

SUMMARY
Human brain size changes dynamically through early development, peaks in adolescence, and varies up to 2-fold among adults. However, the molecular genetic underpinnings of interindividual variation in brain size remain unknown. Here, we leveraged postmortem brain RNA sequencing and measurements of brain weight (BW) in 2,531 individuals across three independent datasets to identify 928 genome-wide significant associations with BW. Genes associated with higher or lower BW showed distinct neurodevelopmental trajectories and spatial patterns that mapped onto functional and cellular axes of brain organization. Expression of BW genes was predictive of interspecies differences in brain size, and bioinformatic annotation revealed enrichment for neurogenesis and cell-cell communication. Genome-wide, transcriptome-wide, and phenome-wide association analyses linked BW gene sets to neuroimaging measurements of brain size and brain-related clinical traits. Cumulatively, these results represent a major step toward delineating the molecular pathways underlying human brain size variation in health and disease.
Graphical abstract
In brief
Seidlitz et al. identify genes differentially expressed in individuals with larger or smaller brains that are enriched in growth signaling pathways and are highly expressed in cell types critical for human development and primate evolution. Translational results in a “real world” clinical biobank demonstrate promise for more informed polygenic scoring.
INTRODUCTION
The size of the cerebral cortex varies approximately 100-fold across primate species1,2 and 2-fold across adult humans.3 While metrics of total brain size have historically been assessed postmortem using tissue weight or volume, highly correlated measurements including volume and surface area (SA) are readily quantifiable with modern non-invasive neuroimaging methods. Distinct from head circumference, which plateaus around 5–7 years, total brain volume peaks during adolescence at around 13–15 years. This growth in absolute brain size is accompanied by an increase in the variability of brain size among humans that also peaks in adolescence, and arises from differential contributions of underlying tissue types with discrete growth trajectories.4 Many studies have demonstrated high twin-based and single-nucleotide polymorphism (SNP) heritability of brain morphology, with many global brain size indices reaching above 50% (twin) and 25% (SNP) of interindividual variance explained by genetic factors.4–7 Brain size has also been implicated in multiple clinically relevant contexts, including case-control differences in both neuropsychiatric and neurodegenerative diseases, as well as dimensional associations with anthropometric and cognitive traits.4,8
Large-scale neuroimaging genetic studies have begun to determine the polygenic architecture of specific components of human brain size, such as total volume and cortical SA, which are highly genetically correlated.6,7,9,10 Collectively, these studies have implicated genes that are involved in critical developmental signaling pathways (e.g., Wnt, PI3K-AKT), are highly expressed during the prenatal period, and are related to neuropsychiatric and neurodegenerative disorders as well as polygenic traits (e.g., cognition). In addition to neuroimaging genetics, progress toward identifying candidate gene sets and related molecular processes underlying brain size has stemmed from convergent evidence across a diverse body of work, including transcriptome-wide association studies (TWAS) of neuroimaging phenotypes in humans,11 blood transcriptomic signatures of neuroimaging-derived brain size metrics in humans,12,13 and in vitro brain size-related effects in rare neurogenetic and cephalic disorders.14–16 In parallel, recent postmortem brain RNA sequencing data aggregation efforts across tissue banks, such as the PsychENCODE (PEC) project,17 have led to the discovery of major transcriptomic signatures of brain evolution and development.18,19 Although not direct examinations of brain size, the phylogenetic and ontogenetic dynamics investigated by these studies are intertwined with pronounced changes in brain size across primate species and developmental epochs. In light of these informative prior results, we aimed to directly investigate the functional molecular correlates of interindividual variation in human brain size.
The current study capitalizes on the fact that a direct and accurate measure of brain size—ex vivo brain weight (BW)—is commonly quantified during postmortem autopsy across tissue banks. We leverage reported BW and associated transcriptomic data to perform a genome-wide analysis of transcriptomic associations with BW, in a sample of 2,531 individuals across multiple widely accessed postmortem brain transcriptomic datasets (PEC discovery n = 1,670, GTEx and ROSMAP replication total n = 861). We discover replicable associations across independent datasets, identifying 928 genes reaching genome-wide significance that are upregulated in individuals with higher BW (BW positive or BW+; n = 442 genes) or upregulated in individuals with lower BW (BW negative or BW−; n = 486 genes). These distinct gene sets show distinct spatiotemporal and relative expression effects (directly comparing expression of BW+ genes versus BW− genes; STAR Methods) across non-human primates, cell types, and neuropsychiatric disease. Moreover, using an integrative multiscale approach, we pinpoint genetic regulatory mechanisms that link transcriptomic signatures to phenotypic variation in both neuroimaging-derived and brain-related traits. Our comprehensive study represents a significant advance in elucidating the molecular landscape underlying variability in human brain size.
RESULTS
Brain size is associated with differential brain gene expression across datasets
We first confirmed the validity of postmortem BW as a proxy for brain size. Using comprehensive lifespan neuroimaging models of total cerebrum volume (TCV)4 and previous measurements of average brain density,20 we found a robust relationship for predicted versus measured BW aggregated across datasets (n = 3,689, Pearson’s r = 0.92, p < 0.0001; Figure S1; STAR Methods). We saw an almost identical relationship using total cortical SA in lieu of TCV to predict BW measurements (r = 0.92, p < 0.0001). In line with many previous studies on brain morphology, we also observed a significant difference in BW between sexes across datasets (males > females, t = 25.772, Cohen’s d = 0.76, p < 0.0001).
Next, we performed a transcriptome-wide differential expression (DE) analysis of interindividual variation in human BW (STAR Methods). Multiple linear regression was used to model the relationship between BW and gene expression (n = 25,774 genes) from RNA sequencing in postmortem brain tissue (sampled from the frontal cortex; STAR Methods). In the discovery dataset (PEC, n = 1,670 subjects), 928 genes showed significant genome-wide DE (pBonferroni < 0.05, i.e., 0.05/25,774, p < 1.94e–6; partial r range = 0.16–0.18, absolute partial r range = 0.05–0.18; Figures 1A and S1; Table S1; STAR Methods). Of these genes, 442 exhibited a positive association with BW (i.e., gene expression tended to be greater in individuals with higher BWs), while 486 genes exhibited a negative association with BW (i.e., gene expression tended to be greater in individuals with lower BWs). These prioritized genes are henceforth referred to as BW+ and BW− genes, respectively. The large majority of both BW+ (n = 379, 86%) and BW− (n = 467, 96%) gene sets had higher than average expression across individuals (Figure S4), with a small percentage of BW genes showing the opposite pattern—BW+ and BW− genes having lower expression across individuals with higher or lower BW (n = 69, 14% and n = 19, 4%, respectively). Restricting our analyses to the top expressed genes across subjects (n = 15,634 genes CPM > 1) also showed consistent results (786 [85%] of significant genes overlapping). Overall, these results suggest that the DE reflected by the BW+ and BW− gene sets can be interpreted as opposing effects of actively expressed genes.
Figure 1. Differential expression of genes associated with an increase or decrease in brain weight across PsychENCODE subjects.
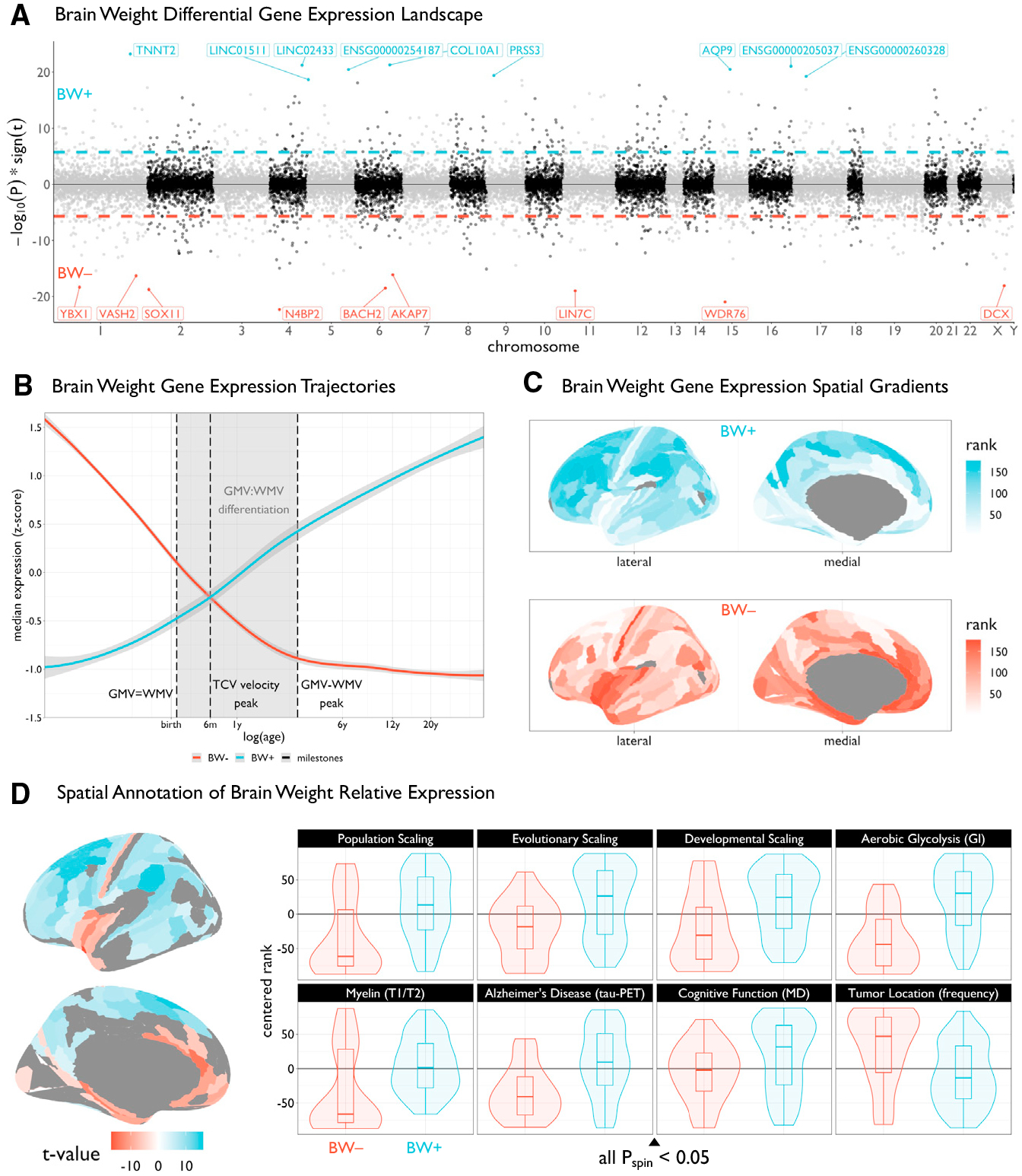
(A) Miami plot showing the relationship between brain weight (BW) and expression across all genes (depicting −log10 scaled p values multiplied by the sign of the t value effect), ordered by chromosome. Dashed lines represent the genome-wide significance threshold (pBonferroni < 0.05). For visualization, the top ten genes are labeled for both BW positive (BW+, blue) and BW negative (BW−, red) sets.
(B) Trajectories of median expression of BW− and BW+ gene sets across development, using the BrainSpan atlas. Black vertical dashed lines represent neuroimaging-derived milestones, highlighting the critical period of gray matter volume (GMV) and white matter volume (WMV) differentiation (shaded area). This period delineates the time when GMV and WMV are equal until the peak difference between GMV and WMV. The BW-associated gene sets show opposing developmental trajectories, intersecting when the rate of total cerebrum volume (TCV) growth peaks, around 6 postnatal months. Shaded area around trajectories denotes 95% confidence intervals.
(C) Plots of the brain surface showing the differential spatial patterns of the suprathreshold genes (from A) in the Allen Human Brain Atlas (AHBA). Colors denote the rank (from lowest to highest) of cortical regions in terms of median expression of genome-wide significant genes. AHBA probes were resampled from native coordinates to a cortical parcellation (STAR Methods).
(D) Left: cortical map of regions showing significant BW+ versus BW− gene expression (pBonferroni < 0.05) in the AHBA, i.e., the statistical comparison of the maps in (B). Right: box-violin plots showing the distributions of diverse multimodal neuroimaging maps across the significant BW+ and BW− regions (see key resources table and Figure S6). For all scaling maps, local/regional surface area was modeled as a function of total cortical surface area across species (evolution), human development, and human subjects (population). All box-violin plots show median and interquartile range (IQR) with whiskers denoting 1.5 × IQR.
We performed multiple sensitivity analyses across the PEC dataset, showing highly convergent results (Figure S4). First, using a Freedman-Lane permutation test21 to assess robustness to a nonparametric hypothesis test where transcriptomic data were permuted across individuals, all 928 BW genes remained significant (all pBonferroni < 0.05, 25,000 permutations). In addition, the total number of significant BW genes was greater than the number of BW genes under the null hypothesis (p < 2e–6, 25,000 permutations). Next, using a cell-type deconvolution approach with combined reference single-cell sequencing datasets,22 we found that interindividual variability in the proportion of neurons (which approximates neuron density) was not significantly related to BW (t = −1.54, p = 0.12). In addition, when including principal-component scores based on genotype (to account for possible ancestry effects) in the BW gene models in the PEC sample, 619 of the BW genes remained significant (pBonferroni < 0.05), with a high correlation to original model coefficients (r = 0.92, p < 0.0001; Figure S2). Furthermore, there are known inter-relationships between brain size and anthropometrics,23 which we observed in our data between BW and height, as well as between BW and body weight (n = 1,206; partial r2 from regression model = 0.09 and 0.03, respectively; both p < 0.0001). Consistent with these significant but relatively low effect sizes, BW models including height and weight as covariates (available only in a subset of individuals) were highly correlated to the original models (cross-gene correlation, r = 0.89, p < 0.0001; Figure S2). Comparing genes with significant associations (pBonferroni < 0.05) across BW and anthropometrics supported the existence of specific associations with BW—507 of 928 genes were not significant for height or weight (Figure S2).
To further examine the role of age in the BW expression results in the PEC dataset (age range = 0–90 years, median = 53 years, standard deviation = 21.2 years), we performed a post hoc association analysis to examine the interaction between BW and age on expression for each gene. An additional 222 genes showed significant (pBonferroni < 0.05) BW-by-age interaction effects (linear or quadratic age terms). There were 130 genes with positive coefficients denoting the effect of BW on expression increasing with age, and 92 genes with negative coefficients denoting the effect of BW on expression decreasing with age (Figure S3). In contrast to the defined BW gene sets, whose expression shows positive (BW+) or negative (BW−) associations with BW across the lifespan, these gene sets identified for having significant age interaction effects show an opposite effect in early life. Specifically, genes with positive interactions initially show a negative relationship between expression and BW in early life, and then positive relationships across older age bins; whereas genes with negative interactions initially show a positive relationship between expression and BW in early life, and then negative relationships across older age bins. Thus, we find genes whose changing expression patterns are differentially related to BW across the lifespan. These genes are detailed in Table S1.
Furthermore, a meta-analytic approach examining the 733/928 significant BW genes available in two replication datasets (GTEx: n = 227 subjects, n = 1,327 samples across 12 brain regions, STAR Methods; ROSMAP: n = 634 subjects and samples, all frontal cortex) revealed highly similar BW gene sets: 329/733 PEC BW genes were nominally significant (uncorrected p < 0.05; weighted Sime’s method24) compared with approximately 37 genes that would be expected to replicate by chance under the null hypothesis of no association.9 In addition, combining the discovery and replication datasets revealed highly similar BW gene sets to those identified by the discovery dataset alone (weighted Sime’s method: 733/733 overlapping genes, pBonferroni < 0.05), with an additional 116 genes meeting significance under the combined framework (pBonferroni < 0.05; Table S1).
We performed additional assessments of replicability in addition to the meta-analytic approach for replicating the PEC results in GTEx and ROSMAP. Rank-rank hypergeometric overlap25 analysis confirmed significant replication in both datasets: GTEx (n = 227 subjects; Spearman r = 0.39, p < 0.0001; 417/846 significant overlapping genes) and ROSMAP (n = 634 subjects; Spearman r = 0.34, p < 0.0001; 525/847 significant overlapping genes).
Brain size-associated genes have distinctive neurodevelopmental and anatomical profiles
Comparing BW+ and BW− genes revealed opposing neurodevelopmental trajectories in tissue samples spanning the prenatal period through young adulthood in the BrainSpan atlas26 (Figure 1B). Moreover, the BW sets showed striking overlap with previously defined gene sets based on developmental trajectory across the perinatal period in an independent dataset with dense prenatal sampling (BW+ and “rising” genes, BW− and “falling” genes; Figure S3,27). For example, OPALIN (BW+ gene) was the highest-ranked rising gene based on the slope of expression, and SOX11 (BW− gene) was the fourth highest-ranked falling gene. The fact that neither BW gene set was relatively highly expressed just after birth, at the peak of overall brain growth and during a critical period of constituent tissue class differentiation (Figure 1B), is consistent with previously established findings of low overall gene expression variation after the late-fetal transition.18
In line with the neurodevelopmental results, BW+ and BW− gene sets revealed opposing spatial topographies of average gene expression in densely sampled postmortem adult human brain tissue (Figure 1C),28 with BW+ genes showing high expression in association cortical regions and BW− genes showing high expression in sensorimotor and paralimbic/limbic cortical regions. Therefore we computed a map of relative expression differences, comparing BW+ versus BW− genes across cortical regions. Out of 177 cortical regions with sufficient samples (STAR Methods), 115 (65%) showed significantly greater relative expression of either BW+ or BW− gene sets (two-tailed t test per region, pBonferroni < 0.05 across regions). Out of 12 subcortical regions, 10 showed significant BW− relative expression differences (Table S2). These differences in BW+/−relative expression demonstrated extreme regional divergence and were far greater than expected by chance given the number of BW+ and BW− genes (ppermutation < 0.0001). Moreover, these regions of significant BW+/− relative expression (Figure 1D, left) showed different patterns of correspondence with multiple maps of brain organization, function, and disease (all pspin < 0.05; Figure 1D, right)—including axes of fine-grained cortical SA expansion,29,30 aerobic glycolysis,31 myelin-related topography,32 cognitive function,33 tau distribution in Alzheimer’s disease (AD) patients,34 and tumor location frequency across patients with glioblastoma and low-grade glioma.35 In addition, the BW+/− relative expression map was robust to an alternative cortical parcellation (Figure S3), and regions with high relative expression of BW genes showed greater morphological effects across diagnostic categories and showed differential patterns of neurodevelopmental growth. Specifically, the spatial topography of BW+/− relative expression was correlated with regional SA alterations across neuropsychiatric disorders (r = 0.52, p = 0.0017, ppermutation < 0.0001, pspin < 0.000136); as well as regional variation in the age at peak gray matter volume during neurodevelopment (r = −0.47, p = 0.0046, ppermutation < 0.0001, pspin < 0.00014).
Cortical regions with BW+/− relative expression differences were also affiliated with specific neurophysiological, functional, and cytoarchitectonic classes. Using available spatially comprehensive positron emission tomography (PET) neuroradiography data,37 areas of high relative expression of BW− genes showed greater densities of dopaminergic and serotonergic receptors compared with areas of high BW+ relative expression (Figure S4). Moreover, there was an abundance of BW− regions in primary sensory cortices containing short distance (<80 mm) white matter fibers as estimated with diffusion MRI, and an abundance of BW+ regions in higher-order association cortices with long distance white matter fibers (Figures S4 and S5). This aligns with previous neuroimaging genome-wide association studies (GWASs) that have identified axon guidance molecules (SEMA3D and ROBO3) associated with diffusion MRI phenotypes of the crossing pontine tract9—a nexus of both intra-cerebellar long-range corticopontine fibers. Relatedly, a 3D-recon-structed Merker-stained postmortem human brain38 revealed a highly diverging laminar architecture of relative cell density, but not relative thickness, in layer IV for BW− regions and layers V/VI for BW+ regions,39 a pattern which was also validated using spatial RNA sequencing data (Figure S5).40 Collectively, these results are aligned with century-old observations on cytoarchitectonic variation across the cerebral cortex,41 demonstrating the convergence of BW− regions with the relatively small number of regions in “heterotypic” cortex and BW+ regions with the more abundant canonical “homotypic” six-layered cortical regions (Figure S5).
Brain size-associated genes show cross-species and cell-type specificity
Genes that are positively associated with variability in brain size among humans may also be implicated in brain size on an evolutionary timescale. Using data from a parallel cross-species study in humans and non-human primates (http://evolution.psychencode.org/; STAR Methods; n = 16 regions; n = 6 humans [H], not included in main analyses; n = 5 chimpanzees [C]; n = 5 macaques [M]),19 we sought to determine whether phylogenetic variability in brain size (PBS+ = H > C > M; PBS− = H < C < M) was associated with DE in BW genes across species. Across brain regions, 726 genes showed patterns of interspecies DE in accordance with PBS (pBonferroni < 0.05), and over 60% of genes previously identified as differentially expressed in humans42 showed specific PBS directional effects (STAR Methods). We found a general consistency between overlapping BW and PBS genes (n = 366 significant gene-by-region effects, n = 110 unique genes) such that BW+ aligned with PBS+ and BW− aligned with PBS-, respectively (73% congruence, ppermutation < 0.0001; Figure 2A). Considering all homologous genes across species, we found that there was a moderate but significant relationship between BW effect size and PBS effect size (Spearman r = 0.19, ppermutation < 0.0001). However, it is interesting to note that, in macaques, there were no regions with significant BW+ relative expression (Table S2),43 which is consistent with previous results showing human-specific expansion of association cortical regions29 (where BW+ genes show greater relative expression differences in humans; Figure 1D).
Figure 2. Genes associated with BW show differential effects across species.
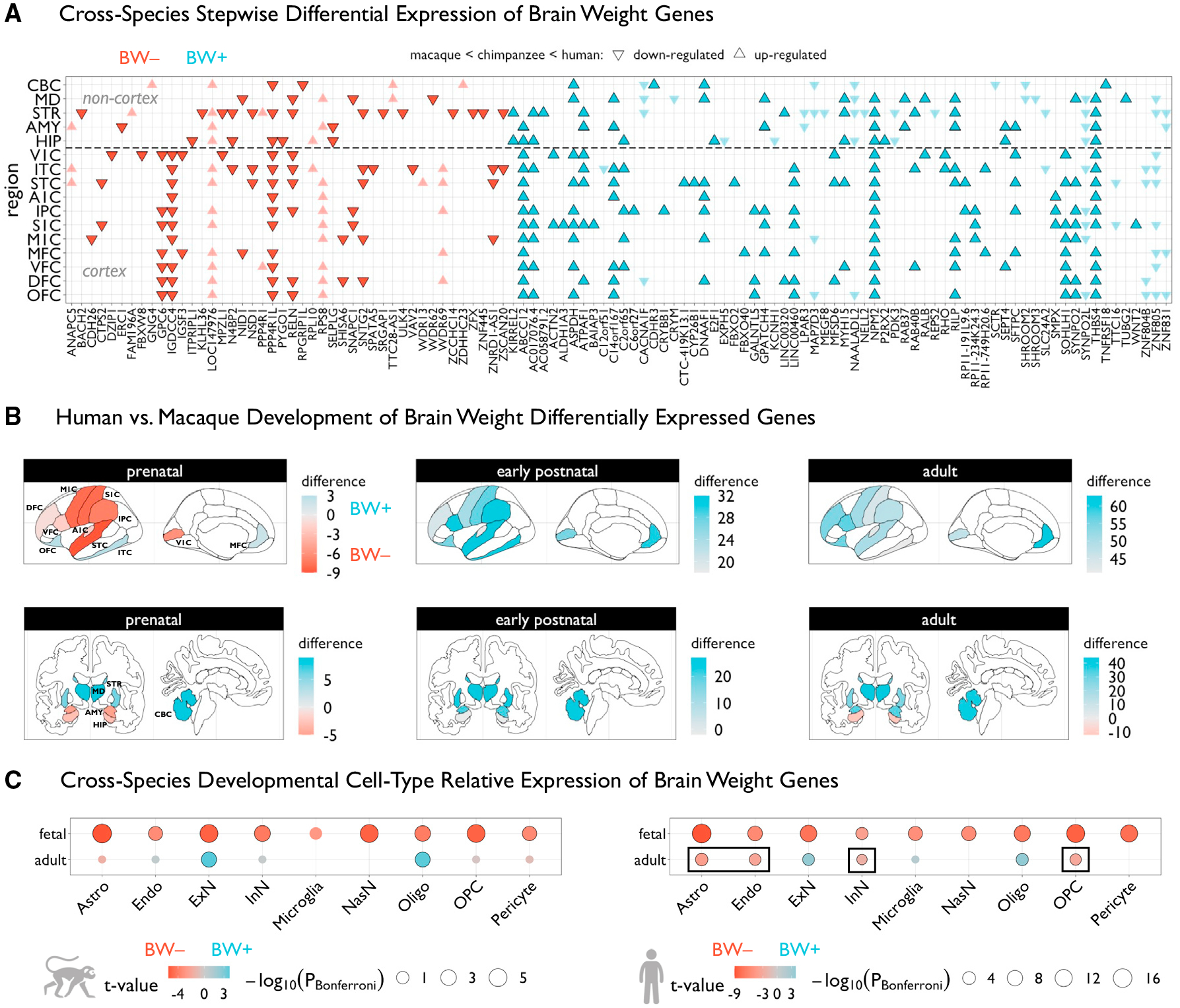
(A) Grid plot showing BW-associated genes that are also significantly differentially expressed in humans relative to chimpanzees and macaques (pBonferroni < 0.05), across 16 brain regions (11 neocortical areas). These are BW-associated genes that were previously defined42 as having higher or lower expression compared with the two species collectively, and reanalyzed to assess stepwise interspecies effects that reflect the absolute differences in brain size between species (i.e., up-regulated implies gene expression humans > chimpanzees > macaques, while downregulated implies gene expression humans < chimpanzees < macaques). Triangles represent directions of effects and colors denote the respective BW gene set. Genes that are highlighted show congruent directional effects in respective BW sets (i.e., BW+ and upregulated in humans, and vice versa).
(B) Brain plots of differences in counts of number of BW+ or BW− genes that were defined to be significantly differentially expressed in humans relative to macaques each of three developmental epochs—a positive regional difference (blue) indicates that BW+ genes tend to be upregulated in humans in that region in a given developmental epoch, while a negative difference (red) indicates that BW− genes tend to be upregulated. The same 16 regions from (A) are shown anatomically, based on manual assignment using a common human atlas.44
(C) Differential expression of BW+ versus BW− genes across individual cell types, using cell-specific RNA sequencing data in fetal and adult samples from macaques (left) and humans (right). BW− relative expression (red) indicates that BW− genes are more highly expressed in that cell type compared with BW+, whereas BW+ relative expression (blue) indicates the opposite effect. Black outlines denote significant effects (pBonferroni < 0.05). Circles are scaled according to Bonferroni-corrected p values. Black rectangles denote human-specific effects relative to macaques. V1C, primary visual cortex; M1C, primary motor cortex; S1C, primary somatosensory cortex; A1C, primary auditory cortex; ITC, inferior temporal cortex; IPC, inferior parietal cortex; STC, superior temporal cortex; OFC, orbitofrontal cortex; VFC, ventrolateral frontal cortex; DFC, dorsolateral frontal cortex; MFC, medial frontal cortex; STR, striatum; MD, medial dorsal thalamus; AMY, amygdala; HIP, hippocampus; CBC, cerebellar cortex. Astro, astrocytes; Endo, endothelial cells; ExN, excitatory neurons; InN, inhibitory neurons; NasN, nascent neurons; Oligo, oligodendrocytes; OPC, oligodendrocyte precursor cells.
If the neurodevelopmental trajectories of BW+ and BW− genes differ between humans and non-human primates, we would expect to see differences between species in BW+/− relative expression. We leveraged established DE statistics between humans and macaques19 to assess whether there were differences in BW+/− relative expression across brain regions and developmental epochs were similar across species. Greater numbers of BW− genes were differentially expressed during the prenatal epoch and greater numbers of BW+ genes were differentially expressed in the postnatal epochs in humans compared with macaques (Figure 2B). Although there were more differentially expressed BW+ genes in the early postnatal period in humans (Figure S6), there was a small number of exceptions to the general pattern observed in the prenatal period (generally BW− predominant) and the adult period (generally BW+ predominant). Specifically, the striatum and thalamus showed more BW+ differentially expressed genes prenatally, and the hippocampus showed more BW− differentially expressed genes in adult humans. This relative heterochronicity in the abundance of BW differentially expressed genes in non-cortical brain regions could reflect the protracted neurogenesis of adult hippocampal compared with thalamic neurons.45 Overall, these results suggest that BW gene sets are enriched for genes that show divergent expression in humans compared with non-human primates, with BW− and BW+ genes showing increased expression in early and late periods, respectively (where each set is most highly expressed across human development).
We hypothesized that BW+/− relative expression differences could be related to the relative abundance of specific cell types among brain regions and across developmental and evolutionary timescales. In single-cell (fetal) and single-nucleus (adult) RNA sequencing data18 in macaques http://evolution.psychencode.org/ and humans http://development.psychencode.org/, there was greater BW− expression compared with BW+ expression across most macaque prenatal cell types and all human prenatal cell types, including human-specific progenitor cell subtypes (Figures 2C and S6). Overall, in both species, there were fewer adult cell types with BW+/− relative expression differences. Whereas BW− genes did not show any relative expression differences in adult macaques, they demonstrated greater relative expression in human endothelial and oligodendrocyte progenitor cells, as in their respective prenatal cell type counterparts. In both species, BW+ genes showed greatest relative expression in excitatory neurons (primarily supragranular layer subtypes) and oligodendrocytes, in contrast to BW− relative expression in those prenatal counterparts (Figures 2C and S6). These results are consistent with spatiotemporal and laminar findings (Figure 1), as well as the differential trajectories of human gene expression across BW+ (low prenatal, high postnatal) and BW− (high prenatal, low postnatal) gene sets. This coincides with observed protracted maturation of cortical myelin4 as well as the relative neuronal density of cortical regions with expanded supragranular layers.41
The BW cell-type divergence is further reflected by the predominance of prenatal BW− differentially expressed cell types within species versus a predominance of postnatal BW+ differentially expressed genes across species. The human-specific neotenic BW− DE in inhibitory neurons and astroglial cell types is in line with the abovementioned findings of BW− relative expression between species in the adult hippocampus—a structure that not only is preferentially enlarged in humans relative to non-human primates46 but is also highly connected (via specified GABAergic inhibitory circuits47) to functionally related areas of significant BW− relative expression (see Figure 1D). Recent work has also demonstrated the convergent roles of inhibitory neuron and astrocyte marker genes (e.g., BW− genes ROBO1 and AQP4, respectively) on synaptic plasticity in the hippocampus, identifying a potential underlying link between brain size and local circuit function.48,49 Independent human fetal RNA sequencing across brain regions further suggest greater BW− relative expression signatures in the subventricular zone (Figure S7), which is expanded in primates50,51 and in line with the BW− enrichment of neuronal precursor subtypes and perseveration of BW− enrichment in adult human inhibitory neuron subtypes.52
Brain size-associated genes are implicated in neuropsychiatric and neurodevelopmental disorders
Given myriad previous studies showing brain volume abnormalities across a range of neuropsychiatric and neurodegenerative disorders, we predicted that BW− expression associations would be related to DE statistics from postmortem brain case-control studies of these conditions. Akin to genetic correlation analysis—where overlap between significant loci from GWAS does not necessarily imply an overall genetic correlation between two traits (or vice versa)—we would expect to observe “transcriptomic correlations” of DE statistics across all genes if relative patterns of DE are similar (or opposing) between these phenotypes.53 Across shared genes in each condition (n = 4,226 genes), there were significant gene-level correlations between PEC BW− expression statistics and DE statistics from previous studies on neuropsychiatric53 and neurodegenerative54,55 disorders (Figure 3A). Specifically, positive correlations were found with alcohol abuse disorder, whereas negative correlations were found with Parkinson’s disease, AD, autism spectrum disorder (ASD), bipolar disorder (BD), and schizophrenia (SCZ), with no significant correlation with inflammatory bowel disease—a non-neural control. The direction of correlation related to the proportion of intersecting significant BW and DE genes (both thresholded pFDR < 0.05) showing either convergent (positive correlation = BW/DE +/+ or −/−) or divergent (negative correlation = BW/DE +/− or −/+) effects, with five disorders showing significant BW-DE alignment scores (all ppermutation < 0.001; Figure 3B). Moreover, we identified a subset of 36 BW-DE genes shared across disorders showing divergent BW-DE alignment (AD, ASD, BD, SCZ; Figure 3C). This subset of BW-DE genes was almost perfectly separated in terms of the direction of effect, such that BW+ genes were downregulated and BW− genes were upregulated in patient tissue (Figure 3C). In supplemental analyses, we also derived a cumulative BW transcriptome score, which showed case-control differences across diagnostic categories (Figure S7). Consistent with the spatial alignment between the map of BW+/− relative expression and tau-PET (Figure 1D), the cumulative transcriptome score was also associated with indices of tau and amyloid neuropathology in patients with dementia in the independent ROSMAP dataset (supplemental information).
Figure 3. BW gene sets are differentially enriched across disease and functional domains.
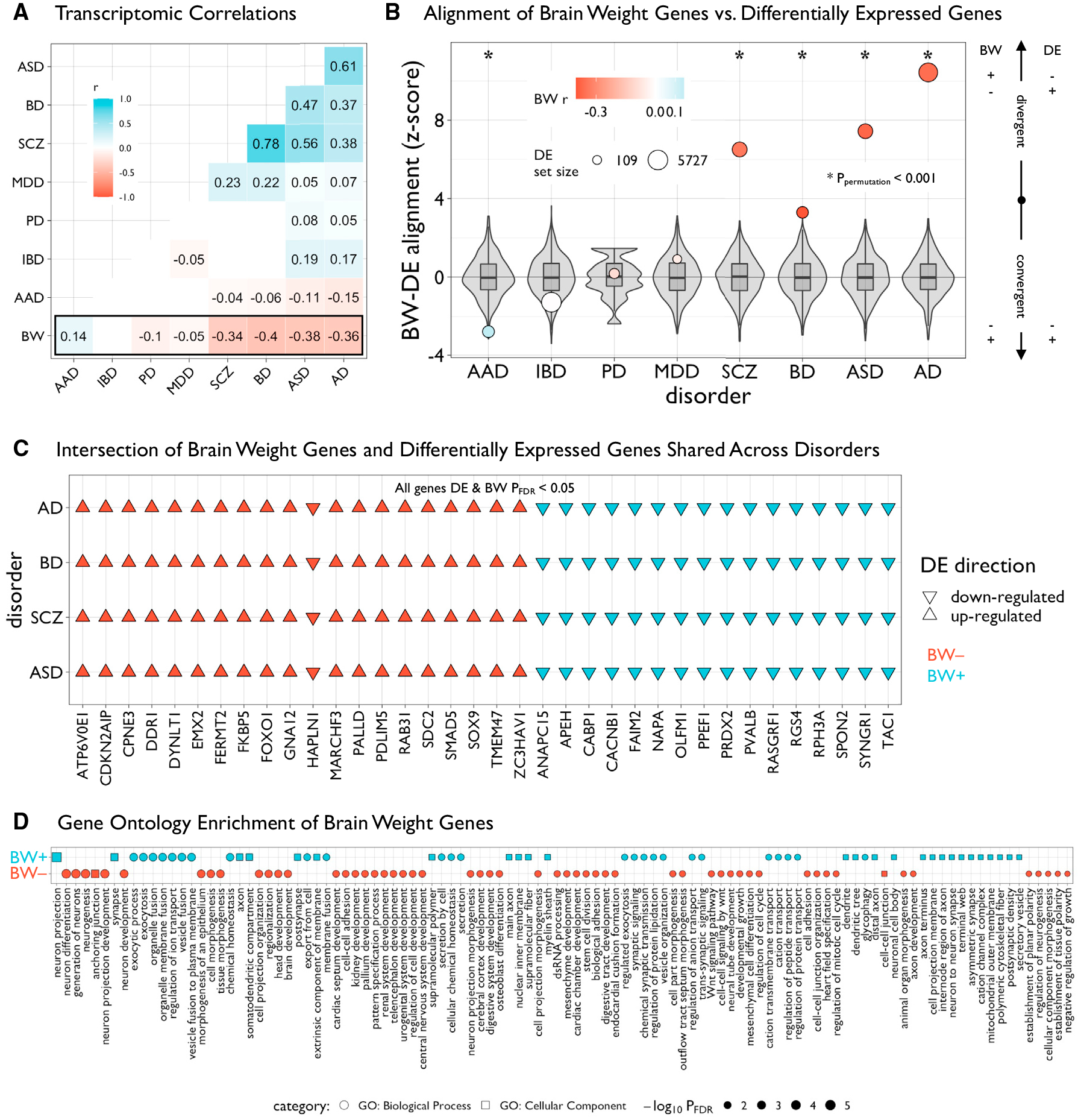
(A) Matrix of transcriptomic correlations of differential expression (DE) statistics in multiple human diseases and BW statistics in the PsychENCODE dataset. Pairwise Pearson’s r coefficients were computed across overlapping genes between datasets (n = 4,226)—only significant (pBonferroni < 0.05) values are plotted. Autism spectrum disorder (ASD), bipolar disorder (BD), schizophrenia (SCZ), major depressive disorder (MDD), Parkinson’s disease (PD), alcohol abuse disorder (AAD), Alzheimer’s disease (AD). Inflammatory bowel disease (IBD) was included as a non-neural control.
(B) Plot showing the alignment of BW and DE genes within each disorder. Intersecting lists of significant BW and DE genes (both pFDR < 0.05) were categorized as “convergent” or “divergent” based on the concordance of the directions of effects, then Z scored according to a null distribution based on 10,000 resamples of BW gene sets of similar size (gray box-violins). Circles are sized according to the number of DE genes and colored according to values in (A). Asterisks denote significant (ppermutation < 0.001) Z scores.
(C) Grid plot showing 36 significant BW and DE genes (DE and BW pFDR < 0.05) in patients with AD, BD, SCZ, and ASD. Triangles represent directions of effects (up- or downregulated in patients compared with controls) and colors denote the respective BW gene set.
(D) Grid plot showing significant (pFDR < 0.05, reduced for visualization, see Table S5) Gene ontology enrichment of BW-associated genes for biological processes (circles) and cellular components (squares) using ToppGene.56 Shapes are sized and ordered (high-to-low) according to adjusted negative log-scaled p values.
All box-violin plots show median and IQR with whiskers denoting 1.5 × IQR.
Furthermore, functional gene ontology enrichment analysis of the BW gene sets aligned with disease DE signatures. BW+ genes showed significant enrichment for axonal and synaptic functions, including ion channel signaling, and BW− genes showed significant enrichment for neurogenesis/organogenesis and abnormal developmental physiology (Figures 3D and S8; Table S3). Additional analysis using the Synaptic GO (SynGO) database57 https://www.syngoportal.org/ yielded specific synapse-related enrichments for both gene sets (cellular component [BW+]: postsynaptic cytoskeleton [pFDR = 0.032]; cellular component [BW−]: postsynaptic ribosome [pFDR = 4.64e–4], presynaptic ribosome [pFDR = 0.021]; biological process [BW+]: synaptic vesicle exocytosis [pFDR = 0.037]; biological process [BW−]: translation at postsynapse [pFDR = 0.037], translation and presynapse [pFDR = 0.037]).
In addition to the gene ontology analyses, BW genes showed enrichment across multiple modules derived from gene co-expression networks in the PEC dataset58 (Figure S9), indicating that BW genes are significant components of transcriptional regulatory programs, some of which may be cell-type specific. In alignment with the cell-type enrichment results described above, we found module-specific enrichments of BW− genes in “geneM3” (related to astrocytes) and of BW+ genes in “geneM1” and “geneM2” (related to neuronal/synaptic signaling and oligodendrocytes, respectively). Using gene co-expression modules derived In the ROSMAP cohort,59 we found module-specific enrichments of BW− genes in “m107” (related to astrocytes) and of BW+ genes in “m23” (related to neuronal/synaptic signaling) (Figure S9; Table S4; STAR Methods). Moreover, we found enrichment of BW− genes in “m109”, which was shown in prior work59 to be linked to cognitive decline and multiple indices of neuropathology (Figure S9). Further gene set enrichment analysis using independent RNA sequencing data across multiple tissues from the Human Protein Atlas60 yielded significant enrichment of BW+ genes in the adult cerebral cortex versus enrichment of BW− genes in female reproductive tissues (Figure S10), which aligns with the high prenatal expression of BW− genes.
To examine the landscape of rare genetic variation in BW gene sets, hypothesized to be associated with neurodevelopmental disorders, we leveraged two metrics of mutational constraint derived from public databases of whole-exome sequencing and de novo mutations.61,62 We found that BW− genes, and not BW+ genes, were significantly enriched for intolerance to loss-of-function (protein truncating) variation (Figure S10). Using previously defined gene sets of high dosage sensitivity (n = 2,987 haploinsufficient; n = 1,559 triplosensitive), we found a significant bidirectional enrichment (χ2 = 6.1534, p = 0.0131, ppermutation = 0.0047; Figure S10), with greater overlap in BW− and haploinsufficient genes and between BW+ and triplosensitive genes.63 Moreover, BW− genes showed greater overlap with documented pathogenic de novo developmental mutations (BW− odds ratio [OR] = 1.46, ppermutation = 0.008; BW+ OR = 0.83, ppermutation = 0.31), which are associated with abnormalities of brain and cognition, head size, facial, gastrointestinal, and reproductive systems (all ppermutation < 0.05; Figure S10).
Brain size-associated transcription is regulated by genetic variants
The genetic regulation of the transcriptome is one factor that could contribute to interindividual variability in brain gene expression and brain size. We used multiple approaches to distinguish the influence of germline genetic variation on BW transcripts from reactive or secondary gene expression changes due to environmental events. First, leveraging individual-level transcriptome and genotype data from subjects in PEC as the reference dataset, we performed a TWAS of structural neuroimaging phenotypes in the UK Biobank (n = 21,936), normalized using BrainChart,4 to assess the degree to which BW gene sets overlapped with significant genes across neuroimaging TWAS. In general, BW genes significantly overlapped with 29 transcriptome-wide significant global (ppermutation < 0.01) and 124 unique transcriptome-wide regional (ppermutation < 0.05) genes associated with multimodal neuroimaging phenotypes (using a TWAS threshold of pBonferroni < 0.05 across regions and phenotypes to establish significant gene sets). Specificity analysis across neuroimaging phenotypes did not reveal any significant differences in BW gene set overlap or enrichment. For TCV and cortical SA, two of the phenotypes whose models accurately predicted BW, 21 genes reached genome-wide significance (pBonferroni < 0.05) with 15 shared between both phenotypes and an additional 6 significant only for SA. Fourteen of these shared genes were not identified in previous TWASs of related phenotypes—intracranial volume and total brain volume.11,64 Of these 15 significant shared genes, 2 genes (PRR13, EGFR) overlapped with BW gene sets (OR = 3.07, ppermutation = 0.0053), and an additional 7/13 genes (LRP11, INPP5F, ERBB3, EML2, SUOX, GINM1, CCT7P1) were members of PEC co-expression modules significantly enriched for BW genes (see previous section). Sensitivity analysis (conducted with an uncorrected p < 0.05 for TWAS genes) also demonstrated significant enrichment of TCV and SA genes, with 40 genes overlapping with BW gene sets (OR = 9.21, ppermutation < 0.0001). Supplemental analyses with a wide range of neuroimaging phenotypes including diffusion MRI measures (Figure S11), at both whole-brain and regional scales, also demonstrated significant overlap with BW genes (pBonferroni < 0.05; see supplemental information; Table S5).
To further evaluate the genetic influence of BW gene sets on other phenotypes, we leveraged >2,300 GWAS summary statistics across micro- and macro-structural neuroimaging phenotypes.10 Using established gene set enrichment methods (MAGMA65), competitive enrichment analyses of multimodal global neuroimaging phenotypes (averaged or summed across regions) revealed a strong and specific link of BW− genes to brain size (cortical SA and volume) but not brain composition measured via diffusion MRI (Figure 4A; Table S6). Interestingly, we did not find enrichment of BW gene sets with recent large-scale GWAS of height,66 body mass index,67 or head size (head circumference and intracranial volume),68–70 which could either indicate insufficient statistical power for current head size GWAS or suggest a specificity to brain parenchyma size (Table S6). Based on BW− gene set enrichment in GWAS of morphological phenotypes (cortical SA and volume), post hoc analysis revealed a spatially patterned enrichment of BW− genes in GWAS of specific cortical regions (Figure 4A; Table S6). Remarkably, cortical regions showing significant BW− gene enrichment in SA GWAS overlapped with regions with high BW− relative expression as shown in Figure 1D (OR = 6.17, p = 1.14e–5, pspin < 0.0001), but did not overlap with regions with high BW+ relative expression (OR = 0.61, p = 0.14, pspin = 0.94). These brain regions with significant BW− gene enrichment in SA GWAS also showed significant overlap with established regions of hypo-allometric scaling (OR = 3.98, pspin < 0.0001; using maps of allometric [non-linear] scaling of regions with total brain size across population, evolutionary, and developmental scales). Collectively, these results integrate neuroimaging maps of local nonlinear scaling with brain size, the genetic regulatory signatures of brain size-related morphology, and the spatial differential gene expression underlying brain size variation.
Figure 4. Functional relevance of genes associated with BW.
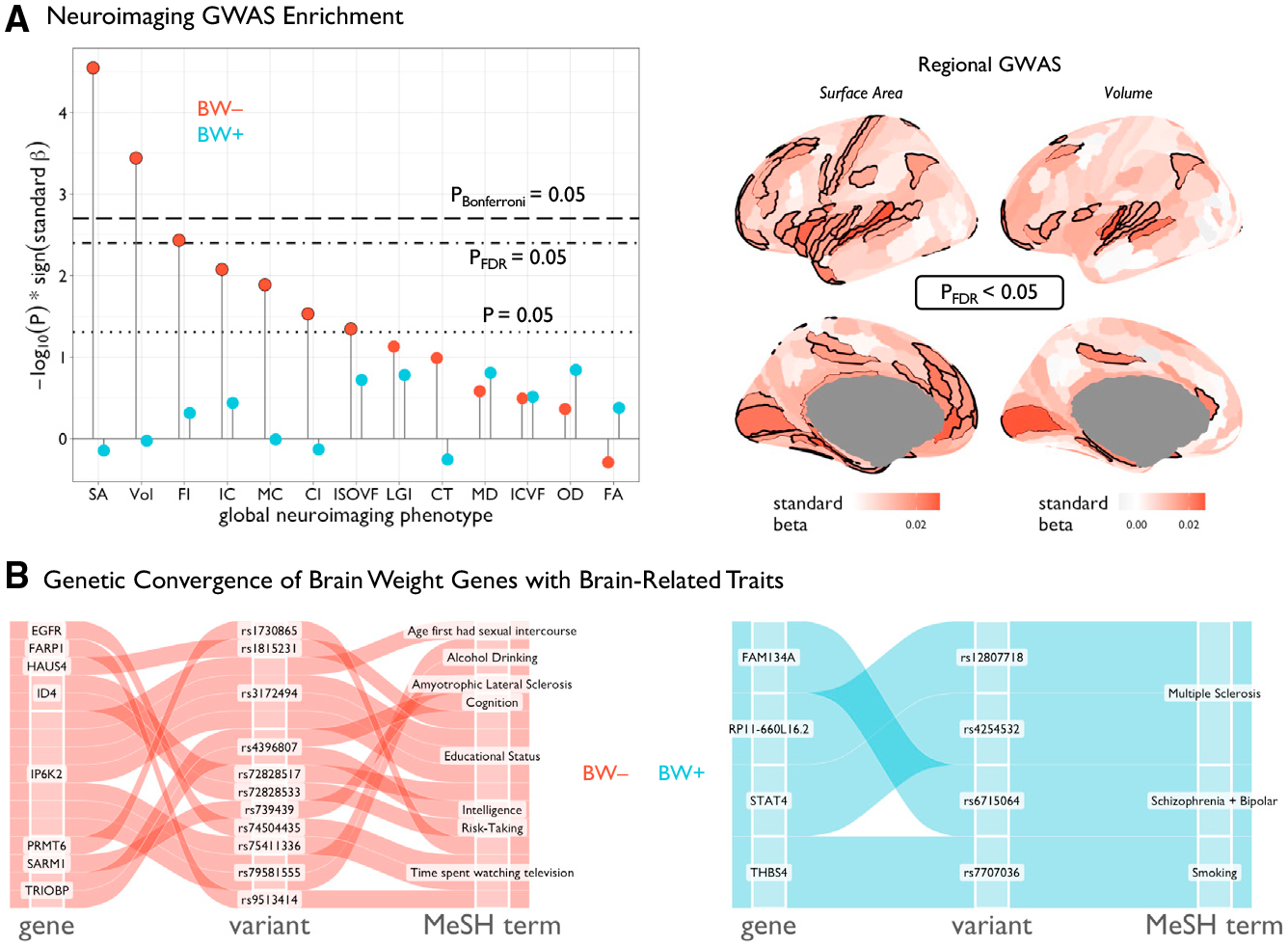
(A) Left: lollipop plot showing MAGMA65 enrichment statistics for BW gene sets across GWASs of 12 multimodal neuroimaging phenotypes we generated in the UK Biobank dataset (STAR Methods). Black lines represent varioussignificance thresholds. Black outlines denote suprathreshold effects (at least uncorrected p < 0.05). No significant effects were observed for the BW+ gene set. Right: enrichment of BW− genes for regional surface area and gray matter volume across 180 cortical regions (the same parcellation as in Figure 1; STAR Methods). Black outlines denote significant areas of enrichment (pFDR < 0.05 shown for visualization, see Table S10). (B) Alluvial diagrams representing the concordance between BW genes (pBonferroni < 0.05) and multi variate multiple quantitative trait loci71 with genome-wide significant association with brain-related traits. SNP, single-nucleotide polymorphism; FDR, false discovery rate.
Finally, we used recently published multivariate multiple quantitative trait loci (mmQTLs) derived from the same three cohorts71 used in the current study to triangulate genetic variants (i.e., SNPs) that may influence brain-related traits via an impact on regulating expression of BW genes. Thus, we were able to identify eight BW− genes (EGFR, FARP1, HAUS4, ID4, IP6K2, PRMT6, SARM1, TRIOBP) and four BW+ genes (FAM134A, RP11–660LI6.2, STAT4, THBS4) with mmQTLs associated with brain-related traits including educational attainment, impulsivity, and psychotic disorders (Figure 4B). Of these, BW− genes ID4, IP6K2, and TRIOBP showed evidence of pleiotropy across multiple brain-related traits, and expanded analysis across all studied traits showed additional pleiotropic associations between both BW gene sets and multiple anthropometric and metabolic domains (Table S7).
A brain size transcriptomic score is associated with disease pathology and clinical outcomes in healthcare system data
Inspired by recent work on the integration of transcriptomic data and polygenic indices,72 and based on the overlap of BW gene sets with differentially expressed genes in patients with neuropsychiatric disease, we developed a BW− transcriptome score—calculated for each individual as the linear combination of weighted gene expression (i.e., sum of expression across genes weighted by the PEC BW model coefficients). Using all available genes, this cumulative PEC BW− transcriptome score was predictive of BW in the replication samples (GTEx partial r = 0.19, p = 0.0266; ROSMAP partial r = 0.17, p = 1.44e–5; Figure S7), demonstrating similar out-of-sample prediction as polygenic scores derived from GWASs of structural neuroimaging phenotypes in much larger cohorts.73 Moreover, we found this BW− transcriptome score to have significant group differences across neuropsychiatric disorders (PEC), as well as patients with a reported history of smoking (GTEx; Figure S12)—a well-documented comorbidity with deleterious effects on brain morphology.74 In addition, considering the spatial alignment between the BW+/− relative expression and neurodegenerative disease pathology (from tau PET), we found significant group differences in the BW transcriptome score in patients with dementia (ROSMAP). In ROSMAP, we implemented a linear mixed effects model framework similar to the per gene BW models (STAR Methods) with the BW− transcriptome score instead as the outcome variable and Braak stage or amyloid as predictor variables in lieu of BW. We found significant relationships between interindividual variation in BW− transcriptome scores and both postmortem neuro-pathological metrics (all gene BW− transcriptome score: Braak stage t = −2.173, p = 0.0171; amyloid t = −4.832, p = 1.71e–6).
To further investigate the clinical correlates of genetic variation influencing BW transcription, we conducted a phenome-wide association study of patient medical outcomes in the Mass General Brigham Biobank, an independent biorepository from a US healthcare system.75 We then used logistic regression models to examine relationships between the BW− transcriptome score and 1,482 case-control disease/disorder phenotypes in up to 37,272 individuals of European ancestry. All models included sex assigned at birth, current age, genotyping batch, and the first 10 genetic principal components as covariates.
Overall, we found that 241 phenotypes were associated with the BW− transcriptome score at pFDR < 0.05, of which 36 were pBonferroni < 0.05, with odds ratios ranging from 0.3 to 3.1 per standard deviation increase in the score (Figure 5; Table S8). The most notable associations were with neurodegenerative disorders (negative association), general mental and behavioral problems (negative), congenital brain abnormalities and neural tube defects (negative), and intellectual disability (positive). Individuals with extreme BW− transcriptome scores in both directions also showed worse health in many other bodily systems. They were more likely to suffer, for example, from infertility or adverse pregnancy outcomes. Collectively, this integrative genomic scoring approach, leveraging observed BW− expression associations and imputed gene expression data, demonstrates a proof-of-concept bridge between functional enrichments and bioinformatic annotations of the BW gene sets with real-world translational potential.
Figure 5. Phenome-wide association study of a BW transcriptomic index in the Mass General Brigham Biobank.

Manhattan plot of the associations between BW transcriptomic index and 1,482 medical outcomes defined by “phecodes” in the Mass General Brigham Biobank, estimated with logistic regression in up to 37,272 patients. Associations are represented as triangles, where an upward-facing triangle denotes a positive relationship, while a downward-facing triangle denotes a negative relationship. The x axis refers to the statistical significance of the association (plotted as −log10 p values), the y axis refers to the category of disease/disorder, and the dashed line denotes the pBonferroni < 0.05 significance threshold (i.e., 0.05/1,482 = 3.37e−5). Legibility necessitated that some condition labels be omitted from the plot. The complete results, including case-control record counts, effect sizes, and standard errors, are reported in Table S14.
DISCUSSION
Human brain size changes dynamically during the lifespan and varies substantially across individuals. Using available postmortem brain RNA sequencing and coupled ancillary data including BW from over 2,000 human brain samples, we identified hundreds of genes that are differentially expressed across the BW continuum. Our results are replicable across independent datasets, show evidence of specificity to BW compared with correlated traits such as height and weight, and reveal many BW genes that have not been implicated previously in imaging-genetic studies of brain size-related phenotypes. Overall, genes upregulated in individuals with larger brains (BW+) or upregulated in individuals with smaller brains (BW−) showed highly diverging spatiotemporal and cell-type-specific patterns of gene expression, and opposing DE patterns across species and in patients with neuropsychiatric disorders. These results pinpoint unreported molecular features of human brain size variation, and highlight the value of integrating measurements of brain morphology with postmortem transcriptomics.
The opposing developmental trajectories of BW gene sets, such that the expression of BW+ genes generally increases with age while that of BW− genes decreases with age, are highly suggestive of functional differences between these genes and their relationship with brain size. The difference in developmental trajectories of BW+ and BW− genes is corroborated by the anatomical convergence between the BW+/− relative expression map in the Allen Human Brain Atlas (AHBA) and the topography of volumetric growth during human development.4 We find that brain areas with higher BW+ relative expression (frontal, parietal, temporal cortices) undergo protracted maturation with peak volume occurring later in life, while brain areas with higher BW− relative expression (insula, sensory cortices) achieve peak volume earlier in development. Moreover, the cross-species relative expression patterns in BW gene sets collectively mirror the expression differences observed in humans relative to non-human primates,19 with greater differences in the prenatal and late postnatal periods. BW− genes showed greater relative expression differences across both brain regions and cell types during the prenatal epoch—and these differences showed more pronounced expression in humans compared with macaques—which may be explained by BW− genes’ significant enrichment for molecular pathways involved in overall organismal growth and neurogenesis in particular. Notably, ID4, a BW− gene that functions as a transcriptional regulator, was previously implicated as part of a human-specific neural progenitor class showing earlier and higher expression in human fetal development compared with mice, and Id4-deficient mice also exhibit decreased brain size and mistimed neurogenesis.76,77 In contrast to BW− genes, BW+ genes showed relative expression differences across brain regions and cell types during postnatal periods (differences that were, again, more pronounced in humans relative to macaques). Of particular note, the BW+ cell membrane protein-encoding gene FREM3 has been shown to have human-specific expression in deep cortical layer III glutamatergic neurons and distinct relationships with morphological and electrophysiological properties relative to other supragranular genes.78 The functions of FREM3 and the BW+ gene MBP, which is critical for myelination of white matter tracts, pinpoint a potential role of BW+ genes in the origination of long-range cortical feedforward projections that emerge during childhood.79
The aforementioned spatiotemporal associations demonstrate the critical link between BW− genes and brain growth during dynamic periods of human neurodevelopment, well before peak brain size is reached. Thus, BW− genes can be interpreted as being both highly expressed in individuals with lower BW and when the brain is smallest. As only postnatal data were available to initially define the BW gene sets, the limited prevalence of high expression of BW− gene transcription in specific cell types later in life suggests that overall brain size is heavily influenced by critical periods of neuronal proliferation and migration when BW− genes are highly expressed. This hypothesis is in line with the finding of BW− relative expression differences in distinct postnatal cell types in humans relative to non-human primates, including inhibitory neurons in cortical layer IV. Notably, the AHBA BW+/− relative expression map and respective bio-annotations—against cytoarchitectonic, connectivity, and spatial expression data—further support the relationship between BW− genes and cortical layer IV. Previous work has demonstrated the potential role of protocadherins in the specification of layer IV identity in mice,80 and eight protocadherin family genes were identified within the BW− gene set (and none in the BW+ set). This gene superfamily is involved with dendritic arborization and synaptic functioning, with evidence of broader developmental roles in programmed cell death and interneuron positioning in the cortex,80,81 and has been implicated in a number of neuropsychiatric and neurodegenerative diseases.82
There was direct evidence that BW genes are relevant to human disease. Intriguingly BW+ genes tended to be downregulated, while BW− genes tended to be upregulated, in individuals with SCZ and AD, two disorders that are robustly associated with decreased brain size.4 This trend was also seen in BD and ASD, informing long-standing debates about the relevance of mechanisms that influence brain size to the pathophysiology of neuropsychiatric disorders in light of many shared genetic risk factors.53,83,84 Although these BW expression associations were identified in postmortem transcriptomic datasets, the translation of the cumulative BW− transcriptome score using imputed expression in the Mass General Brigham Biobank yielded associations to diverse clinical phenotypes that aligned with the bioinformatic annotations of BW gene sets. Notably, the relationship between the BW− transcriptome score and general mental health problems, intellectual disability, and neurodegenerative conditions helps validate the translational potential of this approach.
Of particular interest were phenotypic relationships with congenital brain-related anomalies, which were associated with a negative BW− transcriptome score. While a comprehensive examination of the relationship between gene dosage sensitivity and mechanisms that influence brain size is outside the scope of the current study, we did observe increased loss-of-function intolerance as well as enrichment for known causes of microcephaly in BW− (and not BW+) genes. Two of 12 genes (PHC1, CEP135) involved in primary microcephaly were within the BW− gene set,85 and recent work has also demonstrated the causal role of BW− genes TRIM71 and EGFR in the pathogenesis of hydrocephalus-induced and viral-induced microcephaly, respectively.86,87
Despite the myriad results demonstrating a clear distinction between the BW+/− gene sets, it is important to note exceptions to this trend. For example, we identified multiple gene groupings from both BW+/− sets that are affiliated with prominent growth signaling pathways: IGF2BP2 (BW−) and IGF2BP3 (BW−) versus IGFBP6 (BW+); WNT2 (BW+) versus WNT4 (BW−); BMP4 (BW−) versus BMPR1B (BW+); EGF (BW+) versus EGFR (BW−). This molecular convergence of genes showing opposing associations with brain size points to a possible regulatory model of brain growth whereby BW− genes in these mixed BW+/− gene subgroups are negatively regulated by their complement BW+ genes. Under this model, BW− genes (enriched for neurogenesis) may promote brain growth during early development. Thus, an observed reduction in brain size could result from either (1) overexpression of BW+ genes prenatally (when BW− genes are highly expressed, leading to brain growth faltering) or (2) overexpression of BW− genes postnatally (when BW+ genes are highly expressed, leading to premature degeneration). This simple regulatory model is in line with prior evidence on head size in children with ASD harboring distinct rare genetic mutations.88
Limitations of the study
Some methodological aspects of the present study deserve further consideration in future work. First, most donor samples came from the cerebral cortex (specifically dorsolateral prefrontal cortex). The GTEx dataset is an exception, with samples coming from numerous cortical and non-cortical regions; however, GTEx contains considerably fewer donors than the PEC and ROSMAP datasets. Future work will incorporate additional brain regions to assess variability in differential gene expression with respect to variation in BW. Second, BW was used as a proxy for brain size in transcriptomic analyses. Although future studies could directly measure brain volume in addition to weight, the linear association between BW and volume has been clearly established. Third, given the lack of linked brain size and transcriptomic data in non-human primate datasets, the cross-species analyses herein were restricted to general inter-species differences. Thus, although we found evidence of divergence in regional patterning of BW+/− relative expression, it remains possible that different genes also control brain size in non-human primates. Future work in other non-primate species (e.g., mice) with more comprehensive accompanying phenotypic data will be critical to extend this cross-species assessment and experimentally validate the molecular pathways, cell-type specificity, and causal roles of identified BW genes in humans. Furthermore, given the lack of cell-specific resolution from the bulk tissue transcriptomics and, despite our cell type deconvolution results showing an insignificant relationship between estimated neuronal proportion and BW across individuals, we cannot fully rule out the possible contribution of cell type proportionality and brain size. Further analysis in datasets with both single-cell sequencing and brain size estimates from the same individuals will be required to validate the cell-specific findings herein. Finally, all donors for the transcriptomic analyses were sampled during the postnatal period. Given that many of our results highlight the importance of prenatal epochs, future work using similar transcriptomic data in fetal tissue will be critical for comparative analysis during early periods of neurodevelopment.
Despite these methodological considerations, this study comprehensively outlines the transcriptomic underpinnings of human brain size variation and demonstrates the importance of postmortem measurement of brain size as a quantitative trait to incorporate into transcriptomic analyses. Examined separately or in contrast to one another, the BW+/− gene sets define the spatiotemporal extremes of brain gene expression, charting a molecular segregation of brain regions and developmental epochs. Our results implicate BW genes in the expansion of the human brain relative to non-human primates, as well as gene expression differences observed in brain tissue from patients with neuropsychiatric and neurodegenerative disorders with established differences in brain size. The BW− transcriptome score bridges ex vivo transcriptomics and in vivo genetic risk, highlighting the role of BW genes in both primary and non-primary brain conditions. With ever-increasing data from open science initiatives, parallel innovations in RNA sequencing technology, and widespread utility of clinical genetics, the power of leveraging phenotypic associations from readily accessible transcriptomic datasets holds tremendous promise for future translational studies across human traits and diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jakob Seidlitz (jakob.seidlitz@pennmedicine.upenn.edu; seidlitzj@chop.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original code is available in this paper’s supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| PsychENCODE human brain RNAseq | Li etal.18 | https://psychencode.synapse.org; http://resource.psychencode.org/ |
| GTEx v8 human brain RNAseq | GTEx Consortium89 | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v8.p2 Accession ID: 26317 |
| ROSMAP human RNAseq | De Jager et al.90 | https://www.radc.rush.edu; https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage/StudyDetails?Study=syn3219045 |
| BrainSpan human brain developmental RNAseq | Miller et al.26 | https://www.brainspan.org |
| Allen Human Brain Atlas | Hawrylycz et al.28 and Arnatkeviciute et al.91 | https://figshare.com/articles/dataset/AHBAdata/6852911 |
| PsychENCODE non-human primate brain RNAseq | Zhuetal.19 | http://evolution.psychencode.org |
| Developmental macaque RNA-seq atlas | Yin et al.43 | GEO: GSE128537 |
| UK Biobank (neuroimaging+genetics) | Warrieretal.10 | Application 20904 |
| Neuroimaging GWAS summary statistics | Warrieretal.10 | https://portal.ide-cam.org.uk/overview/483 |
| Neuroimaging lifespan models | Bethlehem et al4 | https://github.com/brainchart/Lifespan |
| Evolution, development, and cross-subject “population” allometric scaling maps | Hill et al.29 and Reardon et al.30 | https://github.com/netneurolab/neuromaps |
| Neurotransmitter/receptor maps | Hansen et al.37 | https://github.com/netneurolab/hansen_receptors |
| Venous density maps (VENAT) | Huck et al.92 | https://figshare.com/articles/dataset/VENAT_Probability_map_nii_gz/7205960 |
| Diffusion MRI fiber length profiling | Bajada et al.93 | https://balsa.wustl.edu/study/1K3l |
| Cognitive function map | Assem et al.33 | https://balsa.wustl.edu/study/B4nkg |
| Alzheimer disease PET maps | Vogel et al.34 | https://neurovault.org/collections/12296/ |
| ENIGMA cross-disorder cortical thickness map | Larivière et al.36 | https://enigma-toolbox.readthedocs.io/en/latest/pages/04.crossdisorder/index.html |
| Glioblastoma and low grade glioma tumor map | Mandal et al.35 | https://neurovault.org/images/785830/ |
| BigBrain | Amunts et al.38 | https://bigbrain.loris.ca/main.php |
| LiBD Spatial RNA-seq | Die cytoarchitektonik der hirnrinde des erwachsenen menschen Google Books.40 | http://research.libd.org/spatialLIBD/ |
| PsychENCODE WGCNA modules | Gandal et al.58 | http://resource.psychencode.org |
| Mutational constraint datasets (gnomAD v2.1.1) | Chen et al.62 | https://gnomad.broadinstitute.org |
| Mutational constraint datasets (DECIPHER) | Firth et al.61, Collins et al.63 | https://www.deciphergenomics.org |
| Meta-analytic multi-ancestry quantitative trait loci | Zeng et al.71 | https://github.com/jxzb1988/MMQTL |
| MGB Biobank | Zeng et al.75 | https://www.massgeneralbrigham.org/en/research-and-innovation/participate-in-research/biobank |
| Software and algorithms | ||
| RStudio (“Ghost Orchid” for macOS) with R (v.4.1.2) | Team, R.S. RStudio: integrated development for R | https://www.rstudio.com |
| RNA-seq preprocessing for PsychENCODE and GTEx | Kim et al.94 | https://github.com/gandallab/C4A-network |
| ToppGene | Chen et al.56 | https://toppgene.cchmc.org |
| MAGMA | de Leeuw et al.65 | https://ctg.cncr.nl/software/magma |
| FUSION | Gusev et al.95 | https://github.com/gusevlab/fusion_twas |
| Genomic quality control | Peterson et al.96 | https://github.com/getian107/MGBB-QC; https://github.com/Annefeng/PBK-QC-pipeline |
| FreeSurfer (v6.0.1) | Fischl et al.97 | https://afni.nimh.nih.gov/ |
| AFNI | Cox et al.98,99 | https://surfer.nmr.mgh.harvard.edu/ |
| ENIGMA toolbox | Larivière et al.36 | https://enigma-toolbox.readthedocs.io/en/latest/ |
| neuromaps | Markello et al.100 | https://github.com/netneurolab/neuromaps |
| Lifespan brain charts | Bethlehem et al.4 | https://github.com/brainchart/Lifespan |
METHOD DETAILS
Validation of brain weight predictions from lifespan neuroimaging models
Brain weight data from the three primary transcriptomic datasets (N = 2,531; PsychENCODE, GTEx, ROSMAP) was combined with additional brain weight data from the literature101 (total N = 3,689). Metrics of brain size (total cerebrum volume and total cortical surface area) for each subject were predicted using life-spanning models of the two neuroimaging phenotypes.4 Subsequently, brain weights for each subject were converted to volumes based on reported estimates of average brain density,20 and correlations were computed with predicted brain size.
Brain weight differential gene expression analysis
All transcriptomic datasets were downloaded from accessible repositories listed in the table above, with methods described previously in the respective source citations. Briefly, the PsychENCODE “freeze 2” dataset consisted of uniformly processed data from six studies: BipSeq, LIBD_szControl, CMC_HBCC, CommonMind, BrainGVEX and UCLA-ASD.94,102 Post-quality-control RNA-seq reads were previously aligned to the hg19 reference genome with STAR 2.4.2a and gene-level quantifications were calculated using RSEM v1.2.29. Genes were filtered to include those with >0.1 TPM in at least 25% of samples. Similarly, GTEx RNA-seq reads were aligned to the hg19 reference genome with STAR 2.4.2a and transcript-level counts quantified with RSEM v1.2.22. Samples from non-brain tissues and tissues with different sample preparation (cortex and cerebellar hemisphere) were removed. Additionally, samples with a history of disease possibly affecting the brain prior to filtering for features with CPM >0.1 in at least 25% of samples were also removed. Gene-level counts were then normalized using TMM normalization in edgeR and log2-transformed to match PsychENCODE. Each brain region was then assessed for outlier samples, defined as those with standardized sample network connectivity Z scores < −3, which were removed. For the ROSMAP cohort, normalized data from previous publications was downloaded from the link provided in the table above based on previous work.90 RNA-seq data was aligned by Tophat v2.0 and v2.1 and transcript enrichments were estimated with RSEM. Quality metrics were provided by Picard, which was also used to mark duplicate reads. Within-batch normalization was conducted through quantile normalization while the between-batches normalization was conducted through ComBat.103 Overall, 25,774 genes were included in the PsychENCODE analyses after filtering, 21,347 in GTEx (19,481 overlapping with PsychENCODE), and 15,083 in ROSMAP (all overlapping with PsychENCODE).
For the PsychENCODE and GTEx cohorts, code for performing the transcriptomic normalization and differential expression analysis followed a previously published approach58,94 with the addition of brain weight as an independent predictor variable (https://github.com/gandallab/C4A-network). Per gene linear models were implemented for PsychENCODE, and linear mixed effects models using the “lme4” package in R were implemented for the GTEx cohort with the addition of a random effect for donor (to account for the multiple brain regions per donor). p-values for the linear mixed effects models were calculated using the likelihood ratio test, as implemented in the “lmerTest” package in R. For ROSMAP, as for the GTEx dataset, per gene linear mixed effects models were implemented, including covariates used for the normalization, brain weight, and a random effect of study. Cross-cohort meta-analyses were performed at the level of gene-brain weight association statistics (beta coefficients) and empirical p-values using the ‘metap’ packing in R.
Neuroimaging data
Structural minimally processed (https://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/brain_mri.pdf) T1 and T2-FLAIR weighted data was downloaded from the UK BioBank, and further preprocessed with Freesurfer (v6.0.1)97 using the T2-FLAIR weighted image to improve pial surface reconstruction. The ‘recon-all’ reconstruction included bias field correction, registration to stereotaxic space, intensity normalization, skull-stripping, and white matter segmentation. When no T2-FLAIR image was available, Freesurfer reconstruction was done using the T1 weighted image only. Given systematic variation related to the inclusion of T2-FLAIR, this was included as a confound variable in downstream analyses. Following reconstruction, the Human Connectome Project multimodal parcellation “HCP-MMP”104 was aligned to each individual freesurfer average image and parcellated values were extracted. Reconstruction reliability was assessed using the Euler index105 and included as a covariate in subsequent analyses.
Structural diffusion weighted imaging was obtained for both datasets. In addition to MD and FA, we ran Accelerated Microstructure Imaging via Convex Optimization (AMICO) to estimate neurite orientation density and dispersion indices.106 The T1 aligned parcellation template was co-registered to the diffusion weighted image using FSL FLIRT and regional values for FA, MD and the three NODDI parameters were extracted using AFNI’s 3dROIstats function. Total sample size of the imaging dataset (to match with imputed expression data for the transcriptome-wide association analyses, described below) was 22,387 subjects.
Neuroimaging transcriptome-wide association studies
To identify genes whose cis-regulated expression is associated with global and regional neuroimaging phenotypes in the UK Biobank in vivo dataset, we performed a series of transcriptome wide association studies (TWAS) leveraging individual-level genotype and postmortem brain expression data from PsychENCODE, described previously.58,95 Briefly, TWAS was implemented using the FUSION software package (https://github.com/gusevlab/fusion_twas95) with custom SNP-brain-expression weights generated using the PsychENCODE dataset of 1321 unique individuals with imputed genotypes. Using the AI-REML algorithm107 implemented in GCTA108 by the FUSION package, we first identified the subset (N = 14,750) of total expressed genes found to have significant cis SNP-heritability in our dataset (cis-h2g uncorrected p < 0.05 within 1 Mb window around the gene body). SNP-expression weights were calculated in a 1Mb region around all heritable genes using expression measurements adjusted for diagnosis, study, age, age,2 RIN, RIN2, sex, tissue, PMI, 20 ancestry PCs, and 100 hidden covariates.58 Accuracy of five expression prediction models were tested (best cis-eQTL, best linear unbiased predictor, Bayesian linear mixed model, Elastic-net regression, LASSO regression) using the most accurate model for final weight calculations as implemented in the FUSION package. TWAS neuroimaging-association statistics were computed using these custom weights, LD structure calculated from our PsychENCODE samples’ genotypes, and neuroimaging data from UK Biobank as described above. For each global and regional neuroimaging phenotype, TWAS association statistics were Bonferroni-corrected for multiple comparisons (final count N = 13,421 genes). At loci (+/−100 kb) with multiple significant associations, joint and conditional association analyses were further performed as implemented in the FUSION.post_process.R script. Gene weights are available from http://resource.psychencode.org/.
Bioinformatics analyses
Spatiotemporal annotation
Spatially-comprehensive cortical microarray gene expression data from the Allen Human Brain Atlas (AHBA) was processed and mapped to the same HCP-MMP atlas (N = 180 regions) as the previously described neuroimaging data28,91 for the purpose of assessing regional expression and relative expression differences in brain weight gene sets. Three regions (“MT”, “a10p”, “RI”) were removed due to inadequate sampling across donors and quality control. AHBA data was also mapped to the Desikan-Killiany atlas (N = 34 regions) depending on the comparative maps used for spatial correlation. Additional multimodal neuroimaging, cytoarchitectonic, and functional maps (each projected to the same abovementioned cortical parcellations) can be found in the key resources table. Developmental RNA-seq data as well as prenatal laser microdissection microarray data from BrainSpan26 was used for mapping age trajectories and assessing early regional relative BW expression differences (see also key resources table).
Cross-species effects
To assess relative expression differences in BW across species, we leveraged a matched human and non-human primate dataset associated with the PsychENCODE project.19 This dataset combines tissue samples from the six adult humans, five adult chimpanzee brains and five adult macaque brains. The dataset can be downloaded from http://evolution.psychencode.org where it is labeled as “Adult human, chimpanzee, macaque data” in the “Processed Data”/”mRNA-seq” tab. The dataset is pre-harmonized to include a consistent set of 16 cortical and subcortical brain regions with mRNA sequencing performed on 11,346 curated homologous genes. Similarly, to examine species-by-development effects, we used a second pre-curated dataset, downloadable from the same link, labeled “Developmental rhesus and human data”. This dataset includes tissue samples from 36 human brains (15 female, ages 8 post-conception weeks to 40 years, mean = 97 ± 147 post-conception months) and 26 macaque brains (8 female, ages 60 post-conception days to 11 years, mean = 36 ± 46 post-conception months) across 16 homologous cortical, subcortical and cerebellar brain regions. Three transient developmental brain regions were excluded (lateral, medial and caudal ganglionic eminence), while other prenatal regions were considered equivalent to their most similar adult brain regions (e.g., dorsal thalamus to mediodorsal thalamus, upper rhombic lip to cerebellum, etc). This was only relevant for two of 62 total brains that possessed these early developmental regions. Each brain region had mRNA sequencing for 27,932 genes. Phylogenetic variability in brain size (PBS) was assessed for each gene across species, modeled as follows: PBS+ = H > C > M and PBS− = H < C < M, with model significance assessed based on likelihood ratio tests for nested models and thresholded (PBonferroni < 0.05) to examine overlap with BW gene sets.
Additionally, to assess the consistency of regional BW +/− relative expression differences across species, we used an independent postmortem brain gene expression dataset (RNAseq; see key resources table) in macaques containing 416 samples from 8 macaques across 52 cortical and non-cortical brain regions.43 Processed data was available (see key resources table), and we included biological (age, sex) and technical (RIN, medTIN) covariates when examining BW +/− relative expression differences for each brain region. Each brain region had mRNA sequencing for 14,898 genes.
Cell-type relative expression differences
To assess relative expression differences in BW across cell types, we leveraged aligned developmental single cell (fetal) and single nucleus (adult) RNA-seq datasets in both humans and non-human primates. Macaque data was downloaded from http://evolution.psychencode.org, where they are labeled as “Fetal expression matrix” and “Adult expression (Rdata)” under the “Processed Data”/“Single cell RNA-seq” or “Processed Data”/”Single nucleus RNA-seq” tabs, respectively. These datasets contain expression data on ~15k genes across various cell types sampled in the dorsolateral prefrontal cortex (DFC) from two (fetal) and three (adult) macaques. More detailed protocol information can be found in previous work.19
Human data were downloaded from http://development.psychencode.org, where they are labeled as “scRNA-seq (Rdata)” and “snRNA-seq (Rdata)” under the “Processed Data”/”Single cell/nucleus RNA-seq” tab. For over 20k genes, the fetal single cell dataset contains expression data sampled across the pallium (labeled as “DFC”) from nine donors, and the adult single nucleus dataset contains expression data in the DFC from three donors. More detailed protocol information can be found in previous work.18
We performed the cell-type deconvolution analysis using the “dtangle” R package to evaluate the relationship between interindividual variation in brain weight and predicted cell-type proportions. We chose this approach because it is a top performing algorithm for brain gene expression,22 and it has the capacity to combine multiple reference single cell datasets for deconvolution of canonical cell-types.
Gene ontology enrichment
Brain weight gene lists were submitted as gene sets to ToppGene’s ToppFun enrichment feature (https://toppgene.cchmc.org/enrichment.jsp). The following term categories were assessed: GO: Molecular Function, GO: Biological Process, GO: Cellular Component, Pathway (all), Mouse Phenotype and Human Phenotype. All other settings were left to their defaults. Note that ToppGene databases are continuously updated; this ToppGene query was last conducted on October 23rd, 2021.
Functional enrichment
The gene set enrichment analysis further nominated associations with brain morphological phenotypes derived from in vivo structural magnetic resonance imaging (MRI). To validate this finding, we used an established gene-set analysis method, MAGMA,65 to evaluate the enrichment of BW gene sets against genome-wide association studies of global and regional neuroimaging phenotypes,10 as well as metrics of head size – head circumference and intracranial volume.68–70
Gene co-expression network modules
For the PsychENCODE and ROSMAP datasets, modules were derived from gene co-expression network analysis using Weighted Gene Co-expression Analysis (WGCNA)58,109 and the SpeakEasy clustering methods,59,110 respectively. WGCNA module assignments and annotations for PsychENCODE are available from http://resource.psychencode.org/) (see also key resources table), and module enrichment results for ROSMAP can be found in Table S6.
Phenome-wide association study in Mass General Brigham Biobank
Phenome-wide association study (PheWAS) analyses were performed in the Mass General Brigham (MGB) Biobank, a biorepository from the MGB healthcare system based in the greater Boston area with patient data on electronic health record, genetic, and lifestyle variables.75 Launched in 2010, the MGB Biobank has enrolled 138,042 participants and generated genotyping microarray data for more than 65,265 participants to date. To reduce the risk of population stratification, we restricted all PheWAS analyses to 37,272 patients of European ancestry (22,232 were genotyped using Illumina MEG, MEGA, or MEGA EX arrays and the remaining 15,040 were genotyped using the Illumina GSA array). The recruitment strategy, genotyping procedures, and quality control procedures of MGB Biobank have been described previously (see key resources table).75 All participants provided written consent upon enrollment. MGB Institutional Review Board approval for the present analyses is covered under protocols #2009P002312 and #2021P003641.
We used the FUSION package in R, as in the TWAS, to impute individual-level gene expression for patients in MGB Biobank, using pre-computed weights from the PsychENCODE consortium as the reference transcriptome dataset (gene weights available from http://resource.psychencode.org/). We then calculated a cumulative brain weight transcriptomic index as a coefficient-weighted sum of imputed gene expression – similar to how polygenic indexes are calculated using single nucleotide polymorphism data. This score was subsequently standardized and linked to electronic health record data from the MGB Biobank participants. Case status for each medical phenotype was assigned using a standard “phecode” approach,111 where the presence of at least two International Classification of Disease (ICD)-10CM codes was required. PheWAS was then conducted using the PheWAS package in R (https://github.com/PheWAS/PheWAS), fitting logistic regression models to each of the 1,482 medical outcomes in order to estimate the odds of each diagnosis given the brain weight transcriptomic index while accounting for sex assigned at birth, current age, genotyping batch, and the first 10 genetic principal components as covariates.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all analyses in this study significance was determined at p < 0.05, after Bonferroni correction for multiple comparisons (PBonferonni). For the brain weight discovery analyses in PsychENCODE this equated to an uncorrected p < 1.94e−6 threshold. For the TWAS analyses, PBonferroni < 0.05 equated to an uncorrected p < 3.73e−6 for global phenotypes, uncorrected p < 2.07e−8 for regional surface-based phenotypes, and uncorrected p < 1.19×10e−8 for regional volume-based phenotypes. False Discovery Rate (FDR) correction was used in lieu of Bonferroni correction to determine significant brain weight gene sets (PFDR < 0.05) in the intersection analyses with the differential expression results across disorders, for consistency with how these data were thresholded in their original publications. Additionally, where applicable, we used two different permutation-based tests for empirical statistics to complement uncorrected p-values based on context-specific robust null models (Ppermutation and Pspin), as implemented previously.112 Ppermutation denotes performing gene-level statistical analyses using 10,000 randomly sampled gene sets (without replacement) of similar size to the empirical sets, drawn from the entire gene list used for the brain weight gene expression analysis in PsychENCODE. Pspin denotes performing spatial correlations between two brain maps using 10,000 “spins” (preserving spatial autocorrelation) of one map (see also key resources table). We report t-values from linear mixed models of brain weight expression association analyses, in addition to partial correlation values of brain weight and expression after regression of the same covariates used in the linear mixed models. For all BW “relative expression” analyses, we used Student t-tests to compare mean expression of BW + genes and BW− genes. Unless otherwise noted, all p-values for linear mixed models were calculated using likelihood ratio tests as implemented in the “lmerTest” package in R. As this was a retrospective study, no statistical methods were used to pre-determine sample sizes, however, collectively, this study makes use of the three largest datasets of postmortem human brain tissue. As such, we comprehensively assessed the main brain weight gene expression results explicitly in a discovery (PsychENCODE) and in two independent replication datasets (GTEx and ROSMAP). Randomization and blinding were not possible due to the study being retrospective and observational. Accordingly, subject-level covariates were used to account for variation in gene expression as well as to remove unwanted confounding effects. Normalized gene expression was assumed to follow normal distribution, but this was not formally tested. Additional details for each analysis are provided in relevant sections above.
Supplementary Material
Highlights.
Transcriptomic analysis of human BW revealed 928 significant genes
BW genes align with predicted developmental and evolutionary changes in brain size
BW genes are cell-type specific and align with structural neuroimaging GWASs and TWASs
BW genes are differentially expressed in psychiatric and neurodegenerative disorders
ACKNOWLEDGMENTS
The authors would like to acknowledge the various open science initiatives, and their associated funding, that provided the necessary datasets used in this manuscript. J.S. and J.W.V. were supported by NIMH T32MH019112. A.F.A.-B. and J.S. were supported by NIMH K08MH120564. T.T.M. was supported by NHGRI T32HG010464. V.W. was supported by St. Catharine’s College Cambridge. R.A.I.B. was supported by the Autism Research Trust. K.W. was supported by the Wellcome Trust (215901/Z/19/Z). T.D.S. was supported by NIH R01MH113550, R01MH120482, R01MH112847, R01EB022573, RF1MH121867, and R37MH125829. G.B. was supported by the Australian National Health and Medical Research Council (NHMRC; Investigator Grant 1194497). We thank Mass General Brigham Biobank for providing genomic and health information data.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113439.
DECLARATION OF INTERESTS
J.S., R.A.I.B., J.D.B., and A.F.A.-B. are directors and hold equity in Centile Bioscience. J.D.B. holds positions/equity in UpFront Diagnostics, Treovir, and NeuroX1. A.F.A.-B. receives consulting income from Octave Bioscience.
REFERENCES
- 1.Heuer K, Gulban OF, Bazin PL, Osoianu A, Valabregue R, Santin M, Herbin M, and Toro R (2019). Evolution of neocortical folding: A phylogenetic comparative analysis of MRI from 34 primate species. Cortex 118, 275–291. [DOI] [PubMed] [Google Scholar]
- 2.Assaf Y, Bouznach A, Zomet O, Marom A, and Yovel Y (2020). Conservation of brain connectivity and wiring across the mammalian class. Nat. Neurosci 23, 805–808. [DOI] [PubMed] [Google Scholar]
- 3.Giedd JN, Raznahan A, Alexander-Bloch A, Schmitt E, Gogtay N, and Rapoport JL (2015). Child Psychiatry Branch of the National Institute of Mental Health Longitudinal Structural Magnetic Resonance Imaging Study of Human Brain Development. Neuropsychopharmacology 40, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bethlehem RAI, Seidlitz J, White SR, Vogel JW, Anderson KM, Adamson C, Adler S, Alexopoulos GS, Anagnostou E, Areces-Gonzalez A, et al. (2022). Brain charts for the human lifespan. Nature 604, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mallard TT, Liu S, Seidlitz J, Ma Z, Moraczewski D, Thomas A, and Raznahan A (2021). X-chromosome influences on neuroanatomical variation in humans. Nat. Neurosci 24, 1216–1224. [DOI] [PubMed] [Google Scholar]
- 6.Grasby KL, Jahanshad N, Painter JN, Colodro-Conde L, Bralten J, Hibar DP, Lind PA, Pizzagalli F, Ching CRK, McMahon MAB, et al. (2020). The genetic architecture of the human cerebral cortex. Science 367, eaay6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams HHH, Hibar DP, Chouraki V, Stein JL, Nyquist PA, Rentería ME, Trompet S, Arias-Vasquez A, Seshadri S, Desrivières S, et al. (2016). Novel genetic loci underlying human intracranial volume identified through genome-wide association. Nat. Neurosci 19, 1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Opel N, Goltermann J, Hermesdorf M, Berger K, Baune BT, and Dannlowski U (2020). Cross-Disorder Analysis of Brain Structural Abnormalities in Six Major Psychiatric Disorders: A Secondary Analysis of Mega- and Meta-analytical Findings From the ENIGMA Consortium. Biol. Psychiatr 88, 678–686. [DOI] [PubMed] [Google Scholar]
- 9.Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, Marchini J, and Smith SM (2018). Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warrier V, Stauffer E-M, Huang QQ, Wigdor EM, Slob EAW, Seidlitz J, Ronan L, Valk SL, Mallard TT, Grotzinger AD, et al. (2022). The genetics of cortical organisation and development: a study of 2,347 neuroimaging phenotypes. Preprint at bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao B, Shan Y, Yang Y, Yu Z, Li T, Wang X, Luo T, Zhu Z, Sullivan P, Zhao H, et al. (2021). Transcriptome-wide association analysis of brain structures yields insights into pleiotropy with complex neuropsychiatric traits. Nat. Commun 12, 2878–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pramparo T, Lombardo MV, Campbell K, Barnes CC, Marinero S, Solso S, Young J, Mayo M, Dale A, Ahrens-Barbeau C, et al. (2015). Cell cycle networks link gene expression dysregulation, mutation, and brain maldevelopment in autistic toddlers. Mol. Syst. Biol 11, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lombardo MV, Eyler L, Pramparo T, Gazestani VH, Hagler DJ Jr., Chen C-H, Dale AM, Seidlitz J, Bethlehem RAI, Bertelsen N, et al. (2021). Atypical genomic cortical patterning in autism with poor early language outcome. Sci. Adv 7, eabh1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Yang S-L, Yang M, Herrlinger S, Shao Q, Collar JL, Fierro E, Shi Y, Liu A, Lu H, et al. (2019). Modeling microcephaly with cerebral organoids reveals a WDR62–CEP170–KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun 10, 2612–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urresti J, Zhang P, Moran-Losada P, Yu N-K, Negraes PD, Trujillo CA, Antaki D, Amar M, Chau K, Pramod AB, et al. (2021). Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol. Psychiatr 26, 7560–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.PsychENCODE Consortium; Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, Jaffe AE, Pinto D, Dracheva S, et al. (2015). The PsychENCODE project. Nat. Neurosci 18, 1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y, Sousa AMM, Gao T, Skarica M, Li M, Santpere G, Esteller-Cucala P, Juan D, Ferrández-Peral L, Gulden FO, et al. (2018). Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362, eaat8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(1979). Report of the task group on reference man. Ann. ICRP 3. [DOI] [PubMed] [Google Scholar]
- 21.Winkler AM, Ridgway GR, Webster MA, Smith SM, and Nichols TE (2014). Permutation inference for the general linear model. Neuroimage 92, 381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutton GJ, Poppe D, Simmons RK, Walsh K, Nawaz U, Lister R, Gagnon-Bartsch JA, and Voineagu I (2022). Comprehensive evaluation of deconvolution methods for human brain gene expression. Nat. Commun 13, 1358–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoenemann PT (2004). Brain size scaling and body composition in mammals. Brain Behav. Evol 63, 47–60. [DOI] [PubMed] [Google Scholar]
- 24.Simes RJ (1986). An improved Bonferroni procedure for multiple tests of significance. Biometrika 73, 751–754. [Google Scholar]
- 25.Plaisier SB, Taschereau R, Wong JA, and Graeber TG (2010). Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res 38, e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. (2014). Transcriptional Landscape of the Prenatal Human Brain. Nature 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Werling DM, Pochareddy S, Choi J, An JY, Sheppard B, Peng M, Li Z, Dastmalchi C, Santpere G, Sousa AMM, et al. (2020). Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep 31, 107489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill J, Inder T, Neil J, Dierker D, Harwell J, and Van Essen D (2010). Similar patterns of cortical expansion during human development and evolution. Proc. Natl. Acad. Sci. USA 107, 13135–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reardon PK, Seidlitz J, Vandekar S, Liu S, Patel R, Park MTM, Alexander-Bloch A, Clasen LS, Blumenthal JD, Lalonde FM, et al. (2018). Normative brain size variation and brain shape diversity in humans. Science 360, 1222–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaishnavi SN, Vlassenko AG, Rundle MM, Snyder AZ, Mintun MA, Raichle ME, and Raichle ME (2010). Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. USA 107, 17757–17762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glasser MF, and Van Essen DC (2011). Mapping human cortical areas in vivo based on myelin content as revealed by T1- and T2-weighted MRI. J. Neurosci 31, 11597–11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Assem M, Glasser MF, Van Essen DC, and Duncan J (2020). A Domain-General Cognitive Core Defined in Multimodally Parcellated Human Cortex. Cerebr. Cortex 30, 4361–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, La Joie R, Aksman LM, Grothe MJ, Iturria-Medina Y, et al. (2021). Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med 27, 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandal AS, Romero-Garcia R, Hart MG, and Suckling J (2020). Genetic, cellular, and connectomic characterization of the brain regions commonly plagued by glioma. Brain 143, 3294–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larivière S, Paquola C, Park B-Y, Royer J, Wang Y, Benkarim O, Vos de Wael R, Valk SL, Thomopoulos SI, Kirschner M, et al. (2021). The ENIGMA Toolbox: multiscale neural contextualization of multisite neuroimaging datasets. Nat. Methods 18, 698–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen JY, Shafiei G, Markello RD, Smart K, Cox SML, Nørgaard M, Beliveau V, Wu Y, Gallezot J-D, Aumont É, et al. (2022). Mapping neurotransmitter systems to the structural and functional organization of the human neocortex. Preprint at bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amunts K, Lepage C, Borgeat L, Mohlberg H, Dickscheid T, Rousseau MÉ, Bludau S, Bazin PL, Lewis LB, Oros-Peusquens AM, et al. (2013). BigBrain: an ultrahigh-resolution 3D human brain model. Science 340, 1472–1475. [DOI] [PubMed] [Google Scholar]
- 39.Wagstyl K, Larocque S, Cucurull G, Lepage C, Cohen JP, Bludau S, Palomero-Gallagher N, Lewis LB, Funck T, Spitzer H, et al. (2020). BigBrain 3D atlas of cortical layers: Cortical and laminar thickness gradients diverge in sensory and motor cortices. PLoS Biol 18, e3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maynard KR, Collado-Torres L, Weber LM, Uytingco C, Barry BK, Williams SR, Catallini JL, Tran MN, Besich Z, Tippani M, et al. (2021). Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci 24, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Die cytoarchitektonik der hirnrinde des erwachsenen menschen Google Books. https://books.google.com/books/about/Die_cytoarchitektonik_der_hirnrinde_des.html?id=2DVBAAAAYAAJ.
- 42.Sousa AMM, Zhu Y, Raghanti MA, Kitchen RR, Onorati M, Tebbenkamp ATN, Stutz B, Meyer KA, Li M, Kawasawa YI, et al. (2017). Molecular and Cellular Reorganization of Neural Circuits in the Human Lineage. Science 358, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin S, Lu K, Tan T, Tang J, Wei J, Liu X, Hu X, Wan H, Huang W, Fan Y, et al. (2020). Transcriptomic and open chromatin atlas of high-resolution anatomical regions in the rhesus macaque brain. Nat. Commun 11, 474–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, et al. (2006). An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest (2006). Neuroimage 31, 968–980. [DOI] [PubMed] [Google Scholar]
- 45.(2020). The Timing of Brain Maturation, Early Experience, and the Human Social Niche. In Evolutionary NeuroscienceEvolutionary Neuroscience (Academic Press; ), pp. 815–843. [Google Scholar]
- 46.Barger N, Hanson KL, Teffer K, Schenker-Ahmed NM, and Semendeferi K (2014). Evidence for evolutionary specialization in human limbic structures. Front. Hum. Neurosci 0. 8, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tremblay R, Lee S, and Rudy B (2016). GABAergic interneurons in the neocortex: From cellular properties to circuits. Neuron 91, 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blockus H, Rolotti SV, Szoboszlay M, Peze-Heidsieck E, Ming T, Schroeder A, Apostolo N, Vennekens KM, Katsamba PS, Bahna F, et al. (2021). Synaptogenic activity of the axon guidance molecule Robo2 underlies hippocampal circuit function. Cell Rep 37, 109828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szu JI, and Binder DK (2016). The Role of Astrocytic Aquaporin-4 in Synaptic Plasticity and Learning and Memory. Front. Integr. Neurosci 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dehay C, Kennedy H, and Kosik K (2015). The Outer Subventricular Zone and Primate-Specific Cortical Complexification (2015). Neuron 85, 683–694. [DOI] [PubMed] [Google Scholar]
- 51.Lui JH, Hansen DV, and Kriegstein AR (2011). Development and evolution of the human neocortex. Cell 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Letinic K, Zoncu R, and Rakic P (2002). Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649. [DOI] [PubMed] [Google Scholar]
- 53.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM, et al. (2018). Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marques-Coelho D, Iohan L.d.C.C., Melo de Farias AR, Flaig A, Brainbank Neuro–CEB Neuropathology Network; Lambert JC, and Costa MR (2021). Differential transcript usage unravels gene expression alterations in Alzheimer’s disease human brains. NPJ Aging Mech. Dis 7, 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nido GS, Dick F, Toker L, Petersen K, Alves G, Tysnes O-B, Jonassen I, Haugarvoll K, and Tzoulis C (2020). Common gene expression signatures in Parkinson’s disease are driven by changes in cell composition. Acta Neuropathol. Commun 8. 55–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Bardes EE, Aronow BJ, and Jegga AG (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, Cornelisse LN, Farrell RJ, Goldschmidt HL, Howrigan DP, et al. (2019). SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 103, 217–234.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein H-U, Patrick E, Komashko V, et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci 21, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 61.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, and Carter NP (2009). Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet 84, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen S, Francioli LC, Goodrich JK, Collins RL, Wang Q, Alföldi J, Watts NA, Vittal C, Gauthier LD, Poterba T, et al. (2022). A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. Preprint at bioRxiv [Google Scholar]
- 63.Collins RL, Glessner JT, Porcu E, Lepamets M, Brandon R, Lauricella C, Han L, Morley T, Niestroj LM, Ulirsch J, et al. (2022). A cross-disorder dosage sensitivity map of the human genome. Cell 185, 3041–3055.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walker RL, Ramaswami G, Hartl C, Mancuso N, Gandal MJ, de la Torre-Ubieta L, Pasaniuc B, Stein JL, and Geschwind DH (2019). Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 179, 750–771.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Leeuw CA, Mooij JM, Heskes T, and Posthuma D (2015). MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yengo L, Vedantam S, Marouli E, Sidorenko J, Bartell E, Sakaue S, Graff M, Eliasen AU, Jiang Y, Raghavan S, et al. (2022). A saturated map of common genetic variants associated with human height. Nature 610, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turcot V, Lu Y, Highland HM, Schurmann C, Justice AE, Fine RS, Bradfield JP, Esko T, Giri A, Graff M, et al. (2018). Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat. Genet 50, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haworth S, Shapland CY, Hayward C, Prins BP, Felix JF, Medina-Gomez C, Rivadeneira F, Wang C, Ahluwalia TS, Vrijheid M, et al. (2019). Low-frequency variation in TP53 has large effects on head circumference and intracranial volume. Nat. Commun 10, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taal HR, Pourcain BS, Thiering E, Das S, Mook-Kanamori DO, Warrington NM, Kaakinen M, Kreiner-Møller E, Bradfield JP, Freathy RM, et al. (2012). Common variants at 12q15 and 12q24 are associated with infant head circumference. Nat. Genet 44, 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith SM, Douaud G, Chen W, Hanayik T, Alfaro-Almagro F, Sharp K, and Elliott LT (2021). An expanded set of genome-wide association studies of brain imaging phenotypes in UK Biobank. Nat. Neurosci 24, 737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zeng B, Bendl J, Kosoy R, Fullard JF, Hoffman GE, and Roussos P (2022). Multi-ancestry eQTL meta-analysis of human brain identifies candidate causal variants for brain-related traits. Nat. Genet 54, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang Y, Pividori M, Manichaikul A, Palmer AA, Cox NJ, Wheeler HE, and Im HK (2022). Polygenic transcriptome risk scores (PTRS) can improve portability of polygenic risk scores across ancestries. Genome Biol 23. 23–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao B, Luo T, Li T, Li Y, Zhang J, Shan Y, Wang X, Yang L, Zhou F, Zhu Z, et al. (2019). Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat. Genet 51, 1637–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boksa P (2017). Smoking, psychiatric illness and the brain. J. Psychiatry Neurosci 42, 147–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boutin NT, Schecter SB, Perez EF, Tchamitchian NS, Cerretani XR, Gainer VS, Lebo MS, Mahanta LM, Karlson EW, and Smoller JW (2022). The Evolution of a Large Biobank at Mass General Brigham. J. Personalized Med 12, 1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, and Kriegstein AR (2021). Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci 24, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bedford L, Walker R, Kondo T, van Crüchten I, King ER, and Sablitzky F (2005). Id4 is required for the correct timing of neural differentiation. Dev. Biol 280, 386–395. [DOI] [PubMed] [Google Scholar]
- 78.Berg J, Sorensen SA, Ting JT, Miller JA, Chartrand T, Buchin A, Bakken TE, Budzillo A, Dee N, Ding S-L, et al. (2021). Human neocortical expansion involves glutamatergic neuron diversification. Nature 598, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Petanjek Z, Judas M, Kostović I, and Uylings HBM (2008). Lifespan alterations of basal dendritic trees of pyramidal neurons in the human prefrontal cortex: a layer-specific pattern. Cerebr. Cortex 18, 915–929. [DOI] [PubMed] [Google Scholar]
- 80.Oishi K, Nakagawa N, Tachikawa K, Sasaki S, Aramaki M, Hirano S, Yamamoto N, Yoshimura Y, and Nakajima K (2016). Identity of neocortical layer 4 neurons is specified through correct positioning into the cortex [DOI] [PMC free article] [PubMed]
- 81.Mancia Leon WR, Spatazza J, Rakela B, Chatterjee A, Pande V, Maniatis T, Hasenstaub AR, Stryker MP, and Alvarez-Buylla A (2020). Clustered gamma-protocadherins regulate cortical interneuron programmed cell death. Elife 9, e55374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peek SL, Mah KM, and Weiner JA (2017). Regulation of Neural Circuit Formation by Protocadherins. Cell. Mol. Life Sci 74, 4133–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brainstorm Consortium; Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, Escott-Price V, Falcone GJ, Gormley P, et al. (2018). Analysis of shared heritability in common disorders of the brain. Science 360, eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grotzinger AD, Mallard TT, Liu Z, Seidlitz J, Ge T, and Smoller JW (2022). Multivariate Genomic Architecture of Cortical Thickness and Surface Area at Multiple Levels of Analysis. Preprint at medRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, Pushparaj P, Ahmed F, Algahtani HA, Al-Qahtani MH, and Saleh Jamal H (2015). Molecular genetics of human primary micro-cephaly: an overview. BMC Med. Genom 8, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duy PQ, Weise SC, Marini C, Li X-J, Liang D, Dahl PJ, Ma S, Spajic A, Dong W, Juusola J, et al. (2022). Impaired neurogenesis alters brain biomechanics in a neuroprogenitor-based genetic subtype of congenital hydrocephalus. Nat. Neurosci 25, 458–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun G, Chiuppesi F, Chen X, Wang C, Tian E, Nguyen J, Kha M, Trinh D, Zhang H, Marchetto MC, et al. (2020). Modeling Human Cytomegalovirus-Induced Microcephaly in Human iPSC-Derived Brain Organoids (2020). Cell Rep. Med 1, 100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Krumm N, O’Roak BJ, Shendure J, and Eichler EE (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.GTEx Consortium (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D, Klein H-U, White CC, Peters MA, Lodgson B, et al. (2018). A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data 5, 180142–180213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arnatkeviciute A, Fulcher BD, and Fornito A (2019). A practical guide to linking brain-wide gene expression and neuroimaging data (2019). Neuroimage 189, 353–367. [DOI] [PubMed] [Google Scholar]
- 92.Huck J, Wanner Y, Fan AP, Jäger AT, Grahl S, Schneider U, Vill-ringer A, Steele CJ, Tardif CL, Bazin PL, and Gauthier CJ (2019). High resolution atlas of the venous brain vasculature from 7 T quantitative susceptibility maps. Brain Struct. Funct 224, 2467–2485. [DOI] [PubMed] [Google Scholar]
- 93.Bajada CJ, Schreiber J, and Caspers S (2019). Fiber length profiling: A novel approach to structural brain organization. Neuroimage 186, 164–173. [DOI] [PubMed] [Google Scholar]
- 94.Kim M, Haney JR, Zhang P, Hernandez LM, Wang L-K, Perez-Cano L, Loohuis LMO, de la Torre-Ubieta L, and Gandal MJ (2021). Brain gene co-expression networks link complement signaling with convergent synaptic pathology in schizophrenia. Nat. Neurosci 24, 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, Jansen R, de Geus EJC, Boomsma DI, Wright FA, et al. (2016). Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peterson RE, Kuchenbaecker K, Walters RK, Chen C-Y, Popejoy AB, Periyasamy S, Lam M, Iyegbe C, Strawbridge RJ, Brick L, et al. (2019). Genome-wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations. Cell 179, 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, et al. (2004). Automatically parcellating the human cerebral cortex. Cerebr. Cortex 14, 11–22. [DOI] [PubMed] [Google Scholar]
- 98.Cox RW (1996). AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput. Biomed. Res 29, 162–173. [DOI] [PubMed] [Google Scholar]
- 99.Cox RW, and Hyde JS (1997). Software tools for analysis and visualization of fMRI data. NMR Biomed 10, 171–178. [DOI] [PubMed] [Google Scholar]
- 100.Markello RD, Hansen JY, Liu Z-Q, Bazinet V, Shafiei G, Suárez LE, Blostein N, Seidlitz J, Baillet S, Satterthwaite TD, et al. (2022). neuromaps: structural and functional interpretation of brain maps. Nat. Methods 19, 1472–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Borzage M, Blüml S, and Seri I (2014). Equations to describe brain size across the continuum of human lifespan. Brain Struct. Funct 219, 141–150. [DOI] [PubMed] [Google Scholar]
- 102.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, et al. (2018). Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- 104.Glasser MF, Coalson TS, Robinson EC, Hacker CD, Harwell J, Yacoub E, Ugurbil K, Andersson J, Beckmann CF, Jenkinson M, et al. (2016). A multi-modal parcellation of human cerebral cortex. Nature 536, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rosen AFG, Roalf DR, Ruparel K, Blake J, Seelaus K, Villa LP, Ciric R, Cook PA, Davatzikos C, Elliott MA, et al. (2018). Quantitative assessment of structural image quality. Neuroimage 169, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Daducci A, Canales-Rodríguez EJ, Zhang H, Dyrby TB, Alexander DC, and Thiran J-P (2015). Accelerated Microstructure Imaging via Convex Optimization (AMICO) from diffusion MRI data. Neuroimage 105, 32–44. [DOI] [PubMed] [Google Scholar]
- 107.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang J, Lee SH, Goddard ME, and Visscher PM (2011). GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
- 110.Gaiteri C, Chen M, Szymanski B, Kuzmin K, Xie J, Lee C, Blanche T, Chaibub Neto E, Huang S-C, Grabowski T, et al. (2015). Identifying robust communities and multi-community nodes by combining top-down and bottom-up approaches to clustering. Sci. Rep 5, 16361–16414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wu P, Gifford A, Meng X, Li X, Campbell H, Varley T, Zhao J, Carroll R, Bastarache L, Denny JC, et al. (2019). Mapping ICD-10 and ICD-10-CM Codes to Phecodes: Workflow Development and Initial Evaluation. JMIR Med. Inform 7, e14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seidlitz J, Nadig A, Liu S, Bethlehem RAI, Vértes PE, Morgan SE, Váša F, Romero-Garcia R, Lalonde FM, Clasen LS, et al. (2020). Transcriptomic and cellular decoding of regional brain vulnerability to neurogenetic disorders. Nat. Commun 11, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original code is available in this paper’s supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| PsychENCODE human brain RNAseq | Li etal.18 | https://psychencode.synapse.org; http://resource.psychencode.org/ |
| GTEx v8 human brain RNAseq | GTEx Consortium89 | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v8.p2 Accession ID: 26317 |
| ROSMAP human RNAseq | De Jager et al.90 | https://www.radc.rush.edu; https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage/StudyDetails?Study=syn3219045 |
| BrainSpan human brain developmental RNAseq | Miller et al.26 | https://www.brainspan.org |
| Allen Human Brain Atlas | Hawrylycz et al.28 and Arnatkeviciute et al.91 | https://figshare.com/articles/dataset/AHBAdata/6852911 |
| PsychENCODE non-human primate brain RNAseq | Zhuetal.19 | http://evolution.psychencode.org |
| Developmental macaque RNA-seq atlas | Yin et al.43 | GEO: GSE128537 |
| UK Biobank (neuroimaging+genetics) | Warrieretal.10 | Application 20904 |
| Neuroimaging GWAS summary statistics | Warrieretal.10 | https://portal.ide-cam.org.uk/overview/483 |
| Neuroimaging lifespan models | Bethlehem et al4 | https://github.com/brainchart/Lifespan |
| Evolution, development, and cross-subject “population” allometric scaling maps | Hill et al.29 and Reardon et al.30 | https://github.com/netneurolab/neuromaps |
| Neurotransmitter/receptor maps | Hansen et al.37 | https://github.com/netneurolab/hansen_receptors |
| Venous density maps (VENAT) | Huck et al.92 | https://figshare.com/articles/dataset/VENAT_Probability_map_nii_gz/7205960 |
| Diffusion MRI fiber length profiling | Bajada et al.93 | https://balsa.wustl.edu/study/1K3l |
| Cognitive function map | Assem et al.33 | https://balsa.wustl.edu/study/B4nkg |
| Alzheimer disease PET maps | Vogel et al.34 | https://neurovault.org/collections/12296/ |
| ENIGMA cross-disorder cortical thickness map | Larivière et al.36 | https://enigma-toolbox.readthedocs.io/en/latest/pages/04.crossdisorder/index.html |
| Glioblastoma and low grade glioma tumor map | Mandal et al.35 | https://neurovault.org/images/785830/ |
| BigBrain | Amunts et al.38 | https://bigbrain.loris.ca/main.php |
| LiBD Spatial RNA-seq | Die cytoarchitektonik der hirnrinde des erwachsenen menschen Google Books.40 | http://research.libd.org/spatialLIBD/ |
| PsychENCODE WGCNA modules | Gandal et al.58 | http://resource.psychencode.org |
| Mutational constraint datasets (gnomAD v2.1.1) | Chen et al.62 | https://gnomad.broadinstitute.org |
| Mutational constraint datasets (DECIPHER) | Firth et al.61, Collins et al.63 | https://www.deciphergenomics.org |
| Meta-analytic multi-ancestry quantitative trait loci | Zeng et al.71 | https://github.com/jxzb1988/MMQTL |
| MGB Biobank | Zeng et al.75 | https://www.massgeneralbrigham.org/en/research-and-innovation/participate-in-research/biobank |
| Software and algorithms | ||
| RStudio (“Ghost Orchid” for macOS) with R (v.4.1.2) | Team, R.S. RStudio: integrated development for R | https://www.rstudio.com |
| RNA-seq preprocessing for PsychENCODE and GTEx | Kim et al.94 | https://github.com/gandallab/C4A-network |
| ToppGene | Chen et al.56 | https://toppgene.cchmc.org |
| MAGMA | de Leeuw et al.65 | https://ctg.cncr.nl/software/magma |
| FUSION | Gusev et al.95 | https://github.com/gusevlab/fusion_twas |
| Genomic quality control | Peterson et al.96 | https://github.com/getian107/MGBB-QC; https://github.com/Annefeng/PBK-QC-pipeline |
| FreeSurfer (v6.0.1) | Fischl et al.97 | https://afni.nimh.nih.gov/ |
| AFNI | Cox et al.98,99 | https://surfer.nmr.mgh.harvard.edu/ |
| ENIGMA toolbox | Larivière et al.36 | https://enigma-toolbox.readthedocs.io/en/latest/ |
| neuromaps | Markello et al.100 | https://github.com/netneurolab/neuromaps |
| Lifespan brain charts | Bethlehem et al.4 | https://github.com/brainchart/Lifespan |