolites) over the first 24 h of LPS intoxication. This experiment addressed the following questions: Does AUDA-BE delay inflammation, promote healing, and/or suppress the inflammatory response? Mice were s.c. injected with AUDA-BE (20 mg/kg), vehicle (olive oil), or saline 24 h before and immediately after LPS (10 mg/kg) exposure. Animals were killed 0, 6, 12, and 24 h after LPS exposure. Pathology was examined in a blind fashion on tissues collected 24 h after LPS injection.
As anticipated, LPS-exposed mice had extensive damage to the liver and kidney (16). However, liver and kidney tissues were not distinguishable from controls in AUDA-BE treated mice. Endotoxemia also produced enlarged spleens with elevated numbers of megakaryocytes, which were decreased by AUDA-BE administration (data not shown).
Endotoxemic hypotension is linked to iNOS expression and excessive NO production (17). As shown in Fig. 2A, the NO metabolites nitrite (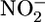 ) and nitrate (
) and nitrate (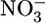 ) were elevated in the plasma of LPS-treated mice. Treatment with AUDA-BE markedly reduced NO production (Fig. 2 A), correlating with blood pressure recovery (Fig. 1B). Western blots also showed a decrease in hepatic iNOS in LPS-exposed mice treated with AUDA-BE (Fig. 6, which is published as supporting information on the PNAS web site). Thus, sEH inhibitors can suppress the induction of iNOS, thereby reducing NO production and the resulting hypotension associated with endotoxemia. In addition, hepatic COX-2 protein levels, a parallel and characteristic index of proinflammatory gene expression, were also suppressed by the sEH inhibitor (Fig. 6).
) were elevated in the plasma of LPS-treated mice. Treatment with AUDA-BE markedly reduced NO production (Fig. 2 A), correlating with blood pressure recovery (Fig. 1B). Western blots also showed a decrease in hepatic iNOS in LPS-exposed mice treated with AUDA-BE (Fig. 6, which is published as supporting information on the PNAS web site). Thus, sEH inhibitors can suppress the induction of iNOS, thereby reducing NO production and the resulting hypotension associated with endotoxemia. In addition, hepatic COX-2 protein levels, a parallel and characteristic index of proinflammatory gene expression, were also suppressed by the sEH inhibitor (Fig. 6).
Fig. 2.

The sEH inhibitor AUDA-BE reduced LPS-induced plasma levels of NOS metabolites, proinflammatory cytokines, and chemokines. Data represent the average ± SD (n = 3) of nitrate/nitrite (A), IL-6 (B), and MCP-5 (C) plasma concentrations in LPS-exposed mice treated with AUDA-BE (gray bars) or vehicle (black bars), depicted as the percentage of controls receiving vehicle without LPS. *, Treated mice are significantly different from mice receiving vehicle (P < 0.05). (Left) Results at various times after LPS exposure. (Right) Dose–response relationships at 24 h, with the x axis indicating the dosing regimen, (x + y), where x = dose before LPS exposure and y = dose after LPS exposures. The TNF-α response was also decreased (data not shown); however, a 2-h time point would best document the magnitude of this change.
Moreover, the characteristic elevations of plasma TNF-α, IL-6, and MCP-5 associated with endotoxemia were also suppressed by AUDA-BE (Fig. 2 B and C). TNF-α, initially produced by resident monocytes and macrophages, is considered an early innate inflammatory mediator. Among its many physiological effects is the induction of vascular cell adhesion molecule-1 on endothelial cells, a critical factor for the transmigration of leukocytes from vascular compartments into inflamed tissues. TNF-α can also induce the formation of IL-6, an ensuing specific immune response. IL-6 plays a significant role in the development of acute inflammatory responses, including endothelial and lymphocyte activation and fever induction (18). Our findings indicate that the AUDA-BE increases EETs and decreases IL-6 production, which is consistent with the antipyretic effects of EETs and pyretic action of epoxygenase inhibitors (6, 19). The chemokine MCP-5 is a potent monocyte chemoattractant (20). The elevation of MCP-5 over control levels in the AUDA-BE-treated animals indicated that, although suppressed relative to endotoxemic mice, lymphocytes still mounted a response in the presence of the sEH inhibitor. Therefore, LPS-exposed mice retain the ability to clear infectious agents. The influence of sEH inhibitor treatment on ≈30 plasma oxylipins was also evaluated by using a mass spectrometric method (12). The result for each analyte was examined individually by Student's t tests. After LPS administration, plasma DHETs and DiHOMEs increased over 24 h (Fig. 3A), revealing activation of the P450 epoxygenase–sEH metabolic pathway during endotoxemia. In contrast and consistent with our theoretical construct, pharmacologic inhibition of sEH with AUDA-BE increased plasma EpOMEs and decreased DiHOMEs. This finding can be depicted more clearly by comparing ratios of EETs to DHETs. With LPS administration alone, the ratio decreases, whereas prophylactic administration of AUDA-BE increases the EET and decreases the DHET concentrations to below the 1 nM detection limit (Fig. 3A). Although AUDA does not directly inhibit phospholipase A2, COX-2, thromboxane synthase, or human 15-LOX-1 in vitro (P. Baecker, personal communication), the sEH inhibitor indirectly altered endotoxemia-induced COX and LOX metabolite production (Fig. 3 B and C).
Fig. 3.

LPS exposure produced temporal changes in plasma oxylipins derived from epoxygenases, LOX, and COX pathways, some of which were reversed by treatment with AUDA-BE. Results at each time point (average ± SD; n = 4) in LPS-exposed mice treated with AUDA-BE (gray bars) or vehicle (black bars) are depicted as the percentage of controls receiving vehicle without LPS. *, Treated mice are significantly different from mice receiving vehicle (P < 0.05). (A) Epoxygenase-dependent metabolites. (Top) ∑ EpOME (∑ linoleate epoxides). (Middle) ∑ DiHOME (∑ linoleate diols). (Bottom) Ratio of ∑ EET to ∑ DHET (arachidonate epoxides/diols). (B) COX-dependent metabolites. From top to bottom, the graphs show PGD2 (prostagladin), PGE2 (prostaglandin), TXB2 (thromboxane), and 6-keto-PGF1α (prostaglandin). (C) LOX-dependent metabolites. (Top) HODE (monohydroxy linoleate). (Middle and Bottom) 15-HETE (Middle) and 5-Hete (Bottom) (monohydroxy arachidonates).
Consistent with the observed decline in hepatic COX-2 expression, AUDA-BE decreased prosta-5,13-dien-1-oic acid, 9,15-dihydroxy-11-oxo-, (5Z,9α,13E,15S) (PGD2) and prosta-5,13-dien-1-oic acid, 11,15-dihydroxy-9-oxo-, (5Z,11α,13E,15S) (PGE2), which peaked 6 and 12 h, respectively, after LPS exposure (Fig. 3B). By comparison LPS-induced changes in 9α,11,15S-trihydroxythromba-5Z,13E-dien-1-oic acid (TXB2) and 6-oxo-9α,11α,15S-trihydroxy-prost-13-en-1-oic acid (6-keto-PGF1α) were modest and consistent with the literature, with concentrations peaking by 6 h (21, 22). With AUDA-BE treatment, small but significant decreases in these metabolites were also observed at all time points.
LPS challenge also produced dramatic changes in products of LOX metabolism, including hydroxyoctadecadienoic acids (HODEs) and midchain HETEs that were modulated by AUDA-BE exposure (Fig. 3C). In vehicle-treated mice, initial HODE concentrations dropped 6 h after LPS exposure, then increased gradually over the study period, with 9- and 13-HODE showing similar behavior. In contrast, mice treated with AUDA-BE showed an oscillating HODE pattern peaking at 12 h. Although the magnitude of the response was lower than that of the HODEs, this temporal pattern was paralleled by the 15-HETE (Fig. 3C) and 12-HETE (data not shown). The 12-HETE was associated with higher variability, most likely because of differential contamination or rupture of platelets in the isolated plasma. Because the enzyme(s) that produces HODEs also catalyzes the formation of 12- and 15-HETE, these parallel patterns are not surprising. As opposed to the changes in these midchain hydroxy fatty acids, 5-HETE concentrations showed a steady increase to 12 h and remained constant through 24 h in untreated LPS-exposed mice (Fig. 3B). 5-HETE is rapidly produced upon inflammatory insult and is a potent mediator of neutrophil function with chemotactic activity and the ability to modulate lysosomal enzyme release (23). The sEH inhibitors blunted the initial 5-HETE increase and lowered concentrations at 24 h. The involvement of 5-LOX in inflammation is complex because its primary metabolite, the 5-hydroperoxyeicosatetraenoic acid, is a precursor not only for 5-HETE but also for leukotrienes (24) and, ultimately, lipoxins (25).
Inflammatory resolution is promoted by the production of lipoxins, which contribute to the counter regulation of leukocyte recruitment, neutrophil adherence to endothelial and epithelial cells, and macrophage clearance of apoptotic neutrophils. Lipoxin formation can occur when 5-LOX products interact with 12-LOX or 15-LOX (25). Decreased 15-HETE levels observed at the middle-to-late phases of endotoxemia might therefore relate to 5S,6R,15S-trihydroxy-7E,9E,11Z,13E-eicosatertraenoic (lipoxin A4) formation. Consistent with this hypothesis, lipoxin A4 was only detected at the 24-h time point in mice treated with AUDA-BE (data not shown). Hence, inhibition of sEH may reduce the initiation phase and promote the resolution phases of the acute inflammatory response.
AUDA-BE Dose Response. Knowing that the AUDA-BE treatment not only directly affected epoxygenase-dependent metabolism but also altered profiles of other inflammatory mediators impacting survival, an experiment was conducted to determine a minimum effective inhibitor dose and dosing regimen. Groups of mice were administered 0, 5, 10, or 20 mg/kg AUDA-BE by s.c. injection 24 h before LPS challenge and another dose of the AUDA-BE immediately after LPS exposure.
The plasma oxylipin profiles 24 h after LPS exposure for each dosing regimen were compared with vehicle control mice and vehicle-plus-LPS endotoxemic mice. The resulting data matrix was analyzed to assess group differences by using two-tailed t tests (P < 0.05) (Fig. 4). Prophylactic administration of AUDA-BE provided the maximum reduction in LPS-induced proinflammatory mediators (Figs. 2 A–C and 4). The AUDA-BE regimens (expressed as the dose before LPS + the dose after LPS) of (10 + 10), (10 + 20), (20 + 10), or (20 + 20) produced similar responses in oxylipin profiles (Fig. 4), as they did for IL-6, MCP-5, and NO metabolites (Fig. 2 A–C). In all cases, the (20 + 10) and (20 + 20) doses were indistinguishable. Without prophylactic AUDA-BE administration and with AUDA-BE doses after LPS of ≤10 mg/kg, (0 + 5) and (0 + 10), treated mice were not different from endotoxemic controls (Fig. 4, V + LPS). Increasing the AUDA-BE dose after LPS to 20 mg/kg produced differences in epoxygenase and LOX metabolites. However, all analytes remained different from controls (Fig. 4, V no LPS), suggesting that, in the current formulation, a single AUDA-BE treatment after LPS does not fully return animals to healthy conditions.
Fig. 4.

Paired t tests (P < 0.05) were used to evaluate the efficacy of sEH inhibitor treatments in shifting endotoxemic oxylipin concentrations toward control values. Mice were administered AUDA-BE (A), compound 950 (950), or vehicle (V) before and after a 10 mg/kg LPS exposure. The inhibitor dose in mg/kg is depicted as (x + y), where x = inhibitor dose before LPS exposure and y = inhibitor dose after LPS exposure. Black squares indicate values not statistically different from those of endotoxemic mice. Gray squares indicate values not statistically different from those of endotoxemic and control mice. White squares indicate values not statistically different from those of control mice. *, Not statistically different from endotoxemic or control mice.
A pathway-specific analysis is used to discuss the remaining trends in this complex data set. The epoxygenase metabolites were the most responsive to AUDA-BE dose, followed by those derived from the LOX and COX pathways. A (5 + 20) dose of AUDA-BE displayed significant differences in all epoxygenase analytes; it increased the epoxides and decreased their corresponding diols, as compared with the endotoxemic mice. In fact, concentrations of the DiHOMEs and DHETs returned to control concentrations with a (10 + 10) AUDA-BE dosing regimen. The arachidonic acid diols actually decreased to control concentrations at even lower AUDA-BE doses: (5 + 10) for 11, 12-DHET and 8,9-DHET and (10 + 0) for 14,15-DHET and 5,6-DHET.
Higher AUDA-BE doses were required to produce differences in the LOX pathway. Although a (5 + 5) AUDA-BE dose decreased the HODEs, trihydroxyoctadecenoic acids and 5-HETE, a (5 + 10) dose was required to reduce the 12- and 15-HETE relative to endotoxemic mice. Increasing the prophylactic dose to 10 mg/kg followed by 10 mg/kg after LPS exposure (i.e., 10 + 10) reduced the trihydroxyoctadecenoic acids and 5-HETE back to concentrations indistinguishable from control mice.
Finally, the COX metabolites depicted minimal but significant changes with the addition of AUDA-BE. Concentrations of (10 + 10) or (20 + 10) were necessary to decrease the concentrations of PGD2 and PGE2, whereas even the highest doses of AUDA-BE did not alter TXB2 or 6-keto-PGF1α at 24 h (Fig. 4).
From a therapeutic perspective, the greatest limitation of AUDA-BE is its solubility in aqueous media (5 μg/ml) and its current formulation. As evidenced by the efficacy of the (0 + 20) regimen (Fig. 4), administering higher amounts of the inhibitor could reduce inflammatory mediators without requiring prophylactic AUDA-BE exposure. To address this issue, we synthesized an analog of AUDA-BE (compound 950) (Fig. 1C) with greater aqueous solubility (95 μg/ml) and evaluated its ability to modulate LPS-induced shifts in oxylipin profiles at 24 h by using the maximum AUDA-BE dosing regimen of (20 + 20). The results with compound 950 were slightly more variable than with AUDA-BE, possibly because of the influence of its increased water solubility on pharmacokinetic parameters. However, compound 950 was comparable to AUDA-BE but had greater effects on COX metabolism. In particular, plasma PGE2 concentrations after LPS exposure were reduced to control levels, and 6-keto-PGF1α and TXB2 were different from endotoxemic mice (Fig. 4).
Conclusion
In summary, the adamantyl alkyl urea-based sEH inhibitors increase fatty acid epoxides, indirectly reduce the production of NO, cytokines, and proinflammatory lipid mediators, minimize systemic hypotension, and prevent mortality in a mouse model of septic shock. Additionally, they appear to accelerate inflammatory resolution by enhancing lipoxin A4 production. Our findings are also consistent with the hypothesis that EETs prevented the amplification of an inflammatory event by inhibition of transcription factor NF-κB and IκB kinase (5). Without NF-κB translocation to the nucleus, resident macrophages would not produce proinflammatory proteins, such as TNF-α, IL6, iNOS, and COX-2. Additionally, as alluded to by Smith et al. (26), it would prevent the surface expression of cell adhesion molecules, which would further dampen infiltration of leukocytes and thus the amplification of proinflammatory mediators, including cytokines and oxylipins. Further studies clarifying the precise molecular mechanisms and mode of action of these inhibitors will increase our understanding of the role of sEH in inflammation and provide a strong foundation for the development of antiinflammatory therapeutics. The ability of these compounds to modulate not only epoxygenase metabolism but also LOX and COX cascades are reminiscent of the new drug licofelone, which affects the 5-LOX and COX-2 in vivo activity. However, unlike the LOX/COX inhibitor, which acts directly to prevent synthesis of proinflammatory lipid mediators, the sEH inhibitors stabilize antiinflammatory oxylipins and elevate proresolution mediators. Therefore, these molecules, which are structurally distinct from other known antiinflammatory drugs, appear to act as multifunctional/multitarget agents that modulate acute inflammatory responses.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Environmental Health Sciences Grant R37 ES02710, Superfund Basic Research Program Grant P42 ES04699, Center for Children's Environmental Health and Disease Prevention Grants P30 ES05707 and 1 P01 ES11269, and Advanced Training in Environmental Toxicology T32 ES0075059 (K.R.S.); and by University of California Systemwide Biotechnology Training Grant 2001-07 (to K.R.S.); a University of California, Davis, Health Systems Research Award (to J.P.E.); and the Paul F. Gulyassy Endowed Professorship (to J.P.E.).
Author contributions: K.R.S. and L.K. performed research; J.W.N. and I.-H.K. contributed new reagents/analytic tools; K.R.S. and L.K. analyzed data; K.R.S. wrote the paper; and J.P.E. supervised research and writing of manuscript.
Abbreviations: AUDA-BE, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid butyl ester; compound 950, 1-adamantan-3-(5-(2-(2-ethylethoxy)ethoxy)pentyl)urea; iNOS, inducible nitric oxide synthase; COX, cyclooxygenase; LOX, lipoxygenase; sEH, soluble epoxide hydrolase; DHET, dihydroxyeicosatrienoic acid; DiHOME, dihydroxyoctadecenoic acid; EET, epoxyeicosatrienoic acid; EpOME, epoxyoctadecenoic acid; HETE, hydroxyeicosatetraenoic acid; HODE, hydroxyoctadecadienoic acid; LPS, lipopolysaccharide; MCP-5, monocyte chemoattractant protein 5; TXB2, 9α,11,15S-trihydroxythromba-5Z,13E-dien-1-oic acid; PGE2, prosta-5,13-dien-1-oic acid,11,15-dihydroxy-9-oxo-,(5Z,11α,13E,15S); PGD2, prosta-5,13-dien-1-oic acid, 9,15-dihydroxy-11-oxo-,(5Z,9α,13E,15S); 6-keto-PGF1α, 6-oxo-9α,11α,15S-trihydroxy-prost-13-en-1-oic acid.
References
- 1.Cohen, J. (2002) Nature 420, 885–891. [DOI] [PubMed] [Google Scholar]
- 2.Campbell, W. B., Brady, M. T., Rosolowsky, L. J. & Falck, J. R. (1991) Endocrinology 128, 2183–2194. [DOI] [PubMed] [Google Scholar]
- 3.Carroll, M. A., Schwartzman, M., Sacerdoti, D. & McGiff, J. C. (1988) Am. J. Med. Sci. 295, 268–274. [DOI] [PubMed] [Google Scholar]
- 4.Campbell, W. B., Gebremedhin, D., Pratt, P. F. & Harder, D. R. (1996) Circ. Res. 78, 415–423. [DOI] [PubMed] [Google Scholar]
- 5.Node, K., Huo, Y., Ruan, X., Yang, B., Spiecker, M., Ley, K., Zeldin, D. C. & Liao, J. K. (1999) Science 285, 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozak, W., Kluger, M. J., Kozak, A., Wachulec, M. & Dokladny, K. (2000) Am. J. Physiol. 279, R455–R460. [DOI] [PubMed] [Google Scholar]
- 7.Falck, J. R., Reddy, L. M., Reddy, Y. K., Bondlela, M., Krishna, U. M., Ji, Y., Sun, J. & Liao, J. K. (2003) Bioorg. Med. Chem. Lett. 13, 4011–4014. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki, M., Ostanin, D., Elrod, J. W., Oshima, T., Jordan, P., Itoh, M., Joh, T., Minagar, A. & Alexander, J. S. (2003) Am. J. Physiol. 284, C422–C428. [DOI] [PubMed] [Google Scholar]
- 9.Moghaddam, M. F., Grant, D. F., Cheek, J. M., Greene, J. F., Williamson, K. C. & Hammock, B. D. (1997) Nat. Med. 3, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morisseau, C., Goodrow, M. H., Newman, J. W., Wheelock, C. E., Dowdy, D. L. & Hammock, B. D. (2002) Biochem. Pharmacol. 63, 1599–1608. [DOI] [PubMed] [Google Scholar]
- 11.Kim, I. H., Morisseau, C., Watanabe, T. & Hammock, B. D. (2004) J. Med. Chem. 47, 2110–2122. [DOI] [PubMed] [Google Scholar]
- 12.Schmelzer, K., Newman, J., Dettmer, K. & Hammock, B. (2005) J. Chrom. B, in press.
- 13.Braman, R. S. & Hendrix, S. A. (1989) Anal. Chem. 61, 2715–2718. [DOI] [PubMed] [Google Scholar]
- 14.Van Der Vliet, A., Nguyen, M. N., Shigenaga, M. K., Eiserich, J. P., Marelich, G. P. & Cross, C. E. (2000) Am. J. Physiol. 279, L537–L546. [DOI] [PubMed] [Google Scholar]
- 15.Krege, J. H., Hodgin, J. B., Hagaman, J. R. & Smithies, O. (1995) Hypertension 25, 1111–1115. [DOI] [PubMed] [Google Scholar]
- 16.Maronpot, R. & Gaul, B. W. (1999) Pathology of the Mouse: Reference and Atlas (Cache River, Vienna, IL).
- 17.Thiemermann, C. (1997) Gen. Pharmacol. 29, 159–166. [DOI] [PubMed] [Google Scholar]
- 18.Machnicki, M. (1995) Postepy. Hig. Med. Dosw. 49, 53–57. [PubMed] [Google Scholar]
- 19.Nakashima, T., Yoshida, Y., Miyata, S. & Kiyohara, T. (2001) Neurosci. Lett. 310, 141–144. [DOI] [PubMed] [Google Scholar]
- 20.Kopydlowski, K. M., Salkowski, C. A., Cody, M. J., van Rooijen, N., Major, J., Hamilton, T. A. & Vogel, S. N. (1999) J. Immunol. 163, 1537–1544. [PubMed] [Google Scholar]
- 21.Filipov, N. M., Thompson, F. N., Stuedemann, J. A., Elsasser, T. H., Kahl, S., Stanker, L. H., Young, C. R., Dawe, D. L. & Smith, C. K. (2000) Proc. Soc. Exp. Biol. Med. 225, 136–142. [DOI] [PubMed] [Google Scholar]
- 22.Cargile, J. L., MacKay, R. J., Dankert, J. R. & Skelley, L. (1995) Am. J. Vet. Res. 56, 1445–1450. [PubMed] [Google Scholar]
- 23.Spanbroek, R., Hildner, M., Steinhilber, D., Fusenig, N., Yoneda, K., Radmark, O., Samuelsson, B. & Habenicht, A. J. (2000) Blood 96, 3857–3865. [PubMed] [Google Scholar]
- 24.Samuelsson, B., Dahlen, S. E., Lindgren, J. A., Rouzer, C. A. & Serhan, C. N. (1987) Science 237, 1171–1176. [DOI] [PubMed] [Google Scholar]
- 25.Serhan, C. N. & Chiang, N. (2002) Sci. World J. 2, 169–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith, K. R., Pinkerton, K. E., Watanabe, T., Pedersen, T. L., Ma, S. J. & Hammock, B. D. (2005) Proc. Natl. Acad. Sci. USA 102, 2186–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.