

Abstract
Carbon stable isotope fractionation of tetrachloroethene (PCE) during reductive dechlorination by whole cells and crude extracts of Sulfurospirillum multivorans and Desulfitobacterium sp. strain PCE-S and the abiotic reaction with cyanocobalamin (vitamin B12) was studied. Fractionation was largest during the reaction with cyanocobalamin with αC = 1.0132. Stable isotope fractionation was lower but still in a similar order of magnitude for Desulfitobacterium sp. PCE-S (αC = 1.0052 to 1.0098). The isotope fractionation of PCE during dehalogenation by S. multivorans was lower by 1 order of magnitude (αC = 1.00042 to 1.0017). Additionally, an increase in isotope fractionation was observed with a decrease in cell integrity for both strains. For Desulfitobacterium sp. strain PCE-S, the carbon stable isotope fractionation factors were 1.0052 and 1.0089 for growing cells and crude extracts, respectively. For S. multivorans, αC values were 1.00042, 1.00097, and 1.0017 for growing cells, crude extracts, and the purified PCE reductive dehalogenase, respectively. For the field application of stable isotope fractionation, care is needed as fractionation may vary by more than an order of magnitude depending on the bacteria present, responsible for degradation.
The chlorinated ethenes tetrachloroethene (PCE) and trichloroethene (TCE) have been used as solvents in the dry-cleaning industry and as metal degreasing agents and are among the most common groundwater contaminants due to spillage and leakage (12). The assessment of in situ biodegradation of groundwater contaminants is difficult since a decrease in the concentration can be a result of many factors, including dilution, sorption, and biological conversion. Monitoring and assessment of in situ microbial degradation activities of contaminants at polluted sites is therefore a major challenge. In the last few years, the application of stable isotope techniques has been suggested for assessment of the in situ biodegradation of contaminants (for a review, see references 17 and 33).
Stable isotope fractionation of PCE and TCE has been observed under field conditions in contaminated aquifers as well as in laboratory studies by mixed microbial cultures and microcosms (2, 9, 39, 40); however, the factors affecting the isotope fractionation have not yet been studied systematically.
Several factors may influence the degree of stable isotope fractionation, including biodegrading microorganisms, properties of the dehalogenating enzymes, and the reaction mechanism. Cobalamins are important cofactors of reductive dehalogenases in organisms capable of dehalorespiration (8, 10, 15, 20, 25, 26). The microbial dehalogenation by cobalamin-containing dehalogenases has been proposed to proceed by alkylating a superreduced corrinoid containing a Co(I) species at the reactive site with the chloroethene (26). The chemical mechanism of the reductive dehalogenation of chlorinated ethenes catalyzed by cobalamin has been the subject of previous studies and has been suggested to occur via a single electron transfer from a reduced cob(I)alamin, leading to the formation of chloride and vinyl radicals as intermediates (3, 7, 36, 41). The cleavage of the carbon-chlorine bond may produce the primary carbon isotope effect during dehalogenation by cobalamin-containing dehalogenases. The solvent kinetic effect of deuterated water in the dehalogenation reaction of aryl substituted chlorine with the cobalamin-containing dehalogenase of Desulfitobacterium chlororespirans is moderate (10). Apparently, the protonation reaction of the substrate is to some extent only a reaction step which controls the overall kinetics of the reaction and may occur after the electron has been transferred and the chlorine cleaved (10). This reaction scheme would explain the carbon isotope and chlorine isotope fractionation typically associated with the dehalogenation of PCE and TCE and halogenated benzenes (4, 28, 34). In addition to the reaction mechanism, a number of other factors such as binding of the chlorinated substrate to the catalytic centre of the enzyme or passive or active transport of the substrate across the membrane may affect carbon isotope fractionation (29, 32).
To our knowledge, only limited information of PCE isotope fractionation upon reductive dehalogenation with pure cultures is available and the effect of microbial membranes on the isotope fractionation of chlorinated alkenes has not yet been studied. Here, we present a study of the carbon stable-isotope fractionation of tetrachloroethene during reductive dechlorination by reduced cyanocobalamin (vitamin B12) and with two bacterial strains, Sulfurospirillum multivorans (21), formerly Dehalospirillum multivorans (14), and Desulfitobacterium sp. strain PCE-S (19). Of both strains, stable carbon isotope fractionation during growth and in crude extracts was analyzed and additionally, carbon fractionation by the purified PCE reductive dehalogenase of S. multivorans and by cyanocobalamin was studied.
MATERIALS AND METHODS
Chemicals.
All chemicals were purchased from Merck (Darmstadt, Germany) or Sigma-Aldrich Chemie (Munich, Germany) at the highest purity available unless stated elsewhere. Gases were purchased from Linde Gas AG (Höllriegelskreuth, Germany).
Cultivation of bacteria and preparation of crude extracts.
S. multivorans (21) and Desulfitobacterium sp. strain PCE-S (19) were cultured as described previously with PCE as the electron acceptor, except as indicated. For S. multivorans, pyruvate and fumarate, and for Desulfitobacterium sp. strain PCE-S, pyruvate was added as the electron donor and carbon source. For preparation of crude extracts, cells were harvested anaerobically by centrifugation at 11,000 × g for 10 min at 4°C. The cells were resuspended in buffer (100 mM Tris-HCl, pH 7.5) and centrifuged again. The pellet was resuspended, and a crude extract was produced by passing the suspension through a French press cell at 20,000 lb/in2. All preparations were performed anaerobically within an anaerobic glovebox (Coy Laboratory Products Inc.) with an N2/H2 (96%/4%) atmosphere or under a stream of nitrogen.
Purification of PCE-reductive dehalogenase.
The PCE-reductive dehalogenase of S. multivorans was purified under strictly anaerobic conditions essentially as described in reference 26. Cells were grown with pyruvate plus fumarate rather than PCE as electron acceptor. Next, 30 g (wet weight) of S. multivorans cells was lysed by resuspension in 90 ml 50 mM Tris-HCl buffer, pH 7.5, with the addition of 300 mg lysozyme and 30 mg DNaseI. The mixture was incubated at 37°C for 1 h. Cell debris was then removed by centrifugation at 35,000 × g for 1 h. The supernatant was applied to a Q-Sepharose column which had been equilibrated with buffer A (50 mM Tris-HCl [pH 7.5] 0.5 mM 1,4-dithiothreitol). The dehalogenase was eluted from the column using a potassium gradient from 0 to 1 M KCl in buffer A and the enzyme eluted at about 0.3 M KCl. Activity of the fractions was assayed as described previously (26). PCE-dehalogenase-containing fractions were applied to a phenyl-Sepharose column which had been equilibrated with buffer A plus 0.4 mM (NH4)2SO4. The enzyme was eluted from the column using buffer A with decreasing concentrations of (NH4)2SO4 and the dehalogenase eluted at a (NH4)2SO4 content between 60 and 0 mM. An additional purification step using a phenyl-Sepharose column was performed and the dehalogenase eluted at a (NH4)2SO4 content between 120 and 80 mM. Until use, the fractions containing the dehalogenase were stored at −20°C.
Analytical methods.
Gas chromatography with flame ionization detection (GC-FID) was applied to analyze ethene and the chlorinated ethenes (gas chromatograph Chrompack CP-3800; Varian, Middelburg, The Netherlands) equipped with a 30-m by 0.53-mm GS-Q column (J&W Scientific, Waldbronn, Germany). The temperature program used was as follows: 1 min at 100°C, 50°C/min to 225°C, hold for 2.5 min. The FID was operated at 250°C, and helium was used as carrier gas (10 lb/in2). This method allowed the separation of ethene; vinyl chloride; and 1,1-, trans-, and cis-DCE, TCE, and PCE. The injection was automated using an HP 7694 headspace autosampler (Hewlett Packard, Palo Alto, Calif.), adding 0.5-ml headspace samples to 10-ml autosampler vials flushed with helium, which were closed with a Teflon-coated butyl rubber septum and crimped. The data were recorded using the Varian STAR software.
Gas chromatography combustion isotope ratio mass spectroscopy (GC-C-IRMS) was applied to determine the stable carbon isotope composition (18). All samples were measured in at least five replicates. Aliquots (100 to 1,000 μl) of headspace samples were injected into a gas chromatograph (model 6890; Agilent, Palo Alto, Calif.) in split mode (split ratio was set to at least 1 to 5) using a split/splitless injector at 250°C. In the case of pentane extraction, 5 μl of extract was injected in split mode (split 1:10 to 1:1) using an autosampler (CTC Combipal; Chromtech, Idstein, Germany). For chromatographic separation, a ZB-1 capillary column (60 m by 0.32 mm, 0.5-μm film; Phenomex Inc.) or a Poraplot Q column (25 m by 0.32 mm inner diameter, 1-μm particle size; Chrompack, Middelburg, The Netherlands) was used.
For the separation on a Poraplot column, the temperature was initially set to 40°C for 2 min and was then increased at a rate of 10°C min−1 to 230°C, where it was held for 5 min. For the separation of compounds on a ZB1 column, the temperature was held at 40°C for 6°C min, increased at a rate of 6°C min−1 to 125°C, and then increased at a rate of 20°C min−1 to a final temperature of 250°C.
Determination of stable-isotope fractionation factors.
Dehalogenation experiments of the halogenated ethenes with vitamin B12 were performed under the reaction conditions described elsewhere (13). Briefly, 27-ml culture tubes were filled with 14 ml 0.1 M Tris-HCl buffer adjusted to pH 8. The solution was flushed with N2 to remove oxygen and sealed with a Teflon-coated butyl rubber stopper and crimped with an aluminum seal. Vitamin B12 (13.2 μM) and PCE (700 μM) were added. To start the dehalogenation reaction, 0, 0.6, 1.2, 1.5, or 1.8 mM Ti(III)citrate dissolved in 0.5 M Tris-HCl buffer adjusted to pH 8 was added to the tubes as an electron donor (42). The solutions were shaken on a rotary shaker at 20°C, and the dehalogenation of PCE was allowed to react at least overnight. The samples were stored at 4°C until headspace analysis.
For the determination of stable-isotope fractionation by growing cultures of S. multivorans and Desulfitobacterium sp. strain PCE-S, several parallel cultures were prepared which were sacrificed for analysis of the concentration and isotope composition of the chlorinated ethenes after the appropriate growth periods. All cultures received PCE (500 μM) and were prepared with a small headspace (100 ml of liquid in 120-ml vials) to limit the isotope fractionation effect of volatilization of the chlorinated ethenes from the liquid phase to the gas phase, although these effects are considered to be negligible (37). At various incubation times, the first 0.5-ml gas phase was removed from one culture and analyzed for reductive dechlorination by GC-FID and the respective culture was then killed by the addition of 5 ml aerobic, saturated Na2SO4, pH 1. For extraction of the chlorinated ethenes, 2 ml pentane was added to each culture, which was incubated on a rotary shaker overnight (25°C). The pentane phase was then removed, transferred to autosampler vials, and analyzed by GC-C-IRMS for isotopic composition of the chlorinated ethenes.
Isotope fractionation by crude extracts and partially purified enzyme was determined as follows. Six 12-ml vials were filled with 9 ml of test buffer [100 mM Tris-HCl (pH 7.5), 0.2 mM (NH4)2SO4] containing methyl viologen (1.6 mM) that acted as the artificial electron donor after reduction with titanium(III). To each vial, 1 mM PCE was added (from a 100 mM solution in ethanol) and then 50 μl of crude extract was added to start the reaction. The reductive dechlorination of PCE to about 20, 40, 60, 80, and 100% of the initial concentration was achieved by adding various amounts of titanium(III) citrate. The vials were incubated at 25°C in a rotary shaker during the reaction. After the complete reaction time, 0.5 ml of headspace was removed and analyzed by GC-FID and then 1 ml pentane was added to each vial to extract the chlorinated ethenes for analysis by GC-C-IRMS.
Calculations and definitions.
The carbon isotope composition (R) is reported in δ-notation (‰) relative to the Vienna Pee Dee Belemnite standard (International Atomic Energy Agency, Vienna, Austria) (1), as follows:
 |
(1) |
The isotope fractionation during the dehalogenation reaction was calculated by applying the Rayleigh-equation (6), as follows:
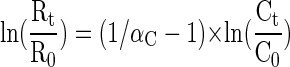 |
(2) |
where the kinetic carbon isotope fractionation factor αC is a constant for the reaction at the given experimental conditions, Rt and R0 are the isotope compositions, and Ct and C0 are the concentrations of the compound at times t and zero, respectively. The isotope fractionation factor αC relates the changes in the isotope composition to changes in the concentration of the residual fraction during the transformation.
RESULTS
Desulfitobacterium sp. strain PCE-S.
Desulfitobacterium sp. strain PCE-S dechlorinates PCE via TCE to cis-DCE (19). During growth, PCE was dechlorinated and the isotope ratio of PCE increased from δ = −24.7 to −16.2 ‰, indicating an enrichment of 13C in the residual PCE (Fig. 1A and B). TCE is formed as an intermediate and is at first lighter than PCE with δ = −27.7‰ but after 4 h becomes more enriched in 13C, to a value of up to δ = −1.8‰. cis-DCE was depleted in 13C from initially δ = −26.3 to −33.1‰ after about 3 h. Toward the end of the experiment, cis-DCE became heavier again, with a final composition near the initial isotope composition of PCE (−28.2‰).
FIG. 1.

Change in concentrations (A, C, and E) and carbon isotope composition (B, D, and F) of PCE (▪) and its products TCE (▴) and cis-DCE (⋄) during reductive dechlorination by Desulfitobacterium sp. strain PCE-S (A and B) and S. multivorans (C and D) and abiotic reactions with reduced cyanocobalamin (E and F). The standard deviation of typically less than 0.5 δ-units for the isotope measurement is much smaller than the symbol. PDB, Pee Dee Belemnite standard.
To assess the effect of the gram-positive cell wall and of the cytoplasmic membrane, experiments using crude extracts of Desulfitobacterium sp. strain PCE-S were performed. The PCE reductive dehalogenase is thought to be located inside the cytoplasm in this organism. Similar patterns of fractionation were found for the dechlorination of PCE by using crude cell extracts compared to the growing cultures of Desulfitobacterium sp. strain PCE-S (data not shown). The data were summarized for the various experiments and treatments as shown in Fig. 2A and, using the Rayleigh equation (equation 2), the carbon stable-isotope fractionation factor was calculated (Table 1). Fractionation by crude extracts of Desulfitobacterium sp. strain PCE-S was higher, αC = 1.0089, compared to that of the growing cells, αC = 1.0052, indicating an effect of the cytoplasmic membrane and/or cell wall on the extent of fractionation.
FIG. 2.

Stable-isotope fractionation of the residual fraction of PCE during reductive dechlorination by growing cells (□) and crude extracts (▴) of Desulfitobacterium sp. strain PCE-S (A), growing cells (□), crude extracts (▴), and the purified reductive dehalogenase (⧫) of S. multivorans (B), and reduced cyanocobalamin (•) (C).
TABLE 1.
Compound-specific carbon stable isotope fractionation factors α and the calculated partitioning factor Part-F for reductive dechlorination of PCE by S. multivorans and Desulfitobacterium sp. strain PCE-S and abiotic reactions with cyanocobalamin (vitamin B12)a
| Parameter |
Sulfurospirillum multivorans
|
Desulfitobacterium sp. strain PCE-S
|
Abiotic dechlorination α | ||
|---|---|---|---|---|---|
| α | Part-F | α | Part-F | ||
| Growth | 1.00042 ± 0.00008 | 30.4 | 1.00521 ± 0.0005 | 1.5 | |
| Crude extract | 1.00097 ± 0.00007 | 12.6 | 1.0089 ± 0.0007 | 0.5 | |
| Enzyme | 1.0017 ± 0.0004 | 6.8 | |||
| Cyanocobalamin | 1.0132 ± 0.00055 | ||||
Sulfurospirillum multivorans.
Carbon fractionation by S. multivorans was studied for reductive dechlorination of PCE by growing cells, crude extracts, and the purified PCE reductive dehalogenase. In Fig. 1 C and D, stable isotope composition and concentrations of chlorinated ethenes during reductive dechlorination of PCE by growing cells is shown. PCE was reduced via TCE to primarily cis-DCE. The isotope signature of PCE barely changed 1δ unit, from about −26.3 to −25.5‰. TCE became clearly enriched in 13C over time, with a change of about 11δ ‰ from −26.5 to −15.8‰ and was subject to much larger isotope discrimination than the parent compound PCE. A similar change was found with cis-DCE; the compound became depleted in 13C by about 10δ units from −25.7 to −35.1‰.
The PCE reductive dehalogenase of S. multivorans is thought to be attached to the cytoplasmic membrane facing the cytoplasm. Therefore, studies on the fractionation effects due to the transport or diffusion of PCE over the cytoplasmic membrane were performed using crude extracts and the purified PCE reductive dehalogenase of this organism and comparing the results to those obtained with whole cells. Similar patterns in the reductive dechlorination of PCE, the product formation, and the isotopic composition of the chloroethenes were observed for the experiments using crude extracts and the purified reductive dehalogenase (data not shown). In Fig. 2B a summary of the results is represented and the respective αC's were calculated from the data (Table 1). Fractionation of PCE by S. multivorans was 1 order of magnitude lower, ranging from αC = 1.00042 to 1.0017, compared to the values obtained for Desulfitobacterium sp. strain PCE-S. Similar to the results with Desulfitobacterium, isotope fractionation increased from growing cells compared to crude extracts, indicating an influence of the cytoplasmic membrane (Table 1; Fig. 2B). The highest fractionation occurred using the purified reductive dehalogenase, suggesting that cell components might affect the fractionation and cause a decrease of the overall αC.
Cyanocobalamin.
To test the isotope fractionation caused by the reactive center, cyanocobalamin (vitamin B12) was used as the model catalyst. Figure 1E and F shows the reductive dechlorination of PCE by reduced cyanocobalamin. TCE was the major product, and minor quantities of cis-DCE were observed. The isotope signature of PCE changed significantly, from δ = −25.2‰ initially to 29.0‰ at the end of the experiment. The isotope composition of TCE changed from −41.0 to −2.3‰, and cis-DCE was relatively depleted in 13C in the samples where it could be detected, ranging from −51.7 to −46.7‰. Figure 2C summarizes the results of the experiments in which cyanocobalamin reduced with Ti(III) was used as the reactant. The carbon fractionation with cyanocobalamin was the highest observed in this study (αC = 1.0132).
DISCUSSION
A carbon isotope fractionation factor of 1.0055 was previously reported for PCE in a mixed culture (34). This value is in the same order of magnitude compared to the fractionation factors determined for the reductive dechlorination of PCE by Desulfitobacterium sp. strain PCE-S. Interestingly, the isotope fractionation by S. multivorans was much lower (αC = 1.0017). One of the primary factors that might cause stable-isotope fractionation is the reaction mechanism of degradation. Cobalamins are important cofactors in microorganisms capable of dehalorespiration (10, 15, 21, 25). In this study, the highest fractionation was observed during reductive dechlorination by cyanocobalamin (αC = 1.0132), which was very similar to previously reported values (38) but higher than that for the microbial systems tested.
Isotope fractionation at the reaction center.
Major carbon and significant chlorine isotope fractionation has been observed during dechlorination of PCE (28). The extent of the carbon isotope effect, which points to a primary kinetic isotope effect, suggests that the rate-limiting step in the dehalogenation reaction is probably controlled by cleavage of a σ bond. The kinetic isotope effect of hydrogen was much lower, suggesting a secondary isotope effect, which indicates that the protonation was not rate limiting during the overall reaction (10) and may occur subsequent to the dehalogenation. Identification of chloroethenylcobalamins as intermediates provides evidence for the formation of a covalent cobalt-carbon bond during dehalogenation (13, 16, 27, 35, 36). The chlorine pattern of the chloroethenylcobalamin (13) indicates that a chlorine has already been released, suggesting an irreversible reaction step. Therefore, the subsequent cleavage of the carbon-cobalt bond is probably not a rate-limiting step in the dehalogenation reaction.
Partitioning factors.
Enzyme-catalyzed reactions involve a sequence of steps which may be represented by binding of the substrate to the enzyme in the first step and dissociation of the product from the enzyme in the last step. Although the number of intermediate steps in unidirectional biochemical reactions may be unknown, the kinetic isotope fractionation should be caused by the first irreversible reaction step in the reaction sequence associated with the conformational change of a chemical bond. Other steps controlling the overall kinetic of the reaction, such as uptake of contaminants by the organism and binding to the enzyme, may affect the extent of isotope effects as represented in Fig. 3 (30).
FIG. 3.

Scheme showing that the resistance of membranes can modify the substrate flow into the cell and may lead to nonisotope equilibrium conditions between substrate in the cell (Sin) and the medium (Sout). Furthermore, the formation of the enzyme substrate complex can be a rate-limiting step in the reaction (ES1, ESn) before the biochemical reaction takes place.
Rate limitations of single steps preceding the biochemical dehalogenation reaction can be quantified calculating the partitioning factor. The partitioning factor is a measure of the rate of the key step in the isotope fractionation relative to the rates of all preceding steps in the reaction sequence. In a two-step reaction, the partitioning factor (P) can be quantified according to O′Leary (30) using equation 3, below, assuming that the preceding reaction steps are rate limiting to some extent but not associated with a significant isotope effect. This assumption is reasonable because preceding reaction steps such as uptake and the binding of a substrate are usually not associated with a change of bond conformation. Equation 3 is as follows:
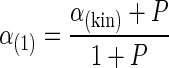 |
(3) |
where α(1) is the fractionation factor of the reaction sequence and αkin is the fractionation factor of the biochemical reaction. The rearrangement of the equation gives equation 4, as follows:
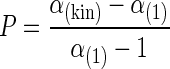 |
(4) |
This model may be used to describe the impact on the isotope fractionation by other factors controlling the overall reaction kinetic and may be used as a simplification to compare the extent of various kinetic steps affecting isotope fractionation in a complex reaction.
Rate limitation affecting isotope fractionation.
The comparison of the fractionation by the enzyme, cobalamin, crude extracts, and growing cultures demonstrates that factors others than the chemical reaction may influence the extent of isotope fractionation which can be quantified by the partitioning factor as summarized in Table 1. With the assumption that the isotope fractionation is caused by the dehalogenation reaction at the cobalamin, significant primary isotope effects may be masked by physiological factors, lowering the extent of the overall observed isotope fractionation. Hence, it is extremely difficult to analyze the transition state and mechanism of the biochemical reaction just by using isotope fractionation factors obtained from culture experiments.
The isotope fractionation by the PCE reductive dehalogenase of S. multivorans was about 1 order of magnitude lower than that observed for cobalamins, although a corrinoid is the reactive center of the enzyme catalyzing the dehalogenation reaction (25). It should, however, be noted that the cobalamin cofactor of the PCE dehalogenase of this organism is a novel and unusual type of corrinoid (11). The cofactor of tetrachloroethene reductive dehalogenase of S. multivorans is norpseudo-B12, a new type of a natural corrinoid (11).
The calculated partitioning factor for carbon isotope fractionation by the enzyme was 6.8, representing a reduction of the isotope effect by 87% compared to that with cobalamin (Table 1). This implies that the protein environment of the cofactor and/or the cofactor's structure affects the fractionation and thus limits the rate of the dehalogenation reaction. The molecular structure of the enzyme is not known, but probably binding of the reactant (PCE) to the enzyme complex and coordination of the reactant in the reactive center affects the rate of the overall reaction. Sorption did not affect the isotope composition significantly (F.-D. Kopinke, personal communication); however, sorption may affect the reaction kinetic of dehalogenation. Alternatively, the coordination of the reactant in the enzyme complex can change activation energy and thus the reaction kinetic and may thereby affect the isotope fractionation. Nevertheless, association of the cofactor and binding of the substrate to the enzyme obviously depresses the extent of isotope fractionation, as was found for the PCE reductive dehalogenase of S. multivorans.
Comparing the carbon fractionation of PCE by the different systems, an increase of αC is observed with a decrease in cell integrity. In Desulfitobacterium sp. strain PCE-S, fractionation by crude extracts was higher than that for the growing cells (Table 1; Fig. 2A), with partitioning factors of 0.5 and 1.5, respectively, corresponding to a reduction of the isotope effect of 33 to 60% compared to those for the cyanocobalamin. The partitioning factors calculated for the reduction of isotope fractionation in cell extracts (12.6) and growing cells (30.4) of S. multivorans were even higher with a reduction of 92 to 97% compared to those for cyanocobalamin (Table 1).
The PCE reductive dehalogenases of S. multivorans and of Desulfitobacterium sp. strain PCE-S are considered to be located inside the cytoplasm (19, 20, 26). Although the order of magnitude of fractionation is different, the effect of the membrane and/or cell wall appears to be similar in both S. multivorans and Desulfitobacterium sp. PCE-S, with reductions in isotope fractionation of 56 and 41%, respectively, comparing growing cells to crude extracts. Therefore, the membrane may act as a barrier separating PCE pools inside and outside of the cell. If there is a transport limitation over the membrane, it can be expected that the isotope discrimination will be decreased due to a nearly complete consumption of the PCE available inside the cell. Similar effects are known from isotope discrimination during CO2 fixation where transport limitations upon uptake of CO2 affect the isotope discrimination in green plants (29). Transport limitation may also account for the differences between crude extracts and purified enzyme in the experiments with S. multivorans. The specific activity of the PCE reductive dehalogenase of S. multivorans was about four times the specific activity of that of the PCE reductive dehalogenase of Desulfitobacterium sp. strain PCE-S (7), correlating with the observed differences in isotope fractionation. It seems worth mentioning, though, that the PCE dehalogenase of Desulfitobacterium sp. strain PCE-S contains a different type of corrinoid cofactor than the PCE dehalogenase of S. multivorans (11; A. Siebert, personal communication). This difference might also account for the different results obtained for the two microorganisms.
Morasch et al. (22) showed that the hydrogen isotope fractionation of toluene (m/z 92) and toluene labeled with eight deuterium atoms (m/z 100) species was about 30% lower in growth experiments than in enzyme assays, also indicating that transport through the membrane may affect isotope fractionation. In our experiments, the transport through the membrane caused a fractionation decrease of about 41 to 56% but the values were in the same order of magnitude as observed before. The relative mass difference between the 13C and 12C isotopes is significantly lower than that in experiments with deuterium-labeled and nonlabeled substrates. Therefore, the decrease in isotope fractionation between crude extract and growing cells is probably not dependent on the molecular mass of the substrate, which may imply that the isotope fractionation upon diffusion is not directly affecting the isotope discrimination significantly. It is more likely that transport limitation, i.e., non-isotope equilibrium conditions between the medium and the active site of the enzyme, may reduce the isotope fractionation.
Fractionation of TCE.
Although fractionation of PCE was small in S. multivorans, fractionation of TCE was obviously larger. This difference in fractionation between PCE and TCE was not observed for Desulfitobacterium sp. strain PCE-S. We cannot explain these observations, and due to fact that TCE was an intermediate and subsequently DCE was formed, the fractionation could not be quantified but will be subject to further detailed studies.
Implication for the assessment of in situ biodegradation using isotope fractionation patterns.
Recently, the compound-specific isotope fractionation has been used to assess the in situ biodegradation of pollutants in aquifers (5, 31, 34). For a quantitative approach, however, selection of an appropriate fractionation factor representative for the active biodegrading microbial community within the aquifer is necessary to relate changes in isotope composition to the concentration. For toluene degradation, a large variation in the extent of fractionation was found depending on the enzyme system used for the initial attack. While no significant fractionation was observed during aerobic toluene degradation, limiting the use of the isotope fractionation concept (23), anaerobic degradation of toluene via the benzylsuccinate pathway, was always associated with a similar significant isotope discrimination, which may allow the quantification of in situ biodegradation under anaerobic conditions (24). The isotope fractionation of S. multivorans was about 1 order of magnitude lower than that observed for Desulfitobacterium sp. strain PCE-S and other reported fractionation factors from mixed cultures (34). This demonstrates that even if similar biochemical degradation pathways are involved, stable isotope fractionation may differ significantly. Therefore, caution is necessary in the application of isotope fractionation factors to quantify microbial PCE degradation in the field. Moreover, systematic work with pure cultures is important to assess the range of isotope fractionation during reductive dehalogenation. The investigation of the microbial consortium in the aquifer may be necessary for the selection of an appropriate fractionation factor for quantitative work. The uncertainty associated in applying isotope fractionation factors for quantification of in situ biodegradation is high; however, in the field of applied science, the selection of a high fractionation factor to calculate in situ degradation using the Rayleigh model will result in an underestimation but not in an overestimation of the in situ biodegradation, which may be sufficient for consulting operations.
Acknowledgments
We thank M. Gehre and U. Günther for assistance in the isotope laboratory of the UFZ. We thank Denitsa Gugova, University of Jena, for providing the purified enzyme and F.-D. Kopinke for discussion.
This project was funded by a European Union Marie Curie Development Host Fellowship (EVK1-CT-2000256120), the Deutsche Forschungsgemeinschaft (grant Di314/8), and the European Union (HPRN-CT-2002-00195).
REFERENCES
- 1.Anonymous. 1995. IAEA-TECHDOC-825. International Atomic Energy Agency, Vienna, Austria.
- 2.Bloom, Y., R. Aravena, D. Hunkeler, E. Edwards, and S. K. Frape. 2000. Carbon isotope fractionation during microbial dechlorination of trichloroethene, cis-dichloroethene, and vinyl chloride: implications for assessment of natural attenuation. Environ. Sci. Technol. 34:2768-2772. [Google Scholar]
- 3.Glod, G., U. Brodmann, W. Angst, C. Holliger, and R. P. Schwarzenbach. 1997. Cobalamin-mediated reduction of cis- and trans-dichloroethene, 1,1-dichloroethene, and vinyl chloride in homogeneous aqueous solution: reaction kinetics and mechanistic considerations. Environ. Sci. Technol. 31:3154-3160. [Google Scholar]
- 4.Griebler, C., L. Adrian, R. U. Meckenstock, and H. H. Richnow. 2004. Stable carbon isotope fractionation during aerobic and anaerobic transformation of trichlorobenzene. FEMS Microbiol. Ecol. 48:313-321. [DOI] [PubMed] [Google Scholar]
- 5.Griebler, C., M. Safinowski, A. Vieth, H. H. Richnow, and R. U. Meckenstock. 2004. Combined application of stable carbon isotope analysis and specific metabolites determination for assessing in situ degradation of aromatic hydrocarbons in a tar oil-contaminated aquifer. Environ. Sci. Technol. 38:617-631. [DOI] [PubMed] [Google Scholar]
- 6.Hoefs, J. 1997. Stable isotope geochemistry, 4th ed. Springer-Verlag, Berlin, Germany.
- 7.Holliger, C., C. Regeard, and G. Diekert. 2002. Dehalogenation by anaerobic bacteria, p. 115-158. In M. M. Häggblom and I. D. Bossert (ed.), Dehalogenation: microbial processes and environmental applications. Kluwer Academic Publishers, Norwell, Mass.
- 8.Holliger, C., G. Wohlfarth, and G. Diekert. 1999. Reductive dechlorination in the energy metabolism of anaerobic bacteria. FEMS Microbiol. Rev. 22:383-398. [Google Scholar]
- 9.Hunkeler, D., R. Aravena, and B. J. Butler. 1999. Monitoring microbial dechlorination of tetrachlorethene (PCE) in groundwater using compound-specific stable isotope ratios: microcosm and field studies. Environ. Sci. Technol. 33:2733-2738. [Google Scholar]
- 10.Krasotkina, J., T. Walters, K. A. Maruya, and S. W. Ragsdale. 2001. Characterization of the B12- and iron-sulfur-containing reductive dehalogenase from Desulfitobacterium chlororespirans. J. Biol. Chem. 276:40991-40997. [DOI] [PubMed] [Google Scholar]
- 11.Kräutler, B., W. Fieber, S. Osterman, M. Fasching, K.-H. Ongania, K. Gruber, C. Kratky, C. Mikl, A. Siebert, and G. Diekert. 2003. The cofactor of tetrachloroethene reductive dehalogenase of Dehalospirillum multivorans is norspseudo-B12, a new type of a natural corrinoid. Helv. Chim. Acta 86:3698-3716. [Google Scholar]
- 12.Lee, M. D., J. M. Odom, and R. J. Buchanan, Jr. 1998. New perspectives on microbial dehalogenation of chlorinated solvents: insights from the field. Annu. Rev. Microbiol. 52:423-452. [DOI] [PubMed] [Google Scholar]
- 13.Lesage, S., S. Brown, and K. Millar. 1998. A different mechanism for the reductive dechlorination of chlorinated ethenes: kinetic and spectroscopic evidence. Environ. Sci. Technol. 32:2264-2272. [Google Scholar]
- 14.Luijten, M. L., J. de Weert, H. Smidt, H. T. Boschker, W. M. de Vos, G. Schraa, and A. J. Stams. 2003. Description of Sulfurospirillum halorespirans sp. nov., an anaerobic, tetrachloroethene-respiring bacterium, and transfer of Dehalospirillum multivorans to the genus Sulfurospirillum as Sulfurospirillum multivorans comb. nov. Int. J. Syst. Evol. Microbiol. 53:787-793. [DOI] [PubMed] [Google Scholar]
- 15.Maillard, J., W. Schumacher, F. Vazquez, C. Regeard, W. R. Hagen, and C. Holliger. 2003. Characterization of the corrinoid iron-sulfur protein tetrachloroethene reductive dehalogenase of Dehalobacter restrictus. Appl. Environ. Microbiol. 69:4628-4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCauley, K. M., S. R. Wilson, and W. A. van der Donk. 2003. Characterization of chlorovinylcobalamin, a putative intermediate in reductive degradation of chlorinated ethylenes. J. Am. Chem. Soc. 125:4410-4411. [DOI] [PubMed] [Google Scholar]
- 17.Meckenstock, R. U., B. Morasch, C. Griebler, and H. H. Richnow. 2004. Stable isotope fractionation analysis as a tool to monitor biodegradation in contaminated aquifers. J. Contam. Hydrol. 75:215-255. [DOI] [PubMed] [Google Scholar]
- 18.Michaelis, W., R. Seifert, K. Nauhaus, T. Treude, V. Thiel, M. Blumenberg, K. Knittel, A. Gieseke, K. Peterknecht, T. Pape, A. Boetius, R. Amann, B. B. Jorgensen, F. Widdel, J. Peckmann, N. V. Pimenov, and M. B. Gulin. 2002. Microbial reefs in the black sea fueled by anaerobic oxidation of methane. Science 297:1013-1015. [DOI] [PubMed] [Google Scholar]
- 19.Miller, E., G. Wohlfarth, and G. Diekert. 1997. Comparative studies on tetrachloroethene reductive dechlorination mediated by Desulfitobacterium sp. strain PCE-S. Arch. Microbiol. 168:513-519. [DOI] [PubMed] [Google Scholar]
- 20.Miller, E., G. Wohlfarth, and G. Diekert. 1998. Purification and characterization of the tetrachloroethene reductive dehalogenase of strain PCE-S. Arch. Microbiol. 169:497-502. [DOI] [PubMed] [Google Scholar]
- 21.Miller, E., G. Wohlfarth, and G. Diekert. 1996. Studies on tetrachloroethene respiration in Dehalospirillum multivorans. Arch. Microbiol. 166:379-387. [DOI] [PubMed] [Google Scholar]
- 22.Morasch, B., H. H. Richnow, B. Schink, and R. U. Meckenstock. 2001. Stable hydrogen and carbon isotope fractionation during microbial toluene degradation: mechanistic and environmental aspects. Appl. Environ. Microbiol. 67:4842-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morasch, B., H. H. Richnow, B. Schink, A. Vieth, and R. U. Meckenstock. 2002. Carbon and hydrogen stable isotope fractionation during aerobic bacterial degradation of aromatic hydrocarbons. Appl. Environ. Microbiol. 68:5191-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morasch, B., H. H. Richnow, A. Vieth, B. Schink, and R. U. Meckenstock. 2004. Stable isotope fractionation caused by glycyl radical enzymes during bacterial degradation of aromatic compounds. Appl. Environ. Microbiol. 70:2935-2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neumann, A., A. Siebert, T. Trescher, S. Reinhardt, G. Wohlfarth, and G. Diekert. 2002. Tetrachloroethene reductive dehalogenase of Dehalospirillum multivorans: substrate specificity of the native enzyme and its corrinoid cofactor. Arch. Microbiol. 177:420-426. [DOI] [PubMed] [Google Scholar]
- 26.Neumann, A., G. Wohlfarth, and G. Diekert. 1996. Purification and characterization of tetrachloroethene reductive dehalogenase from Dehalospirillum multivorans. J. Biol. Chem. 271:16515-16519. [DOI] [PubMed] [Google Scholar]
- 27.Nonnenberg, C., W. A. van der Donk, and H. Zipse. 2002. Reductive dechlorination of trichloroethylene: a computational study. J. Phys. Chem. A 106:8708-8715. [Google Scholar]
- 28.Numata, M., N. Nakamura, H. Koshikawa, and Y. Terashima. 2002. Chlorine isotope fractionation during reductive dechlorination of chlorinated ethenes by anaerobic bacteria. Environ. Sci. Technol. 36:4389-4394. [DOI] [PubMed] [Google Scholar]
- 29.O'Leary, M. H. 1981. Review: Carbon isotope fractionation in plants. Phytochemistry 20:553-567. [Google Scholar]
- 30.O'Leary, M. H., and C. J. Yapp. 1978. Equilibrium carbon isotope effect on a decarboxylation reaction. Biochem. Biophys. Res. Commun. 80:155-160. [DOI] [PubMed] [Google Scholar]
- 31.Richnow, H. H., E. Annweiler, W. Michaelis, and R. U. Meckenstock. 2003. Microbial in situ degradation of aromatic hydrocarbons in a contaminated aquifer monitored by carbon isotope fractionation. J. Contamin. Hydrol. 65:101-120. [DOI] [PubMed] [Google Scholar]
- 32.Roeske, C. A., and M. H. O'Leary. 1984. Carbon isotope effects on the enzyme-catalyzed carboxylation of ribulose bisphosphate. Biochemistry 23:6275-6284. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt, T. C., L. Zwank, M. Elsner, M. Berg, R. U. Meckenstock, and S. B. Haderlein. 2004. Compound-specific stable isotope analysis of organic contaminants in natural environments: a critical review of the state of the art, prospects, and future challenges. Anal. Bioanal. Chem. 378:283-300. [DOI] [PubMed] [Google Scholar]
- 34.Sherwood Lollar, B., G. F. Slater, B. Sleep, M. Witt, G. M. Klecka, M. Harkness, and J. Spivack. 2001. Stable carbon isotope evidence for intrinsic bioremediation of tetrachloroethene and trichloroethene at area 6, Dover Air Force Base. Environ. Sci. Technol. 35:261-269. [DOI] [PubMed] [Google Scholar]
- 35.Shey, J., C. M. McGinley, K. M. McCauley, A. S. Dearth, B. T. Young, and W. A. van der Donk. 2002. Mechanistic investigation of a novel vitamin B-12-catalyzed carbon-carbon bond forming reaction, the reductive dimerization of arylalkenes. J. Org. Chem. 67:837-846. [DOI] [PubMed] [Google Scholar]
- 36.Shey, J., and W. A. van der Donk. 2000. Mechanistic studies on the vitamin B-12-catalyzed dechlorination of chlorinated alkenes. J. Am. Chem. Soc. 122:12403-12404. [Google Scholar]
- 37.Slater, G. F., H. S. Dempster, B. S. Lollar, and J. Ahad. 1999. Headspace analysis: A new application for isotopic characterization of dissolved organic contaminants. Environ. Sci. Technol. 33:190-194. [Google Scholar]
- 38.Slater, G. F., B. Sherwood Lollar, S. Lesage, and S. Brown. 2003. Carbon isotope fractionation of PCE and TCE during dechlorination by vitamin B12. Ground Water Monit. R. 23:59-67. [Google Scholar]
- 39.Song, D. L., M. E. Conrad, K. S. Sorenson, and L. Alvarez-Cohen. 2002. Stable carbon isotope fractionation during enhanced in situ bioremediation of trichloroethene. Environ. Sci. Technol. 36:2262-2268. [DOI] [PubMed] [Google Scholar]
- 40.Vieth, A., J. Müller, G. Strauch, M. Kästner, M. Gehre, R. U. Meckenstock, and H. H. Richnow. 2003. In-situ biodegradation of tetrachloroethene and trichloroethene in contaminated aquifers monitored by stable isotope fractionation. Isot. Environ. Health Stud. 39:113-124. [DOI] [PubMed] [Google Scholar]
- 41.Wohlfarth, G., and G. Diekert. 1999. Reductive dehalogenases, p. 871-893. In R. Banerjee (ed.), Chemistry and biochemistry of B12. John Wiley & Sons, New York, N.Y.
- 42.Zehnder, A. J. B., and K. Wuhrmann. 1976. Titanium (III) citrate as a nontoxic oxidation-reduction buffering system for the culture of obligate anaerobes. Science 194:1165-1166. [DOI] [PubMed] [Google Scholar]