

Abstract
Previous studies of genes involved in the production of sakacin P by Lactobacillus sakei Lb674 revealed the presence of an inducible promoter downstream of the known spp gene clusters. We show here that this promoter drives the expression of an operon consisting of a bacteriocin gene (sppQ), a cognate immunity gene (spiQ), another gene with an unknown function (orf4), and a pseudoimmunity gene containing a frameshift mutation (orf5). The leader peptide of the new one-peptide bacteriocin sakacin Q contains consensus elements that are typical for so-called “double-glycine” leader peptides. The mature bacteriocin shows weak similarity to the BrcA peptide of the two-peptide bacteriocin brochocin C. Sakacin Q has an antimicrobial spectrum that differs from that of sakacin P, thus expanding the antimicrobial properties of the producer strain. The genes encoding sakacin Q and its cognate immunity protein showed strong translational coupling, which was investigated in detail by analyzing the properties of a series of β-glucuronidase fusions. Our results provide experimental evidence that production of the bacteriocin and production of the cognate immunity protein are tightly coregulated at the translational level.
Many lactic acid bacteria are able to inhibit the growth of related bacterial species by secreting ribosomally synthesized antimicrobial peptides called bacteriocins. These small peptides usually contain between 20 and 60 residues, they have a net positive charge, and parts of their sequences have a tendency to contain hydrophobic and/or amphiphilic stretches of amino acids (8, 15, 30). Lactic acid bacterium bacteriocins are divided into three classes, and class I bacteriocins (lantibiotics) and class II bacteriocins (nonmodified heat-stable bacteriocins) are the most thoroughly investigated (8, 18, 26, 43). Lantibiotics are characterized by posttranslational modifications, and the most widely studied bacteriocin in this class is nisin. Class II bacteriocins are usually separated into three subcategories. Class IIa (“pediocin-like”) bacteriocins have a strong inhibitory effect on Listeria and contain a conserved YGNG sequence motif in the N-terminal half of the mature protein. Class IIb bacteriocins require the complementary action of two peptides for full activity. All other nonlantibiotics that do not belong to class IIa and class IIb are classified as class IIc.
Sakacin P is a class IIa bacteriocin produced by Lactobacillus sakei strains Lb674 and LTH673, which contain identical spp gene clusters (13, 40). The production of sakacin P in these strains is regulated by a typical peptide pheromone-based quorum-sensing system (25). Huehne et al. (14) and Brurberg et al. (6) have shown that production of sakacin P requires the following three gene clusters, all of which are preceded by typical inducible promoters: (i) a regulatory operon encoding the peptide pheromone (sppIP), a histidine kinase (sppK) sensing the pheromone, and a response regulator (sppR) that, when activated by the histidine kinase, induces spp promoters; (ii) an operon consisting of the sakacin P structural gene (sppA) and a cognate immunity gene (spiA); and (iii) an operon encoding a dedicated ABC transporter (sppT) for transport and processing of the bacteriocin and pheromone precursors, as well as an accessory protein for transport (sppE). The promoters preceding these operons are designated PsppIP, PsppA, and PsppT, respectively. Both the bacteriocin and the pheromone precursor contain a typical “double-glycine” leader peptide (11) which is recognized by the transport machinery.
There are strong indications that L. sakei Lb674 and LTH673 produce at least one bacteriocin in addition to sakacin P (6, 9). Eijsink et al. (9) noted that during purification of sakacin P from L. sakei LTH673 inhibitory activity toward some strains was lost, while activity toward other strains (sakacin P) was preserved. The sequence of the spp gene cluster (14) contained the start of an open reading frame (ORF) downstream of the sppTE operon encoding a typical “double-glycine” leader peptide. Brurberg et al. (6) showed that this ORF was preceded by a fourth inducible and seemingly very strong promoter that gave rise to two transcripts that were approximately 0.5 and 1.2 kb long.
In this paper, we describe the DNA sequence and a functional analysis of the operon downstream of the sppTE operon, and we show that two of the genes in this operon encode a new one-peptide bacteriocin and an unusually small cognate immunity protein. During the course of this work, strong translational coupling between the new bacteriocin gene (sppQ) and the gene encoding the cognate immunity protein (spiQ) became apparent. We used a series of β-glucuronidase (GUS) fusions to study this phenomenon in detail and found that, indeed, the production of bacteriocin and immunity proteins is tightly coregulated at the translational level.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli DH5α and TOP10 (both obtained from Invitrogen, Carlsbad, CA) were grown in brain heart infusion medium (Oxoid Ltd., Basingstoke, United Kingdom) at 37°C with shaking. Lactobacillus strains were grown in MRS medium (Oxoid) without shaking at 30°C. Solid media were prepared by adding 1.5% (wt/vol) agar to the broth. The antibiotic concentrations used for E. coli were 100 μg/ml ampicillin, 200 μg/ml erythromycin, and 50 μg/ml kanamycin. The antibiotic concentrations used for lactobacilli were 5 μg/ml erythromycin and 5 μg/ml chloramphenicol.
TABLE 1.
Plasmids and strains used in this study
| Bacterial strain or plasmid | Characteristicsa | Source or reference(s) |
|---|---|---|
| Strains | ||
| Escherichia coli DH5α | Host strain | Invitrogen |
| E. coli TOP10 | Host strain for TOPO cloning | Invitrogen |
| Lactobacillus sakei Lb674 | Sakacin P and Q producer | 13,36 |
| L. sakei LTH673 | Sakacin P and Q producer | 40 |
| L. sakei Lb790 | Host strain: Bac− | 34 |
| L. sakei Lb790/pSak20 | Lb790 containing pSAK20; host strain; Bac−; sakacin Q sensitive; Cmr | 4 |
| L. sakei NCDO 2714 | Indicator strain for sakacin Q | |
| Plasmids | ||
| pBluescript KS(+) | 3.0-kb cloning vector; Ampr | Stratagene |
| pCR4 Blunt II-TOPO | 3.5 kb; vector for cloning PCR fragments; Kanr | Invitrogen |
| pSAK20 | 11.8 kb: contains the orf4-sapKRTE operon; necessary for activation of PsapA; Cmr | 3 |
| pBLS7 | 5.3 kb; pBluescript KS(−) derivative with a 2.35-kb HindIII insert containing part of sppE, sppQ, spiQ, and orf4, orf5; Ampr | This study |
| pLPV111 | 4.2-kb E. coli-L. sakei shuttle vector; Emr | 3 |
| pSPP2 | pLPV111 derivative containing the PsapA promoter translationally fused to the sppA-spiA operon; used for overproduction of sakacin P; Emr | 4 |
| pSIP403 | Source of E. coli gusA gene; expression vector with PsppA (the regulated promoter preceding sppA) translationally fused to gusA and a terminator downstream of gusA; Emrb | 38 |
| pSIP407 | pSIP403 derivative; pepN gene replaces gusAc | 38a |
| pSIP410 | pSIP407 derivative; PsppQ replaces PsppA | 38a |
| pSIP413 | pSIP407 derivative; Porf4 replaces PsppA | This study |
| pGM11 | Expression vector; PsapA translationally fused to sppQ, spiQ, and orf4, orf5; 5.7 kbd | This study |
| pGM112, pGM112B, pGM112C, pGM112D, pGM112E, pGM112F, pGM112G, pGM112I | pGM11 derivativese | This study |
| pGM200 | Backbone vector for the pGM200-seriesd | This study |
| pGM201, pGM202, pGM203, pGM204, pGM206, pGM207 | Derivatives of pGM200f | This study |
DNA manipulation and transformation.
General molecular biological techniques were performed essentially as described by Sambrook et al. (32). PCRs were performed with a PTC programmable thermal controller (MJ Research, Inc., Waltham, MA) using Pfu polymerase (Promega Corp., Madison, WI) and standard procedures. PCR fragments were purified with a QIAquick PCR purification kit or a QIAquick gel extraction kit (both obtained from QIAGEN, Venlo, The Netherlands). Purified PCR fragments were not used directly but were subcloned using the TOPO system (Invitrogen) and the protocol provided by the supplier. Primers used for PCR amplification and for creating site-directed mutations were purchased from Medprobe (Oslo, Norway) and are listed in Table 2. The sequences of all PCR-derived DNA fragments were checked using a Big Dye sequencing kit obtained from Applied Biosystems and an ABI Prism 377 DNA sequencer (PE Applied Biosystems, Foster City, CA).
TABLE 2.
Primers used in this study
| Primer | Sequencea | Direction | Constructed plasmid(s) |
|---|---|---|---|
| sak10-1 | CACACAGGAAACAGCTATGACCAT | Forward | pGM11b |
| sak10-2 | GTATTTTGCATAACTAAGAATCTCCTCTGTTATTATTTTTTGT | Reverse | pGM11 |
| BLS7-1 | GATTCTTAGTTATGCAAAATACAAAAGAACTAAGCGT | Forward | pGM11 |
| BLS7-3 | GGTTGATTCGTTAAATCCTCATTGT | Reverse | pGM11 |
| orf3B | GTCTAGACTATTTAAGTTGACGCCTT (XbaI) | Reverse | pGM112B, pGM112I |
| orf3C | GGTCTAGACGACAACGTTATTAATAATT (XbaI) | Reverse | pGM112C |
| orf3D | GGTCTAGATTAGAGACACCCGTT (XbaI) | Reverse | pGM112D |
| gm115A | TTTAGAGACACAACTAAGAATCTCCTCTGT | Reverse | pGM112I |
| gm115B | GAGATTCTTAGTTGTGTCTCTAAACAAGCGTGTT | Forward | pGM112I |
| gus1c | CTCTAAACAAGGGTGTACGTCCTGTAGAAACCC | Forward | pGM201, pGM202, pGM203 |
| gus2c | GGACGTACACCCTTGTTTAGAGACACCCGTT | Reverse | pGM201, pGM202, pGM203 |
| gus3 | CCTTGTCCAGTTGCAACCACCT | Reverse | pGM201, pGM202, pGM203 |
| mut1 | CGGAGAATTAGCTTACATAGGAGCTAACGGATGTCTCTAAAC | Forward | pGM206 |
| mut2 | CTTGTTTAGAGACATCCGTTAGCTCCTATGTAAGCTAATTCTC | Reverse | pGM206 |
| 413F | GACCGGTGGCGATTTGAAT (AgeI) | Forward | pSIP413 |
| 413R | GCCATGGTCCCCAACTCCTTT (NcoI) | Reverse | pSIP413 |
| gus5F | GTCACGCCGTATGTTATTGCC | Forward | gusA-specific probe |
| gus6R | CTGCCCAGTCGAGCATCTCTT | Reverse | gusA-specific probe |
Bases that are complementary to the template are indicated by italic type; introduced restrictions sites are indicated by boldface type; and start codons and the optimized RBS in mut1 and mut2 are underlined. Mutated nucleotides in mut1 are indicated by italics and underlining.
The sak10-1 primer is complementary to pLPV111 just upstream of the MluI site at position 135 in Fig. 1. This primer was also used in the construction of pGM112B, pGM112C, pGM112D, pGM112I, pGM201, pGM202, and pGM203.
The start codon of the gusA gene was changed to GGT (underlined).
Chemically competent E. coli DH5α or TOP10 cells were transformed by using the supplier's protocols. L. sakei Lb790 was transformed by electroporation as described by Aukrust et al. (2).
Isolation of chromosomal DNA from L. sakei was performed as described previously (14). Plasmid DNA from E. coli and Lactobacillus was isolated by using a QIAprep miniprep kit (QIAGEN). Lactobacillus cells were pretreated by 25 min of incubation at 37°C with 20 mg/ml lysozyme, 15 U/ml mutanolysin, and 100 μg/ml RNase (all obtained from Sigma, St. Louis, MO) before the lysis buffer from the QIAprep kit was added.
Gene cloning and construction of plasmids.
Southern hybridization was performed as described previously (14), using total DNA from L. sakei Lb674. The probe was a synthetic oligonucleotide complementary to the truncated ORF previously detected downstream of the sppTE operon (14) (GenBank accession number Z48542; 5′-GGTGCTTGGGGAGCTGG-3′). It was labeled with digoxigenin-dUTP from a DIG oligonucleotide 3′ end labeling kit (Boehringer GmbH, Mannheim, Germany). This probe detected a 2.35-kb HindIII fragment, which was cloned into the HindIII site of the pBluescript KS(+) vector, resulting in plasmid pBLS7, which was used as a template for determining the sequence of the insert.
It has previously been shown that the spp operons from L. sakei Lb674 and L. sakei LTH673 have identical sequences (6, 14). Sequence analysis of the corresponding DNA fragment in LTH673 encompassing the sppQ operon also revealed no differences between Lb674 and LTH673, thus confirming observations concerning the identical behavior of the two strains in terms of bacteriocin production and inducibility (M. B. Brurberg, I. F. Nes, and V. G. H. Eijsink, unpublished observations).
All functional analyses of genes in the sequenced DNA were performed using a constitutive two-plasmid expression system for lactobacilli, which was developed by Axelsson et al. (4). This system consists of plasmid pSAK20 (Cmr), which contains regulatory genes and transport genes involved in production of the bacteriocin sakacin A. The second plasmid is a derivative of pLPV111 (Emr), which contains the sakacin A promoter translationally fused to the gene or operon of interest. The bacteriocin and the promoter (PsapA) are different from sakacin P and Pspp promoters. Plasmid pGM11 (Fig. 1) is a pLPV111 variant with an MluI-XbaI insert in which the PsapA promoter is coupled to the gene cluster containing sppQ, spiQ, orf4, and orf5 (Fig. 1 and 2) (see below). This and other inserts (see below) were generated by recombinant PCR as described by Higuchi (12). The PsapA part of the fragment was amplified with primers sak10-1 and sak10-2 (Table 2) using pSPP2 (Table 1) as the template, while a 0.61-kb fragment containing sppQ-spiQ was generated with primers BLS7-1 and BLS7-3 (Table 2) using pBLS7 as the template. The subsequent fusion PCR was performed with primers sak10-1 and BLS7-3 (Table 2), and the product was subcloned into the pCR4 Blunt II-TOPO vector. The resulting plasmid was digested with MluI-NcoI to excise a 0.54-kb promoter fusion fragment.
FIG. 1.

Schematic overview of plasmids derived from pGM11. In pGM11, open reading frames are indicated by open arrows. The nucleotide sequence from position 257 to position 1602 in pGM11 is accessible in GenBank (accession numbers AJ844595 and AJ844594; same numbering in Fig. 1 and 2); the solid lines below pGM11 indicate nucleotides retained from this sequence in pGM11 derivatives (the numbers in parentheses are nucleotide numbers). Nonsense mutations (asterisk) and in-frame (▵) and out-of-frame (▴) deletions are indicated below the lines. Relevant restriction sites used for plasmid construction in this study are indicated. In most constructs indicated by the solid lines PsapA is translationally fused to the start codon (ATG) of sppQ; the only exception is pGM112I, in which the promoter is fused to the start codon of spiA (GTG). The data on the right indicate the phenotypes observed with the various plasmids in terms of bacteriocin production (SppQ +) and immunity to sakacin Q (SppQ imm.) [+, full immunity; (+), strongly reduced but detectable immunity]. Strains containing the pGM112C and pGM112D constructs were not viable, from which it was concluded that these two plasmids would yield a Bac+ Imm− phenotype. See text for details. nt, nucleotides.
FIG. 2.

Part of the nucleotide sequence downstream of sppE (14), with the open reading frames sppQ and spiQ and the start of orf4. Amino acid sequences are indicated below the nucleotide sequence (in one-letter code). Putative −35 and −10 sites and RBS are underlined. Repeats (LR and RR) which are known (the region from nucleotide 118 to nucleotide 148, previously called PorfX and now called PsppQ) or putative (the region from nucleotide 697 to nucleotide 728; Porf4) parts of regulated promoters are indicated by arrows with two arrowheads. The dashed arrows indicate a possible transcription terminator. The vertical arrow indicates the processing site of presakacin Q.
Various derivatives of pGM11 were obtained from different deletions and mutations (Fig. 1). Plasmid pGM112 was constructed by digesting pGM11 with BlpI and EcoRV. The resulting 5.0-kb fragment was treated with the Klenow fragment (Promega) and self-ligated. A nonsense mutation was introduced into sppQ in pGM112 by cutting the unique NheI site, followed by treatment with the Klenow fragment and self-ligation, yielding plasmid pGM112G.
To construct pGM112B, the PsapA-sppQ-spiQ fragment was amplified with primers sak10-1 and orf3B (Table 2) (introducing an XbaI site immediately downstream of the stop codon of spiQ), using pGM11 as the template, and the resulting fragment was subcloned into pCR4 Blunt II-TOPO. The latter plasmid was digested with MluI and XbaI, and the 0.5-kb fragment was used to replace the 1.5-kb MluI-XbaI fragment of pGM11 (Fig. 1) to obtain pGM112B. pGM112C and pGM112D, both containing deletions in the C-terminal part of spiQ, were generated in the same way by replacing primer orf3B with primers orf3C and orf3D (Table 2), respectively.
Plasmid pGM112I containing a translational fusion between PsapA and spiQ was constructed using a recombinant PCR step. The primers used in the PCRs were sak10-1 and gm115A (for the PsapA fragment), gm115B and orf3B (for the spiQ fragment), and sak10-1 and orf3B (for the final fusion reaction) (Table 2). The resulting fragment was subcloned into pCR4 Blunt II-TOPO. The latter plasmid was digested by MluI and XbaI, and the 0.3-kb fragment was used to replace the 1.5-kb MluI-XbaI fragment of pGM11 (Fig. 1).
To construct pGM112E and pGM112F, pGM112 was digested with NheI (which is unique in pGM112), followed by Klenow treatment and digestion with BsgI to excise a 0.06-kb fragment from sppQ. The BsgI overhang was treated with mung bean nuclease (New England Biolabs Ltd., Hitchin, Hertfordshire, England), and the resulting plasmid fragment was self-ligated. In pGM112E both nucleotides were removed from the BsgI overhang, which resulted in a 57-nucleotide (in-frame) deletion. In pGM112F the nuclease reaction was incomplete, which led to removal of only one nucleotide, which resulted in a 56-nucleotide (out-of-frame) deletion.
As a first step in the construction of gusA fusions, a 1.9-kb fragment containing the gusA gene (excised from pSip403 [38] with NcoI and HindIII) was used to replace the 0.92-kb NcoI-HindIII fragment of pGM11(Fig. 1). The resulting plasmid was digested with XhoI (downstream of gusA, on the inserted fragment) and ZraI (Fig. 1) for insertion of a fragment containing the pepN terminator (GenBank accession number M87840; nucleotides 2781 to 3004), which was excised from pSip403 by cutting with BspHI, followed by Klenow treatment and digestion with XhoI. The resulting plasmid was designated pGM200. To construct plasmids pGM201, pGM202, and pGM203, the N-terminal end of gusA was amplified with primers gus1 and gus3 (Table 2) using pSIP403 as the template, and PsapA-sppQ-spiQ-containing fragments (only 15 nucleotides of spiQ) were generated by using primers sak10-1 and gus2 (Table 2) with pGM112G, pGM112E, and pGM112F as the templates, respectively. In the final fusion PCR, primers sak10-1 and gus3 (Table 2) were used. After subcloning into pCR4 Blunt II-TOPO, the fusion PCR fragments were excised by digestion with NsiI (located in front of PsapA [Fig. 1]) and SnaBI (located 0.39 kb downstream of the gusA start codon) and used to replace the 0.94-kb NsiI-SnaBI fragment of pGM200, yielding pGM201, pGM202, and pGM203, respectively. Thus, in these constructs the gusA gene was fused in-frame to the fifth codon of spiQ.
pGM206, a pGM201 derivative with an optimized ribosome binding site (RBS) and start codon in front of spiQ, was constructed by site-directed mutagenesis using a Quick-change kit (Stratagene, La Jolla, CA) according the protocol provided by the manufacturer, primers mut1 and mut2 (Table 2), and pGM201 as the template. To construct pGM207, a 1.6-kb BsaI fragment in pGM206 (derived from pGM201) replaced the corresponding 1.6-kb BsaI fragment (containing an in-frame deletion in sppQ) from pGM202.
Plasmid pGM204, in which the gusA gene was fused in frame to the 22nd codon of sppQ, was constructed by digestion of pGM200 with NheI (position 317 in Fig. 1) and NcoI (start codon of gusA), followed by Klenow treatment and self-ligation.
To test the strength of the putative promoter in front of the orf4 promoter, a Porf4 fragment (containing nucleotides 682 to 785 [Fig. 2]) was generated by PCR using primers 413F and 413R (Table 2). After subcloning in pCR4 Blunt II-TOPO, the fragment was excised with AgeI and NcoI and used to replace a similar PsppQ-containing fragment in the pepN expression vector pSIP410 (38a) (Table 1). The resulting plasmid, pSIP413, was transformed into L. sakei Lb790.
Bacteriocin purification and characterization.
Sakacin Q was purified from the supernatant of an early-stationary-phase culture (optical density at 600 nm [OD600], 2.3) of L. sakei Lb790 harboring pSAK20/pGM11. The bacteriocin was purified by ammonium sulfate precipitation, followed by three chromatography steps (ion exchange, hydrophobic interaction, and reversed phase), essentially as described previously (13, 40). The bacteriocin was precipitated by addition of ammonium sulfate (400 g/liter, pH 5.8) and was pelleted by centrifugation at 10,000 × g for 30 min (4°C). The pellet was dissolved overnight and then was dissolved with agitation in 0.1 M sodium acetate buffer, pH 4.4, containing 7 M urea (Sigma) overnight and subsequently applied to a 14-ml SP Sepharose Fast Flow column (Amersham Biosciences, Uppsala, Sweden) equilibrated with 0.1 M sodium acetate, 7 M urea (pH 4.4). The column was washed with 50-ml mixtures containing three different concentrations of urea (7, 4, and 1 M in succession) in 0.1 M sodium acetate buffer and finally with 50 ml of 0.1 M sodium acetate buffer (pH 4.4). The bacteriocin was eluted with 50 ml of 1 M NaCl in 20 mM sodium phosphate buffer (pH 5.8). Subsequent purification steps based on hydrophobic interaction chromatography with an Octyl Sepharose 4 Fast Flow column (Amersham Biosciences) followed by reversed-phase chromatography using a PepRPC HR 5/5 column (Amersham Biosciences) and a fast protein liquid chromatography system were performed as described previously (40).
Mass spectrometry was performed with a Voyager-DE RP matrix-assisted laser desorption ionization-time of flight mass spectrometer from PerseptiveBiosystems using α-cyano-4-hydroxycinnamic acid as the matrix. N-terminal amino acid sequencing was carried out with a 477A protein sequencer (PE Applied Biosystems).
Bacteriocin activity and immunity assay.
Bacteriocin activities were determined using a microtiter plate dilution assay, essentially as described previously (10). One bacteriocin unit was defined as the amount of bacteriocin required to inhibit the growth of the indicator strain by 50%. L. sakei NCDO 2714 was utilized as the indicator strain unless indicated otherwise.
Assay for PepN activity.
Overnight cultures of the producer strains were diluted to an OD600 of ∼0.06 in MRS broth containing erythromycin. The cultures were incubated further until the OD600 was ∼0.3 before addition of 25 ng/ml pheromone peptide (IP-673; Molecular Biology Unit, University of Newcastle-upon-Tyne, Newcastle-upon-Tyne, United Kingdom). The bacterial cells were harvested at an OD600 1.8 to 2.0 for determination of PepN activity as described previously (20). PepN activity was measured three times using three independent cultures. Protein concentrations were measured by the Bio-Rad protein assay (Bio-Rad, Hercules, CA) with bovine serum albumin as the standard.
Assay for GUS activity.
β-Glucuronidase activities in L. sakei Lb790(pSAK20) strains harboring various additional plasmids containing a β-glucuronidase gene (Table 1) were determined using a modified β-galactosidase assay (23), as recently described (5). Overnight cultures of Lactobacillus cells were diluted to an OD600 of ∼0.1 in MRS broth, and cells were harvested every hour (until 7 h after dilution) for determination of GUS activity. GUS activities were expressed in Miller unit equivalents, and the data given below are the average results for three independent cultures.
RNA isolation and Northern analysis.
Cultures of L. sakei Lb790(pSAK20) harboring various additional plasmids were started by diluting fresh overnight cultures. After 5 h, 2 volumes of RNAprotect bacterial reagent (QIAGEN) was added, and the cells were harvested by using the RNAprotect bacterial reagent protocol. Total RNA was isolated by using an RNeasy kit (QIAGEN). Lactobacillus was pretreated by incubation for 5 min at 37°C in the presence of 20 mg/ml lysozyme and 250 U/ml mutanolysin (both obtained from Sigma) before RTL buffer from the RNeasy kit was added. For transcript analysis, 2 μg RNA was separated on a 1.2% agarose denaturing gel containing formaldehyde and transferred to a nylon membrane (Amersham Biosciences). A gusA-specific probe (obtained by PCR with primers gus5F and gus6R [Table 2], using pGM201 as the template) was labeled with [α- 32P]dCTP using a RediprimeII kit (RPN1633; Amersham Biosciences) and was subsequently hybridized to the membrane at 65°C overnight. The Northern blot was transferred to a phosphor screen cassette (Molecular Dynamics Inc., Sunnyvale, CA) and was subsequently visualized with a Typhoon 8600 PhosphorImager (Molecular Dynamics Inc.).
DNA sequence analysis.
Database searches were performed by using FASTA (28) and BLAST (1) using default settings. Secondary structures of mRNA were predicted with the Mfold program (47) using default settings.
Nucleotide sequence accession numbers.
The nucleotide sequences described in this study have been deposited in the GenBank database under accession numbers AJ844594 (L. sakei Lb674) and AJ844595 (L. sakei LTH673).
RESULTS
Cloning and sequence analysis.
The 2.35-kb HindIII fragment was isolated from L. sakei Lb674 and cloned into a pBluescript KS(+) vector. The sequence of the 5′ end of this fragment (forward to nucleotide 356 in Fig. 2) was identical to the sequence published previously (14) (accession number Z48542) containing the 3′ end of the sppE gene and the start of a new gene, which previously was called orfX (6). The rest of the fragment revealed three putative ORFs, (orfX [orf2], orf3, and orf4) and a pseudo-ORF (orf5), all preceded by potential ribosome binding sites (Fig. 1). As demonstrated and discussed below, orfX (orf2) and orf3 encode a bacteriocin called sakacin Q and its cognate immunity protein; therefore, these two ORFs are referred to below as sppQ and spiQ, respectively. The spiQ sequence overlaps the sequence encoding the last two amino acids encoded by sppQ and starts with the less frequently used start codon GTG. Downstream of spiQ is a putative rho-independent transcription termination structure with a calculated ΔG of −17.6 kcal/mol. There is another putative rho-independent transcription terminator after orf5 (ΔG, −23.3 kcal/mol). sppQ is preceded by a typical regulated spp promoter (previously designated PorfX and referred to below as PsppQ), which is under control of the regulatory genes in the sakacin P gene cluster (6, 31). It has previously been shown that induction of this promoter gives rise to 0.5-kb and 1.2-kb transcripts (6), which would correspond to transcripts encompassing sppQ plus spiQ and sppQ plus spiQ plus orf4 plus orf5, respectively. The region upstream of orf4 also shows some features of a regulated spp promoter (Fig. 2; also see below).
The N-terminal 18 residues of the putative 67-residue gene product of sppQ have typical features of a double-glycine-type leader peptide often used for bacteriocins (Fig. 3A). BLAST searches with the sequence of the putative mature bacteriocin, called sakacin Q, did not reveal clear sequence similarities with other bacteriocins, except for some similarity to the BrcA peptide of the two-peptide bacteriocin brochocin C (Fig. 3B) (22, 37).
FIG. 3.

(A) Alignment of selected double-glycine leader sequences. Residues that are conserved in at least six of the seven sequences are shown in the consensus line and are indicated by boldface type in the leader peptide of sakacin Q. (B) Alignment of sakacin Q and the A peptide of the two-component bacteriocin brochocin C (22). Conserved residues are indicated by asterisks.
The spiQ gene putatively encodes a protein with 60 amino acids. We could not detect any similarity between the putative gene product and proteins in the database.
orf4, downstream of spiQ, starts with the initiation codon TTG and may encode a protein consisting of 108 amino acids. Database searches revealed no significant similarities. However, a search with the N-terminal half (54 amino acids) resulted in 36% identity to BacB, the immunity protein of bacteriocin 31 from Enterococcus faecalis (42). The region upstream of orf4 has features resembling those of strongly regulated spp promoters, such as PsppQ and PsppA (the regulated promoter preceding the sakacin P structural gene) (6, 7, 31). To test the hypothesis that Porf4 is under control of the spp regulatory genes, we exploited recently developed vectors (38, 38a) in which the orf4 promoter was cloned in front of the reporter gene encoding PepN (Table 1; also see Materials and Methods). These vectors contain the genes (sppK and sppR) that are required for activation of spp promoters, and expression is induced by adding the appropriate peptide pheromone. Table 3 shows that Porf4 indeed is an inducible promoter, but it is approximately 10 times weaker than the two bacteriocin promoters in the spp gene cluster, PsppQ and PsppA.
TABLE 3.
Expression of PepN under control of spp promoters in L. sakei Lb790
| Plasmid | Promoter | Specific PepN activitya
|
|
|---|---|---|---|
| Noninduced cells | Induced cells | ||
| pSIP407 | PsppA | 0.020 ± 0.003 | 3.2 ± 0.3 |
| pSIP410 | PsppQ | 0.010 ± 0.001 | 2.0 ± 0.3 |
| pSIP413 | Porf4 | NDb | 0.23 ± 0.06 |
PepN activity is expressed as μmol p-nitroanilide min/mg protein. The values are means ± standard deviations.
ND, no detectable activity.
Immediately downstream of orf4 is orf5, which could code for a peptide containing 35 amino acids (GenBank accession numbers AJ844595 and AJ844594, nucleotides 1133 to 1237). However, orf5 seems to be a pseudogene, which is inactivated by a one-nucleotide frameshift mutation at about position 1209. Without this mutation, orf5 would encode a protein consisting of 92 amino acids which is almost 50% identical to both the sakacin P immunity protein SpiA and the immunity protein LisB from Listeria innocua (16).
Analysis of sppQ and spiQ.
To analyze the function of the new ORFs and to pinpoint ORFs responsible for bacteriocin production and immunity, various subsets of wild-type and mutant genes were expressed using the two-plasmid expression system described by Axelsson et al. (4) (see Materials and Methods). Various constructs (Table 1) were made and introduced into L. sakei Lb790(pSAK20). Subsequently, bacteriocin production and immunity were examined (Fig. 1). Transformants harboring pGM11 (Fig. 1, all ORFs) produced bacteriocin (Bac+) and had a bacteriocin resistance (Imm+) phenotype. The results obtained with deletion variants of pGM11 (pGM112 and pGM112B) showed that orf4 and orf5 do not contribute to bacteriocin production and immunity. Strains containing plasmids with a deletion in the 3′ end of the spiQ gene (pGM112C) or lacking the spiQ gene completely (pGM112D) did not grow, indicating that they still produced the bacteriocin (encoded by sppQ) but had lost the immunity gene (spiQ) needed to protect them. Indeed, strains containing pGM112I (expressing spiQ only) were immune, confirming that only spiQ is necessary to obtain immunity to sakacin Q.
Constructs in which sppQ had been knocked out by introduction of a TAG stop codon (pGM112G) or an out-of-frame deletion (pGM112F) not only lost the Bac+ phenotype but also lost immunity to sakacin Q. These polar effects indicate that there is translational coupling between the sppQ and spiQ genes. This issue is addressed in more detail below. Strains containing a construct (pGM112E) with an in-frame mutation in sppQ had a Bac− Imm+ phenotype, supporting the roles of the sppQ and spiQ genes and the occurrence of translational coupling.
Sakacin Q purification and characterization.
So far, it has not been possible to purify any bacteriocin other than sakacin P from the culture broth of L. sakei Lb674 or LTH673, but studies with culture supernatants have clearly shown that these strains do produce bacteriocins in addition to sakacin P (9). Our engineered strain for heterologous expression of sakacin Q, L. sakei Lb790 (pSAK20, pGM11), was used in a new attempt to purify the putative sakacin Q bacteriocin using an adapted standard protocol (see Materials and Methods). This resulted in purification of sakacin Q, although the yields were low. The purified peptide displayed antimicrobial activity against many strains, including strains previously identified as strains that were much more sensitive to the (then) unknown second bacteriocin than to sakacin P (9) (see below). Matrix-assisted laser desorption ionization-time of flight mass spectrometry analyses and N-terminal amino acid sequence analysis confirmed that sakacin Q had the sequence and molecular mass (4,486 Da) expected from the gene sequence and confirmed that the putative double-glycine leader peptide (Fig. 3A) was indeed cleaved off during processing of the prebacteriocin (Fig. 2).
The antimicrobial activity of sakacin Q was compared to that of sakacin P and to the total bacteriocin activity in a wild-type producer strain by using supernatants of L. sakei Lb790(pSAK20, pGM11) (producing only sakacin Q), L. sakei Lb790(pSAK20, pSPP2) (producing only sakacin P) (4), and L. sakei LTH673 (producing both sakacin P and Q). Table 4 shows that sakacin Q had the greatest activity against L. sakei NCDO 2714 and Lactobacillus sp. strain LTH469, while sakacin P had the greatest activity against Lactobacillus coryneformis. The results also show that sakacin Q has a narrower inhibitory spectrum than sakacin P, which also has high activity against more distantly related genera, such as Carnobacterium and Listeria.
TABLE 4.
Inhibitory spectra of bacteriocins in cell-free supernatants with different indicator strainsa
| Indicator strainb | Activity
|
||
|---|---|---|---|
| L. sakei Lb790 (pSAK20, pSPP2) Sakacin P | L. sakei LTH673 Sakacin P and sakacin Q | L. sakei Lb790 (pSAK20, pGM11) Sakacin Q | |
| Lactobacillus sakei strains | |||
| Lb790(pSAK20, pLPV111) | 0 | 0.11 | 0.10 |
| Lb790(pSAK20, pGM11) | 0 | 0 | 0 |
| NCDO 2714 | 0.47 | 0.83 | 1 |
| No. 5 | 0.01 | 0 | 0.03 |
| Lb706 | 0.01 | 0.06 | 0.04 |
| Lactobacillus coryneformis subsp. torquens NCDO 2740 | 1 | 1 | 0.07 |
| Lactobacillus plantarum strains | |||
| C11 | 0 | 0 | 0 |
| TMW1.25 | 0 | 0 | 0 |
| NCDO 1867 | 0 | 0 | 0.02 |
| Lactobacillus sp. strain LTH469 | 0.01 | 0.17 | 0.31 |
| Streptococcus faecalis NCDO 581 | 0.71 | 0.80 | 0.12 |
| Pediococcus pentosaceus strains | |||
| LMGT 2722 | 0 | 0 | 0 |
| LMGT 2001 | 0 | 0 | 0.02 |
| Pediococcus acidolactis LMGT 2351 | 0 | 0 | 0 |
| Carnobacterium pisicola U149 | 0.90 | 0.62 | 0.06 |
| Listeria innocua 4202 | 0.95 | 0.79 | 0.04 |
| Leuconostoc mesenteroides No. 6 | 0.07 | 0 | 0.02 |
| Enterococcus faecium LMGT 2749 | 0 | 0 | 0 |
Bacteriocin activities were calculated in terms of bacteriocin units and are expressed as relative values. The activity with the most sensitive indicator strain was defined as 1.
LMGT, Laboratory of Microbial Gene Technology, Department of Chemistry Biotechnology and Food Science, Agricultural University of Norway.
Regulation of immunity gene expression.
To study the strong polar effect of nonsense and frameshift mutations in sppQ, we constructed a series of pGM11 derivatives in which the gusA gene was fused to sppQ or spiQ (Tables 1 and 5). We then monitored GusA activity in L. sakei Lb790 harboring pSAK20 and these constructs. Table 5 shows that a construct with a sppQ-gusA fusion (pGM204) had considerable GUS activity (244 ± 17 Miller units), and so did the construct containing an spiQ fusion and an in-frame deletion in the preceding sppQ gene (pGM202). However, constructs with the spiQ fusion in which the preceding sppQ gene contained a nonsense mutation (pGM201) or a frameshift mutation (pGM203) had much lower activities (18.5- and 5.5- fold lower than the activity observed with pGM202, respectively). The stop codon of the changed sppQ reading frame in pGM203 comprised nucleotides 445 to 447 (Fig. 2) and started two nucleotides downstream of the predicted RBS of spiQ. Thus, in pGM203, the stop codon is located 13 nucleotides upstream of the native stop codon of sppQ (nucleotides 458 to 460 in Fig. 2).
TABLE 5.
Translational activity in the sppQ-spiQ operon
| Plasmid | Description | GUS activity (Miller units)b |
|---|---|---|
| pGM112G | Negative control | 4 ± 3 |
| pGM204 | Analogous to pGM112D; in-frame fusion of gusA to the 22nd amino acid of presakacin Q | 244 ± 17 |
| pGM201 | Analogous to pGM112G; in-frame fusion of gusA to spiQ; nonsense mutation in sppQ | 6 ± 2 |
| pGM202 | Analogous to pGM112E; in-frame fusion of gusA to spiQ; in-frame (57-nucleotide) deletion in sppQa | 111 ± 9 |
| pGM203 | Analogous to pGM112F; in-frame fusion of gusA to spiQ; out-of-frame (56-nucleotide) deletion in sppQ | 20 ± 3 |
| pGM206 | Like pGM201; optimized RBS and start codon in spiQ | 7 ± 1 |
| pGM207 | Like pGM202; optimized RBS and start codon in spiQ | 163 ± 14 |
Note that constructs without any mutation in sppQ and with gusA fused to spiQ were not viable, presumably because of the resulting bacteriocin production.
GUS activity was measured 5 h after dilution of a fresh overnight culture.
Northern blot analysis using a gusA-specific probe demonstrated that transformants harboring pGM201, pGM202, or pGM203 contained roughly the same amount of hybridizing mRNA (Fig. 4). This showed that the differences in GUS levels were not due to different transcription levels but were due to effects on the translational coupling between spiQ and sppQ.
FIG. 4.
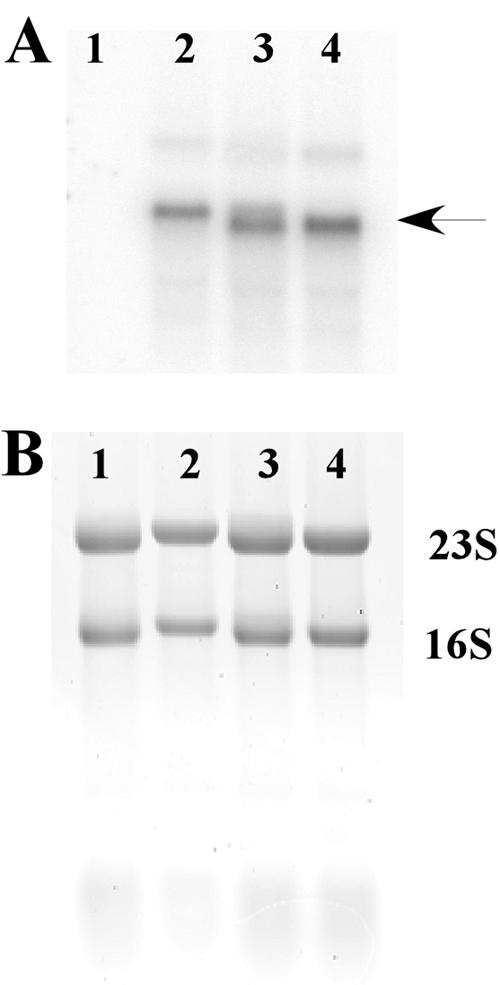
Northern blot analyses of L. sakei Lb790(pSAK20) harboring various gusA-containing constructs. Lane 1, pGM112G (negative control); lane 2, pGM201 (nonsense mutation in sppQ); lane 3, pGM202 (in-frame mutation in sppQ); lane 4, pGM203 (out-of-frame mutation in sppQ). (A) Blot after hybridization with a gusA-specific probe. (B) Amount of total RNA. The results indicate that there was approximately the same amount in all samples.
The RBS preceding spiQ is suboptimal (29) and part of a stem-loop structure (Fig. 5), which may be unfavorable for translation initiation (21). As shown in Table 5, improvement of these sequences in the construct with the nonsense mutation had no effect (compare pGM201 with pGM206). However, the same improvement in pGM202 (in-frame deletion) led to an almost 50% increase in GusA activity (pGM207).
FIG. 5.

Predicted secondary structures of the mRNA for the overlapping area of sppQ and spiQ. (A) The diagram shows the wild-type sequence and the sequence harboring an optimized RBS and start codon (St.c.) (B) The stop codon of sppQ in pGM202 is indicated by Sto.c. The structural analyses were performed using Mfold (47). The predicted stabilities of the structures are ΔG = −11.8 kcal/mol (A) and ΔG = −6.6 kcal/mol (B).
DISCUSSION
Mature sakacin Q is a hydrophobic, cationic peptide consisting of 49 amino acids, and it has a calculated pI of 8.9. These are common features for class II bacteriocins (24). The mass spectrometry analyses did not indicate any modifications, meaning that sakacin Q belongs to class II. Sakacin Q does not contain conserved sequence motifs that are characteristic of class IIa bacteriocins, whereas the genetic organization of its gene and its activity in the absence of other peptides indicate that it is a one-peptide bacteriocin. Thus, sakacin Q seems to belong to class IIc of the bacteriocins.
Table 4 shows that sakacin Q expands the potential of the sakacin P-producing strain for inhibition of other bacteria. Additional immunity genes may also contribute in the competition that goes on in microbial communities, and it is conceivable that orf4 is such a gene since it shows weak similarity to a gene encoding the immunity protein of bacteriocin 31 (42). Interestingly, orf4 is preceded by a weak variant of a typical regulated spp promoter (Fig. 2 and Table 3). Since the activity of the Porf4 promoter is much weaker than the activity of PsppQ (which also drives expression of orf4 and orf5 [6]), it is questionable whether Porf4 plays an important role in vivo. orf4 may simply be nonfunctional “junk DNA,” as suggested by the fact that the next gene, orf5, seems to be nonfunctional.
SpiQ is an unusually short immunity protein and consists of only 60 amino acids. Most reported immunity proteins for nonlantibiotics are larger (range, 100 to 150 residues) (8); the exceptions include the immunity proteins for divergicin A (56 residues) (46) and the two-peptide bacteriocin brochocin C (53 residues) (22). (Note that the original publications on sakacin P and curvacin A [= sakacin A] erroneously suggested small immunity proteins for these bacteriocins too [41]; these suggestions were the result of DNA sequence errors which were later corrected but which persist to some extent in the literature.)
The sakacin Q gene and its cognate immunity gene are organized in overlapping reading frames (Fig. 2), which is not uncommon for bacteriocin operons (17, 19, 22) or for other operons (21, 27, 33). Overlapping genes suggest translational coupling, which indeed has been suggested to occur in bacteriocin operons (17, 22). It would not be surprising if the production of a bacteriocin and the production of its cognate immunity protein were strictly coregulated. However, to the best of our best knowledge, this has never been proven experimentally.
The introduction of a stop codon in sppQ abolished the immunity of the host cells to sakacin Q, whereas a frameshift mutation leading to a ribosomal stop codon 6 to 4 nucleotides upstream of the spiQ start codon (instead of 5 to 7 nucleotides downstream) severely reduced immunity. In both cases, the translational initiation region (TIR) (the ribosome binding site, the start codon, and adjacent up- and downstream regions [21]) of spiQ is intact. Mutations in sppQ that did not influence the reading frame and allowed the ribosomes to terminate at the normal sppQ stop codon resulted in full immunity. The results of the studies with spiQ-β-glucuronidase gene fusions (Table 5) yielded further insight in these polar effects. Introduction of a nonsense mutation (pGM201) and introduction of a frameshift mutation (pGM203) into sppQ reduced the GUS levels 18.5- and 5.5-fold, respectively, compared to a mutation that did not lead to a change in the reading frame (pGM202). It is important to note that pGM202 and pGM203 differ by only one nucleotide and that the Northern blot analyses showed approximately equal transcription levels. Thus, the difference between pGM202 and pGM203 must be solely due the frameshift effect, which in turn affects the presence of ribosomes in the TIR of spiQ.
The stem-loop in the TIR of spiQ (Fig. 5A) may prevent access of the ribosomes to the RBS (27, 45). Optimization of the RBS and start codon of spiQ was not predicted to resolve the stem-loop (Fig. 5B) and indeed had no influence in a situation where ribosomes were not arriving from the preceding sppQ gene (compare pGM206 to pGM201 in Table 5). The results obtained with pGM207 show that the potentially better TIR of spiQ did improve translation in a situation where translation of the preceding genes proceeded all the way to the normal stop codon (compare pGM207 and pGM202 in Table 5).
Three partially interrelated mechanisms for translational coupling have been proposed previously: (i) movement of ribosomes along upstream mRNA may remove a structural barrier for translation (a stem-loop structure) while simultaneously creating a high local concentration of ribosomes that reinitiate easily at the neighboring RBS; (ii) removal of a structural barrier gives “fresh” ribosomes access to the usually occluded RBS; and (iii) ribosomes that translate the first ORF do not fully dissociate from the transcript but move backward and then restart at the RBS of the downstream gene (27, 44). The experimental results show that termination of translation of the upstream gene at a stop codon consisting of nucleotides 6 to 4 upstream of the spiQ start codon (instead of nucleotides 5 to 7 downstream) largely abolishes immunity (pGM112F) and results in relatively low GUS activity (pGM203). It is conceivable that the 13-nucleotide difference in the position of the stop codons (Fig. 5A) affects the ability of a bound ribosome to remove a structural barrier for translation of spiQ. On the other hand, both stop codons are in the stem-loop structure (Fig. 5A), and both are downstream of the RBS for spiQ, indicating that a ribosome should affect local secondary structure in both situations. Clearly, the 13-nucleotide difference does not affect the local concentration of ribosomes.
The third, so-called “restart” mechanism for translational coupling (27) was proposed to occur in lactic acid bacteria by van de Guchte et al. (44), based on the observation that a gradually decreasing distance between the stop codon of an upstream gene and the start codon of a downstream gene had a dramatic influence on the expression of the latter gene. Most importantly, van de Guchte et al. (44) showed that translation became much more effective after changing from a situation like that in pGM203 (stop codon upstream of the start codon) to a situation like that in pGM202, where the stop codon of the upstream gene is located downstream of the start codon of spiQ. It is thus conceivable that the translational coupling observed in this study was due to a restart mechanism. In the case of the sppQ and spiQ genes, translational coupling may be used to ensure that the immunity gene is expressed only when the bacteriocin is expressed, possibly in stoichiometric amounts. Interestingly, expression of GUS could be increased by our limited attempt to optimize the TIR, but only in the case of pGM207 (that is, with the wild-type reading frame). This may indicate that restarting of the ribosomes is more efficient with a more optimized TIR.
In conclusion, the present results show that the spp regulon is involved in the production of at least two bacteriocins, sakacin P and sakacin Q. In addition, our results provide experimental evidence for the often suggested but sparsely analyzed coregulation of the production of bacteriocin and immunity proteins at the translational level.
Acknowledgments
This work was supported by grant 134314/140 from the Research Council of Norway.
We thank Knut Sletten and Dimitris Mantzilas, both of the University of Oslo, for N-terminal amino acid sequencing and mass spectrometry, respectively. We thank Eric Johansen and Per Strøman at Chr. Hansen A/S for supplying the pepN gene. We are grateful to Birgitta Baardsen for technical assistance during the Northern blot analysis.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. H. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aukrust, T. W., M. B. Brurberg, and I. F. Nes. 1995. Transformation of Lactobacillus by electroporation. Methods Mol. Biol. 47:201-208. [DOI] [PubMed] [Google Scholar]
- 3.Axelsson, L., and A. Holck. 1995. The genes involved in production of and immunity to sakacin A, a bacteriocin from Lactobacillus sake Lb706. J. Bacteriol. 177:2125-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axelsson, L., T. Katla, M. Bjørnslett, V. G. H. Eijsink, and A. Holck. 1998. A system for heterologous expression of bacteriocins in Lactobacillus sake. FEMS Microbiol. Lett. 168:137-143. [DOI] [PubMed] [Google Scholar]
- 5.Axelsson, L., G. Lindstad, and K. Naterstad. 2003. Development of an inducible gene expression system for Lactobacillus sakei. Lett. Appl. Microbiol. 37:115-120. [DOI] [PubMed] [Google Scholar]
- 6.Brurberg, M. B., I. F. Nes, and V. G. H. Eijsink. 1997. Pheromone-induced production of antimicrobial peptides in Lactobacillus. Mol. Microbiol. 26:347-360. [DOI] [PubMed] [Google Scholar]
- 7.Diep, D. B., L. S. Håvarstein, and I. F. Nes. 1996. Characterization of the locus responsible for the bacteriocin production in Lactobacillus plantarum C11. J. Bacteriol. 178:4472-4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eijsink, V. G. H., L. Axelsson, D. B. Diep, L. S. Håvarstein, H. Holo, and I. F. Nes. 2002. Production of class II bacteriocins by lactic acid bacteria; an example of biological warfare and communication. Antonie Leeuwenhoek 81:639-654. [DOI] [PubMed] [Google Scholar]
- 9.Eijsink, V. G. H., M. Skeie, P. H. Middelhoven, M. B. Brurberg, and I. F. Nes. 1998. Comparative studies of class IIa bacteriocins of lactic acid bacteria. Appl. Environ. Microbiol. 64:3275-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geis, A., J. Singh, and M. Teuber. 1983. Potential of lactic streptococci to produce bacteriocin. Appl. Environ. Microbiol. 45:205-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Håvarstein, L. S., D. B. Diep, and I. F. Nes. 1995. A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol. Microbiol. 16:229-240. [DOI] [PubMed] [Google Scholar]
- 12.Higuchi, R. 1990. Recombinant PCR, p. 177-183. In M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (ed.), PCR protocols: a guide to methods and applications. Academic Press Inc., San Diego, CA.
- 13.Holck, A. L., L. Axelsson, K. Huehne, and L. Kroeckel. 1994. Purification and cloning of sakacin 674, a bacteriocin from Lactobacillus sake Lb674. FEMS Microbiol. Lett. 115:143-149. [DOI] [PubMed] [Google Scholar]
- 14.Huehne, K., L. Axelsson, A. Holck, and L. Kroeckel. 1996. Analysis of the sakacin P gene cluster from Lactobacillus sake Lb674 and its expression in sakacin-negative Lb. sake strains. Microbiology 142:1437-1448. [DOI] [PubMed] [Google Scholar]
- 15.Jack, R. W., J. R. Tagg, and B. Ray. 1995. Bacteriocins of gram-positive bacteria. Microbiol. Rev. 59:171-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalmokoff, M. L., S. K. Banerjee, T. Cyr, M. A. Hefford, and T. Gleeson. 2001. Identification of a new plasmid-encoded sec-dependent bacteriocin produced by Listeria innocua 743. Appl. Environ. Microbiol. 67:4041-4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kemperman, R., M. Jonker, A. Nauta, O. P. Kuipers, and J. Kok. 2003. Functional analysis of the gene cluster involved in production of the bacteriocin circularin A by Clostridium beijerinckii ATCC 25752. Appl. Environ. Microbiol. 69:5839-5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleerebezem, M., L. E. Quadri, O. P. Kuipers, and W. M. de Vos. 1997. Quorum sensing by peptide pheromones and two-component signal-transduction systems in Gram-positive bacteria. Mol. Microbiol. 24:895-904. [DOI] [PubMed] [Google Scholar]
- 19.Maldonado, A., J. L. Ruiz-Barba, and R. Jimenez-Diaz. 2003. Purification and genetic characterization of plantaricin NC8, a novel coculture-inducible two-peptide bacteriocin from Lactobacillus plantarum NC8. Appl. Environ. Microbiol. 69:383-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathiesen, G., H. M. Namløs, P. A. Risøen, L. Axelsson, and V. G. H. Eijsink. 2004. Use of bacteriocin promoters for gene expression in Lactobacillus plantarum C11. J. Appl. Microbiol. 96:819-827. [DOI] [PubMed] [Google Scholar]
- 21.McCarthy, J. E. G., and C. Gualerzi. 1990. Translational control of prokaryotic gene expression. Trends Genet. 6:78-85. [DOI] [PubMed] [Google Scholar]
- 22.McCormick, J. K., A. Poon, M. Sailer, Y. Gao, K. L. Roy, L. M. McMullen, J. C. Vederas, M. E. Stiles, and M. J. Van Belkum. 1998. Genetic characterization and heterologous expression of brochocin C, an antibotulinal, two-peptide bacteriocin produced by Brochothrix campestris ATCC 43754. Appl. Environ. Microbiol. 64:4757-4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller, K. M. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 24.Nes, I. F., D. B. Diep, L. S. Håvarstein, M. B. Brurberg, V. G. H. Eijsink, and H. Holo. 1996. Biosynthesis of bacteriocins in lactic acid bacteria. Antonie Leeuwenhoek 70:113-128. [DOI] [PubMed] [Google Scholar]
- 25.Nes, I. F., and V. G. H. Eijsink. 1999. Regulation of group II peptide bacteriocin synthesis by quorum-sensing mechanisms, p. 175-192. In G. M. Dunny and S. C. Winans (ed.), Cell-cell signaling in bacteria. American Society for Microbiology, Washington, D.C.
- 26.Nes, I. F., and H. Holo. 2000. Class II antimicrobial peptides from lactic acid bacteria. Biopolymers 55:50-61. [DOI] [PubMed] [Google Scholar]
- 27.Oppenheim, D. S., and C. Yanofsky. 1980. Translational coupling during expression of the tryptophan operon of Escherichia coli. Genetics 95:785-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearson, W. R., and D. J. Lipman. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85:2444-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pouwels, P. H., and R. J. Leer. 1993. Genetics of lactobacilli: plasmids and gene expression. Antonie Leeuwenhoek 64:85-107. [DOI] [PubMed] [Google Scholar]
- 30.Quadri, L. E. N. 2002. Regulation of antimicrobial peptide production by autoinducer-mediated quorum sensing in lactic acid bacteria. Antonie Leeuwenhoek 82:133-145. [PubMed] [Google Scholar]
- 31.Risøen, P. A., M. B. Brurberg, V. G. H. Eijsink, and I. F. Nes. 2000. Functional analysis of promoters involved in quorum sensing-based regulation of bacteriocin production in Lactobacillus. Mol. Microbiol. 37:619-628. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 33.Scherbakov, D. V., and M. B. Garber. 2000. Overlapping genes in bacterial and phage genomes. Mol. Biol. 34:485-495. [PubMed] [Google Scholar]
- 34.Schillinger, U., and F. K. Lücke. 1989. Antibacterial activity of Lactobacillus sake isolated from meat. Appl. Environ. Microbiol. 55:1901-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlaman, H. R. M., E. Risseeuw, M. E. I. Franke-van Dijk, and P. J. J. Hooykaas. 1994. Nucleotide sequence corrections of the uidA open reading frame encoding beta-glucuronidase. Gene 138:259-260. [DOI] [PubMed] [Google Scholar]
- 36.Shaw, B. G., and C. D. Harding. 1984. A numerical taxonomic study of lactic acid bacteria from vacuum-packed beef, pork, lamb and bacon. J. Appl. Bacteriol. 56:25-40. [DOI] [PubMed] [Google Scholar]
- 37.Siragusa, G. R., and C. N. Cutter. 1993. Brochocin C, a new bacteriocin produced by Brochothrix campestris. Appl. Environ. Microbiol. 59:2326-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sørvig, E., S. Grönqvist, K. Naterstad, G. Mathiesen, V. G. H. Eijsink, and L. Axelsson. 2003. Construction of vectors for inducible gene expression in Lactobacillus sakei and L. plantarum. FEMS Microbiol. Lett. 229:119-126. [DOI] [PubMed] [Google Scholar]
- 38a.Sørvig, E., G. Mathiesen, K. Naterstad, V. G. H. Eijsink, and L. Axelsson. High-level, inducible gene expression in Lactobacillus sakei and Lactobacillus plantarum using versatile expression vectors. Microbiology, in press. [DOI] [PubMed]
- 39.Strøman, P. 1992. Sequence of a gene (lap) encoding a 95.3-kDa aminopeptidase from Lactococcus-lactis ssp. cremoris Wg2. Gene 113:107-112. [DOI] [PubMed] [Google Scholar]
- 40.Tichaczek, P. S., J. Nissen-Meyer, I. F. Nes, R. F. Vogel, and W. P. Hammes. 1992. Characterization of the bacteriocins curvacin A from Lactobacillus curvatus LTH1174 and sakacin P from L. sake LTH673. Syst. Appl. Microbiol. 15:460-468. [Google Scholar]
- 41.Tichaczek, P. S., R. F. Vogel, and W. P. Hammes. 1993. Cloning and sequencing of curA encoding curvacin A, the bacteriocin produced by Lactobacillus curvatus LTH1174. Arch. Microbiol. 160:279-283. [DOI] [PubMed] [Google Scholar]
- 42.Tomita, H., S. Fujimoto, K. Tanimoto, and Y. Ike. 1996. Cloning and genetic organization of the bacteriocin 31 determinant encoded on the Enterococcus faecalis pheromone responsive conjugative plasmid pYI17. J. Bacteriol. 178:3585-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Twomey, D., R. P. Ross, M. Ryan, B. Meaney, and C. Hill. 2002. Lantibiotics produced by lactic acid bacteria: structure, function and applications. Antonie Leeuwenhoek 82:165-185. [PubMed] [Google Scholar]
- 44.van de Guchte, M., J. Kok, and G. Venema. 1991. Distance-dependent translational coupling and interference in Lactococcus lactis. Mol. Gen. Genet. 227:65-71. [DOI] [PubMed] [Google Scholar]
- 45.van de Guchte, M., T. van der Lende, J. Kok, and G. Venema. 1991. A possible contribution of mRNA secondary structure to translation initiation efficiency in Lactococcus lactis. FEMS Microbiol. Lett. 65:201-208. [DOI] [PubMed] [Google Scholar]
- 46.Worobo, R. W., M. J. Van Belkum, M. Sailer, K. L. Roy, J. C. Vederas, and M. E. Stiles. 1995. A signal peptide secretion-dependent bacteriocin from Carnobacterium divergens. J. Bacteriol. 177:3143-3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuker, M., D. H. Mathews, and D. H. Turner. 1999. Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide, p. 11-43. In J. Barciszewski and B. F. C. Clark (ed.), RNA biochemistry and biotechnology, vol. 70. Kluwer Academic Publishers, Dordrecht, The Netherlands. [Google Scholar]