

Abstract
Using a combination of various enrichment techniques, the strictly anaerobic, gram-positive, endospore-forming bacterium Sedimentibacter hongkongensis strain KI as revealed by 16S rRNA analysis and the gram-negative enterobacterium Citrobacter amalonaticus strain G as revealed by physiological tests were isolated from an anaerobic cyanophycin (CGP)-degrading bacterial consortium. S. hongkongensis strain KI is the first anaerobic bacterium with the ability to hydrolyze CGP to β-Asp-Arg and β-Asp-Lys dipeptides, as revealed by electrospray ionization-mass spectrometry and reversed-phase high-performance liquid chromatography analysis. However, these primary accumulated hydrolysis products were only partially used by S. hongkongensis strain KI, and significant growth on CGP did not occur. On the other hand, C. amalonaticus strain G did not degrade CGP but grew on the β-linked iso-dipeptides formed in vitro by enzymatic CGP degradation or in vivo by metabolic activity of S. hongkongensis strain KI. Dipeptide utilization occurred at the highest rate if both strains were used in cocultivation experiments with CGP, indicating that cooperation between different bacteria occurs in anaerobic natural environments for complete CGP turnover. The amino acids obtained from the cleavage of dipeptides were fermented to ethanol, acetic acid, and succinic acid, as revealed by gas chromatographic analysis and by spectrophotometric enzyme assays.
Cyanophycin (cyanophycin granule polypeptide) (CGP) is a branched, nonribosomally synthesized natural occurring polyamide consisting of a polymer backbone of α-linked aspartic acid residues [poly(α-aspartic acid)] along with arginine residues covalently linked to the β-carboxyl groups via their α-amino groups (47; for reviews see references 34 and 35). CGP was discovered in 1887 during microscopic studies (6), but its structure and physicochemical properties were described about 100 years later (43, 44, 46, 47). CGP is water insoluble under physiological conditions and occurs in membrane-less granules in the cytoplasm of most cyanobacteria (2, 3, 4, 25, 26, 27, 43, 44, 45, 53). Only recently have bacteria that do not belong to the cyanobacteria (like Acinetobacter calcoaceticus strain DSM 587 or Desulfitobacterium hafniense) and that also possesses CGP biosynthesis genes been discovered, and at least some of these bacteria actually synthesize CGP (23, 55). Biosynthesis of CGP is now well understood, and the cyanophycin synthetase genes (cphA) of many cyanobacteria have been cloned and the corresponding synthetases (CphA) have been characterized biochemically (1, 5, 16, 54). Only recently has a cyanophycin synthetase of a noncyanobacterial strain (A. calcoaceticus) been purified, and kinetic parameters like Km values for the amino acid constituents and for ATP and characteristics of binding of CphA to CGP have been described (22).
The intracellular degradation of the transiently accumulated storage polymer CGP is catalyzed by cyanophycinases (CGPases) (CphB) and proceeds via an α-cleavage mechanism that results in the formation of β-Asp-Arg dipeptides (15, 39). These enzymes are highly specific for hydrolysis of CGP and do not degrade other polypeptide substrates (39). Furthermore, CGP proved to be highly resistant to many proteases and arginase (45, 47). Therefore, CGPases probably evolved as specialized enzymes for the purpose of degrading CGP under certain environmental conditions (30, 32, 39).
CGP is a widespread biopolymer that represents a valuable source of nitrogen, carbon, and energy and probably occurs in many habitats (see the spectrum of CGP-producing organisms mentioned above). Therefore, it is likely that CGP is released into the environment quite frequently from biomass. As a consequence of this, a variety of different bacteria that possess extracellular enzymes which are specialized to hydrolyze CGP are expected to occur in the habitats. An extracellular CGPase exhibiting an α-cleavage mechanism for CGP degradation like intracellular CGPases was detected for the first time by Obst et al. (32), who isolated from pond sediment the gram-negative aerobic bacterium Pseudomonas anguilliseptica BI, which was able to use CGP as a sole carbon source for growth and degraded CGP completely to β-Asp-Arg dipeptides. Later, a similar enzyme was purified from a culture supernatant of the gram-positive organism Bacillus megaterium strain BAC19, which was isolated from soil and also had the ability to degrade CGP completely to small molecules via an α-cleavage mechanism. In addition to β-Asp-Arg dipeptides this enzyme also formed (β-Asp-Arg)2 tetrapeptides as primary degradation products (31). Furthermore, the gene coding for the CGPase of P. anguilliseptica strain BI (cphEPa) was identified and characterized at the nucleotide sequence level (32). By comparing the amino acid sequences of CphEPa and cyanobacterial CGPases (CphA), a conserved catalytic center with a catalytic triad consisting of a histidine, an aspartic acid (or a glutamic acid), and a serine residue as a nucleophile in a lipase box motif (Gly-Xaa-Ser-Xaa-Gly) was found. Analysis of the N terminus of CphEPa revealed the presence of a leader peptide (32), which confirmed the assumption that the extracellular enzyme is probably secreted via a type II secretion mechanism (36). Radioactive labeling experiments employing l-[U-14C]arginine showed that degradation of CGP by CphEPa proceeds via an exomechanism that results in continuous release of β-Asp-Arg dipeptides from the C-terminal region of CGP, to which CphEPa remains attached during degradation (32).
The extracellular degradation of CGP by aerobic gram-negative and gram-positive bacteria is now well understood (for a review see reference 30). However, CGP also occurs in anaerobic habitats and may also be formed as a storage compound by anaerobic bacteria which harbor CGP biosynthesis genes, like Clostridium botulinum ATCC 3502 and D. hafniense (23, 55). In these bacteria, active enzymes which encode CGPases of the CphB type must be present for mobilization of intracellularly accumulated CGP (23). In addition, the occurrence of bacteria that synthesize extracellular CGPases (CphE) can be expected in anaerobic habitats. The aims of this study were to demonstrate the occurrence of anaerobic CGP-degrading bacteria and to characterize the degradation products in cultures of such bacteria.
MATERIALS AND METHODS
Bacterial strains, preparation of media, and growth of bacteria.
Anaerobic CGP-degrading enrichment culture AK15 was obtained by direct application of an environmental sample taken from the sediment of a pond located close to Borkenwirthe (Germany) to low-salt Pelobacter liquid medium (33) containing 0.2% (wt/vol) CGP and subsequent incubation at 30°C. Subsequently, two bacterial strains, Sedimentibacter hongkongensis strain KI and Citrobacter amalonaticus strain G, were isolated from this enrichment culture and were deposited in the culture collection of the Institute for Molecular Microbiology and Biotechnology (Münster, Germany). These strains and Clostridium hydroxybenzoicum DSMZ 7310, which was used as a closely related anaerobic reference strain, were grown in low-salt Pelobacter liquid medium during CGP degradation experiments. The following other media were employed for isolation and cultivation experiments: standard 1 complex medium (Merck, Darmstadt, Germany), glucose yeast extract agar (DSMZ medium 54), M9 mineral medium (40), mineral medium B (9), and mineral salt medium (42). During isolation experiments on agar-solidified media, gas atmospheres having different compositions were added to the 3.5-liter anaerobic jars (Oxoid, Wesel, Germany) employed for cultivation. The concentrations of the gas constituents of the artificial atmospheres used are indicated below. For most cultivations of S. hongkongensis strain KI and C. hydroxybenzoicum DSMZ 7310, which was used as a reference strain on agar plates, C. hydroxybenzoicum complex medium (DSMZ medium 643) was used. The concentrations of CGP and of other carbon sources added to the media are indicated below. C. amalonaticus strain G was cultivated on Luria-Bertani (LB) medium (40) for strain maintenance. All isolates were grown at 30°C for CGP degradation experiments in liquid media. Cultures of C. amalonaticus were routinely incubated at 37°C. The increase in the optical density in Hungate tubes as a function of growth of C. amalonaticus was recorded with a Klett-Summerson photometer.
Production and isolation of cyanophycin.
To obtain CGP as a carbon source, a recombinant Escherichia coli DH1 strain harboring plasmid pMa/c5-914::cphA (cphA from Synechocystis sp. strain PCC6803) with an inducible temperature-sensitive promoter was employed, and cells were cultivated at 37°C in terrific broth complex medium as described previously (14). After CGP-containing recombinant E. coli DH1 cells were harvested, the polymer was isolated and purified by the acid extraction method (14). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis with subsequent Coomassie blue staining (51) and high-performance liquid chromatography (HPLC) analysis after acid hydrolysis of 1- to 2-mg aliquots of CGP samples (see below) were employed to analyze the purity of the isolated CGP and its amino acid composition. The CGP employed had a comparably low degree of dispersity (range, 25 to 40 kDa), and the CGP molecules had an average molecular mass of about 30 kDa.
Enrichment of anaerobic CGP-degrading microorganisms.
Samples from the AK15 enrichment culture were spread on solid basic inorganic medium B (9) supplemented with trace element solution SL 7 (52) and overlaid with 0.5% (wt/vol) agar containing 0.2% (wt/vol) CGP as described previously (32). Due to the CGP these overlay agar plates were turbid. CGP-degrading bacteria or CGPase activities were detected by the presence of degradation halos. Staining of CGP-containing agar plates with Serva Blue R was performed as described previously for the detection of proteins in sodium dodecyl sulfate-polyacrylamide gels (51). To avoid staining that was too strong, the staining solution was applied to CGP overlay agar plates for only a short time (2 to 4 min) and was subsequently replaced with a destaining solution. For preparation of CGP-containing Pelobacter liquid medium, diethyl ether-sterilized CGP was first dissolved in 0.1 N HCl and then injected into sterile, butyl rubber-sealed Hungate tubes containing 10 ml of Pelobacter liquid medium. For readjustment of the initial pH of the medium (pH 7.4) and for complete CGP precipitation, an equal volume of 0.1 N NaOH was added. Dilutions of the AK15 mixed culture were also prepared by using butyl rubber-sealed Hungate tubes containing 10 ml Pelobacter liquid medium. For enrichment of endospore-forming bacteria, Hungate tubes were pasteurized by incubation at 80°C for 30 min.
Isolation, manipulation, and analysis of DNA.
Total genomic DNA of the enriched anaerobic mixed culture and of S. hongkongensis strain KI was isolated by the method of Rao et al. (38). The 16S rRNA genes were amplified from total DNA using oligonucleotide primers as described previously (37). After purification of PCR products with a NucleoTrapCR kit (Macherey-Nagel, Düren, Germany), the amplified 16S rRNA genes were cloned into plasmid pGEM-T Easy (Promega, Madison, WI). The nucleotide sequences were determined as described below. The 16S rRNA gene sequence of S. hongkongensis obtained was aligned with previously published sequences of representative clostridia and other bacteria available from the National Center for Biotechnology Information database.
Competent cells of E. coli TOP10 (Invitrogen, San Diego, CA) were prepared by the method of Hanahan (17) and were used as recipients of plasmid DNA. For identification of E. coli clones harboring hybrid plasmids, cells were grown at 37°C in LB medium containing 75 μg/ml ampicillin and 0.004% (wt/vol) 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). Culture supernatants and cell pellets were obtained by centrifugation (15 min, 2,800 × g, 4°C).
For DNA sequencing by the “primer-hopping strategy” (49), 5′-IRD 800-labeled synthetic oligonucleotides (MWG-Biotech, Ebersberg, Germany) were used as primers, and a SequiTherm EXCEL TM II long-read cycle sequencing kit was employed (Epicentre Technologies, WI). The analysis was performed with 6% (wt/vol) acrylamide gels using Sequagel XR (acrylamide/urea), Complete (buffer reagent) solutions (National Diagnostics, Sommerville, NJ), and buffer containing 89 mM Tris, 89 mM boric acid, and 2 mM EDTA with a LI-COR 4000L automatic sequencing apparatus (MWG-Biotech, Ebersberg, Germany). Nucleic acid sequence data were analyzed with the CAP (Contig Assembly Program) sequence analysis software (21), and Genamics Expression 1.1, ClustalX 1.8 (50), was used for sequence alignment and construction of a phylogenetic tree.
Taxonomic determination with physiological tests.
Motility, Gram behavior, oxidase (Bactident oxidase test strips from Merck, Darmstadt, Germany), and catalase tests were performed according to standard protocols. Further physiological determinations were done by using an API 20E test kit according to the instructions of the manufacturer (BioMérieux, Marcy-l'Etoile, France).
Detection of CGP degradation products.
Degradation products were generally detected in cell-free supernatants obtained by centrifugation, which were then filtered through polyethersulfone filters with a pore size of 0.2 μm. Reversed-phase HPLC was used to determine the products of enzymatic CGP degradation and to create standard calibration curves for quantitative determination of amino acid constituents (aspartic acid, arginine, and lysine) of the CGP fractions employed (see above) and for quantification of β-Asp-Arg dipeptides. The procedure was performed as described by Aboulmagd et al. (1) and was based on the fluorescence detection of derivatives of amino groups containing substances after precolumn derivatization with the o-phthaldialdehyde reagent.
Electrospray ionization -mass spectrometry-mass spectrometry (ESI-MS-MS) was used for identification of primary CGP degradation products by mass determination and structural analysis (10). All measurements were obtained with a Quattro LCZ system (Micromass, Manchester, United Kingdom) with a nanospray inlet.
Gas chromatography (GC) was used for identification of fermentation products produced during anaerobic degradation of primary accumulated CGP degradation products (iso-dipeptides). A GC-9A chromatograph (Shimadzu, Kyoto, Japan), which was equipped with a Porapak QS-packed column (length, 2 m; diameter, 2 mm), was used, and nitrogen was used as the carrier gas at a flow rate of 50 ml/min. A flame ionization detector was used for quantitative measurement of small polar molecules like short-chain fatty acids and short-chain fatty alcohols.
Succinic acid was determined spectrophotometrically by an enzyme assay by coupling the reactions of succinyl coenzyme A (succinyl-CoA) synthetase (EC 6.2.1.4), pyruvate kinase (EC 2.7.1.40), and lactate dehydrogenase (EC 1.1.1.27) as described by the manufacturer (R-Biopharm, Darmstadt, Germany).
All degradation experiments were performed at least in duplicate, and catabolic products were measured in duplicate or triplicate. The maximum deviations of single values from average values for a set of experiments were less than 5%.
Nucleotide sequence accession number.
The 16S rRNA gene sequence data for S. hongkongensis strain KI have been deposited in the National Center for Biotechnology Information database under accession number AY571338.
RESULTS
Isolation of an anaerobic CGP-degrading mixed culture.
Culture AK15 was originally isolated from a pond sediment using Pelobacter liquid medium which contained CGP as the sole carbon source. This stable mixed culture was transferred several times to fresh CGP-containing medium, and it retained the ability to degrade CGP for months. Two bacterial strains were isolated from the anaerobic consortium in this mixed culture and were identified as the strictly anaerobic organism S. hongkongensis strain KI and the facultative anaerobic organism C. amalonaticus strain G (Fig. 1). These strains were finally obtained from AK15 by sequential use of different enrichment strategies (see below). Degradation of CGP was detected simply by observing the disappearance of sedimented water-insoluble CGP at the bottom of the Hungate tubes employed for anaerobic cultivation (Fig. 2). Mixed culture AK15, as well as an axenic culture of S. hongkongensis strain KI, remained viable and stable after they were frozen in the presence of 20% (wt/vol) glycerol.
FIG. 1.

Halo formation on CGP overlay agar plates caused by extracellular enzymes of S. hongkongensis strain KI and cell morphology of strain KI and C. amalonaticus strain G. After 14 days of incubation of S. hongkongensis strain KI on CGP-containing agar plates under a nitrogen atmosphere, a faint diffuse clear zone occurred around the inoculation spot (spot 1 in panel A); the spot became much clearer and appeared to be much more extended after staining of the remaining CGP with Serva Blue R (spot 1 in panel B). C. hydroxybenzoicum DSMZ 7310 was used as a reference strain and did not degrade CGP (spot 2 in panels A and B [inoculation spots are indicated by arrows]). C. hydroxybenzoicum medium containing 0.2% (wt/vol) CGP in the upper layer was used as the basic medium for cultivation experiments on agar plates. The light micrograph in panel C shows pairs of curved vegetative cells of S. hongkongensis strain KI which grew at very low cell densities in CGP-containing liquid medium. (D) One cell of strain KI exhibiting a typical terminal endospore. (E) Light micrograph of the straight, rod-shaped cells of C. amalonaticus strain G grown on solid LB medium.
FIG. 2.

Anaerobic degradation of CGP by S. hongkongensis strain KI and anaerobic growth of C. amalonaticus G on β-Asp-Arg dipeptides. After 10 days of incubation, S. hongkongensis strain KI showed almost complete degradation of CGP in liquid Pelobacter medium containing 0.2% (wt/vol) CGP (right Hungate tubes in panel A before shaking and in panel B after shaking). The controls (left tubes in panels A and B) contained only CGP (white sediment or suspension) and were kept sterile. (C) Growth of C. amalonaticus strain G on solid M9 medium containing 0.1% (wt/vol) dipeptides which were produced by enzymatic digestion of CGP employing the extracellular CGPase of P. anguilliseptica BI (CphEPa) (32). (D) Growth of C. amalonaticus strain G on liquid Pelobacter medium containing 0.2% (wt/vol) dipeptides.
Isolation and taxonomic affiliation of isolate G.
The first attempts to isolate CGP-degrading microorganisms from mixed culture AK15 using simple purification strategies, like plating of small sample volumes on agar plates, failed. Probably because of the rapid growth of other unidentified bacteria on CGP degradation products during these isolation experiments, it was not possible to detect CGP-degrading, halo-forming strains or to differentiate between degrading and nondegrading bacterial colonies. In addition, no useful strain discrimination based on differences in utilization of substrates like aspartic acid, arginine, glucose, or intermediates of the tricarboxylic acid cycle or based on morphological characteristics on the different complex and mineral media employed (see above) was possible. CGP-degrading organisms were detected neither on standard 1 medium or glucose yeast extract agar nor on M9 or mineral salt medium; none of the separate colonies occurring on the agar plates employed was able to degrade CGP after transfer onto CGP overlay agar plates or to CGP-containing liquid medium. Furthermore, supplementation of the medium with vitamins, different trace element solutions, and yeast extract or variations in the composition of the atmosphere used (5% [vol/vol] H2 and 95% [vol/vol] N2; 76% [vol/vol] N2, 4% [vol/vol] H2, and 20% [vol/vol] CO2; 100% N2; 82% [vol/vol] argon and 18% [vol/vol] CO2 [vol/vol]) did not result in significant differences between the isolated bacteria (data not shown). Only when mineral medium B was used in combination with a mixture of arginine and aspartate (each amino acid at a concentration of 0.1% [wt/wt]) did colonies exhibiting certain differences in size appear on the agar plates after 10 days. One bacterial strain (isolate G) formed comparatively large colonies; however, it did not grow on CGP agar plates or degrade CGP in Pelobacter liquid medium. This isolate was determined to be a gram-negative, facultatively anaerobic, motile, oxidase-negative, catalase-positive, rod-shaped bacterium that occurred as single cells or pairs of cells that were about 1.5 to 5 μm long and 1 μm in diameter (Fig. 1E). The strain had a temperature optimum of 37°C on LB agar plates. The API 20E test system identified this isolate as a strain of C. amalonaticus; it exhibited an excellent identification profile with reactions typical only of this bacterium. Typical test results for the genus Citrobacter or results related to amino acid or anaerobic metabolism are as follows: positive arginine dihydrolase, positive ornithine decarboxylase, and positive citrate utilization assays and acid production from glucose (fermentation-oxidation). In addition, lysine decarboxylase and urease assays were negative, and no H2S or acetoin (Voges-Proskauer test) were formed. The strain was cytochrome oxidase negative and also negative for tryptophan deiminase, gelatinase, and acid formation (fermentation-oxidation) from inositol, sucrose, and melibiose.
The cell size (see above), which was close to the typical dimensions described for C. amalonaticus (diameter, 1 μm; length, 2 to 6 μm), motility, temperature optimum, and results of physiological tests indicated that strain G was a C. amalonaticus strain (compare the description of the genus Citrobacter in reference 20). Later, it was shown that this bacterium utilized dipeptides released from CGP by another bacterium (see below).
Isolation of CGP-degrading isolate KI.
To isolate the primary CGP-degrading bacterium, the original AK15 mixed culture was diluted stepwise to eliminate contaminants. A 107-fold dilution of AK15 was the most diluted culture with the ability to utilize CGP for growth. Plating of this enriched culture onto different solid media (see above) still revealed the presence of C. amalonaticus strain G, which was the only visually detectable organism present in the culture broth. To obtain more information about the “invisible” primary CGP-degrading microorganism(s), total DNA of the enriched culture was isolated, and 16S rRNA gene sequencing was employed to gain insight into the species composition. PCR primers with different specificities were employed. Whereas archaeon-specific primers (ArcF and a reverse primer [7]) produced only nonspecific PCR amplification products, indicating the absence of archaea in the degrading consortium, standard 16S rRNA gene primers yielded a mixture of PCR amplification products. These products were cloned, and nucleotide sequence data were obtained for several recombinant E. coli TOP10 16S rRNA gene clones. Of the 12 clones sequenced, 6 were clones of bacterial strains belonging to the genus Sedimentibacter (formerly Clostridium). Therefore, the partially enriched CGP-degrading mixed culture was pasteurized, and an aliquot was used to inoculate fresh CGP-containing liquid medium. After 10 days of incubation at 30°C, the CGP at the bottom of the tube had disappeared, indicating that CGP was degraded by germinated cells. In contrast to previous experiments, in which mixed cultures were used, the medium was not brownish but remained colorless. Although CGP disappeared visibly in the Hungate tubes, no significant increase in the turbidity of the medium was observed, indicating that growth of the cells was poor (Fig. 2). For further cultivation, bacteria enriched by heat treatment were cultivated on C. hydroxybenzoicum agar plates, because C. hydroxybenzoicum (synonym, Sedimentibacter hydroxybenzoicus) was the most closely related species (Fig. 3) for which a defined medium had been described. After aliquots of the pasteurized mixed culture were spread on C. hydroxybenzoicum agar plates, small rigid colonies appeared after about 1 week of incubation at 30°C. These colonies were transferred onto fresh agar plates, and one isolate representing the first anaerobic pure culture with the ability to slowly degrade CGP was obtained. Although CGP degradation by the AK15 mixed culture was faster than CGP degradation by a pure culture of isolate KI, this bacterium was capable of hydrolyzing CGP completely and retained this ability even after it was transferred several times to fresh medium.
FIG. 3.

16S rRNA gene dendrogram showing the phylogenetic position of the strictly anaerobic CGP-degrading bacterium S. hongkongensis strain KI. S. hongkongensis strain KI (enclosed in a box), the most closely related strains of the Clostridiales (including members of clostridial clusters I, XI, and XII (nomenclature of Collins et al. [11]), and some members of the Bacillales were included in the analysis The two CGP-degrading bacteria (underlined), gram-positive B. megaterium strain BAC19 and gram-negative P. anguilliseptica strain BI described previously (31, 32), were also included, demonstrating the wide distribution of CGP degradation genes among bacteria. Nucleotide, partial genome, and whole genome accession numbers are indicated in parentheses. The phylogenetic tree was constructed using ClustalX 1.8. Bar = 0.1 nucleotide change per site.
The isolated strain was transferred onto CGP overlay agar plates prepared with C. hydroxybenzoicum medium. In contrast to all aerobic CGP-degrading bacteria described previously, which formed degradation halos (31, 32) with clear borders, the newly isolated strain KI produced only very faint, translucent, diffuse degradation halos which became visible only after about 14 days of incubation (Fig. 1A, spot 1), indicating that the enzyme activity was low. After staining of the plate with Serva Blue R, the degradation halo appeared to be much more extended (Fig. 1B, spot 1), demonstrating that there was partial degradation of CGP in areas at some distance from the colonies as a result of diffusion of the extracellular enzyme and comparatively weak enzyme activity. The reference strain of C. hydroxybenzoicum did not degrade CGP (Fig. 1A and B, spot 2). Furthermore, no formation of degradation halos was observed if small volumes of sterile filtered culture supernatants were applied to CGP overlay agar plates in CGPase activity tests (data not shown) (31, 32), again indicating that the activity of the CGP-degrading enzyme of strain KI was low or the CGPase was removed by filtration.
Taxonomic affiliation of isolate KI.
Total genomic DNA of purified strictly anaerobic isolate KI was isolated from cells grown on C. hydroxybenzoicum agar plates, and the complete 16S rRNA gene sequence was determined. This sequence was used for identification of the most closely related bacterial strains in a database search to determine the taxonomic position of isolate KI, as shown in the phylogenetic tree in Fig. 3. Only the 16S rRNA gene sequence of S. hongkongensis HKU2 had a high level of identical nucleotides (99% over 1,475 bp). Therefore, isolate KI is referred to here as S. hongkongensis strain KI. The most closely related other species belonging to the genus Sedimentibacter, like Sedimentibacter saalensis, S. hydroxybenzoicus (formerly C. hydroxybenzoicum according to Breitenstein et al. [8]), and Sedimentibacter sp. strain BRS22, exhibited levels of sequence identity to isolate KI between 94 and 95%. Much lower levels of homology to bacilli (89 to 90%) (Fig. 3, right branch) were found. Other related bacteria were Clostridiaceae belonging to clostridial cluster XI (for example, Clostridium felsineum DSM 794) and clostridial cluster XII (e.g., Clostridium purinolyticum ATCC 33906 and Clostridium acidurici ATCC 7906) according to the taxonomic classification proposed by Collins et al. (11). The phylogenetic tree also demonstrates that strain KI exhibits a lower level of relatedness to clostridia belonging to cluster I, like C. botulinum type E Iwanai, Clostridium butyricum ATCC 43755, Clostridium puniceum DSM 2619, and Clostridium beijerinckii NCIMB9362.
Primary CGP degradation products and formation of dipeptides by axenic cultures of S. hongkongensis strain KI.
The primary CGP degradation products occurring during cultivation of S. hongkongensis strain KI in CGP liquid medium were identified by ESI-MS-MS as β-Asp-Arg and β-Asp-Lys dipeptides (Fig. 4). This analysis did not detect higher-order dipeptide oligomers [(β-Asp-Arg)n] like (β-Asp-Arg)2 tetrapeptides, which were observed during previous CGP degradation studies with CphEBm of Bacillus megaterium strain BAC19 (31), or free amino acids in the degraded CGP samples. The β-Asp-Lys dipeptide was identified for the first time in a CGP degradation experiment in which mass spectrometric analysis was used. This was due to the use of CGP produced in recombinant E. coli strains in which about 6 mol% of the arginine side chains were replaced by lysine. The fragmentation patterns of the β-Asp-Arg and β-Asp-Lys dipeptides were in good agreement with the structures of both molecules and confirmed their identity (Fig. 4). For example, fragmentation of the dipeptides at the β-amide bond during analysis was indicated by the peaks at m/z 147 ([lysine+H]+) in the case of β-Asp-Lys and at m/z 175 ([arginine+H]+) in the case of β-Asp-Arg (Fig. 4, peaks IIIa and IIIb). The ESI-MS-MS results clearly showed that S. hongkongensis strain KI degrades CGP via an α-cleavage mechanism.
FIG. 4.

Identification of β-Asp-Arg (Mr, 289) and β-Asp-Lys (Mr, 261) dipeptides from a culture supernatant of S. hongkongensis KI grown on CGP by ESI-MS-MS. The structural analysis of ions at m/z 290 and m/z 262 of a positive ion spectrum revealed a characteristic fragmentation pattern and confirmed the presence of β-Asp-Lys ([M1+H]+) (panel Α, peak Ia) and β-Asp-Arg ([M2+H]+) (panel B, peak Ib), respectively. Peak IIa represents the β-Asp-Lys dipeptide after the loss of NH3, peak IIIa at m/z 147 ([lysine+H]+) indicates fragmentation of β-Asp-Lys at the β-amide bond, and peak IVa at m/z 130 represents the same ion after the loss of NH3 ([lysine+H-17]+). In the spectrum in panel B a peak at m/z 175 ([arginine+H]+) (peak IIIb) was found. Peak IIb represents the β-Asp-Arg dipeptide after the loss of NH3, peak IVb at m/z 158 corresponds to the arginine ion after the loss of NH3 [arginine+H−17]+, and peak Vb represents the residual part of the aspartic acid molecule after fragmentation of the β-Asp-Arg dipeptide at the β-amide bond.
The concentration of dipeptides in the cultures was determined by reversed-phase HPLC after precolumn derivatization with the o-phthaldialdehyde reagent, a method used mostly for identification and quantification of free amino acids. HPLC calibration curves (data not shown) were obtained with dipeptides produced by total hydrolysis of CGP employing purified extracellular CGPase of P. anguilliseptica BI (CphEPa) isolated from recombinant E. coli (32).
The degradation of CGP in the medium (initial concentration, about 2 g/liter) by cells of S. hongkongensis strain KI correlated with the formation of up to 1.5 g/liter of dipeptides after cultivation for about 30 days, as shown in Fig. 5. We therefore concluded that CGP was almost completely hydrolyzed to dipeptides by this strain. Hydrolysis was enhanced and occurred much earlier in the presence of low concentrations of yeast extract. If the incubation period was extended, no further accumulation of β-Asp-Arg dipeptides was observed. The dipeptide concentration remained constant in the presence or absence of yeast extract, indicating that the dipeptides produced were not utilized during a later stage of cultivation (>25 days). Light microscopy showed that the cell density of the culture grown on CGP was extremely low, and sporulation started after only after 10 to 14 days of incubation, indicating that the life cycle of S. hongkongensis KI was short under the conditions employed (Fig. 1C and D). Because most cells of strain KI formed endospores at the early stage of cultivation, we concluded that this strain can utilize only a small fraction of the dipeptides produced (maximum, about 0.5 g/liter, corresponding to 1.7 mmol/liter) for growth before completion of its life cycle.
FIG. 5.

Release of β-Asp-Arg dipeptides during cultivation of axenic S. hongkongensis strain KI on CGP and subsequent degradation of the dipeptides by C. amalonaticus strain G. Cultivation was performed in 10 ml Pelobacter liquid medium. All cultures (▪, □, ▴, and ▵) contained approximately 2 g/liter CGP and were inoculated with 200 μl of an S. hongkongensis KI preculture grown on CGP. Two of the cultures (□ and ▵) also contained 0.01% (wt/vol) yeast extract. After 30 days of incubation (arrow), two of the cultures (▴ and ▵) were inoculated with a suspended colony of C. amalonaticus strain G grown on LB agar plates.
Degradation of dipeptides by an axenic culture of C. amalonaticus strain G.
To analyze degradation of dipeptides by the rapidly growing organism C. amalonaticus strain G, an axenic culture of this bacterium was grown on dipeptides, which were produced by treatment of sterile anaerobic CGP-containing Hungate tubes with isolated CphEPa of P. anguilliseptica strain BI, and growth was monitored for 18 days (Fig. 6). In three independent experiments it was found that axenic cultures of C. amalonaticus strain G degraded only about 13 to 15% [wt/wt] of the dipeptides (0.8 mmol/liter) in the medium during long-term incubation. Nevertheless, C. amalonaticus strain G was able to grow well under anaerobic conditions on dipeptides added to solid or liquid media. For example, on dipeptide-containing (0.1%, wt/vol) M9 agar plates C. amalonaticus strain G grew in about 36 h under a nitrogen atmosphere to a high cell density, and visible colonies were detected on the agar plates (Fig. 2C). On M9 agar plates with the same amount of free arginine and aspartate that was in the dipeptide-supplemented medium, colonies were slightly larger (data not shown), indicating that uptake and subsequent hydrolysis of dipeptides consume more energy than uptake of the free amino acids. In dipeptide-containing liquid medium, an optical density that was about 80 Klett units greater than the inoculation level was obtained (Fig. 2D). This increase may have been partially due to the slightly brownish color of the medium that occurred during growth of C. amalonaticus. Nevertheless, comparably good growth on dipeptides was clearly demonstrated.
FIG. 6.

Degradation of β-Asp-Arg dipeptides by axenic C. amalonaticus strain G. The dipeptides were produced by using heterologously synthesized CphEPa of P. anguilliseptica strain BI which was heat inactivated before inoculation (32). The degradation experiment was reproduced three times to confirm the observed low degradation rates.
CGP degradation by mixed cultures of S. hongkongensis strain KI and C. amalonaticus strain G.
To simulate simultaneous production and utilization of dipeptides in a defined anaerobic mixed culture, C. amalonaticus strain G was used in cocultivation experiments and was added to two of the Hungate tubes employed after 30 days of CGP degradation by S. hongkongensis strain KI (Fig. 5). The latter organism slowly degraded CGP and poorly utilized the released dipeptides, and 75% (wt/vol) of the hydrolyzed CGP was accumulated as dipeptides in the medium over a period of 30 days in axenic cultures (Fig. 5). After addition of cells of strain G the concentration of β-Asp-Arg dipeptides decreased rapidly during the next 48 h. In the case of the culture containing yeast extract, which markedly promoted CGP degradation during the initial degradation phase, about 50% (wt/wt) of the dipeptides present in the medium was degraded (about 2.25 mmol dipeptides/liter).
When S. hongkongensis strain KI and C. amalonaticus strain G were cultivated together in CGP-containing liquid medium, free dipeptides were detected after about 5 days of incubation; however, the final dipeptide concentration of the medium was significantly lower (about 0.65 g/liter) than the concentration in degradation experiments employing only S. hongkongensis KI (Fig. 7). Because this culture originally contained 2 g of CGP per liter, about 1.35 g/liter of dipeptides (4.7 mmol/liter) must have been degraded during total hydrolysis of CGP. Assuming that there was complete CGP hydrolysis to dipeptides in all degradation experiments, the initial dipeptide concentrations were significantly reduced over time if the two bacterial strains were cultivated together. In the absence of yeast extract, cocultivation led to a 68% reduction in the dipeptide concentration (1.35 g/liter) (see above), and sequential cultivation of the two strains also led to a significant reduction (to 0.25 g/liter) in only 48 h (Fig. 5 and 7).
FIG. 7.

Simultaneous release and degradation of β-Asp-Arg dipeptides during cocultivation of S. hongkongensis strain KI and C. amalonaticus strain G in liquid Pelobacter medium containing 0.2% (wt/vol) CGP.
Degradation of aspartate and arginine.
From these data we concluded that S. hongkongensis degrades CGP by employing an extracellular CGPase but cannot use large quantities of the dipeptides for growth. C. amalonaticus strain G, however, can grow on dipeptides in pure culture but utilizes only about 15% of the dipeptides provided at an initial concentration of about 1.55 g/liter. If both strains were cultivated together, up to 68% of the released dipeptides were consumed before another factor became growth limiting.
GC analysis of CGP degradation samples revealed that during formation of dipeptides by S. hongkongensis KI about 1 mmol/liter of ethanol and traces of acetate were formed. After addition of C. amalonaticus cells to an S. hongkongensis culture, the concentrations of ethanol and acetate increased rapidly (Fig. 8). During cocultivation of the two strains, the concentrations of ethanol and acetic acid paralleled each other for about 33 days, and the concentrations of both compounds were about 1 to 1.2 mmol/liter (Fig. 9). After 60 days of incubation the ethanol concentration decreased to 0.25 mmol/liter, whereas the acetate concentration reached a maximum of about 1.3 mmol/liter. If C. amalonaticus strain GI was cultivated alone on dipeptides, very low concentrations of ethanol and acetate were produced over a period of about 15 days (data not shown). Only traces of other fermentative products, like propionate or butanol, were detected during degradation experiments, and no other short-chain length alcohols or fatty acids were detected by GC.
FIG. 8.

Formation of ethanol and acetic acid as fermentation products of S. hongkongensis strain KI cultivated in CGP-containing Pelobacter liquid medium under anaerobic conditions. The samples analyzed were collected from the cultures used for the experiments whose results are shown in Fig. 5, and the same sampling times were used. Ethanol (A) and acetic acid (B) were the only secondary CGP degradation products occurring in significant amounts, as detected by gas chromatography. Two cultures (□ and ▵) also contained 0.01% yeast extract, and two cultures (▪ and ▴) were not supplemented with complex compounds. The time of inoculation of the cultures with C. amalonaticus strain G cells is indicated by arrowheads. Sharp increases in the ethanol concentration and the acetic acid concentration were observed after the addition of C. amalonaticus at 30 days.
FIG. 9.

Formation of ethanol (▵), acetic acid (▪), and succinate (□) during cocultivation of S. hongkongensis strain KI and C. amalonaticus strain G in Pelobacter liquid medium containing 0.2% (wt/vol) CGP.
It was calculated that the molar concentrations of ethanol and acetate approximately equaled the molar concentration of the degraded dipeptides at each sampling time and, accordingly, equaled the concentrations of the released aspartate and arginine. At all sampling times during the early phase of the first degradation experiment only cells of S. hongkongensis strain KI were present, and later, after addition of C. amalonaticus G cells, such a molar proportion of the concentrations of dipeptides and fermentation products was observed (compare Fig. 5 and 8). Furthermore, during cocultivation of the two organisms almost equal concentrations of ethanol and acetate were detected for all samples except the sample taken after 60 days of incubation.
Succinate formation was observed in all degradation experiments after long-term incubation. During cultivation of C. amalonaticus strain G on dipeptides (Fig. 6) about 0.5 mmol/liter of succinate was produced (data not shown), and during cocultivation of the two strains investigated in this study a final concentration of about 2 mmol/liter was obtained (after 60 days) (Fig. 9). In contrast, during degradation of CGP by S. hongkongensis strain KI only traces of succinate (0.1 to 0.2 mmol/liter) were released into the medium. Because the number of samples collected during the experiments was limited by the restricted cultivation volumes, degradation experiments and determination of metabolite concentrations were done at least in duplicate or triplicate to ensure reproducibility of the results.
DISCUSSION
Genes for CGP biosynthesis were recently also detected in anaerobic bacteria (23, 55). Among these bacteria are members of the proteolytic clostridia (for example, C. botulinum type A) (23), indicating that CGP may serve as a transient intracellular storage material in anaerobic habitats. In addition, CGP-containing biomass produced by cyanobacteria may arrive in anaerobic habitats. Since especially bacteria of anaerobic consortia are often metabolically dependent on each other (19, 41), it was not surprising that during this study many stable bacterial mixed cultures, like AK15, were easily isolated in CGP-containing media from several different anaerobic habitats (data not shown). Sequential application of several elaborated enrichment strategies finally led to isolation of the strictly anaerobic bacterium S. hongkongensis strain KI, which is the first known anaerobic CGP-degrading microorganism. A second bacterium, the facultative anaerobic enterobacterium C. amalonaticus strain G, was isolated from the same consortium, which degraded the dipeptides formed by the former organism. It was found that in liquid media the overall effectiveness of the CGP degradation process was highest during cocultivation of the two bacteria, and the presence of S. hongkongensis strain KI strongly promoted dipeptide degradation by C. amalonaticus strain G (Fig. 5 and 7).
Most likely, dipeptides are hydrolyzed to their amino acid constituents by intracellular enzymes (see equation 2 below) (18) after uptake into bacterial cells. Because of the low overall turnover and the use of arginine and aspartate as building blocks (for example, in protein biosynthesis), it was impossible to estimate on the basis of the data obtained exact turnover rates for the conversion of dipeptides to fermentation products with respect to energy yield and redox balance. However, useful suggestions about how a major fraction of the dipeptides was converted to ethanol, acetate, and succinate are described below. On the basis of known catabolic enzymes of clostridia (for example, Clostridium thermosuccinogenes [48] or C. botulinum [12]), the following pathways could be involved in amino acid utilization. Aspartate could be converted to pyruvate by aspartate ammonia lyase (EC 4.3.1.1) in combination with fumarase (EC 4.2.1.2.), malate dehydrogenase (EC 1.1.1.37), and oxaloacetate decarboxylase (EC 4.1.1.3). Pyruvate could subsequently be oxidized to acetyl-CoA by pyruvate-ferredoxin oxidoreductase (EC 1.2.7.1). The reducing equivalents produced by pyruvate-ferredoxin oxidoreductase could be transferred to NAD+ (via NADH2:ferredoxin-oxidoreductase [EC 1.18.1.3]). Together with reducing equivalents obtained from the malate dehydrogenase reaction (a total of 4 [H]), acetyl-CoA could be reduced subsequently to ethanol by a combination of acetaldehyde dehydrogenase (acetylating; EC 1.2.1.10) and alcohol dehydrogenase (EC 1.1.1.1) (48). One molecule of ethanol would be produced from one molecule of aspartate (see equation 3 below). This is a plausible explanation for the initial formation of the comparatively large amounts of only ethanol observed during CGP degradation of axenic S. hongkongensis (Fig. 8) without the production of any reducing equivalents. However, these reactions do not yield ATP, which is a prerequisite for growth and which could be easily obtained from acetyl-CoA via phosphotransacetylase (EC 2.3.1.8) and acetate kinase (EC 2.7.2.1) reactions; however, acetate was not observed in the initial degradation phase, and its formation would require another sink for the reducing equivalents produced, which were obviously mainly employed for ethanol production (see above). Therefore, it seems likely that during the initial degradation phase energy is obtained mainly by arginine degradation, which does not yield reducing equivalents. Degradation of arginine under anaerobic conditions is most likely to occur via the arginine deiminase (ADI) (arginine dihydrolase; EC 3.5.3.6) pathway, which is widespread in anaerobic bacteria (12, 13) and probably is present also in C. amalonaticus strain G, as indicated by a positive arginine dihydrolase test. One product of the ADI pathway reactions is carbamoyl phosphate, which yields ATP by the carbamate kinase (EC 2.7.2.2) reaction. Ornithine, the second product of the ADI pathway, was not detected in significant amounts in the media. Clostridium sticklandii and C. botulinum convert ornithine to proline by means of ornithine cyclase (12), and this product may have been formed but was not detectable by the HPLC system employed. A combination of the two pathways yields equation 4 (see below).
Ethanol, which is produced during aspartate degradation, could serve as an overflow product or metabolic intermediate which is reused in the late degradation phase, presumably when arginine is almost completely utilized. Reoxidation of ethanol to acetyl-CoA and conversion to acetate by phosphotransacetylase and acetate kinase would subsequently yield additional energy (1 ATP/ethanol) (48), thus explaining the disappearance of a fraction of ethanol and the occurrence of comparatively large amounts of acetate in the late degradation phase (Fig. 9). Furthermore, reducing equivalents obtained by reoxidation of ethanol (4 [H]) could be transferred to fumarate formed from aspartate, yielding succinate, by fumarate reductase (EC 1.3.1.6) (24). This would explain formation of comparatively large amounts of succinate, as observed after long-term incubation of defined cocultures on CGP (60 days) (Fig. 9). The x in equation 5 indicates that the amount of ATP formed via fumarate respiration cannot be stated exactly because less than one ATP molecule is obtained during reoxidation of one molecule of NADH2 (24).
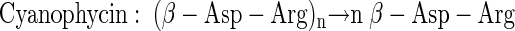 |
(1) |
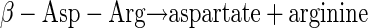 |
(2) |
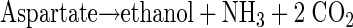 |
(3) |
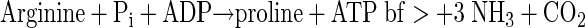 |
(4) |
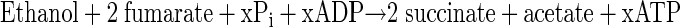 |
(5) |
A positive ornithine decarboxylase (EC 4.1.1.17) test for C. amalonaticus strain G indicated that putrescine is formed from ornithine; however, putrescine was not detected in the medium. It was shown previously that putrescine can be fermented to acetate, butyrate, molecular hydrogen, and ammonia by species related to Clostridium or Eubacterium (28, 29). However, butyrate was not detected by GC, indicating that this type of fermentation does not contribute on a larger scale to production of acetate from CGP.
Both strains isolated in this study were dependent on the other strain for increased utilization of about 68% (wt/vol) of the CGP employed and may be dependent on other unidentified consortium partners in the AK15 mixed culture for complete fermentative utilization of β-Asp-Arg dipeptides. For a more detailed analysis of enzyme activity measurements, product formation, and metabolic cooperation under anaerobic conditions, it would be useful to analyze CGP utilization by bacteria of other anaerobic consortia which exhibit better growth characteristics and a lesser tendency to sporulate almost immediately after germination, as observed for S. hongkongensis strain KI.
Acknowledgments
This study was supported by a collaborative research grant provided by Fachagentur Nachwachsende Rohstoffe e.V. (FNR) (FKZ: 00NR125) and by Bayer AG (Leverkusen, Germany).
REFERENCES
- 1.Aboulmagd, E., F. B. Oppermann-Sanio, and A. Steinbüchel. 2000. Molecular characterization of the cyanophycin synthetase from Synechocystis sp. strain PCC6308. Arch. Microbiol. 174:297-306. [DOI] [PubMed] [Google Scholar]
- 2.Allen, M. M., F. Hutchinson, and P. J. Weathers. 1980. Cyanophycin granule polypeptide formation and degradation in the cyanobacterium Aphanocapsa 6308. J. Bacteriol. 141:687-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, M. M. 1984. Cyanobacterial cell inclusions. Annu. Rev. Microbiol. 38:1-25. [DOI] [PubMed] [Google Scholar]
- 4.Allen, M. M. 1988. Inclusions: cyanophycin. Methods Enzymol. 167:207-213. [Google Scholar]
- 5.Berg, H., K. Ziegler, K. Piotukh, K. Baier, and W. Lockau. 2000. Biosynthesis of the cyanobacterial reserve polymer multi-l-arginyl-poly-l-aspartic acid (cyanophycin): mechanism of the cyanophycin synthetase reaction studied with synthetic primers. Eur. J. Biochem. 267:5561-5570. [DOI] [PubMed] [Google Scholar]
- 6.Borzi, A. 1887. Le comunicazioni intracellulari delle Nostochinee. Malpighia 1:74-203. [Google Scholar]
- 7.Brambilla, E., H. Hippe, A. Hagelstein, B. J. Tindall, and E. Stackebrandt. 2001. 16S rDNA diversity of cultured and uncultured prokaryotes of a mat sample from Lake Fryxell, McMurdo Dry Valleys, Antartica. Extremophiles 5:23-33. [DOI] [PubMed] [Google Scholar]
- 8.Breitenstein, A., J. Wiegel, C. Haertig, N. Weiss, J. R. Andreesen, and U. Lechner. 2002. Reclassification of Clostridium hydroxybenzoicum as Sedimentibacter hydroxybenzoicus gen. nov., comb. nov., and description of Sedimentibacter saalensis sp. nov. Int. J. Syst. Evol. Microbiol. 52:801-807. [DOI] [PubMed] [Google Scholar]
- 9.Claus, D., and N. Walker. 1964. The decomposition of toluene by soil bacteria. J. Gen. Microbiol. 36:107-122. [DOI] [PubMed] [Google Scholar]
- 10.Cole, R. B. 1997. Electrospray ionization mass spectrometry. John Wiley & Sons, Inc., New York, N.Y.
- 11.Collins, M. D., P. A. Lawson, A. Willems, J. J. Cordoba, J. Fernandez-Garayzabal, P. Garcia, H. Hippe, and J. A. E. Farrow. 1994. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 44:812-826. [DOI] [PubMed] [Google Scholar]
- 12.Cunin, R., N. Glansdorff, A. Piérard, and V. Stalon. 1986. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50:314-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dyer, J. K., and R. N. Costilow. 1968. Fermentation of ornithine by Clostridium sticklandii. J. Bacteriol. 96:1617-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey, K. M., F. B. Oppermann-Sanio, H. Schmidt, and A. Steinbüchel. 2002. Technical scale production of cyanophycin with recombinant strains of Escherichia coli. Appl. Environ. Microbiol. 68:3377-3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta, M., and N. G. Carr. 1981. Enzyme activities related to cyanophycin metabolism in heterocysts and vegetative of Anabaena spp. J. Gen. Microbiol. 125:7-13. [Google Scholar]
- 16.Hai, T., F. B. Oppermann-Sanio, and A. Steinbüchel. 2002. Molecular characterization of a thermostable cyanophycin synthetase from the thermophilic cyanobacterium Synechococcus sp. strain MA19 and in vitro synthesis of cyanophycin and related polyamides. Appl. Environ. Microbiol. 68:93-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 18.Hejazi, M., K. Piotukh, J. Mattow, R. Deutzmann, R. Volkmer-Engerts, and W. Lockau. 2002. Isoaspartyl dipeptidase activity of plant-type asparaginases. Biochem. J. 364:129-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmeister, M., and W. Martin. 2003. Interspecific evolution: microbial symbiosis, endosymbiosis and gene transfer. Environ. Microbiol. 5:641-649. [DOI] [PubMed] [Google Scholar]
- 20.Holt, J. G., N. R. Krieg, P. H. A. Sneath, J. T. Staley, and S. T. Williams. 1994. Genus Citrobacter, p. 174. In Bergey's manual of determinative bacteriology. Williams & Wilkins, Baltimore, Md.
- 21.Huang, X. 1992. A contig assembly program based on sensitive detection of fragment overlaps. Genomics 14:18-25. [DOI] [PubMed] [Google Scholar]
- 22.Krehenbrink, M., and A. Steinbüchel. 2004. Partial purification and characterization of a non-cyanobacterial cyanophycin synthetase from Acinetobacter calcoaceticus strain ADP1 with regard to substrate specificity, substrate affinity and binding to cyanophycin. Microbiology 150:2599-2608. [DOI] [PubMed] [Google Scholar]
- 23.Krehenbrink, M., F. B. Oppermann-Sanio, and A. Steinbüchel. 2002. Evaluation of non-cyanobacterial genome sequences for occurrence of genes encoding proteins homologous to cyanophycin synthetase and cloning of active cyanophycin synthetase from Acinetobacter sp. strain DSM 587. Arch. Microbiol. 177:371-380. [DOI] [PubMed] [Google Scholar]
- 24.Kröger, A., V. Geisler, E. Lemma, F. Theis, and R. Lenger. 1992. Bacterial fumarate respiration. Arch. Microbiol. 158:311-314. [Google Scholar]
- 25.Lawry, N. H., and R. D. Simon. 1982. The normal and induced occurrence of cyanophycin inclusion bodies in several blue-green algae. J. Phycol. 18:391-399. [Google Scholar]
- 26.Liotenberg, S., D. Campbell, R. Rippka, J. Hourmard, and N. T. de Marsac. 1996. Effect of the nitrogen source on phycobiliprotein synthesis and cell reserves in a chromatically adapting filamentous cyanobacterium. Microbiology 142:611-622. [DOI] [PubMed] [Google Scholar]
- 27.Mackerras, A. H., N. M. De Chazal, and G. D. Smith. 1990. Transient accumulation of cyanophycin in Anabaena cylindrica and Synechocystis 6308. J. Gen. Microbiol. 136:2057-2065. [Google Scholar]
- 28.Matthies, C., F. Mayer, and B. Schink. 1989. Fermentative degradation of putrescine by new strictly anaerobic bacteria. Arch. Microbiol. 151:498-505. [DOI] [PubMed] [Google Scholar]
- 29.Matthies, C., S. Evers, W. Ludwig, and B. Schink. 2000. Anaerovorax odorimutans gen. nov., a putrescine-fermenting, strictly anaerobic bacterium. Int. J. Syt. Evol. Microbiol. 50:1591-1594. [DOI] [PubMed] [Google Scholar]
- 30.Obst, M., and Steinbüchel, A. 2004. Microbial degradation of poly(amino acid)s. Biomacromolecules 5:1166-1176. [DOI] [PubMed] [Google Scholar]
- 31.Obst, M., A. Sallam, H. Luftmann, and A. Steinbüchel. 2004. Isolation and characterization of Gram-positive cyanophycin degrading bacteria—kinetic studies on cyanophycin depolymerase activity in aerobic bacteria. Biomacromolecules 5:153-161. [DOI] [PubMed] [Google Scholar]
- 32.Obst, M., F. B. Oppermann-Sanio, H. Luftmann, and A. Steinbüchel. 2002. Isolation of cyanophycin degrading bacteria, cloning and characterization of an extracellular cyanophycinase gene (cphE) from Pseudomonas anguilliseptica strain BI. J. Biol. Chem. 277:25096-25105. [DOI] [PubMed] [Google Scholar]
- 33.Oppermann, F. B., B. Schmidt, and A. Steinbüchel. 1991. Purification and characterization of acetoin:2,6-dichlorophenolindophenol oxidoreductase, dihydrolipoamide dehydrogenase, and dihydrolipoamide acetyltransferase of the Pelobacter carbinolicus acetoin dehydrogenase system. J. Bacteriol. 173:757-767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oppermann-Sanio, F. B., and A. Steinbüchel. 2002. Occurrence, functions and biosynthesis of polyamides in microorganisms and biotechnological production. Naturwissenschaften 89:11-22. [DOI] [PubMed] [Google Scholar]
- 35.Oppermann-Sanio, F. B., and A. Steinbüchel. 2003. Cyanophycin, p. 83-106. In S. R. Fahnestock and A. Steinbüchel (ed.), Biopolymers, vol. 7, Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 36.Pugsley, A. P. 1993. The complete general secretory pathway in Gram-negative bacteria. Microbiol. Rev. 57:50-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rainey, F. A., N. Ward-Rainey, R. M. Kroppenstedt, and E. Stackebrandt. 1996. The genus Nocardiopsis represents a phylogenetically coherent taxon and a distinct actinomycete lineage: proposal of Nocardiopsaceae fam. nov. Int. J. Syst. Bacteriol. 46:1088-1092. [DOI] [PubMed] [Google Scholar]
- 38.Rao, R. N., M. A. Richardson, and S. Kuhstoss. 1987. Cosmid shuttle vectors for cloning and analysis of Streptomyces DNA. Methods Enzymol. 153:166-198. [DOI] [PubMed] [Google Scholar]
- 39.Richter, R., M. Hejazi, R. Kraft, K. Ziegler, and W. Lockau. 1999. Cyanophycinase, a peptidase degrading the cyanobacterial reserve material multi-l-arginyl-poly-l-aspartic acid (cyanophycin): molecular cloning of the gene of Synechocystis sp. PCC 6803, expression in Escherichia coli, and biochemical characterization of the purified enzyme. Eur. J. Biochem. 263:163-169. [DOI] [PubMed] [Google Scholar]
- 40.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 41.Schink, B. 2000. Synergistic interactions in the microbial world. Antonie Leeuwenhoek 81:257-261. [DOI] [PubMed] [Google Scholar]
- 42.Schlegel, H. G., H. Kaltwasser, and G. Gottschalk. 1961. Ein Submersverfahren zur Kultur wasserstoffoxidierender Bakterien: Wachstumsphysiologische Untersuchungen. Arch. Mikrobiol. 38:209-222. [PubMed] [Google Scholar]
- 43.Simon, R. D. 1971. Cyanophycin granules from blue-green alga Anabaena cylindrical—reserve material consisting of copolymers of aspartic and arginine. Proc. Natl. Acad. Sci. USA 68:265-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simon, R. D. 1976. The biosynthesis of multi-l-arginyl-poly(l-aspartic acid). Biochim. Biophys. Acta 422:407-418. [DOI] [PubMed] [Google Scholar]
- 45.Simon, R. D. 1987. Inclusion bodies in the cyanobacteria: cyanophycin, polyphosphate, polyhedral bodies, p. 199-225. In P. Fay and C. Van Baalen (ed.), The cyanobacteria. Elsevier, Amsterdam, The Netherlands.
- 46.Simon, R. D., and P. Weathers. 1973. Measurement of the cyanophycin granule polypeptide contained in the blue-green alga Anabaena cylindrica. J. Bacteriol. 114:1213-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simon, R. D., and P. Weathers. 1976. Determination of the structure of the novel polypeptide containing aspartic acid and arginine which is found in cyanobacteria. Biochim. Biophys. Acta 420:165-176. [DOI] [PubMed] [Google Scholar]
- 48.Sridhar, J., M. A. Eiteman, and J. W. Wiegel. 2000. Elucidation of enzymes in fermentation pathways used by Clostridium thermosuccinogenes growing on inulin. Appl. Environ. Microbiol. 66:246-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strauss, E. C., J. A. Kobori, G. Siu, and L. E. Hood. 1986. Specific-primer-directed DNA sequencing. Anal. Biochem. 154:353-360. [DOI] [PubMed] [Google Scholar]
- 50.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weber, K., and M. Osborn. 1969. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244:4406-4412. [PubMed] [Google Scholar]
- 52.Widdel, F., and N. Pfennig. 1981. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. I. Isolation of new sulfate-reducing bacteria enriched with acetate from saline environments. Description of Desulfobacter postgatei gen. nov., sp. nov. Arch. Microbiol. 129:395-400. [DOI] [PubMed] [Google Scholar]
- 53.Wingard, L. L., S. R. Miller, J. M. Sellker, E. Stenn, M. M. Allen, and A. M. Wood. 2002. Cyanophycin production in phycoerythrin-containing marine Synechococcus strain of unusual phylogenetic affinity. Appl. Environ. Microbiol. 68:1772-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ziegler, K., A. Diener, C. Herpin, R. Richter, R. Deutzmann, and W. Lockau. 1998. Molecular characterization of cyanophycin synthetase, the enzyme catalyzing the biosynthesis of cyanobacterial reserve material multi-l-arginyl-poly-l-aspartate (cyanophycin). Eur. J. Biochem. 254:154-159. [DOI] [PubMed] [Google Scholar]
- 55.Ziegler, K., R. Deutzmann, and W. Lockau. 2002. Cyanophycin synthetase-like enzymes of non-cyanobacterial eubacteria: characterization of the polymer produced by a recombinant synthetase of Desulfitobacterium hafniense. Z. Naturforsch. Sect. C 57:522-529. [DOI] [PubMed] [Google Scholar]