

Abstract
Xylella fastidiosa is a pathogen that causes leaf scorch and related diseases in over 100 plant species, including Pierce's disease in grapevines (PD), phony peach disease (PP), plum leaf scald (PLS), and leaf scorch in almond (ALS), oak (OAK), and oleander (OLS). We used a high-resolution DNA sequence approach to investigate the evolutionary relationships, geographic variation, and divergence times among the X. fastidiosa isolates causing these diseases in North America. Using a large data set of 10 coding loci and 26 isolates, the phylogeny of X. fastidiosa defined three major clades. Two of these clades correspond to the recently identified X. fastidiosa subspecies piercei (PD and some ALS isolates) and X. fastidiosa subsp. multiplex (OAK, PP, PLS, and some ALS isolates). The third clade grouped all of the OLS isolates into a genetically distinct group, named X. fastidiosa subsp. sandyi. These well-differentiated clades indicate that, historically, X. fastidiosa has been a clonal organism. Based on their synonymous-site divergence (∼3%), these three clades probably originated more than 15,000 years ago, long before the introduction of the nonnative plants that characterize most infections. The sister clades of X. fastidiosa subsp. sandyi and X. fastidiosa subsp. piercei have synonymous-site evolutionary rates 2.9 times faster than X. fastidiosa subsp. multiplex, possibly due to generation time differences. Within X. fastidiosa subsp. multiplex, a low level (∼0.1%) of genetic differentiation indicates the recent divergence of ALS isolates from the PP, PLS, and OAK isolates due to host plant adaptation and/or allopatry. The low level of variation within the X. fastidiosa subsp. piercei and X. fastidiosa subsp. sandyi clades, despite their antiquity, suggests strong selection, possibly driven by host plant adaptation.
Bacterial systematics is based on identifying species that reflect historical phenotypic clusters, while recognizing that clear species groups can be obscured by the action of genetic processes uncommon among eukaryotes that facilitate horizontal gene transfer from unrelated sources (11). Named bacterial species are often further subdivided by the recognition of distinct pathovars and strains that typically infect different hosts. This strain recognition has been facilitated by the availability of whole-genome DNA sequence data for the phylogenetic comparison of an increasing number of species. Within species, isolates can be compared across multiple loci, enabling strain diversity and genetic diversity to be quantified.
Many species exhibit a high degree of genetic diversity among strains that infect the same host, and this pattern is typical of the gamma subdivision proteobacteria, e.g., Escherichia coli (17, 27), Vibrio cholerae (5), Pseudomonas stutzeri (36), Xanthomonas axonopodis pv. manihotis (30), and Xanthomonas fragariae (31). Xylella fastidiosa, another gamma subdivision proteobacterium, offers an opportunity to study genetic variability both between and within plant-host strains (pathovars) of a single species that infects multiple hosts. Currently two complete genomes (41, 37) and two draft genomes (7) of strains from four different plant hosts have been sequenced.
X. fastidiosa infects the xylem of a range of plant species and is typically transmitted by xylem-feeding leaf hoppers (28). Xylem blockage by X. fastidiosa causes the plant scorch diseases of several economically important plants, such as grapevine, almond, and citrus (28). It also infects ornamentals, such as oleander (29). The known host range continues to expand, with over 100 host species identified (35) and with the most symptomatic hosts being plants nonnative to North or South America (23). Because of its potentially serious economic effect, X. fastidiosa has been the subject of intensive study, and several host plant strains have been identified, including three different pathovars causing Pierce's disease of grapevine (PD), almond leaf scorch (ALS), and oleander leaf scorch (OLS) in California (1, 4, 8-10, 12, 14, 20, 25, 26, 32).
Early DNA-DNA hybridization studies of several X. fastidiosa isolates indicated at least 70% similarity (24) which, combined with studies of nutritional fastidiousness and reciprocal transmission tests, placed the different host strains into groups (23). The greater than 70% similarity indicated that X. fastidiosa was a single species (11); however, no meaningful phylogenetic relationships could be defined. Randomly amplified polymorphic DNA analysis (RAPD) led to a more comprehensive phylogenetic classification of a larger variety of X. fastidiosa plant-host pathovars distributed across its range, such as grapevine (western United States), mulberry and oak (eastern United States), and coffee (South America) (1, 4, 10, 14, 20, 26). Similarity indices of the RAPDs indicated that bacteria isolated from the same plant-host species were more similar than bacteria from different hosts (20). The amount of diversity within these plant-host strains, however, was contradictory, with one study indicating limited diversity (12) and another indicating moderate amounts (20). Several single-gene trees and noncoding regions have also been analyzed, including 16S rRNA (25, 32), 16S-23S intergenic spacer sequences (20, 25, 35), and gyrB (32). The resulting trees had conflicting topologies, due to a lack of informative sites and phylogenetic signal.
A recent DNA-DNA hybridization study (35) again supported that the different pathovars of X. fastidiosa formed a single species. The North American strains were divided into two subspecies, X. fastidiosa subsp. piercei (PRC) and X. fastidiosa subsp. multiplex (MULT), each subspecies showing at least 84% internal similarity (35). In this study, our aim was to reexamine the relationship among the three major plant-host pathovars found in California, PD, ALS, and OLS, and examine the genetic and evolutionary relationships between these California groups and strains from the eastern United States isolated from oak, peach, and plum. For this purpose we used a high-resolution approach using DNA sequence data from genes distributed throughout the genome. We sequenced 10 chromosomal genes, including genes involved in such varied functions as aerobic respiration (nuoN and nuoL) and cell surface structures (pilU). We used these sequence data to establish a robust phylogeny, which is an important prerequisite for understanding how this bacterial species evolves and how genetic change has allowed it to adapt to new host plants across the United States. We used phylogeny as the basis for estimating the divergence times of the North American clades and to determine if these phylogenetic groupings showed geographical differentiation.
MATERIALS AND METHODS
X. fastidiosa isolates.
Twenty-six isolates of X. fastidiosa from the six host plants of grape, oleander, almond, oak, peach, and plum were obtained for analysis (Table 1). These strains included those used for genome sequencing of PD (Temecula), OLS (Ann-1), and ALS (Dixon) (41, 6). Other isolates were chosen to represent the geographic distribution of PD and ALS within California and to represent some of the North American variation in the PD (Florida) and OLS (Texas) strains. Three isolates of the oak leaf scorch (OAK), two isolates of phony peach (PP), and one isolate of plum leaf scald (PLS) from the eastern United States were also examined. The sequenced citrus variegated chlorosis (CVC) strain (37) from South America was used as the outgroup.
TABLE 1.
X. fastidiosa isolates used in this study
| ID | Strain | Host of origin | Geographic origin | Source or reference |
|---|---|---|---|---|
| PD1a | Temecula | Grape | Temecula, S. Calif.b | 41 |
| PD10 | I03 | Grape | Temecula, S. Calif.b | 13 |
| PD7 | Traver | Grape | Tulare, S. Calif.b | 20 |
| PD14 | Douglas | Grape | San Luis Obispo, S. Calif.b | 20 |
| PD4 | STL | Grape | Napa, N. Calif.b | 20 |
| PD6a | Conn Creek | Grape | Napa, N. Calif.b | 20 |
| PD16a | 95-2 | Grape | Florida | 20 |
| OLS2 | Ann-1 | Oleander | Palm Springs, Calif. | 29 |
| OLS9a | Riverside | Oleander | Riverside, Calif. | 13 |
| OLS8a | Texas | Oleander | Texas | 13 |
| OLS19 | Cathedral City | Oleander | Palm Springs, Calif. | 29 |
| OLS20 | TR2 | Oleander | Orange, Calif. | A. Purcell |
| OLS21 | TS5 | Oleander | Orange, Calif. | A. Purcell |
| ALS3a | Dixon | Almond | Solano, N. Calif.b | 20 |
| ALS15 | ALS-2 | Almond | San Joaquin, N. Calif.b | 20 |
| ALS5 | Tulare | Almond | Tulare, S. Calif.b | 20 |
| ALS11 | 237 | Almond | Temecula, S. Calif.b | 13 |
| ALS12 | 276 | Almond | Temecula, S. Calif.b | 13 |
| ASL13a | 187 | Almond | Temecula, S. Calif.b | 13 |
| ALS22 | ALS-6 | Almond | San Joaquin, N. Calif.b | 20 |
| OAK17a | Stucky | Oak | Georgia | 20 |
| OAK23a | 92-3 | Oak | Florida | 20 |
| OAK24 | Oak#2 | Oak | Georgia | 20 |
| PP27a | 5S2 | Peach | Georgia | 20 |
| PP28a | 5R1 | Peach | Georgia | 20 |
| PLS26 | 2#4 | Plum | Georgia | 20 |
| CVC18 | 9a5c | Citrus | Sao Paulo, Brazil | 37 |
Isolates used to determine divergence times and percent differences.
Those isolates occurring north of Santa Clara and Merced Counties were considered to be from northern California. This demarcation was used for evaluating geographic differentiation in PRC and non-PRC ALS isolates.
The strains for PD, ALS, OLS, PP, and PLS were grown on PD3 agar medium and OAK on PW (15). Bacterial DNA was extracted using the Chelex preparation (42) or by scraping bacterial cells and lysing the cells with distilled water.
Amplification and sequence determination of 10 genes.
The genes sequenced were chosen from among the genes that occur in all four genomes, ALS strain Dixon (NC_002723), OLS strain Ann-1 (NC_002722), PD strain Temecula (AE009442), and CVC strain 9a5c (AE003849). The 10 genes chosen are distributed throughout the CVC genome, represent a variety of biochemical functions, and have KA/KS values of <1, where KA and KS are, respectively, the rate of the nonsynonymous and synonymous base substitutions (Table 2).
TABLE 2.
Position, name, function, PCR primers, and length sequenced for the 10 X. fastidiosa genes
| Gene positiona | Gene IDa | Gene namea | Biochemical functiona | Primer sequence (forward, reverse) | KA/KSb | Gene length (bp) |
|---|---|---|---|---|---|---|
| XF0136 | holC | DNA polymerase III holoenzyme chi subunit | Replication | 5′-GATTTCCAAACCGCGCTTTC-3′, 5′-TCATGTGCAGGCCGCGTCTCT-3′ | 0.08 | 342 |
| XF0257 | rfbD | dTDP-4-dehydrorhamnose 3,5-epimerase | Surface polysaccharides | 5′-TTTGGTGATTGAGCCGAGGGT-3′, 5′-CCATAAACGGCCGCTTTC-3′ | 0.29 | 429 |
| XF0316 | nuoL | NADH-ubiquinone oxidoreductase, NQO12 subunit | Aerobic respiration | 5′-CATTATTGCCGGATTGTTAGG-3′, 5′-GCGGGAAACATTACCAAGC-3′ | 0.14 | 1,821 |
| XF0318 | nuoN | NADH-ubiquinone oxidoreductase, NQO14 subunit | Aerobic respiration | 5′-GGGTTAAACATTGCCGATCT-3′, 5′-CGGGTTCCAAAGGATTCCTAA-3′ | 0.14 | 1,311 |
| XF0656 | gltT | Glutamate symport protein | Transport, amino acids | 5′-TTGGGTGTGGGTACGTTGCTG-3′, 5′-CGCTGCCTCGTAAACCGTTGT-3′ | 0.09 | 951 |
| XF0832 | cysG | Siroheme synthase | Biosynthesis, heme and porphyrin | 5′-GGCGGCGGTAAGGTTG-3′, 5′-GCGTATGTCTGTGCGGTGTGC-3′ | 0.32 | 1,170 |
| XF0910 | petC | Ubiquinol cytochrome c oxidoreductase, cytochrome c1 subunit | Electron transport | 5′-CTGCCATTCGTTGAAGTACCT-3′, 5′-CGTCCTCCCAATAAGCCT-3′ | 0.32 | 531 |
| XF1632 | pilU | Twitching motility protein (pilU) | Surface structures | 5′-CAATGAAGATTCACGGCAATA-3′, 5′-ATAGTTAATGGCTCCGCTATG-3′ | 0.17 | 873 |
| XF1818 | leuA | 2-Isopropylmalate synthase (leuA) | Amino acid biosynthesis | 5′-GGGCGTAGACATTATCGAGAC-3′, 5′-GTATCGTTGTGGCGTACACTG-3′ | 0.08 | 1,218 |
| XF2447 | lacF | ABC transporter sugar permease (lacF) | Transport, carbohydrates | 5′-TTGCTGGTCCTGCGGTGTTG-3′, 5′-CCTCGGGTCATCACATAAGGC-3′ | 0.15 | 642 |
Gene position ID, name, and function are based on those reported in reference 37.
KA/KS calculated across all N. American isolates.
Primers were designed using sequences available from the four genomes with the Oligo6 program (34) (Table 2). Each reaction mixture contained 5 to 15 ng/μl of DNA template, 1× buffer solution (Promega), 1 mM deoxynucleotide triphosphates, 1 μM of each primer, and 5 U of Taq polymerase (Promega) for a total 30-μl reaction volume. The thermocycler (Eppendorf Mastercycler) reaction conditions were an initial denaturation step at 94°C for 3 min, followed by 30 cycles of 94°C for 30 seconds, 60°C for 30 seconds for primer annealing, and an extension period at 72°C for 1 min. The final step was an extension period at 72°C for 5 min. For genes over 1,000 bp, the extension time was lengthened from 1 min to 90 seconds. Purification of the amplification product was completed with the Wizard PCR Preps DNA purification system. Sequencing was performed by the UCR Genome Institute using the Big Dye Terminator kit with an ABI Prism 377 DNA sequencer.
Sequence analysis.
The sequences were aligned with BioEdit v5.0.9 (19) and ClustalX (40). A maximum likelihood (ML) tree of the 10-gene concatenated data set for 25 of the isolates was constructed with PAUP* v4 (39), using the following options: the general time-reversible model of molecular evolution, which includes six separate rates for each nucleotide substitution type, and a gamma distribution and an invariant sites parameter to account for evolutionary rate variation among sites. A second ML tree with only seven genes and all 26 isolates was similarly constructed to determine the position of the PLS isolate. Bootstrap (n = 200 replicates) and Bayesian analyses were only performed on the 10-gene tree. Bayesian analyses were performed with MrBayes 2.01 (22) with a general time-reversible model plus gamma distribution plus invariant sites model of molecular evolution, 1,000,000 generations with burn-in after 200,000, four simultaneous Markov chains (three heated, one cold), and random starting trees.
To estimate clonal diversity, the genetic differentiation between populations relative to within-population variation, or KST (21), was calculated using DNAsp v. 4.00 (33) and ProSeq (18). The isolates were divided to test if the variation was different in five comparisons: (i) northern California PRC (isolates PD4 and -6) versus southern California PRC (isolates PD1, -7, -10, and -14 and ALS5 and -11); (ii) northern California ALS isolates (ALS3, -15, and -22) versus southern California ALS isolates (ALS12 and -13); (iii) PP isolates (PP27 and -28) versus OAK isolates (OAK17, -23, and -24); (iv) ALS isolates (ALS3, -13, and -15) versus eastern United States isolates (OAK17, -23, and -24 and PP27 and -28); (v) PRC isolates (all PD isolates and ALS5 and -11) versus OLS isolates (all OLS); and (vi) PRC/OLS (all PD, ALS5 and -11, and all OLS) versus MULT isolates (all OAK, all PP, and ALS3, -12, -13, -15, and -22). In tests ii and iv, ALS5 and -11 were excluded since, unlike the other ALS isolates, they are subspecies of PRC. In tests iv and vi, ALS12 and ALS22 were excluded since they carry several recombinant genes (M. Scally et al., submitted for publication).
To determine the divergence times, the concatenated data set of PD1 (southern California), PD6 (northern California), PD16 (Florida), OLS8 (Texas), OLS9 (California), ALS3 (northern California), ALS13 (southern California), OAK17 (Georgia), OAK 23 (Florida), PP27 (Georgia), and PP28 (Georgia) were used with CVC18 as the outgroup. These isolates represented the available geographic range of each plant host and showed no evidence of recombination. The codeml application of PAML (44) with the Fcodon substitution model was used to estimate the number of synonymous and nonsynonymous substitutions along each branch (without imposing a clock). Using these estimates of synonymous substitution (K), plus an estimated mutation rate (μ) of 5.4 × 10−10 changes per site per generation (16) and a division rate of 1,000 generations per year (G), the time since divergence can be calculated assuming selective neutrality at synonymous sites with the following equation:
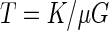 |
(1) |
In order to calculate standard errors for the divergence times, Tukey's jackknife method (38) was employed with 10 replications. We also used the average divergence estimates given by the 10 replications as the divergence times reported for each branch. The percent difference for nonsynonymous and synonymous sites was also calculated from the nonsynonymous and synonymous substitution rates for each branch.
Nucleotide sequence accession numbers.
The gene sequences are available under GenBank accession numbers AY876648 to AY876897 and AY887139 to AY887145.
RESULTS
An ML tree using 9,288 bp was constructed for the 25 North American X. fastidiosa isolates, with the South American CVC strain as the outgroup (Fig. 1). Three major clades are supported by 100% bootstrap support and 1.00 posterior probability. Two of these clades correspond to X. fastidiosa subsp. piercei (PRC clade) and X. fastidiosa subsp. multiplex (MULT clade) previously identified based on DNA hybridization studies (35). As in that previous work, the PRC clade includes all PD isolates plus a subset of the ALS isolates, and the MULT clade includes PP, PLS, and another subset of ALS isolates. The PLS isolate was not included in the statistical evaluation of the phylogenetic tree (Fig. 1) since only 7 of the 10 genes were sequenced (holC, rfbD, gltT, cysG, petC, pilU, and leuA) due to limited DNA. The ML tree based on these seven genes (data not shown) placed PLS within the PP isolates, and this placement was included in Fig. 1. Our analysis also included the three OAK isolates, and these grouped within the MULT clade.
FIG. 1.

Maximum-likelihood phylogeny of 9,288 bp including 25 isolates across five plant host strains of Xylella fastidiosa using a general time-reversible model with a gamma distribution and invariant sites. Bootstrap values with 200 replicates for the major division are reported above each branch, and Bayesian posterior probabilities are reported below. The PLS isolate was not included in the full maximum-likelihood analysis since only 7 of the 10 genes were sequenced; however, its position in the phylogeny based on the maximum-likelihood analysis using those 7 genes is marked (*).
The third clade groups all of the OLS isolates. The branch uniting this OLS group to the PRC clade is fully supported by bootstrap and Bayesian analyses, placing X. fastidiosa subsp. piercei and the group of OLS isolates as sister clades, more closely related to each other than to MULT.
The only isolates failing to group within these major groupings were ALS12 and ALS22. However, examination of all single-gene trees (data not shown) showed that these isolates group within the MULT strain for 7 of 10 trees. Visual inspection and multilocus sequence typing analysis (Scally et al., submitted) of the three remaining genes (holC, cysG, and pilU) indicated that the alleles carried by ALS12 and ALS22 were the result of recombination with other pathovars. This same interpretation applies to the cysG gene in the PD14 isolate. No other signs of recombination were apparent among the sequences, and their single-gene trees showed the same broad patterns as the concatenated tree (Fig. 1).
Geographic variation within plant-host pathovars was not apparent (Table 3). Within the PD clade, the comparison of subpopulations of isolates from northern and central/southern California showed no indication of genetic differentiation (Table 3, test i). There was also no differentiation between northern and southern California subpopulations among the non-PRC ALS isolates (Table 3, test ii). Within the MULT clade subspecies, the ALS clade isolates (isolates 3, 13, and 15) were significantly different (P < 0.05) from the eastern U.S. isolates from peach and oak (Table 3, test iv); however, this effect confounds geography and plant host. Within the East Coast group, there was no significant plant-host differentiation found between the OAK and PP isolates (Table 3, test iii). Finally, this KST approach confirms the highly significant genetic differentiation of the three major clades (Table 3, tests v and vi).
TABLE 3.
Geographic differentiation within and between strains
| Test type and no. | Comparison | KSTa |
|---|---|---|
| Geographical variation within groups | ||
| i | N. California PRC vs S. California PRC | 0.000 |
| ii | N. California ALS vs S. California ALSb | −0.257 |
| Plant host variation within MULT | ||
| iii | OAK vs PP | 0.652 |
| iv | ALSc vs OAK/PP | 0.428* |
| Variation between the major groups | ||
| v | PD vs OLS | 0.923*** |
| vi | PD/OLS vs MULT | 0.542*** |
*, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001.
All non-PRC ALS isolates.
ALS isolates within MULT (see Fig. 1).
We used synonymous-site divergence to estimate the time of origin of each of the clades in the phylogeny (equation 1). For this analysis we used a subset of 11 isolates representing the phylogenetic plant-host groupings (PD, OLS, ALS, OAK, and PP) (Table 1) plus CVC. Note that this subset excluded the isolates ALS12, ALS22, and PD14 implicated in recombination.
The average estimate for the divergence time of the MULT clade from the PRC/OLS clade is 30,000 years, with the PRC and OLS clades separating about 23,000 years ago (Fig. 2). The rate of evolution at synonymous sites in the OLS and PRC clades has been very similar, based on the two estimates of their divergence time (22,400 versus 24,000 years); however, the combined PRC/OLS clade has been evolving at almost three times the rate of the MULT clade based on the estimated divergence times of 43,700 and 16,900 years ago. The estimated split of the ALS (1,800 years), OAK (1,400 years), and PP (700 years) plant-host strains within the MULT clade averages 1,300 years.
FIG. 2.

Maximum-likelihood phylogeny of 12 isolates from six plant hosts of Xylella fastidiosa. The branch lengths are proportional to their estimated substitution rates (nonsynonymous plus synonymous). The divergence times were estimated from the synonymous component of these rates of evolution, so that time is only weakly correlated to branch length. The divergence times reported are the average values for that branch from Tukey's jackknife analysis. The standard errors are reported after the divergence time estimates for those branches that were directly estimated. The estimates shown at the nodes of the two major divisions are averages from the component branches. CVC18 was the South American outgroup.
Despite many thousands of years of separation, we found little sequence divergence within each of the three major clades. The six isolates from three plant hosts within MULT differ at synonymous sites by 0.13%, which is little more than the within-strain percent difference of PRC (0.11%) and OLS (0.05%). The nonsynonymous percent difference within the three clades is also comparable: MULT (0.07%), PRC (0.03%), and OLS (0.02%). Between the three clades, MULT differs at synonymous and nonsynonymous sites from PRC/OLS by 3.21% and 0.75%, respectively (Table 4). The PRC and OLS clades, being more closely related, are 2.60% and 0.40% different at synonymous and nonsynonymous sites. The North American strains differ on average from the South American strain, CVC, by 6.85% at synonymous sites and 1.22% at nonsynonymous sites.
TABLE 4.
Nonsynonymous and synonymous DNA sequence differences between the five plant host strainsa
| Group | % Difference with:
|
||||
|---|---|---|---|---|---|
| PD | OLS | ALS | OAK | PP | |
| PD | 0.42 | 0.71 | 0.69 | 0.70 | |
| OLS | 2.59 | 0.83 | 0.81 | 0.78 | |
| ALS | 3.27 | 3.20 | 0.07 | 0.08 | |
| OAK | 3.25 | 3.18 | 0.16 | 0.05 | |
| PP | 3.22 | 3.14 | 0.12 | 0.11 | |
Nonsynonymous (above the diagonal) and synonymous (below the diagonal) DNA sequence differences, based on the isolates indicated in Table 1.
DISCUSSION
X. fastidiosa is considered a single species (24), but one that is subdivided into distinct groups (8). It has recently been suggested that it can be divided into three subspecies (35), one in South America and two in North America. Our results confirm the view that these are distinct subspecies that are largely clonal, although there is evidence of interstrain recombination (Scally et al., submitted).
We identified three genetically divergent phylogenetic clades, PRC, MULT, and OLS, that differ by 2.6 to 3.3% at synonymous sites (Table 4). We estimate that these differences have taken more than 15,000 years to accumulate. The PRC and MULT clades correspond to the two named North American subspecies, X. fastidiosa subsp. piercei and X. fastidiosa subsp. multiplex, respectively (35). The OLS clade is most closely related to PRC (Fig. 1), but it has a high degree of synonymous-site differentiation from PRC (2.6%) and has very little internal variability (0.05%). In addition, the OLS and PRC strains cannot infect each other's major host plant, oleander and grapevine, respectively (29). For these reasons, placing the old and genetically distinct OLS clade within the X. fastidiosa subsp. piercii subspecies cannot be justified. We suggest that the OLS isolates should be recognized as a new subspecies with the name X. fastidiosa subsp. sandyi; the type specimen Ann-1 (Table 1) is deposited as ATCC 700598 (29), and the genome of this strain has been sequenced (7).
The relatively close relationship of PD and OLS pathovars was first suggested by Chen et al. (10), whose RAPD analysis placed them as genetically similar relative to plum and periwinkle pathovars. However, Hendson et al. (20), also using RAPDs, placed OLS as the outgroup to PD, ALS, PP, and OAK isolates and single-gene trees of 16S rRNA and gyrB (32) recovered different tree topologies for the PD, ALS, and OLS pathovars, but in both cases, ALS was closer to PD. With our larger data set and an appropriate outgroup (CVC), the PRC subspecies, which included all of the PD isolates, was shown to be most closely related to the sandyi clade (SAN), which includes all of the OLS isolates, a relationship with 100% bootstrap support and 1.00 posterior probability on the branch between the two clades. The MULT isolates formed a clade with 100% support outside the PRC/SAN clade.
The PD isolates from grapevines together with a subset of ALS isolates from almonds had previously been placed together based on RAPDs (20, 26), 23S-16S intergenic spacer sequence region (20, 35), and DNA-DNA hybridization (35). Our results agree with these previous findings and support this division into X. fastidiosa subsp. piercei. Initially, it was suggested that all ALS-causing isolates were identical to the PD-causing bacteria, but that these were different from the pathovars causing disease in oaks and peaches (23). PD and the Dixon-like ALS (i.e., like ALS3) have since been distinguished as separate groups based on RAPDs (20), but evidence defining the relationship between these ALS isolates and OAK, PP, and PLS was conflicting. RAPDs supports a division between ALS and PP/PLS (20), while DNA-DNA hybridization supports a single group (35). We confirmed the association between ALS and PP/PLS within the X. fastidiosa subsp. multiplex isolates and added OAK to this group.
Our finding that the major North American X. fastidiosa clades diverged about 16,000 years ago (Fig. 2) means that they predate the introduction of nonnative plants by European colonists. For example, X. fastidiosa subsp. sandyi has only been isolated from oleander, but oleander is a relatively recently introduced plant; yet, the SAN clade has a history of many thousands of years as separated from the sister taxon X. fastidiosa subsp. piercei (PRC clade). There is no evidence that X. fastidiosa occurs in Europe, apart from a single isolation from an infected grapevine in Croatia, which probably represented an invasion from the New World (6). Thus, it appears that the SAN subspecies must have evolved in North America within native plant hosts that have yet to be identified.
Our divergence time estimates raise two important issues. First, how reliable are such estimates. Two of the parameters used to estimate our divergence times, mutation rate and generation time, could be over- or underestimated. If mutation rate is overestimated, this could lead to our estimates being an order of magnitude or more too high. Underestimation of generation time would also lead to our divergence time being too large. The second issue raised is what factors might lead to the almost-threefold difference in the estimated divergence time of the MULT lineage and the PRC/SAN lineage.
Our calculation of divergence times using equation 1 required estimates of generation time and mutation rate for X. fastidiosa. We assumed a generation time of 9 h, i.e., 1,000 generations per year, and a mutation rate of 5.4 × 10−10. Generation times for X. fastidiosa have been estimated at between 9 and 60 h (43), i.e., a range between about 1,000 and 150 generations per year. Our use of the top of this range provides a minimum estimate of divergence times. The mutation rate is based on an estimate from Escherichia coli (16), which has a genome that is twice as large as X. fastidiosa (37). The mutation rate, in general, is inversely proportional to genome size (16), suggesting that our estimate may be low, which would overestimate the divergence times. If the mutation rate of E. coli is extrapolated to X. fastidiosa on the basis of this proportionality, the estimated mutation rate is 1.0 × 10−9, which roughly halves the estimates shown in Fig. 2. However, the three major groups are still estimated to have been distinct for more than 12,000 years, placing the split between MULT and PRC/SAN at 16,000 years ago. Given that the long-term average generation time is very unlikely to be as short as 9 hours, these divergence time estimates are likely to be significantly less than the actual values.
The divergence times and percent differences also indicate that the PRC and SAN clades have had an overall evolutionary rate that is 2.9 times faster than MULT. The effect is long term, since the rate difference is seen both before and after the PRC/SAN split (Fig. 2). This is indicative of a historically shorter generation time in PRC and SAN bacteria than MULT. Alternatively, an increased effective population size in the ancestral MULT strain could have increased the efficiency of selection acting on codon usage, thus slowing the rate of synonymous substitutions (3); however, this effective size difference would have to have been maintained for many thousands of years to account for the observed pattern.
An important characteristic of the X. fastidiosa phylogeny (Fig. 1) is the long basal branches leading to very short terminal branches. Thus, the three major groups show highly significant genetic differentiation (Table 3, tests v and vi), but there is little variation within subspecies. MULT shows the most variability, but much of this variation is explained by host plant and/or geographical differentiation (test iv), so that across the three major clades the plant-host isolates show minimal genetic variation, with synonymous-site variation of less than 0.12%. This lack of variation, despite plenty of time for its accumulation, suggests that the strains experience strong selective pressures from their host plants, eliminating all but the best-adapted clones. The lack of geographical differentiation among the PD strains and among the OLS strains is consistent with this hypothesis. In both cases, strains were sampled from five widely separated sites, and yet the genetic differences among the pathovars was minimal. This sampling strategy is adequate to assess levels of genetic diversity, provided that the culturing technique does not itself select for specific genotypes. We are planning to sample some fresh isolates directly (i.e., without culturing) to eliminate this possibility. The adaptive hypothesis has been further supported by a direct test of plant-host adaptation using isolates that are symptomatic in almonds (2). ALS5 and -11, which are PRC bacteria isolated from almonds, exhibited a slower growth rate and lower survivorship compared to the MULT isolates ALS3 and -22 when tested in almonds.
An alternative explanation for the loss of variability within the plant host strains would be recurrent population bottlenecks. At each bottleneck, much of each clade's genetic variation would be lost. However, given the wide geographical range and the wide range of host plants of this species, this demographic pattern appears unlikely.
Within the MULT clade, there are three groupings. The ALS isolates from California form a separate clade from the eastern U.S. isolates, which form two (nonsignificant) groupings of the OAK isolates and the PP/PLS isolates. However, it is not known if these isolates of MULT from plum, peach, oak, and almond are host specific.
The isolates ALS12 and ALS22 cluster outside of the MULT clade, apparently due to recombination (Scally et al., submitted). It is notable that ALS22 has been found to have different nutritional requirements and differentiation of RAPDs from ALS3 (2, 20). Since ALS22 is found in northern California and ALS12 in southern, this may indicate that the recombinant clade of ALS-causing bacteria is becoming well established as a new strain.
We found little evidence of geographical substructure within the major clades. Within PRC, we found no evidence of geographic differentiation between northern and southern California (Table 4). These results contrast with previous work using RAPDs, where geographic variation was found between PD strains from northern California and central California (2, 20). Within SAN, the single non-Californian isolate (OLS8 from Texas) showed no sign of genetic differentiation. In the non-PRC ALS isolates, we again found that isolates from northern and southern California showed no signs of geographical differentiation (Table 4). There was, however, differentiation within MULT between the western ALS isolates and the eastern OAK and PP isolates, but identifying causation confounds geography and plant hosts. However, even this genetic differentiation is minor (0.14% at synonymous sites) (Table 4) compared to the strain differences of about 3%. ALS is not known to occur in the east and OAK and PP are not known to occur in California, suggesting the biogeography of these closely related forms may involve several intermediates across the United States.
The biogeographic history of the North American strains of X. fastidiosa remains poorly understood. The origins of the PRC, SAN, and MULT clade subspecies predate the human introduction of many of their host plants. Since there has been only one isolation of X. fastidiosa from the Old World (6), these subspecies must have differentiated in hosts native to the New World, hosts that may be largely asymptomatic and that are, at present, unknown.
Acknowledgments
This study was supported by a USDA-CREES grant under the UC Pierce's Disease program to L.N. and R.S.
We gratefully acknowledge Donald Cooksey, Rufina Hernandez-Martinez, Alexander Purcell, and Christina Wistrom for providing the strains used in this study.
REFERENCES
- 1.Albibi, R., J. Chen, O. Lamikanra, D. Banks, R. L. Jarret, and B. J. Smith. 1998. RAPD fingerprinting Xylella fastidiosa Pierce's disease strains isolated from a vineyard in North Florida. FEMS Microbiol. Lett. 165:347-352. [Google Scholar]
- 2.Almeida, R. P. P., and A. H. Purcell. 2003. Biological traits of Xylella fastidiosa strains for grapes and almonds. Appl. Environ. Microbiol. 69:7447-7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagnoli, F., and P. Liò. 1995. Selection, mutation and codon usage in a bacterial model. J. Theor. Biol. 173:271-281. [DOI] [PubMed] [Google Scholar]
- 4.Banks, D., R. Albibi, J. Chen, O. Lamikanra, R. L. Jarret, and B. J. Smith. 1999. Specific detection of Xylella fastidiosa Pierce's disease strains. Curr. Microbiol. 39:85-88. [DOI] [PubMed] [Google Scholar]
- 5.Beltrán, P., G. Delgado, A. Navarro, F. Trujillo, R. K. Selander, and A. Cravioto. 1999. Genetic diversity and population structure of Vibrio cholerae. J. Clin. Microbiol. 37:581-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berisha, B. Y., D. Chen, G. Y. Zhang, B. Y. Xu, and T. A. Chen. 1998. Isolation of Pierce's disease bacteria from grapevine in Europe. Eur. J. Plant Pathol. 104:427-433. [Google Scholar]
- 7.Bhattacharyya, A., et al. 2002. Whole-genome comparative analysis of three phytopathogenic Xylella fastidiosa strains. Proc. Natl. Acad. Sci. USA 99:12403-12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, J., C. J. Chang, R. L. Garret, and N. Gawal. 1992. Genetic variation among Xylella fastidiosa strains. Phytopathology 82:973-977. [Google Scholar]
- 9.Chen, J., O. Lamikanra, C. J. Chang, and D. L. Hopkins. 2000. 16S rDNA analysis of Xylella fastidiosa strains. Syst. Appl. Microbiol. 23:349-354. [DOI] [PubMed] [Google Scholar]
- 10.Chen, J., O. Lamikanra, C. J. Chang, and D. L. Hopkins. 1995. Randomly amplified polymorphic DNA analysis of Xylella fastidiosa Pierce's disease and oak leaf scorch pathotypes. Appl. Environ. Microbiol. 61:1688-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohan, F. M. 2002. What are bacterial species? Annu. Rev. Microbiol. 56:457-487. [DOI] [PubMed] [Google Scholar]
- 12.Coletta-Filho, H. D., and M. A. Machado. 2002. Evaluation of the genetic structure of Xylella fastidiosa populations from different Citrus sinensis varieties. Appl. Environ. Microbiol. 68:3731-3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costa, H. S., E. Raetz, T. R. Pinckard, C. Gispert, R. Hernandez-Martinez, C. K. Dumenyo, and D. A. Cooksey. 2004. Plant hosts of Xylella fastidiosa in and near southern California vineyards. Plant Dis. 88:1255-1261. [DOI] [PubMed]
- 14.DaCosta, P. I., C. F. Franco, V. S. Miranda, D. C. Teixeira, and J. S. Hartung. 2000. Strains of Xylella fastidiosa rapidly distinguished by arbitrarily primed-PCR. Curr. Microbiol. 40:279-282. [DOI] [PubMed] [Google Scholar]
- 15.Davis, M. J., W. J. French, and N. W. Schaad. 1981. Axenic culture of the bacteria associated with phony peach disease and plum leaf scald. Curr. Microbiol. 6:309-314. [Google Scholar]
- 16.Drake, J. W., B. Charlesworth, D. Charlesworth, and J. F. Crow. 1998. Rates of spontaneous mutation. Genetics 148:1667-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Escobar-Páramo, P., C. Guidicelli, C. Parsot, and E. Denamur. 2003. The evolutionary history of Shigella and enteroinvasive Escherichia coli revised. J. Mol. Evol. 57:140-148. [DOI] [PubMed] [Google Scholar]
- 18.Filatov, D. A. 2002. ProSeq: a software for preparation and evolutionary analysis of DNA sequence data sets. Mol. Ecol. Notes 2:621-624. [Google Scholar]
- 19.Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95-98. [Google Scholar]
- 20.Hendson, M., A. H. Purcell, D. Chen, C. Smart, M. Guilhabert, and B. Kirkpatrick. 2001. Genetic diversity of Pierce's disease strains and other pathotypes of Xylella fastidiosa. Appl. Environ. Microbiol. 67:895-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudson, R. R., D. D. Boos, and N. L. Kaplan. 1992. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9:138-151. [DOI] [PubMed] [Google Scholar]
- 22.Huelsenbeck, J. P., and F. Ronquist. 2001. MRBAYES: Bayesian inference of phylogeny. Bioinformatics 17:754-755. [DOI] [PubMed] [Google Scholar]
- 23.Hopkins, D. L. 1989. Xylella fastidiosa: xylem-limited bacterial pathogen of plants. Annu. Rev. Phytopathol. 27:271-290. [Google Scholar]
- 24.Kamper, S. M., W. J. French, and S. R. deKloet. 1985. Genetic relationships of some fastidious xylem-limited bacteria. Int. J. Sys. Bacteriol. 35:185-188. [Google Scholar]
- 25.Mehta, A., and Y. B. Rosato. 2001. Phylogenetic relationships of Xylella fastidiosa strains from different hosts, based on 16S rDNA and 16S-23S intergenic spacer sequences. Int. J. Syst. Evol. Microbiol. 51:311-318. [DOI] [PubMed] [Google Scholar]
- 26.Pooler, M. R., and J. S. Hartung. 1995. Genetic relationships among strains of Xylella fastidiosa from RAPD-PCR data. Curr. Microbiol. 31:134-137. [DOI] [PubMed] [Google Scholar]
- 27.Prager, R., A. Liesegang, W. Voigt, W. Rabsch, A. Fruth, and H. Tschäpe. 2002. Clonal diversity of Shiga toxin-producing Escherichia coli O103:H2/H− in Germany. Infect. Genet. Evol. 1:265-275. [DOI] [PubMed] [Google Scholar]
- 28.Purcell, A. H., and D. L. Hopkins. 1996. Fastidious xylem-limited bacterial pathogens. Annu. Rev. Phytopathol. 34:131-151. [DOI] [PubMed] [Google Scholar]
- 29.Purcell, A. H., S. R. Saunders, M. Hendson, M. E. Grebus, and M. J. Henry. 1999. Causal role of Xylella fastidiosa in oleander leaf scorch. Phytopathology 89:53-58. [DOI] [PubMed] [Google Scholar]
- 30.Restrepo, S., M. Duque, J. Tohme, and V. Verdier. 1999. AFLP fingerprinting: an efficient technique for detecting genetic variation of Xanthomonas axonopodis pv. manihotis. Microbiology 145:107-114. [DOI] [PubMed] [Google Scholar]
- 31.Roberts, P. D., N. C. Hodge, H. Bouzar, J. B. Jones, R. E. Stall, R. D. Berger, and A. R. Chase. 1998. Relatedness of strain of Xanthomonas fragariae by restriction fragment length polymorphism, DNA-DNA reassociation, and fatty acid analyses. Appl. Environ. Microbiol. 64:3961-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodrigues, J. L. M., M. E. Silva-Stenico, J. E. Gomes, J. R. S. Lopes, and S. M. Tsai. 2003. Detection and diversity assessment of Xylella fastidiosa in field-collected plant and insect samples by using 16S rRNA and gyrB sequences. Appl. Environ. Microbiol. 69:4249-4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozas, J., and R. Rozas. 1999. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15:174-175. [DOI] [PubMed] [Google Scholar]
- 34.Rychlik, W. 2002. OLIGO: primer analysis software, version 6.65. Molecular Biology Insights Inc., Cascade, Colo.
- 35.Schaad, N. W., E. Pastnikova, G. Lacey, M. Fatmi, and C. J. Chang. 2004. Xylella fastidiosa supspecies: X. fastidiosa subsp. piercei, subsp. nov., X. fastidiosa subsp. multiplex subsp. nov., and X. fastidiosa subsp. pauca subsp. nov. Syst. Appl. Microbiol. 27:290-300. [DOI] [PubMed] [Google Scholar]
- 36.Sikorski, J., M. Möhle, and W. Wackernagel. 2002. Identification of complex composition, strong strain diversity and directional selection in local Pseudomonas stutzeri populations from marine sediment and soils. Environ. Microbiol. 4:465-476. [DOI] [PubMed] [Google Scholar]
- 37.Simpson, A. J., et al. 2000. The genome sequence of the plant pathogen Xylella fastidiosa. Nature 406:151-157. [DOI] [PubMed] [Google Scholar]
- 38.Sokal, R. R., and F. J. Rohlf. 1995. Biometry, 3rd ed. W.H. Freeman and Company, New York, N.Y.
- 39.Swofford, D. L. 2002. PAUP*. Phylogenetic analysis using parsimony (* and other methods), version 4. Sinauer Associates, Sunderland, Mass.
- 40.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 24:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Sluys, M. A., et al. 2003. Comparative analyses of the complete genome sequences of Pierce's disease and citrus variegated chlorosis strains of Xylella fastidiosa. J. Bacteriol. 185:1018-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh, P. S., D. A. Metzger, and R. Higuchi. 1991. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. BioTechniques 10:506-513. [PubMed] [Google Scholar]
- 43.Wells, J. M., B. C. Raju, H. Y. Hung, W. G. Weisburg, L. Mandelco-Paul, and D. J. Brenner. 1987. Xylella fastidiosa gen. nov., sp. nov.: gram-negative, xylem-limited, fastidious plant bacteria related to Xanthomonas spp. Int. J. Syst. Bacteriol. 37:136-143. [Google Scholar]
- 44.Yang, Z. 1998. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15:568-573. [DOI] [PubMed] [Google Scholar]