

Abstract
Based on previous screening for keratinolytic nonpathogenic fungi, Paecilomyces marquandii and Doratomyces microsporus were selected for production of potent keratinases. The enzymes were purified and their main biochemical characteristics were determined (molecular masses, optimal temperature and pH for keratinolytic activity, N-terminal amino acid sequences). Studies of substrate specificity revealed that skin constituents, such as the stratum corneum, and appendages such as nail but not hair, feather, and wool were efficiently hydrolyzed by the P. marquandii keratinase and about 40% less by the D. microsporus keratinase. Hydrolysis of keratin could be increased by the presence of reducing agents. The catalytic properties of the keratinases were studied and compared to those of some known commercial proteases. The profile of the oxidized insulin B-chain digestion revealed that both keratinases, like proteinase K but not subtilisin, trypsin, or elastase, possess broad cleavage specificity with a preference for aromatic and nonpolar amino acid residues at the P-1 position. Kinetic studies were performed on a synthetic substrate, succinyl-Ala-Ala-Pro-Phe-p-nitroanilide. The keratinase of P. marquandii exhibited the lowest Km among microbial keratinases reported in the literature, and its catalytic efficiency was high in comparison to that of D. microsporus keratinase and proteinase K. All three keratinolytic enzymes, the keratinases of P. marquandii and D. microsporus as well as proteinase K, were significantly more active on keratin than subtilisin, trypsin, elastase, chymotrypsin, or collagenase.
Keratins are the most abundant proteins in epithelial cells of vertebrates and represent the major constituents of skin and its appendages such as nail, hair, feather, and wool. The protein chains are packed tightly either in α-chain (α-keratins) or in β-sheet (β-keratins) structures. Keratins belong to the superfamily of intermediate filament proteins. Their high degree of cross-linking by disulfide bonds, hydrophobic interactions, and hydrogen bonds stabilizes keratin filament structure (11). Therefore, keratinous material is water insoluble and extremely resistant to degradation by proteolytic enzymes such as trypsin, pepsin, and papain.
A group of proteolytic enzymes which are able to hydrolyze insoluble keratins more efficiently than other proteases are called keratinases (29). They are produced by some insects and mostly by microorganisms. The best studied are keratinases from the dermatophytic genera Microsporum (26, 37) and Trichophyton (30, 38) as well as from bacteria of the genera Bacillus (4, 14, 20, 24, 34, 35) and Streptomyces (2, 3, 27). There are relatively few reports on characterization of the keratinases from nondermatophytic fungi (5, 7, 12, 25, 32, 33).
Most keratinases have some common characteristics despite their different origins. They belong mainly to the extracellular serine proteases, with the exception of keratinases from yeasts, which belong to the aspartic proteases (23, 28). The molecular masses of the enzymes range from 20 kDa to 60 kDa. They are mostly active in alkaline environments, with optimal activity at temperatures up to 50°C. Thermostable keratinases with optimal temperatures of around 85°C and a higher molecular mass have been reported (5, 8, 31). The potential use of keratinases is in different applications where keratins should be hydrolyzed, such as the leather and detergent industries, textiles, waste bioconversion, medicine, and cosmetics for drug delivery through nails and degradation of keratinized skin.
In our previous studies potent keratinase producers, among them Aspergillus flavus (9) and Doratomyces microsporus (12), were selected out of 300 nonpathogenic fungi which are preferred for biotechnological applications. The keratinases were produced by cultivating strains in optimized media under submerged aerobic conditions. The enzymes were isolated and purified and their main biochemical characteristics were determined. A keratinase produced by the fungus Paecilomyces marquandii promised to be still more active than the previously studied keratinolytic enzymes. In the present report, we investigated its characteristics. We examined in detail the catalytic properties as well as the specificity of the P. marquandii and D. microsporus keratinases for both natural and synthetic substrates. A comparison of the enzyme properties with those of some commercial proteases is presented. Our aim was to elucidate specific characteristics which distinguish keratinases from nonkeratinolytic proteases.
MATERIALS AND METHODS
Production and purification of keratinases.
Fungal strains Paecilomyces marquandii (MZKI B639) and Doratomyces microsporus (MZKI B399) were used as producers of the keratinolytic enzymes. Both strains have been isolated and identified in the Laboratory of Botany, Faculty of Medicine and Pharmacy, University of Franche-Comté, Besançon, France, and are deposited in the Culture Collection of the National Institute of Chemistry (MZKI), Ljubljana, Slovenia. The fungi are maintained on potato dextrose agar (Fluka) slants.
For keratinase production, strains of P. marquandii and D. microsporus were cultivated in shaken flasks. Each strain was grown in a liquid medium containing soy flour as an enzyme inducer and (g/liter): KH2PO4, 1.5; K2HPO4, 1.0; MgSO4 · 7H2O, 0.2; CaCl2 · 2H2O, 0.2; NaCl, 0.2; ZnSO4 · 7H2O, 0.002; peptone, 0.4; malt extract, 1.0; glucose, 1.0; soy flour, 5.0; and glycerol, 2 ml/liter. The pH of the medium was adjusted to 6, and aliquots of 100 ml were distributed into 500-ml Erlenmayer flasks and autoclaved. After inoculation with a spore suspension (106 spores/ml medium), the fermentation was carried out at 30°C and 100 rpm on a rotary shaker until the keratinolytic activity reached its maximum, i.e., for 5 days with P. marquandii and for 4 days with D. microsporus.
Crude enzymes were prepared by concentration of the broth filtrate (PLCC 5K membrane, Minitan system, Millipore, Bedford, MA) followed by overnight dialysis against deionized water and subsequent lyophilization. For enzyme purification, the lyophilized powder was dissolved in 50 mM phosphate buffer (pH 7.0) containing 1 M ammonium sulfate and the enzyme solution was applied onto a preparative column for hydrophobic interaction chromatography (HiPrep 16/10 Phenyl FF, Pharmacia), previously equilibrated with the same buffer. The enzyme was eluted by stepwise decreasing salt concentrations from 1 M to 0 M at a flow rate of 3.0 ml/min. The keratinolytic active fraction was pooled and concentrated by ultrafiltration (YM10 membrane, Amicon). Subsequently the enzyme solution was applied on a gel filtration column (Superose 12 HR 10/30, Pharmacia) equilibrated with a 50 mM phosphate buffer (pH 7.5) containing 0.2 M NaCl. The purified enzyme was stored in aliquots at −20°C.
Soluble protein determination.
The concentration of soluble protein was measured at 562 nm by the bicinchoninic acid assay according to the manufacturer's instructions (Sigma). The protein concentration was determined by using a calibration curve that was established with known concentrations of bovine serum albumin.
The purity of the keratinases and their molecular masses were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) according to the method of Laemmli (19). The MiniProtean II system was used according to the manufacturer's instructions (Bio-Rad). The proteins were separated on a 12% gel and stained with a 0.1% (wt/vol) solution of Coomassie brilliant blue R (Sigma). Low-molecular-mass markers (Dalton mark VII-L, Sigma) were used as protein standards for determination of molecular masses.
N-terminal amino acid sequencing of keratinases and sequence analysis.
The N-terminal sequences of the purified keratinases were determined by automated Edman degradation using a pulsed-liquid sequence analyzer (Procise 492A protein sequencing system, Applied Biosystems). Samples were purified by reversed-phase high-performance liquid chromatography (RP-HPLC), dried on a SpeedVac concentrator (Savant), dissolved in 20% (vol/vol) acetonitrile/water containing 0.001% (vol/vol) trifluoroacetic acid (TFA), and sequenced. The sequences obtained were compared to sequences in the TrEMBL/Swissprot database using BLASTp algorithm (http://www.expasy.org/tools/BLAST/).
Determination of keratinase activity.
Keratinolytic activity was examined using keratin from the stratum corneum of the human sole as a substrate. The preparation of keratin powder was described previously (12). Briefly, scrapings of human sole were defatted, well rinsed, dried, ground to a powder, and sifted through a 0.2-mm screen. The previously described activity assay (12) was slightly modified and performed as follows: the reaction mixture comprised 4.0 ml of 28 mM Tris-HCl buffer (pH 8.0), 1.0 ml of the enzyme solution, and 20 mg of the keratin powder. Incubation was carried out in a water bath at 45°C for 30 min with constant agitation of 160 rpm. The enzyme reaction was terminated by the addition of 2.0 ml 10% (wt/vol) trichloroacetic acid (TCA) and then allowed to stand at 4°C for 30 min. After centrifugation (10,000 rpm, 15 min) in a cooled centrifuge (Sorvall), the absorbance of the supernatant was measured spectrophotometrically at 280 nm (spectrophotometer from Beckman). The control was treated the same way except that TCA was added before the incubation. One unit of keratinolytic activity was defined as an increase of corrected A280 for 0.100 under the conditions described. The data presented are mean values of three parallel determinations.
Further, more native keratins (keratin from human nail and hair, porcine nail, chicken feather, sheep wool, and commercial bovine keratin [ICN Biomedicals Inc.]) and nonkeratinous fibrillar proteins (collagen and elastin) as well as bovine serum albumin and casein were used as substrates to examine keratinase activity on different native proteins. The preparation of keratinous materials was described in our previous publication (12). The incubation procedure was the same as described for keratinolytic activity determination. The extent of proteolysis was determined spectrophotometrically at 280 nm by measuring of TCA-soluble peptides released from the substrate during the incubation.
In addition, the keratinase activity assay was used to compare the activities of selected proteases (Sigma). The enzymes tested were keratinases from P. marquandii and D. microsporus, chymotrypsin, collagenase, elastase, proteinase K, subtilisin, and trypsin. Keratin of the stratum corneum and casein were chosen as model substrates for enzyme activity determination. Stock solutions in concentration of 1 mg/ml were prepared in 28 mM Tris-HCl buffer (pH 8.0) for each enzyme. They were diluted adequately with the same buffer and then the substrate was added. Incubation and determination conditions were the same as described above.
Effects of pH, temperature, proteinase inhibitors, organic solvents, detergents, and reducing agents on keratinolytic activity.
The effect of pH on the keratinolytic activity of the keratinases was assayed at 45°C using 28 mM buffers of various pH values: citrate buffer (pH 4 to 6), phosphate buffer (pH 6 to 8), Tris-HCl (pH 7 to 9), and glycine-NaOH (pH 9 to 11). The optimum temperature for the enzyme activity was determined by performing the enzyme reaction at various temperatures between 20°C and 80°C at pH 8.0 (28 mM Tris-HCl). All other conditions were the same as described in the keratinolytic activity assay.
The effects of protease inhibitors (EDTA, phenylmethylsulfonyl fluoride [PMSF], and iodoacetamide) and other chemicals, dithiothreitol (DTT), β-mercaptoethanol (β-ME), sodium dodecyl sulfate (SDS), dimethyl sulfoxide (DMSO), and isopropanol, on keratinolytic activity were determined (for concentrations, see Table 1). The purified keratinases were preincubated with each compound in 28 mM Tris-HCl buffer (pH 8.0) at 30°C for 10 min. The control was preincubated without the compound. Keratin of the stratum corneum was then added to each preparation, and the residual keratinolytic activity was measured at 45°C as described above for determination of keratinase activity.
TABLE 1.
Effect of protease inhibitors and additives on activity of P. marquandii and D. microsporus keratinases
| Compound | Concn | Residual activity (%)
|
|
|---|---|---|---|
| Keratinase of P. marquandii | Keratinase of D. microsporus | ||
| Control | 100 | 100 | |
| PMSF | 1 mM | 0 | 0 |
| EDTA | 5 mM | 70 | 75 |
| Iodoacetamide | 0.05 mM | 98 | 99 |
| DTT | 0.5 mM | 143 | 175 |
| 1 mM | 230 | 322 | |
| 5 mM | 197 | 266 | |
| β-ME | 5 mM | 108 | 160 |
| 25 mM | 115 | 215 | |
| SDS | 0.1% | 29 | 23 |
| 0.5% | 14 | 16 | |
| DMSO | 1% | 103 | 100 |
| 5% | 105 | 104 | |
| 10% | 88 | 90 | |
| Isopropanol | 1% | 88 | 85 |
Proteolytic specificity of keratinases.
The proteolytic specificity was determined by measuring the ability of the keratinases to hydrolyze selected synthetic substrates and the oxidized B-chain of insulin.
The synthetic substrates used in this study (all purchased from Bachem) were prepared as stock solutions in DMSO: 200 mM N-succinyl-Ala-Ala-Ala-pNA (AAA), Ac-Tyr-OEt (ATEE), Bz-Arg-pNA · HCl (L-BAPA), 100 mM N-succinyl-Ala-Ala-Pro-Phe-pNA (AAPF), FA-Leu-Gly-Pro-Ala-OH (FALGPA), or in isopropanol (100 mM Bz-Tyr-pNA). The enzymatic reactions were carried out in 28 mM Tris-HCl buffer (pH 8.0) at 45°C. The substrate concentration was 1 mM, while the keratinase was added at an amount adequate to be able to follow the initial reaction velocity. The hydrolysis of peptides was monitored spectrophotometrically at 237 nm for ATEE, at 324 nm for FALGPA, and at 405 nm for p-NA peptides. Then the best substrate was chosen for kinetic studies of keratinase activity. At least five concentrations of the chosen synthetic peptide were assayed under the same conditions. The final concentration of organic solvent in the reaction mixture never exceeded 5% (vol/vol). The hydrolysis was followed continuously and the initial velocities were determined. The values of Michaelis-Menten constant (Km), maximal velocity (Vmax), and catalytic constant (kcat) were calculated by nonlinear regression using Michaelis-Menten equation.
To determine the hydrolysis sites, each keratinase (0.2 μg) was incubated with the oxidized insulin B-chain (0.1% [wt/vol]) in 28 mM Tris-HCl buffer (pH 8.0), and the final volume was 100 μl. The reaction mixture was incubated at 30°C for 20 min and 12 h. The reaction was stopped by adding 1 volume of 5% (vol/vol) acetonitrile in 0.05% (vol/vol) TFA. An aliquot of 100 μl of the sample was applied on RP-HPLC column C18 (Chromospher, HPLC system; Knauer). The peptides were eluted with an increasing gradient of acetonitrile from 5% to 95% (vol/vol) in 0.05% (vol/vol) TFA. The detection was performed at 215 nm. The molecular masses of the peptide products were determined by electrospray ionization mass spectroscopy (ESI-MS). The cleavage sites were defined by using a computer program, FindPept (http://ca.expasy.org/tools/findpept/), which enables peptide identification after protein cleavage on the basis of the experimentally determined size of the products.
RESULTS AND DISCUSSION
Keratinases which are able to degrade extremely resistant keratins are mostly isolated from pathogenic dermatophytes or bacteria. We searched for prospective keratinase producers among the nonpathogenic filamentous fungi. The most outstanding keratinolytic activity was found in keratinases from Paecilomyces marquandii, Doratomyces microsporus, and Aspergillus flavus strains. Due to the reputation of A. flavus as the potential producer of aflatoxins, we have focused our research on the other two strains.
Production and purification of keratinases.
Fungal strains of D. microsporus and P. marquandii were cultivated in a submerged fermentation. Keratinolytic activity of the culture filtrate appeared after 40 h for both strains and increased until reached its maximum, i.e., 49.5 U/ml after 95 h for D. microsporus culture filtrate and 230.6 U/ml after 110 h for that of P. marquandii. The culture filtrates were collected and then concentrated, dialyzed, and lyophilized; 0.25 g of the crude keratinase from D. microsporus and 0.4 g of that of P. marquandii were obtained from 1 liter of fermentation broth. The keratinases were purified, as described in the Materials and Methods section, 3.8-fold to a specific activity of 1,005 U/mg and 4.9-fold to a specific activity of 326 U/mg, respectively. By SDS-PAGE, a single protein band was obtained in each case (Fig. 1).
FIG. 1.
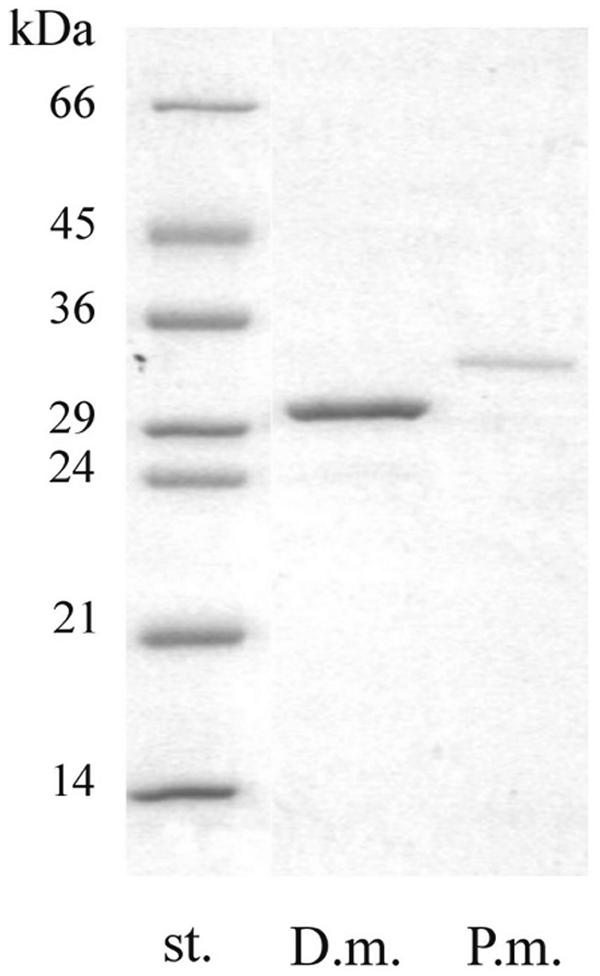
SDS-PAGE of the purified keratinases of P. marquandii (P.m.) and D. microsporus (D.m.). The positions of low-molecular-mass markers (st.) from Sigma are shown. The gel (12%) was stained with Coomassie brilliant blue.
Characterization of keratinases.
The basic biochemical characteristics of D. microsporus keratinase were described in our previous report (12). In this study, the molecular mass of the keratinase of P. marquandii was estimated to be 33 kDa by both SDS-PAGE and gel filtration on Superose 12. The enzyme seemed to be a monomer. The molecular masses of the purified keratinases, 33 kDa for P. marquandii, 30 kDa for D. microsporus (12), and 22 kDa for A. flavus keratinase (not published), fit into the range between 20 kDa and 60 kDa reported for other keratinases. All three keratinases are serine proteases, as are the majority of keratinases. The keratinolytic activity of the keratinase of P. marquandii on stratum corneum keratin was detected in a broad range of pH values (pH 6 to 11), with the maximum activity at pH 8 (Fig. 2A). The optimal pH of keratinolytic activity for both the P. marquandii and D. microsporus enzymes is around pH 8, while the keratinase of A. flavus has its optimum at pH 11, which is very high among the keratinases reported before. The P. marquandii keratinase is special for its temperature optimum, which was determined to be 60 to 65°C (Fig. 2B). Other reported keratinases, except thermostable ones, were mostly active up to 50°C.
FIG. 2.

pH (A) and temperature (B) optima for the keratinase of P. marquandii. ▴, citrate buffer; •, phosphate buffer; ▴, Tris-HCl; ▾, glycine-NaOH buffer.
Since enzyme assays were performed at 45°C, a slightly high temperature, the enzyme stability of keratinases at that temperature was examined. The enzyme solution of each keratinase (0.06 mg/ml) was incubated at 45°C. Every 5 min the residual activity was determined spectrophotometrically by following hydrolysis of 1 mM AAPF under the conditions described for activity measurement in Materials and Methods. The reaction rate decreased for 50% in 15 min with D. microsporus keratinase and in 150 min with P. marquandii keratinase, so the P. marquandii keratinase was stable 10 times longer than that of D. microsporus. SDS-PAGE showed complete degradation of the enzyme and appearance of smaller peptides due to enzyme autolysis (data not shown).
The addition of the inhibitor PMSF prevents autodegradation of keratinases. Since stabilization of the native conformation with calcium ions is commonly observed with extracellular enzymes, the effect of Ca2+ ions on the stability of D. microsporus keratinase was studied in our previous work. It was shown that Ca2+ ions at 1 mM concentration did not significantly influence enzyme stability (15). However, autolysis did not represent a problem when other proteins were present as a substrate. If the stability of the P. marquandii keratinase was compared to the data for keratinases from the literature, only a keratinase from Streptomyces pactum (2) and protease D-1 from Stenotrophomonas sp. (39) proved to be similarly stable, in addition to keratinolytic enzymes of thermophilic microorganisms (5, 31).
We have analyzed the effect of protease inhibitors and reducing agents on enzyme activity (Table 1). Both purified keratinases were totally inhibited by the serine protease inhibitor PMSF and partially by EDTA (around 30%), while iodoacetamide did not cause any inhibition. The maximal increase of enzyme activity on stratum corneum keratin was observed by the presence of the reducing agent DTT. The activity was increased twofold for the P. marquandii keratinase and threefold for the D. microsporus keratinase. The reducing agent β-ME only slightly increased the activity of the P. marquandii keratinase but significantly increased the D. microsporus keratinase activity.
Since DTT and β-ME are known to cleave disulfide bridges, an influence either on enzyme or on keratin substrate was possible. Thus, the enzyme activity was measured with 1 mM AAPF, which does not contain disulfide bridges. The activity on the synthetic substrate remained unchanged in the presence of DTT and β-ME for both keratinases (data not shown). The result confirmed that the reducing agents acted on the keratin substrate and not on the enzymes. The influence of some additives which were present in some assays was also measured (Table 1). DMSO did not influence the activity of keratinases up to a concentration of 5%, while isopropanol decreased the activity about 13% at a concentration of 1%. SDS inhibited both enzymes at a concentration of just 0.1%.
Thirteen-amino-acid-residue sequences of the N termini of the purified keratinases from P. marquandii and D. microsporus were determined, and a sequence similarity search was performed in the SwissProt/TrEMBL database. The comparisons between determined sequences and 13-residue sequences of the N termini of various proteases are presented in Table 2. The sequence of the P. marquandii keratinase and that of the D. microsporus keratinase vary by five amino acid residues. The highest similarity with the sequence of the P. marquandii enzyme, 12 out of 13 residues identical, was found in the sequence of a serine protease from another species of the same genus, Paecilomyces lilacinus, while the sequence most homologous to the D. microsporus enzyme, 10 out of 13 residues identical, was found at the N terminus of a serine protease from Acremonium chrysogenum. Among the proteases from Table 2, only proteinase K from Tritirachium album is described as keratinolytic.
TABLE 2.
Alignment of the N-terminal sequences of P. marquandii and D. microsporus keratinases with N-terminal sequences of similar proteasesa
| Enzymeb | Sequence |
|---|---|
| Keratinase of P. marquandii | ALTQQPGAPWGLG |
| Serine protease of P. lilacinus (Q01471) | AYTQQPGAPWGLG |
| Keratinase of D. microsporus | ATVTQNNAPWGLG |
| Alkaline proteinase of A. chrysogenum (P29118) | ALVTQNGA-WGLG |
| Proteinase K (P06873) | --AAQTNAPWGLA |
| Subtilisin (SUBSCL) | ALA-QTV-PYGIP |
Underlined amino acid residues are identical to corresponding residues in the P. marquandii sequence.
The SwissProt/TrEMBL data base number is in parentheses.
Proteolytic specificity of keratinases.
The peptide cleavage specificity of the enzymes was tested using synthetic substrates and oxidized insulin B-chain. A variety of synthetic oligopeptides were examined as a substrate for keratinases. The most favored substrate for both enzymes was AAPF; cleavage of the substrate AAA was 100-fold lower than cleavage of AAPF. The keratinases also possessed an esterase activity, as they were able to hydrolyze ATEE. The other substrates, FALGPA, L-BAPA, and Bz-Tyr-pNA, were not hydrolyzed (not shown). This experiment demonstrated that keratinases preferentially cleave the peptide bond at hydrophobic aromatic (AAPF) and aliphatic (AAA) amino acids at the P-1 site of synthetic oligopeptides. Several researchers also found AAPF to be the best substrate for their keratinase (6, 17, 26, 32, 38). Therefore, the tetrapeptide AAPF was chosen to determine kinetic parameters. Besides the two keratinases, proteinase K was also tested for comparison.
The initial velocities of hydrolysis for different AAPF concentrations in the range from 0.1 mM to 5.0 mM were determined. Then, kinetic parameters were calculated using the molar absorption coefficient for p-nitroaniline determined under our experimental conditions (ɛ = 8,800 l mol−1 cm−1). The results are summarized in Table 3. The highest affinity towards the substrate AAPF was exhibited by the keratinase of P. marquandii. Its catalytic efficiency (kcat/Km) of 241 mM−1 s−1 was higher than those of the D. microsporus keratinase and of proteinase K, 9 mM−1 s−1 and 95 mM−1 s−1, respectively. Our measurements on AAPF showed that the kinetic constants for the P. marquandii keratinase are comparable to the constants given in the literature for other microbial keratinases (2, 3, 6, 26, 32). Among them, the Michaelis-Menten constant Km for AAPF was the lowest for the keratinase of P. marquandii.
TABLE 3.
Kinetic parameters for hydrolysis of AAPF with P. marquandii and D. microsporus keratinases and with proteinase K
| Enzyme | Km (mM) | Vmax (107 mmol/s) | kcat (s−1) | kcat/Km (mM−1s−1) |
|---|---|---|---|---|
| Keratinase of P. marquandii | 0.17 ± 0.02 | 7.3 ± 0.1 | 41.0 | 241 |
| Keratinase of D. microsporus | 1.03 ± 0.17 | 11.7 ± 0.4 | 8.8 | 9 |
| Proteinase K | 1.49 ± 0.30 | 8.2 ± 0.4 | 142.0 | 95 |
Further, the proteolytic specificity of both keratinases was examined by hydrolysis of the oxidized insulin B-chain. The cleavage sites were compared with the cleavage sites of some other proteases (Fig. 3). The two fungal keratinases exhibited very broad specificity, with preferential selectivity for hydrophobic and aromatic amino acids on the P-1 site for Leu (L), Val (V), Tyr (Y), and Phe (F). The profile of cleavage sites for the keratinase of D. microsporus was more similar to that of proteinase K, while the keratinase of P. marquandii revealed a smaller number of cleavage sites. However, a large number of cleavage sites for the keratinases of P. marquandii and D. microsporus as well as for proteinase K in the oxidized B-chain of insulin distinguished these enzymes from subtilisin and especially from trypsin and elastase.
FIG. 3.

Cleavage sites on oxidized insulin B-chain for the keratinases of P. marquandii and D. microsporus. For comparison, the cleavage sites for proteinase K (14) as well as subtilisin, trypsin, and elastase (1) are presented. Thick arrow, major cleavage site, 20 min of hydrolysis; thin arrow, minor cleavage site, 12 h of hydrolysis.
Substrate specificity of keratinases.
To investigate the keratinase substrate specificity, we prepared different keratinous substrates from natural human and animal keratinous materials. Skin keratins, called soft keratins, possess up to 10% of cysteine residues. Hard keratins, which are constituents of skin appendages, possess more than 15% of cysteine residues and are more resistant to proteolysis than soft keratins. The activity of the P. marquandii and D. microsporus keratinases on keratins and on other proteins is presented in Fig. 4.
FIG. 4.

Hydrolysis of different keratinous and nonkeratinous substrates by keratinases of P. marquandii and D. microsporus after 30 min of incubation. The activity of D. microsporus keratinase on stratum corneum was defined as 100%.
Among keratins, the soft keratins of the stratum corneum were preferably hydrolyzed. The keratinases also showed the ability to degrade commercial bovine keratin and human and porcine nail, but to a much lesser degree. Human hair, sheep wool, and chicken feathers were not hydrolyzed in short incubation times of 30 min. With prolonged incubation of up to 24 h, the D. microsporus (10) and P. marquandii keratinases also hydrolyzed hair and wool keratin (data not shown). Thus, keratinases are able to hydrolyze α-keratins from skin, nail, and hair but not β-keratins from chicken feathers.
It was shown that the presence of reducing agents stimulated enzyme hydrolysis of keratin. At a 1 mM concentration of DTT, the activity of the D. microsporus keratinase was three times higher and that of the P. marquandii keratinase was two times higher than the activity of the enzymes without DTT addition. The stimulating effect of reducing agents has been reported by many authors (16, 18, 20, 22, 36). They agreed that reducing agents reduced disulfide bonds of keratinous filaments and allowed access of the enzymes to the substrate for proteolytic attack. The amounts of soluble products formed during casein hydrolysis were comparable to the product amount after stratum corneum hydrolysis, whereas the globular protein bovine serum albumin was not hydrolyzed to such a high level.
The keratinases of P. marquandii and D. microsporus as well as keratinases reported by other authors, with one single exception (5), in addition to keratin hydrolyzed the nonkeratinous substrates casein and bovine serum albumin. Interestingly, elastin and collagen are also fibrillar constituents of skin, but only slight activity of the tested keratinases on these substrates was detected. The same observations were reported for the Streptomyces albidoflavus (3) and Stenotrophomonas sp. keratinases (39). However, on the majority of substrates tested, the keratinase of P. marquandii proved to be about twice as active as the D. microsporus keratinase. A quantitative comparison to other keratinases reported in the literature is practically impossible because keratinous substrates were not identical and methods of activity measurements and definitions of keratinolytic units are not unified.
Hydrolysis of model substrates with keratinases and other proteases.
We have chosen two model substrates (keratin of the stratum corneum and casein) and compared the keratinase activities on these substrates with the activities of some proteases of fungal (proteinase K), bacterial (collagenase and subtilisin), and animal (chymotrypsin, elastase, and trypsin) origin. Proteinase K is a keratinolytic protease with a well-known broad specificity. Except for the collagenase, which is a metalloprotease, all other enzymes are serine-type proteases, as are the two keratinases studied. The keratinolytic and caseinolytic activities of the proteolytic enzymes are shown in Fig. 5.
FIG. 5.

Amount of soluble products during hydrolysis of (A) keratin and (B) casein by keratinases and selected proteases.
As expected, the P. marquandii and D. microsporus keratinases, as well as proteinase K, hydrolyzed keratin more extensively than the other proteases. The keratinase of P. marquandii was about 20% and the keratinase of D. microsporus was about 50% less active on stratum corneum keratin than was proteinase K. However, all the other enzymes tested were only slightly active or not active at all, as was the case with the collagenase. When caseinolytic activity was measured, the keratinases and proteinase K were closely followed by elastase, chymotrypsin, and subtilisin.
The ratio between the velocity of keratin hydrolysis and the velocity of casein hydrolysis for each enzyme was a criterion for the enzyme specificity for keratinous substrates (Table 4). The keratinase of P. marquandii and proteinase K revealed the highest specificity, with a ratio of around 0.7, and the specificity of the D. microsporus keratinase was lower, with a ratio of 0.52. However, with the other enzymes the values were lower than 0.3. The value for trypsin was 0.42 due to the weak hydrolysis of casein and not due to the significant degradation of keratin. A ratio higher than 0.5 for the keratinases and proteinase K confirms that these enzymes degrade keratins more efficiently than other proteases do. Similar results were reported by Cheng et al. (4) for the bacterial keratinase of Bacillus licheniformis. Generally, the keratinases reported in the literature were 5- to 20-fold more active on keratinous substrates than other proteases (2, 3, 21, 39).
TABLE 4.
Ratio of velocities of keratin to casein hydrolysis as a criterion of enzyme specificity for keratinous substrates
| Enzyme | Velocity (U/mg/min)
|
Ratio, keratin/casein | |
|---|---|---|---|
| Keratin | Casein | ||
| Proteinase K | 35.1 | 50.4 | 0.70 |
| Keratinase of P. marquandii | 24.8 | 36.3 | 0.69 |
| Keratinase of D. microsporus | 14.6 | 27.9 | 0.52 |
| Elastase | 8.2 | 32.0 | 0.25 |
| Subtilisin | 6.6 | 21.3 | 0.30 |
| Trypsin | 4.2 | 10.0 | 0.42 |
| Chymotrypsin | 3.0 | 25.8 | 0.12 |
| Collagenase | 0 | 4.8 | 0 |
In summary, the keratinases are enzymes which catalyze the degradation of keratins. All of them are proteases, but not all proteases hydrolyze keratins. Taking into account that in keratins a high percentage of the molecule represents hydrophobic and aromatic amino acids (approximately 50%) (13), it could be concluded that keratinases are successful in hydrolysis of keratinous materials due to the specific amino acid composition of keratins as well as to their broad specificity.
Acknowledgments
This work was partially supported by the Ministry of Higher Education, Science and Technology of Slovenia.
We thank J. P. Chamount from the Faculty of Medicine and Pharmacy in Besançon, France, for the donation of fungal strains. We also thank B. Kralj from the Jožef Stefan Institute in Ljubljana, Slovenia, for ESI-MS analyses.
REFERENCES
- 1.Beynon, R. J., and J. S. Bond. 1989. Proteolytic enzymes, a practical approach. Oxford University Press, Oxford, United Kingdom.
- 2.Böckle, B., B. Galunsky, and R. Müller. 1995. Characterization of a keratinolytic serine proteinase from Streptomyces pactum DSM 40530. Appl. Environ. Microbiol. 61:3705-3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bressollier, P., F. Letourneau, M. Urdaci, and B. Verneuil. 1999. Purification and characterization of a keratinolytic serine proteinase from Streptomyces albidoflavus. Appl. Environ. Microbiol. 65:2570-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng, S. W., H. M. Hu, S. W. Shen, H. Takagi, M. Asano, and Y. C. Tsai. 1995. Production and characterization of keratinase of a feather-degrading Bacillus licheniformis PWD-1. Biosci. Biotechnol. Biochem. 59:2239-2243. [DOI] [PubMed] [Google Scholar]
- 5.Dozie, I. N. S., C. N. Okeke, and N. C. Unaeze. 1994. A thermostable, alkaline-active, keratinolytic proteinase from Chrysosporium keratinophilum. World J. Microbiol. Biotechnol. 10:563-567. [DOI] [PubMed] [Google Scholar]
- 6.Evans, K. L., J. Crowder, and E. S. Miller. 2000. Subtilisins of Bacillus spp. hydrolyze keratin and allow growth on feathers. Can. J. Microbiol. 46:1004-1011. [DOI] [PubMed] [Google Scholar]
- 7.Farag, A. M., and M. A. Hassan. 2004. Purification, characterization and immobilization of a keratinase from Aspergillus oryzae. Enzyme Microb. Technol. 34:85-93. [Google Scholar]
- 8.Friedrich, A., and G. Antranikian. 1996. Keratin degradation by Fervidobacterium pennavorans, a novel thermophilic anaerobic species of the order Thermotogales. Appl. Environ. Microbiol. 62:2875-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedrich, J., H. Gradišar, D. Mandin, and J. P. Chaumont. 1999. Screening fungi for synthesis of keratinolytic enzymes. Lett. Appl. Microbiol. 28:127-130. [Google Scholar]
- 10.Friedrich, J., and S. Kern. 2003. Hydrolysis of native proteins by keratinolytic protease of Doratomyces microsporus. J. Mol. Catal. B Enzymol. 21:35-37. [Google Scholar]
- 11.Fuchs, E. 1995. Keratins and the skin. Annu. Rev. Cell Dev. 11:123-153. [DOI] [PubMed] [Google Scholar]
- 12.Gradišar, H., S. Kern, and J. Friedrich. 2000. Keratinase of Doratomyces microsporus. Appl. Microbiol. Biotechnol. 53:196-200. [DOI] [PubMed] [Google Scholar]
- 13.Gregg, K., S. D. Wilson, D. A. D. Parry, and G. E. Rogers. 1984. A comparison of genomic coding sequences for feather and scale keratins: Structural and evolutionary implications. EMBO J. 3:175-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han, X. Q., and S. Damodaran. 1998. Purification and characterization of Protease Q: a detergent- and urea-stable serine endopeptidase from Bacillus pumilus. J. Agric. Food Chem. 46:3596-3603. [Google Scholar]
- 15.Hublin, A., H. Gradišar, J. Friedrich, and D. Vasić-Raćki. 2002. Stability and stabilization of Doratomyces microsporus keratinase. Biocatal. Biotransform. 20:329-336. [Google Scholar]
- 16.Kunert, J. 1992. Effect of reducing agents on proteolytic and keratinolytic activity of enzymes of Microsporum gypseum. Mycoses 35:343-348. [DOI] [PubMed] [Google Scholar]
- 17.Kunert, J., and E. Kasafirek. 1988. Preliminary characterization of extracellular proteolytic enzymes of dermatophytes by chromogenic substrates. J. Med. Vet. Mycol. 26:187-194. [DOI] [PubMed] [Google Scholar]
- 18.Kunert, J., and Z. Stransky. 1988. Thiosulfate production from cystine by keratinolytic prokaryote Streptomyces fradiae. Arch. Microbiol. 150:600-601. [Google Scholar]
- 19.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 20.Lee, H., D. B. Suh, J. H. Hwang, and H. J. Suh. 2002. Characterization of a keratinolytic metalloprotease from Bacillus sp. SCB-3. Appl. Biochem. Biotechnol. 97:123-133. [DOI] [PubMed] [Google Scholar]
- 21.Letourneau, F., V. Soussotte, P. Bressollier, P. Branland, and B. Verneuil. 1998. Keratinolytic activity of Streptomyces sp. S.K1-02: a new isolated strain. Lett. Appl. Microbiol. 26:77-80. [DOI] [PubMed] [Google Scholar]
- 22.Lin, X., C. G. Lee, E. S. Casale, and J. C. H. Shih. 1992. Purification and characterization of a keratinase from a feather-degrading Bacillus licheniformis strain. Appl. Environ. Microbiol. 58:327-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin, X., J. Tang, G. Koelsch, M. Monod, and S. Foundling. 1993. Recombinant Canditropsin, an extracellular aspartic protease from yeast Candida tropicalis. J. Biol. Chem. 268:20143-20147. [PubMed] [Google Scholar]
- 24.Lin, X., D. W. Kelemen, E. S. Miller, and J. C. H. Shih. 1995. Nucleotide sequence and expression of kerA, the gene encoding a keratinolytic protease of Bacillus licheniformis PWD-1. Appl. Environ. Microbiol. 61:1469-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malviya, H. K., R. C. Rajak, and S. K. Hasija. 1992. Purification and partial characterization of two extracellular keratinases of Scopulariopsis brevicaulis. Mycopathologia 119:161-165. [DOI] [PubMed] [Google Scholar]
- 26.Mignon, B., M. Swinnen, J. P. Bouchara, M. Hofinger, A. Nikkles, G. Pierard, C. Gerday, and B. Losson. 1998. Purification and characterization of a 31.5 kDa keratinolytic subtilizin-like serine protease from Microsporum canis and evidence of its secretion in naturally infected cats. Med. Mycol. 36:395-404. [PubMed] [Google Scholar]
- 27.Mukhopadhyay, R. P., and A. L. Chandra. 1990. Keratinase of a streptomycete. Indian J. Exp. Biol. 28:575-577. [PubMed] [Google Scholar]
- 28.Negi, M., R. Tsuboi, T. Matsui, and H. Ogawa. 1984. Isolation and characterization of proteinase from Candida albicans: substrate specificity. J. Investig. Dermatol. 83:32-36. [DOI] [PubMed] [Google Scholar]
- 29.Onifade, A. A. 1998. A review: potentials for biotechnological applications of keratin-degrading microorganisms and their enzymes for nutritional improvement of feathers and other keratins as livestock feed resources. Bioresour. Technol. 66:1-11. [Google Scholar]
- 30.Qin, L. M., S. Dekio, and J. Jidoi. 1992. Some biochemical characteristics of a partially purified extracellular keratinase from Trichophyton schoenleinii. Zentralbl. Bakteriol. 277:236-244. [DOI] [PubMed] [Google Scholar]
- 31.Riessen, S., and G. Antranikian. 2001. Isolation of Thermoanaerobacter keratinophilus sp. nov., a novel thermophilic, anaerobic bacterium with keratinolytic activity. Extremophiles 5:399-408. [DOI] [PubMed] [Google Scholar]
- 32.Rojanavanich, V., T. Yoshiike, R. Tsuboi, K. Takamori, and H. Ogawa. 1990. Purification and characterization of an extracellular proteinase from Hendersonula toruloidea. Infect. Immun. 58:2856-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santos, R. M. D. B., A. A. P. Firmino, C.M. de Sá, and C. R. Felix. 1996. Keratinolytic activity of Aspergillus fumigatus fresenius. Curr. Microbiol. 33:364-370. [DOI] [PubMed] [Google Scholar]
- 34.Suh, H. J., and H. K. Lee. 2001. Characterization of a keratinolytic serine protease from Bacillus subtilis KS-1. J. Protein Chem. 20:165-169. [DOI] [PubMed] [Google Scholar]
- 35.Suntornsuk, W., and L. Suntornsuk. 2003. Feather degradation by Bacillus sp. FK 46 in submerged cultivation. Bioresour. Technol. 86:293-243. [DOI] [PubMed] [Google Scholar]
- 36.Takami, H., F. Nakamura, R. Aono, and K. Hirokoshi. 1992. Degradation of human hair by a thermostable alkaline proteinase from Bacillus sp. no. AH 101. Biosci. Biotechnol. Biochem. 56:1667-1669. [Google Scholar]
- 37.Takiuchi, I., Y. Sei, H. Takagi, and M. Negi. 1984. Partial characterization of the extracellular keratinase from Microsporum canis. Sabouraudia: J. Med. Vet. Mycol. 22:219-224. [PubMed] [Google Scholar]
- 38.Tsuboi, R., I. J. Ko, K. Takamori, and H. Ogawa. 1989. Isolation of a keratinolytic proteinase from Trichophyton mentagrophytes with enzymatic activity at acidic pH. Infect. Immun. 57:3479-3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamura, S., Y. Morita, Q. Hasan, K. Yokoyama, and E. Tamiya. 2002. Keratin degradation. A cooperative action of two enzymes from Stenotrophomonas sp. Biochem. Biophys. Res. Commun. 294:1138-1143. [DOI] [PubMed] [Google Scholar]