

SUMMARY
We previously described that the KDM5B histone H3 lysine 4 demethylase is an oncogene in estrogen-receptor-positive breast cancer. Here, we report that KDM5A is amplified and overexpressed in basal breast tumors, and KDM5 inhibition (KDM5i) suppresses the growth of KDM5-amplified breast cancer cell lines. Using CRISPR knockout screens in a basal breast cancer cell line with or without KDM5i, we found that deletion of the ZBTB7A transcription factor and core SAGA complex sensitizes cells to KDM5i, whereas deletion of RHO-GTPases leads to resistance. Chromatin immunoprecipitation sequencing (ChIP-seq) and RNA sequencing (RNA-seq) revealed co-localization of ZBTB7A and KDM5A/B at promoters with high histone H3K4me3 and dependence of KDM5A chromatin binding on ZBTB7A. ZBTB7A knockout altered the transcriptional response to KDM5i at NF-κB targets and mitochondrion-related pathways. High expression of ZBTB7A in triple-negative breast cancer is significantly associated with poor response to neoadjuvant chemotherapy. Our work furthers the understanding of KDM5-mediated gene regulation and identifies mediators of sensitivity to KDM5i.
Graphical abstract
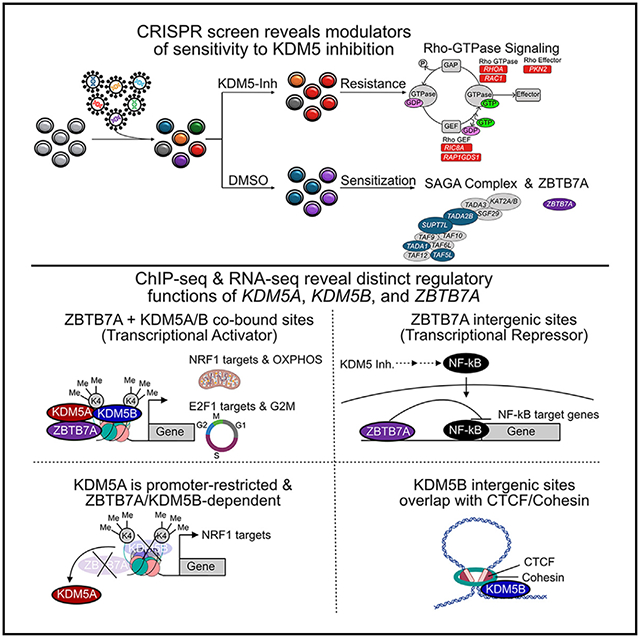
In brief
DiCiaccio et al. conducted a CRISPR viability screen to characterize mechanisms of response and resistance to KDM5 inhibition in basal breast cancer and identified the ZBTB7A transcription factor as a key mediator of KDM5A chromatin binding. Deletion of ZBTB7A alters NF-κB and mitochondrial signaling following KDM5 inhibition.
INTRODUCTION
Histone modifications regulate chromatin structure and transcription, and abnormalities in this process are involved in cancer.1,2 Genes encoding histones and chromatin modifiers are frequently mutated in human cancers,1,3 yet the role of these in tumorigenesis remains poorly defined. Epigenetic regulators define cell states, and cellular phenotypic heterogeneity is a driver of tumor progression and therapeutic resistance.4,5
The KDM5 family of histone 3 lysine 4 (H3K4) demethylases has been implicated as oncogenes in breast cancer. Breast cancer is a heterogeneous disease clinically classified based on the presence of estrogen receptors (ERs) and progesterone receptors (PRs) and HER2 into ER+, HER2+, and triple-negative (ER−/PR−/HER2−; TNBC) subtypes, while gene expression profiling has demonstrated luminal, basal, and mesenchymal molecular subtypes.6 We previously identified KDM5B as a luminal lineage-driving oncogene frequently amplified in ER+ tumors and associated with endocrine resistance.7 In line with this, deletion of Kdm5b in mice delays mammary gland development by perturbing luminal epithelial cell differentiation.8 Subsequently, both KDM5A and KDM5B paralogs were found to contribute to endocrine therapy resistance, indicating redundancy as oncogenes in ER+ breast cancer.9,10 In addition, inhibition of KDM5 catalytic activity decreased cellular transcriptomic heterogeneity, overcoming endocrine resistance.10
The role of KDM5 demethylases in TNBC is less clear, with KDM5A and KDM5B demonstrating divergent results. KDM5A is significantly associated with metastatic relapse in ER− breast cancer patients, and KDM5A knockdown significantly reduced lung metastasis in vivo.11 However, studies of KDM5B in TNBC models implicate KDM5B as a tumor suppressor.12,13 KDM5B inhibits the expression of CCL14 through interaction with the LSD1/NuRD complex, thus suppressing angiogenesis.13 In addition, KDM5B overexpression reduced in vitro migration and invasion of TNBC cell lines, depending on the interaction between KDM5B’s PHD1 domain and unmethylated H3K4.12 Therefore, the function of KDM5 demethylases and their therapeutic potential in TNBC are still unclear.
To investigate the functional relevance of KDM5 in TNBC and basal breast cancer, we analyzed genetic alterations in the KDM5 family and found the KDM5A paralog to be specifically amplified and overexpressed in basal breast cancer. We next analyzed KDM5 inhibition (KDM5i) sensitivity in breast cancer cell lines and identified the SUM149 KDM5A-amplified basal TNBC line with high sensitivity to KDM5i. Using this line and its KDM5i-resistant derivative, we performed genome-wide CRISPR screens to identify modulators of KDM5 inhibitor sensitivity. We followed up top hits and integrated these data with RNA sequencing (RNA-seq) and chromatin immunoprecipitation sequencing (ChIP-seq) to understand the signaling pathways associated with KDM5 activity in basal breast cancer.
RESULTS
KDM5A is commonly amplified and overexpressed in basal breast cancer
Both KDM5A and KDM5B were reported to be amplified and overexpressed in breast cancer.7,14 However, paralog-specific differences between luminal and basal breast cancer have not been analyzed. Thus, we assessed the mutational landscape of KDM5 paralogs in luminal A (LumA) and basal breast tumors in the TCGA cohort. We previously reported that KDM5B is specifically amplified in LumA breast cancer (Figure S1A).7,10 However, KDM5A was specifically amplified in basal breast cancer to a similar degree (Figures S1A and S1B), and KDM5A expression was also the highest in the basal subtype (Figure S1C).15 Although KDM5A had been reported in the 12p13.3 amplicon detected in ~15% of breast cancers,14,15 we demonstrate that this amplification is basal subtype specific, with KDM5A being amplified in over 50% of basal breast cancers (Figure S1B). Given that basal breast cancers tend to be ER−, this could explain why KDM5A expression correlates with metastatic progression only in ER− tumors.11
To address if KDM5A and KDM5B are co-amplified passenger genes and not drivers of their amplicons, we assessed all regions of chromosomes 1 (KDM5B amplicon) and 12 (KDM5A amplicon), respectively, to identify the most frequent amplifications (Figures S1D and S1E). Amplification of chromosome 1q (KDM5B locus) is frequent in LumA breast cancer (Figure S1D), while chromosome 12p13.33 (KDM5A locus) gain is frequent in basal tumors (Figure S1E). In addition, among all genes on chromosome 12, KDM5A is the second most frequently amplified gene next to ETV6. However, the oncoPrint plot shows that KDM5A amplification commonly occurs in the absence of ETV6 amplification (Figure S1F), indicating it is not a passenger associated with ETV6.
The effect of the C70 KDM5 inhibitor on breast cancer cell lines
To determine subtype-specific difference in response to KDM5i, we tested the sensitivity of breast cancer cell lines to the pan-KDM5 inhibitor C70.16 HER2+ and some TNBC cell lines (MDA-MB-436 mesenchymal and SUM149 basal) were the most sensitive (Figures S2A and S2B). Sensitivity to C70 did not significantly correlate with the expression levels of KDM5 family members (Figure S2C), potentially due to small sample size. While high sensitivity of HER2+ breast cancer has been shown before,17 similar sensitivity in a subset of TNBC lines has not been described. Thus, we chose the SUM149 basal KDM5A-amplified TNBC cell line for further studies.
To study mechanisms of acquired resistance to KDM5i, we generated C70-resistant derivatives of SUM149 cells, SUM149CR, by prolonged culture with 10 μM C70 (Figure 1A). We then performed RNA-seq and gene set enrichment analysis (GSEA) on untreated SUM149 and SUM149CR cells. GSEA of differentially expressed genes (DEGs) revealed oxidative phosphorylation as the top enriched pathway in SUM149CR, while inflammatory response and interleukin-10 (IL-10) and nuclear factor κB (NF-κB) signaling were enriched in SUM149 (Figure 1B; Table S1). To test potential differences in mitochondrial function between SUM149 and SUM149CR cells, we performed the Mito Stress Test ± 10 μM C70. SUM149CR showed significantly higher basal respiration compared to SUM149, validating the RNA-seq data, while C70 treatment decreased respiration in both cell lines (Figure 1C).
Figure 1. Characterization of the SUM149CR cell line.
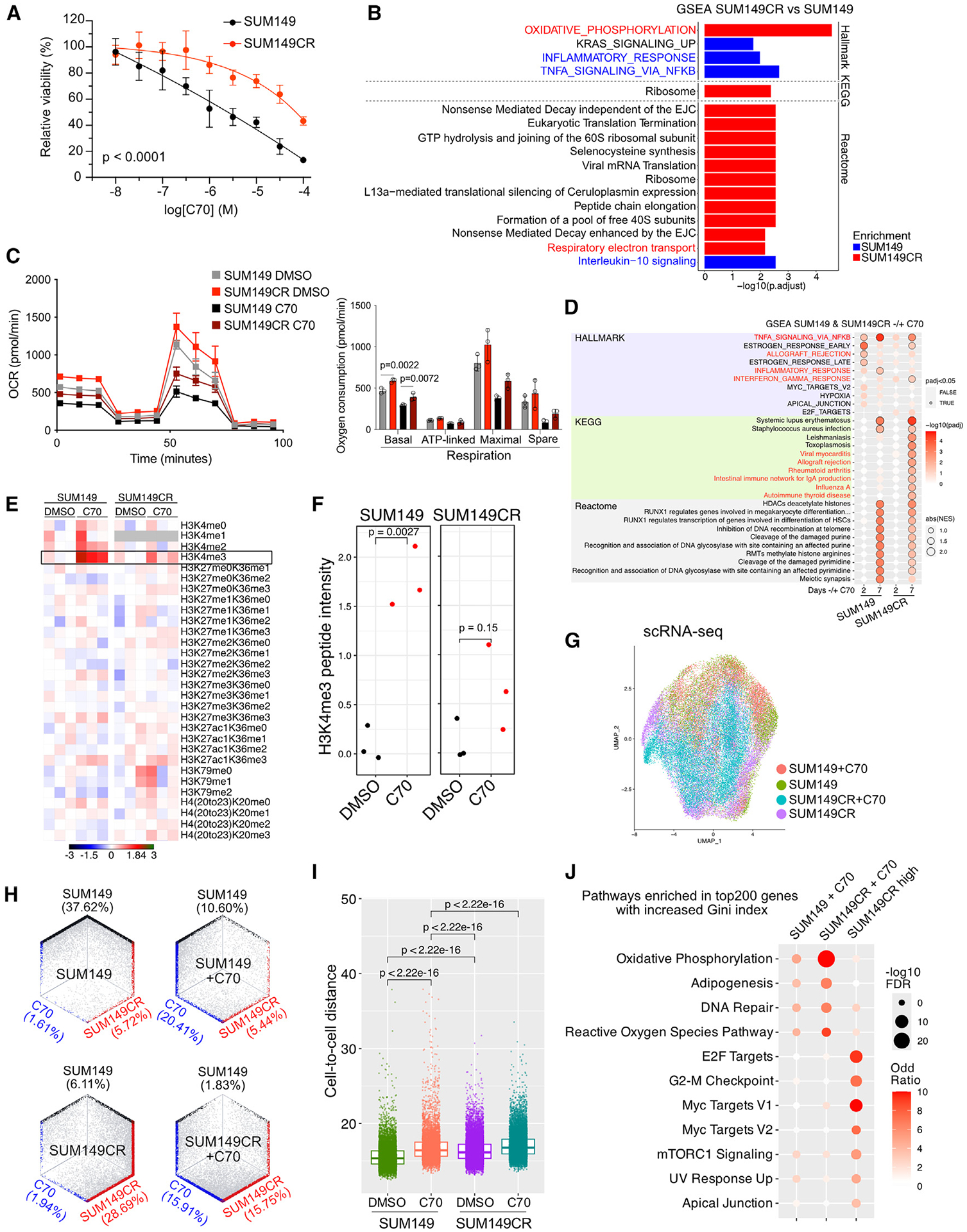
(A) Viability of SUM149 and SUM149CR cells after 7 days of treatment across C70 concentrations. p = t test comparing area under the curve. Data are the mean ± standard deviation (n = 6).
(B) Gene Set Enrichment Analysis (GSEA) comparing RNA-seq profiles of SUM149CR and SUM149 cells.
(C) Oxygen consumption rate (OCR) in SUM149 and SUM149CR cells ± pre-treatment with 10 μM C70 for 6 days; plot (left) and bar graph depicting quantification of differences (right). Values are the mean ± standard deviation. n = 3 for all conditions. One-way ANOVA with multiple comparison within either DMSO- or C70-treated groups for each respiration phase test was used.
(D) GSEA comparing RNA-seq profiles of SUM149 ± 10 μM C70 and SU149CR ± 10 μM C70. Top 10 most significant gene sets from each database are shown.
(E) Heatmap of normalized peptide intensities from mass spectrometry analysis of histone modifications in SUM149 and SUM149CR ± 10 μM C70 treatment for 48 h. Lysine residues that can be methylated are shown. Peptide intensities were normalized to the DMSO control within each cell line, in which there were three processing replicates.
(F) Boxplot depicting quantification of H3K4me3 peptide intensities. The p values are based on the t test.
(G) Uniform Manifold Approximation and Projection (UMAP) of scRNA-seq in SUM149 and SUM149CR ± 10 μM C70 for 7 days.
(H) Hexagonal plots showing classification of single cells as parental (black), parental C70-treated (C70; teal), or C70-resistant (SUM149CR; red) populations.
(I) Boxplot showing transcriptomic cell-to-cell Euclidean distance from PCA dimension reduction in the indicated groups. Mann Whitney U test was used.
(J) Dot plot illustrating the enrichment of hallmark pathways in the top 200 genes with increased Gini index in the indicated groups. Pathways with false discovery rate (FDR) < 0.05 in at least one groups are selected.
To identify C70 resistance-associated gene expression changes, we performed RNA-seq on SUM149 and SUM149CR cells ± 10 μM C70 for 2 or 7 days. SUM149CR had a delayed transcriptional response to C70, indicated by a smaller shift in RNA-seq principal-component analysis (PCA) coordinates at 2 days, but this difference diminished by 7 days of treatment (Figure S2E). Quantification using Euclidian distances confirmed this observation (Figure S2F). In line with this, SUM149CR cells had attenuated enrichment of signaling pathways after C70 treatment (Figure 1D). In SUM149 cells, we found enrichment for tumor necrosis factor α (TNF-α) signaling via NF-κB and inflammatory response upon C70 treatment (Figure 1D). However, in SUM149CR, these pathways were either delayed (TNF-α signaling via NF-κB) or absent (inflammatory response). Quantitative histone mass spectrometry demonstrated muted response of SUM149CR cells to C70 after 2 days of treatment compared to SUM149 based on changes in H3K4me3 (Figures 1E and 1F; Table S2).
To explore resistance and response to C70 at the single-cell level, we performed single-cell RNA-seq (scRNA-seq) on SUM149 and SUM149CR cell lines ± C70 for 7 days. Both SUM149 and SUM149CR cells tended to occupy different regions of Uniform Manifold Approximation and Projection (UMAP) space (Figure 1G). To determine if these cell states are acquired or pre-existing, we next generated cell state signatures from DEGs between SUM149, SUM149CR, and C70-treated SUM149 + C70 from bulk RNA-seq and classified each individual cell into these states. The resistant state was present in a subset of SUM49 cells prior to treatment, while the C70 state was quite distinct, and very few cells occupied this state (Figure 1H). The transition to this C70 state after 7 days of treatment was similar between SUM149 and SUM149CR, indicating that SUM149CR maintains a similar, yet delayed, transcriptional response to C70 treatment.
Previously, we found that C70 treatment reduced cellular transcriptomic heterogeneity of luminal ER+ cell lines but had the opposite effect in the SUM159 TNBC cell line.10 To explore C70 effects in SUM149 and SUM149CR lines, we calculated cell-to-cell transcriptomic distances ± C70 treatment. We found that C70 treatment increased cell-to-cell transcriptomic distances in both SUM149 and SUM149CR cells and that SUM149CR cells had higher baseline heterogeneity compared to SUM149 (Figure 1I). To investigate transcriptomic heterogeneity changes, we performed GSEA on genes with increasing Gini index after C70 treatment or resistance (i.e., more heterogeneously expressed genes). Pathways related to oxidative phosphorylation and reactive oxygen species (ROS) were top enriched in both cell lines after C70 treatment, while proliferation-related pathways were enriched in SUM149CR compared to SUM149 (Figure 1J).
These results imply that C70 has the largest impact on genes related to mitochondrial function and proliferation.
CRISPR viability screen to identify modulators of KDM5 inhibitor sensitivity
To identify synthetic lethal targets and mechanisms of resistance to C70 in basal breast cancer, we performed a genome-wide CRISPR-Cas9 knockout (KO) viability screen ± C70 in SUM149 and SUM149CR. In SUM149 we identified 69 and 138 genes that when deleted made cells more sensitive or resistant to C70, respectively (p < 0.001) (Figure 2A; Table S3). Although KDM5A was a resistance hit (Figure 2A), it had a low robust rank aggregation (RRA) score, and KDM5A and KDM5B KO cell lines did not reveal significant differences in C70 response in cell growth or histone H3K4me3 levels (Figures S2G–S2I). SUM149CR produced fewer hits (24 resistance and 39 sensitizers; p < 0.001) and displayed limited overlap with SUM149 cells (Figures 2A and 2B; Table S3). Only three targets increased sensitivity to C70 in both the SUM149 and the SUM149CR cells: XPR1 (retrovirus receptor), TADA2B (transcriptional adaptor), and MTCH2 (mitochondrial carrier homolog) (Figure 2B; Table S2). We also integrated our RNA-seq data with the CRISPR hits. SOX7, IL7, KIAA1257, MUC6, and DLG5 were differentially enriched in the SUM149 CRISPR screen (p < 0.001) and differentially expressed (padj < 0.05 and abs(log2(FC)) > 1) between SUM149CR and SUM149, while ROS1 and FUT3 were CRISPR hits and differentially expressed between C70- and DMSO-treated SUM149 cells (Table S3).
Figure 2. CRISPR screen results and validation.
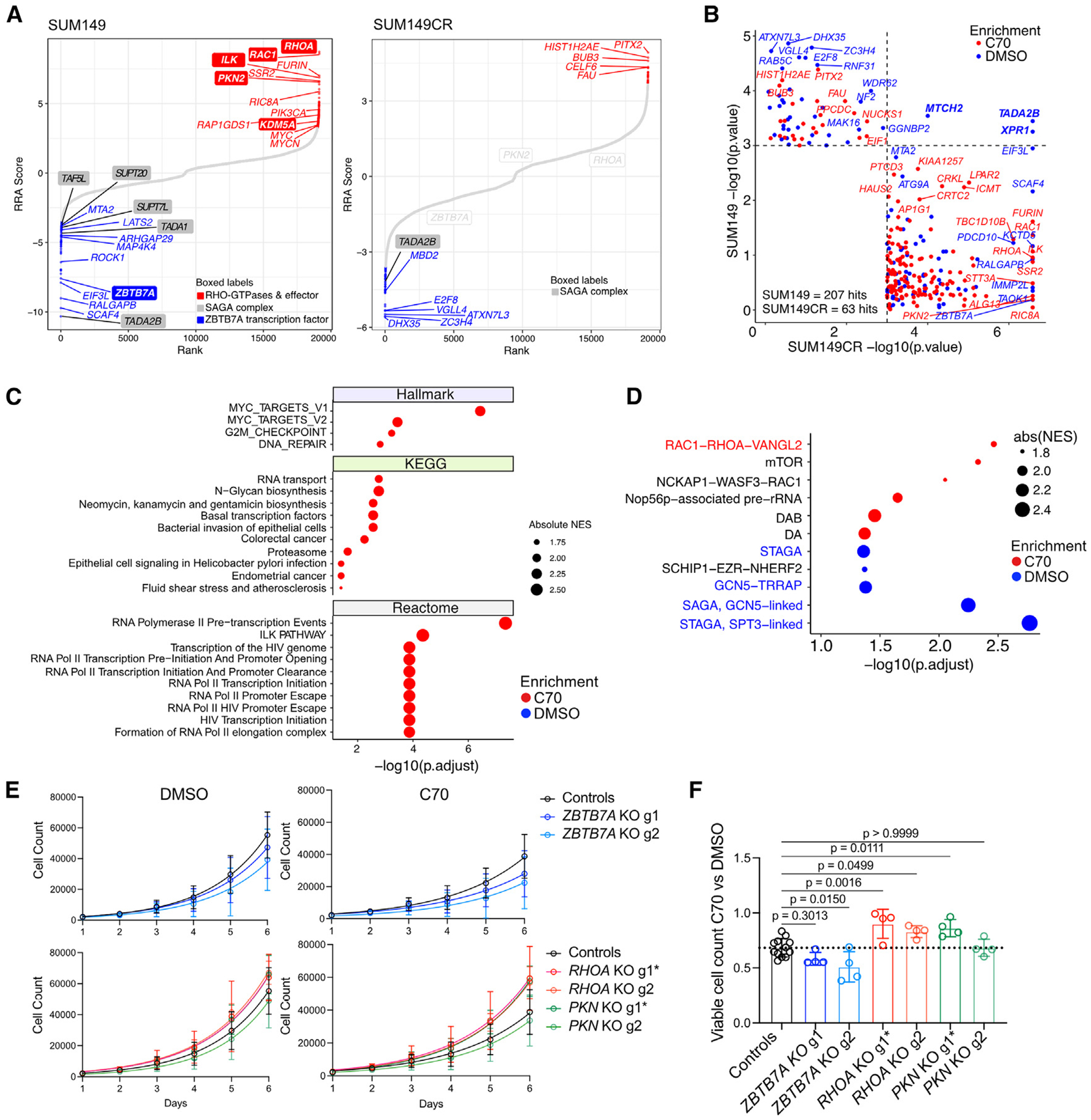
(A) Rank plots of CRISPR KO viability screens in SUM149 and SUM149CR cells after 10 doublings ± 10 μM C70. Genes are ranked based on the computed RRA score from MaGECK RRA, which indicates the essentiality of each gene. Positive RRA scores indicate enriched in C70. Negative RRA scores indicate enriched in DMSO. Differentially enriched hits (p < 0.001) are marked in blue and red for DMSO-enriched and C70-enriched hits, respectively.
(B) Comparison of CRISPR hits with p < 0.001 in either the SUM149 or the SUM149CR CRISPR screens ± C70.
(C and D) GSEA on RRA-ranked CRISPR screen results in SUM149. Top 10 most significant gene sets with padj < 0.05 from the indicated databases (C) and CORUM protein complexes (D) are shown.
(E) Cell growth assays of SUM149 cells expressing constitutive Cas9 and guide RNAs (gRNAs) targeting ZBTB7A, RHOA, PKN2, or non-targeting controls. gRNAs with more efficient KO efficiency are marked with an asterisk (see also Figure S2L). Data are the mean ± standard deviation (n = 4, controls are merged ROSA26 and NonTargeting cells with n = 4 each).
(F) Bar plot depicting quantification of ratios in viable cell numbers upon DMSO vs. C70 treatment at day 6. Data are the mean ± standard deviation, one-way ANOVA followed by Dunnett’s multiple comparisons test comparing to control group only (n = 4, controls are merged ROSA26 and cells expressing non-targeting gRNAs with n = 4 each).
See also Figure S2.
Among the most significantly depleted hits in C70-treated SUM149 was the transcription factor ZBTB7A (Figure 2A; Table S3).18 ZBTB7A is thought to act as a transcriptional repressor and associates with DNA-repressive complexes (e.g., NuRD, Sin3a, and NCoR).19,20 Among the most significantly enriched hits in C70-treated SUM149 were members of the RHOA/RAC1 GTPase cycle (Figures 2A, S2J, and S2K; Table S3). Four of the top six resistance hits included the RHOA and RAC1 GTPases in addition to the RHOA-effector proteins PKN2 and ILK (integrin-linked kinase). Because ILK and RHO/RAC regulate extracellular matrix attachment and cell shape,21 their identification as top C70 resistance hits implies that cell shape might influence response to KDM5i.
We then performed GSEA for Hallmark, KEGG, and Reactome gene sets and CORUM protein complexes within the differentially enriched CRISPR screen hits. Within SUM149, loss of MYC targets was associated with increased resistance (Figure 2C), consistent with prior reports identifying KDM5A required for MYC-driven transcription in multiple myeloma.22 In addition, the RAC1-RHOA-VANGL2 and the SAGA complex were significantly enriched among gRNA targets associated with resistance (Figure 2D). The SAGA complex is a transcriptional co-activator with both histone acetyltransferase (H3K9ac and H3K14ac) and deubiquitinase (H2BK120ub) enzymatic activity.23 Four of the eight SAGA subunits were hits in the SUM149 screen (TADA1, SUPT7L, SPT20H, and TAF5L), and one member of the SAGA histone acetyltransferase (TADA2B) was a hit in both SUM149 and SUM149CR screens (Figures 2A and 2B). This implies that a decrease in SAGA complex activity may increase sensitivity to KDM5i.
To validate the role of ZBTB7A, RHOA, and PKN2 deletion in KDM5i sensitivity, we generated SUM149 cell lines expressing constitutive Cas9 and individual gRNAs (Figure S2L). ZBTB7A deletion reduced growth in the presence of C70, but not DMSO, compared to control guides targeting the ROSA26 locus as well as a non-targeting control guide, confirming the screen results (Figures 2E and 2F). For RHOA and PKN2, only one guide per gene induced a sufficient decrease in protein levels (RHOA-g1 and PKN2-g1, Figure S2L). Consequently, only these effective guides led to increased growth in the presence of C70 compared to controls (Figures 2E and 2F). In addition, RHOA/PKN2 KO had a larger effect on growth in C70 than in DMSO, in agreement with our CRISPR screen.
To investigate how RHO-GTPase signaling could alter sensitivity to KDM5i, we assessed if cell density affects C70-mediated growth arrest and performed RHOA/RAC1-GTP pull-down assays. We found that cells plated at higher densities had diminished sensitivity to C70 (Figure S2M). Pull-downs for GTP-bound RHOA and RAC1 ± C70 showed that under sparse conditions, KDM5i led to a reduction in GTP-bound RHOA (Figure S2N). Therefore, RHO-GTPase signaling might modify KDM5i and vice versa, but delineating these interactions requires further studies.
Because our eventual goal is to evaluate the therapeutic potential of KDM5 inhibitors in breast cancer, we focused on sensitizing hits. We selected ZBTB7A for further studies as it was a top hit sensitizing cells to C70 and the ZBTB7A DNA binding motif was the second most enriched motif in our prior KDM5B ChIP-seq data,7 implying that ZBTB7A and KDM5 family members may interact at common loci.
ZBTB7A, KDM5A/B, repressive chromatin complexes, and response to KDM5i
ZBTB7A and KDM5s are considered repressive factors that associate with multiple histone deacetylase (HDAC)-containing complexes.24,25 Cross-referencing the BioGrid database, we found that ZBTB7A shared 15 interactors with KDM5A and/or KDM5B, 7 within the SWI/SNF, NuRD, and core HDAC chromatin complexes (Figure S3A).26,27 The STRING database of protein-protein interaction networks also predicted functional interactions with chromatin-repressive complexes, in which KDM5A associates with SIN3B and ZBTB7A with NuRD and nuclear co-repressor NCOR1 (Figure S3B).28 Guides targeting the MTA2 and MBD2 subunits of NuRD were significantly depleted upon C70 treatment in SUM149 and SUM149CR CRISPR screens, respectively (Figures 2A and 2B). Therefore, perturbation of the NuRD complex may also sensitize SUM149 cells to KDM5i.13,29,30
To delineate these protein interaction complexes we performed qPLEX-RIME (quantitative multiplexed rapid immunoprecipitation mass spectrometry)31 for KDM5B in luminal ER+ (MCF-7 and T47D) and TNBC (SUM149 and SUM159) cell lines (Table S4). Components of the NuRD, SWI/SNF, and other chromatin complexes were detected in both the luminal and the basal cell lines (Figure S3C). Immunoprecipitation of KDM5B revealed association with many of the core NuRD subunits, MTA1, MTA2, MBD2, MBD3, CHD3, and CHD4, while ZBTB7A pull-downs showed only weak bands for CHD4 and MTA1 (Figure S3D). These data imply that ZBTB7A may sensitize to C70 treatment by modulating the interaction of KDM5A and KDM5B with repressive chromatin complexes.
The STRING database also predicted functional interactions between KDM5A/KDM5B and members of the mitochondrial ATP synthase complex V based on co-expression (Figure S3E) corresponding to C70-resistant cells having higher expression of genes related to oxidative phosphorylation, many of which are subunits of the mitochondrial respiratory chain complexes I–IV or enzymes within the tricarboxylic acid cycle (TCA) cycle (Figures 1B and S3E) and imply that KDM5A/B may regulate mitochondrial activity.
ZBTB7A and KDM5A/B interact and co-localize on chromatin with high H3K4me3 levels
In our previous KDM5B ChIP-seq data ZBTB7A was among the most enriched DNA binding motifs.7 To test if ZBTB7A and KDM5A/B are in the same protein complexes, we performed immunoprecipitation followed by immunoblot using H3K4me3 and CCND1 as positive and negative controls, respectively. We detected ZBTB7A in both KDM5A and KDM5B immunoprecipitants, and both KDM5 family members were present in ZBTB7A pull-downs (Figure 3A). Furthermore, both KDM5A and KDM5B were detected in ZBTB7A immunoprecipitants in both SUM149 and SUM149CR cells regardless of C70 treatment (Figure S3F), indicating that C70 does not disrupt these protein complexes.
Figure 3. ZBTB7A and KDM5A/B interact and co-localize on DNA with high H3K4me3 levels.
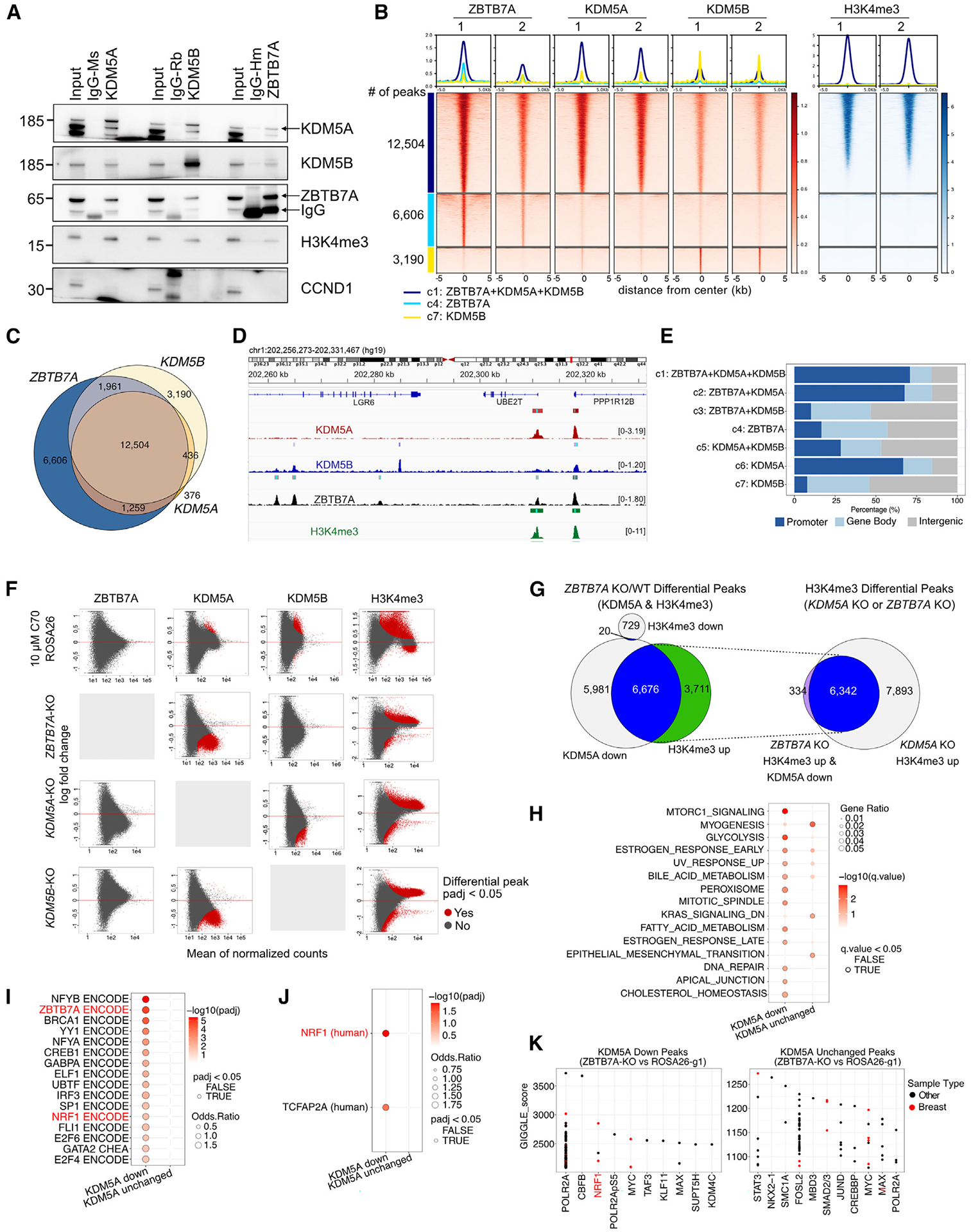
(A) Immunoblot analysis of ZBTB7A, KDM5A, and KDM5B in total cell lysates (input), control IgG, and the indicated immunoprecipitants in SUM149 cells.
(B) Heatmap of ChIP-seq for ZBTB7A, KDM5A, KDM5B, and H3K4me3. Peaks are clustered based on the intersection of peak calls among the three proteins.
(C) Venn diagram illustrating overlap of ChIP-seq peaks.
(D) Example ChIP-seq bigwig tracks with the hg19 genome as a reference.
(E) Genomic feature distribution of peaks within clusters.
(F) MA plots showing differential peak enrichment for the indicated proteins (columns) after the indicated perturbations (rows). Each perturbation is compared to SUM149-ROSA26-g1 in DMSO. Differential peaks are indicated in red (padj < 0.05; default output from CoBRA (Containerized Bioinformatics Workflow for Reproducible ChIP/ATAC-seq Analysis) using the Wald test from DEseq2). The y axis shows log fold change; x axis shows mean of normalized counts.
(G) Venn diagrams showing overlap between KDM5A down and H3K4me3 up peaks in ZBTB7A KO cells. The intersect of these peaks is then compared with H3K4me3 up peaks in KDM5A KO cells.
(H–J) Overlap of the top 500 predicted target genes of KDM5A down/unchanged peaks in the ZBTB7A KO with the described gene sets. (H) Hallmark pathways, (I) consensus target genes for transcription factors present in ENCODE and ChEA, (J) position weight matrices from TRANSFAC and JASPAR at the gene promoters. The top 500 target genes were identified via the regulatory potential score from BETA.32
(K) Overlap of the entire set of KDM5A down/unchanged peaks in the ZBTB7A KO with public ChIP-seq tracks available on CISTROME.33 The top 10–11 enriched transcription factors are shown ranked by GIGGLE score (−log10(p) * odds ratio).34
See also Figure S3.
To assess if ZBTB7A and KDM5A/B co-localize at the same chromatin regions, we performed ChIP-seq for each protein. We found a significant overlap between ZBTB7A, KDM5A, and KDM5B peaks, especially at sites with high levels of H3K4me3 (Figures 3B–3D and S3G; Table S4). We clustered the peaks based on overlap and found that 47% of all high-confidence peaks (i.e., peaks called across replicates) are co-occupied by ZBTB7A, KDM5A, and KDM5B and coincide with high levels of H3K4me3 (12,504 of 26,332) (Figures 3B, 3C, and S3G). Interestingly, KDM5A primarily bound promoter regions with high H3K4me3 signal, while ZBTB7A and KDM5B were found at both promoter and non-promoter regions (Figures 3E and S3H). This suggests that KDM5A and KDM5B may have distinct preference for genomic loci and thus functions, although differences due to antibodies used for ChIP cannot be excluded.
To determine correlations of each peak cluster with other chromatin binding factors, we explored overlap with public ChIP-seq data using Cistrome DB’s toolkit33 and compared the top 10 factors identified for each cluster based on the maximum GIGGLE score (−log10(padj) * odds ratio from Fisher’s exact test)34 (Figure S3I). ZBTB7A binding in non-promoter regions (clusters 3 and 4) had specific overlap for SWI/SNF (SMARCC1 and SMARCA4) and NuRD (MBD3) chromatin complexes. It was also enriched for STAT3, which regulates the NF-κB signaling pathway.35 The KDM5B-specific cluster 7 overlapped with factors related to the cohesin/CTCF complex (STAG1, SMC3, SMC1A, RAD21, ESCO2, and CTCF), in line with our prior data demonstrating that KDM5B physically interacts and co-localizes with CTCF.7 Factors enriched across KDM5A-containing clusters were mostly associated with active promoters (e.g., H3K4me3, H3K9ac, and POLR2A), in agreement with KDM5A being mainly restricted to promoter regions. Enrichment analysis of the ZBTB7A consensus sequence (GACCC)36 showed significant enrichment only in overall ZBTB7A peaks and in clusters where ZBTB7A and KDM5A peaks overlapped (Figure S3J), strengthening the importance of the ZBTB7A/KDM5A interactions.
To gain functional insights into each cluster, we assessed overlap of their predicted target genes with Hallmark gene signatures (Figure S3K) using the top 500 predicted target genes based on regulatory potential scores (Table S5).32 Cluster 1 (co-bound by KDM5A, KDM5B, and ZBTB7A) was significantly enriched for Hedgehog signaling, G2M checkpoint, glycolysis, and hypoxia. Clusters 3 and 4 (mostly intergenic regions bound by ZBTB7A) showed enrichment for apical junction and coagulation. Finally, cluster 7 (mostly intergenic KDM5B peaks) was significantly enriched for downregulated genes in response to UV. There is little overlap between the top 500 target genes of each cluster (Figure S3L), implying both shared and unique functions for each of the three proteins.
ZBTB7A KO reduces chromatin-bound KDM5A
To test if the co-localization of ZBTB7A, KDM5A, and KDM5B is due to direct recruitment mechanisms, we performed ChIP-seq in SUM149 ROSA26g control and in each of the three individual KO cells. ZBTB7A binding was not affected by deletion of KDM5A and KDM5B or by C70 treatment (Figure 3F). However, deletion of ZBTB7A led to decreased KDM5A signal in 43% of KDM5A peaks with no effect on KDM5B binding (Figure 3F). Since total KDM5A protein levels were not reduced in the ZBTB7A KO (Figure S2L), ZBTB7A may help recruit and/or stabilize KDM5A but not KDM5B at specific loci. KDM5A peaks were also decreased in the KDM5B KO, and conversely, we observed diminished KDM5B peak intensities in the KDM5A KO, implying mutual stabilization of KDM5A and KDM5B at a subset of binding sites (Figures 3F and S4A). However, potential cross-reactivity of the KDM5A and KDM5B antibodies used for ChIP cannot be excluded.
Next, we assessed whether KDM5A peaks that were decreased in ZBTB7A KO cells were associated with changes in H3K4me3. We found that 53% of these peaks were associated with an increase in H3K4me3 (Figures 3G and S4B). To determine whether the increase in H3K4me3 signal at these loci is a consequence of diminished KDM5A binding, we analyzed H3K4me3 at these loci in KDM5A KO cells and found that 95% of these peaks also had increased H3K4me3 upon KDM5A KO (Figures 3G and S4B). Therefore, loss of ZBTB7A likely leads to a decrease in KDM5A chromatin binding and a subsequent increase in H3K4me3.
To identify differences between changed and unchanged KDM5A peaks in ZBTB7A KO cells, we first identified the top 500 predicted gene targets based on regulatory potential scores (Table S5) and quantified overlap with Hallmark gene sets. Genes associated with diminished KDM5A peaks had stronger enrichment for mTORC1 signaling and glycolysis, whereas those associated with unchanged KDM5A peaks had stronger enrichment with myogenesis (Figure 3H). Second, overlap with consensus transcription factor targets from ENCODE/ChEA and promoter motifs from TRANSFAC/JASPAR identified nuclear respiratory factor 1 (NRF1) transcription factor as consistently enriched among KDM5A down-target genes (Figures 3I and 3J). NRF1 is a regulator of nuclear genes encoding mitochondrial respiratory complex subunits and the transcription and replication of mitochondrial DNA.37 We also saw enrichment for ZBTB7A among the KDM5A down-target genes (Figure 3I), supporting that these are ZBTB7A-specific peaks. Finally, we assessed the entire KDM5A decreased and unchanged peak sets for overlap with public ChIP-seq data in CISTROME.38 Again, NRF1 was significantly enriched among the KDM5A peaks diminished in the ZBTB7A KO cells, implicating its importance as a putative downstream target of ZBTB7A and KDM5A in basal breast cancer (Figure 3K). To test if the enrichment in NRF1 binding sites in KDM5A peaks lost in ZBTB7A KO cells is due to differences in NRF1 expression or nuclear localization, we performed immunoblot analysis for NRF1 in fractionated cell lysates from wild-type (WT) and KDM5A or ZBTB7A KO cell lines but found no differences (Figure S4C). Thus, the enrichment for NRF1 targets might indicate overlap between the downstream targets of NRF1 and the KDM5A-ZBTB7A complex rather than its direct activation.
ZBTB7A activation/repressive regulatory function depends on the chromatin context
We next investigated gene expression changes induced by KDM5A, KDM5B, or ZBTB7A KO. Overall, KDM5A and KDM5B KOs had minimal impact on gene expression compared to the ZBTB7A KO at the high stringency we used as cut off (padj < 0.05 and |log2(FC)| > 1) (Figures 4A and 4B; Table S1). A possible explanation is the redundancy of KDM5A and KDM5B, and the deletion of only one is not sufficient to cause major transcriptomic changes. Supporting this idea is the observation that C70 treatment that inhibits all KDM5s led to significant changes in gene expression in both WT and KO cells (Figure 4A; Table S1). It is also possible that the acute deletion of the gene has an effect, but this is diminished by the time the cells are expanded due to compensatory mechanisms. However, we do see significant changes in the expression of mitochondria-encoded genes in the KDM5A KO cells; 11 of the 13 most downregulated genes (padj < 0.001) in the KDM5A KO were mitochondrial encoded (Figure 4C; Table S1), which was not observed in KDM5B KO cells (Figure S4D). This result is consistent with our findings of ZBTB7A-dependent KDM5A peaks being enriched in targets of the mitochondrial biogenesis transcription factor NRF1.
Figure 4. Gene expression changes induced by ZBTB7A-KO and its associations with ZBTB7A and KDM5A/B peak sets.
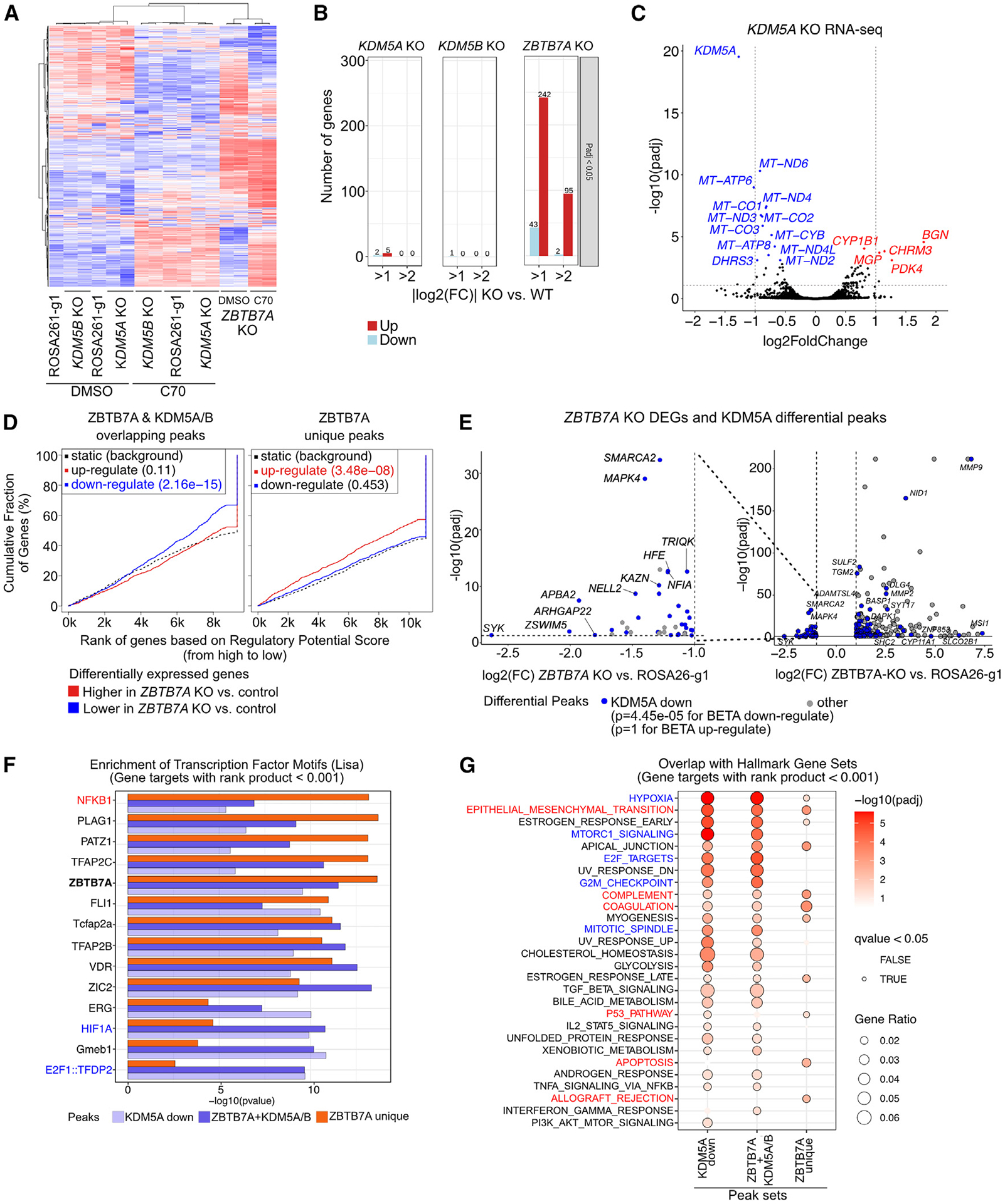
(A) Heatmap of RNA-seq in SUM149 cells expressing the indicated gRNAs treated with DMSO or 10 μM C70 for 7 days. Rows and columns are ordered based on hierarchical clustering. Values are row-normalized Z scores.
(B) Number of DEGs for KOs in SUM149 cells compared to the ROSA26-g1 control.
(C) Volcano plot of RNA-seq in the SUM149 KDM5A KO compared to the ROSA26-g1 control. Dashed gray lines indicate adjusted p value (padj) and fold change (FC) cutoff used for (B).
(D) Output from BETA testing for association between the indicated peak sets and the up-/downregulated genes upon ZBTB7A KO.32 For promoter-enriched peaks, the distance from the transcription start site (TSS) for within which peaks were considered to contribute to the gene regulatory potential score was set to 3 kb. For non-promoter-enriched peak sets, the default parameter of 100 kb was used.
(E) Volcano plot comparing DEGs and decreased KDM5A peak enrichment in the ZBTB7A KO cells. Only significant DEGs are shown (padj < 0.05 and |log2(FC)| > 1). The p values are based on BETA, indicating if the differential peak set is significantly associated with up- or downregulated genes. Nearest genes to each peak are annotated.
(F and G) Overlap of the predicted target genes for each peak set with (F) CISTROME LISA transcription factor motifs (top 5 motifs per cluster are shown) and (G) Hallmark gene sets. Genes with a rank product score <0.001 from the BETA output were used as predicted target genes for each peak set.
See also Figure S4.
We next integrated RNA-seq from the ZBTB7A KO with our ChIP-seq data to determine if ZBTB7A binding was significantly associated with changes in gene expression. Based on binding and expression target analysis (BETA),32 we found that ZBTB7A peaks overlapping with KDM5A/B, which are enriched at promoters, were associated with decreased gene expression in the ZBTB7A KO (p = 2.16e−15 for BETA downregulated) (Figure 4D). Conversely, ZBTB7A-unique sites, which are enriched in intronic/intergenic regions, were associated with increased gene expression in the ZBTB7A KO cells (p = 3.48−08 for BETA upregulated) (Figure 4D). Therefore, non-promoter ZBTB7A may be acting as a canonical repressor, whereas promoter-bound ZBTB7A may be acting as a transcriptional activator.
We next investigated if changes in chromatin were associated with differential gene expression. As expected, increased H3K4me3 signal was associated with increased gene expression (p = 3.48e−07 for BETA upregulated) (Figure S4E). Surprisingly, sites with increased H3K4me3 that coincide with decreased KDM5A binding had no correlation with gene expression (Figure S4E). In fact, diminished KDM5A signal intensity was significantly associated with a decrease in gene expression (Figures 4C and 4E, p = 4.45e−05 for BETA downregulated), implying that the KDM5A-ZBTB7A complex is a positive regulator of gene expression.
We next analyzed the predicted targets of the ZBTB7A and KDM5A/B overlapping, ZBTB7A unique, and KDM5A down peaks for enrichment of specific transcription factors or pathways. We defined direct targets as genes with a rank product <0.001 from the BETA output in Figures 4D and 4E.32 The ZBTB7A transcription factor motif was evenly enriched between ZBTB7A unique and ZBTB7A and KDM5A/B overlapping peaks (Figure 4F). However, several transcription factors showed a bias toward one peak set. For example, NFKB1 was more significantly enriched among the ZBTB7A unique peak target genes, whereas HIF1A and E2F1::TFDP2 were more significantly enriched among the ZBTB7A and KDM5A/B overlapping target genes (Figure 4F). These observations support our analysis of the Hallmark gene set pathways (Figure 4G), in which HIF1A (e.g., hypoxia) and E2F-driven (e.g., E2F targets, G2M checkpoint, and mitotic spindle) pathways were specifically enriched among the ZBTB7A and KDM5A/B overlapping and KDM5A down gene targets, whereas immune pathways (e.g., allograft rejection, coagulation, and complement) were specifically enriched in the ZBTB7A unique peaks.
We also created ZBTB7A KOs in additional breast cancer cell lines (Figure S4F). MDA-MB-436 is another BRCA1-mutant TNBC cell line, like SUM149, with similar sensitivity to C70 (Figures S2A and S2B), and MCF7 is an ER+, luminal breast cancer cell line. Similar to SUM149, deletion of ZBTB7A predominantly increased transcription in both MDA-MB-436 and MCF7 cells (Figure S4G). DEGs between parental and ZBTB7A KO cells in the SUM149 line showed similar trends in both MDA-MB-436 and MCF7 cell lines (Figures S4H and S4I). Upregulated DEGs from the SUM149 ZBTB7A KO cells were significantly enriched in both MDA-MB-436 (padj = 2.7e–19, GSEA) and MCF7 (padj = 7.8e–13, GSEA) ZBTB7A KOs (Figure S4J). However, downregulated DEGs from SUM149 cells were enriched only in the MDA-MB-436 ZBTB7A KO cells (padj = 5.3e−3, GSEA) (Figure S4J).
To determine if ZBTB7A KO alters TNBC subtype-specific transcriptional states, we performed GSEA on the TNBC subtypes we previously described.39 Mesenchymal and basal-specific genes were upregulated in the luminal MCF7 line, while the MDA-MB-436 mesenchymal-TNBC line showed upregulation of genes repressed in mesenchymal cells and downregulation of mesenchymal genes (Figure S4K). Finally, the basal-TNBC line SUM149 reactivated genes repressed in the basal state and turned on genes specific to mesenchymal and luminal-like TNBC (Figure S4K). Therefore, ZBTB7A may be an important regulator of cell state fidelity in breast cancer. Last, the most significantly upregulated pathways by C70 in the TNBC lines were related to immunity (e.g., allograft rejection, complement, inflammatory response), myogenesis, epithelial-to-mesenchymal transition, and IL-6/JAK/STAT3 in SUM149 cells and interferon-α response in the MDA-MB-436 cell line (Figure S4L).
These data show that ZBTB7A is an important regulator of epithelial cell differentiation-related processes and that some of its function is via modulation of KDM5A chromatin binding.
ZBTB7A and KDM5i co-regulate oxidative phosphorylation and NF-κB targets
To explore how loss of ZBTB7A alters the transcriptional response to KDM5i, we performed RNA-seq in WT and ZBTB7A KO cells ± 10 μM C70 for 7 days. We first observed that C70 treatment upregulates more DEGs (padj < 0.05, |log2(FC)| > 1) in the ZBTB7A KO cells compared to WT controls across all three cell lines (SUM149, MDA-MB-436, and MCF7) (Figure S5A; Table S1). In addition, ZBTB7A KO did not change the gene targets of C70, since there was pronounced overlap in the DEGs induced in both WT and KO cells, at least in the two TNBC lines with a more limited overlap in MCF7 ER+ luminal cells (Figure S5A). Therefore, loss of ZBTB7A may amplify response to C70 without completely rewiring its target genes in TNBC.
We next examined if C70 treatment modulated different pathways in the ZBTB7A KO cells (Figure 5A). GSEA in both SUM149 and MDA-MB-436 cells upon C70 treatment identified a reduction in oxidative phosphorylation specifically in the ZBTB7A KO and not in the WT cells (Figure 5A). Analysis of mitochondrial respiration in SUM149 WT and ZBTB7A KO cells ± 10 μM C70 showed that ZBTB7A KO and C70 diminished basal and maximum respiratory potential, and this decrease was saturated with C70 treatment and not further augmented by ZBTB7A KO (Figure 5B). Since dysfunctional mitochondria can be a source of ROS, we assessed total ROS levels and mitochondrial cardiolipins. We found higher ROS levels in both WT and ZBTB7A KO C70-treated cells compared to vehicle controls. However, ROS levels were higher within the ZBTB7A KO cells treated with C70 compared to WT (Figure 5C). In addition, nonyl acridine orange (NAO) staining to measure the abundance and redox status of mitochondrial cardiolipins showed no effect of C70 treatment in the WT cells but a decrease signal in the ZBTB7A KO cells, implying an increase in oxidized cardiolipins (Figure 5C).
Figure 5. Effects of ZBTB7A KO on transcriptional response to KDM5 inhibition.
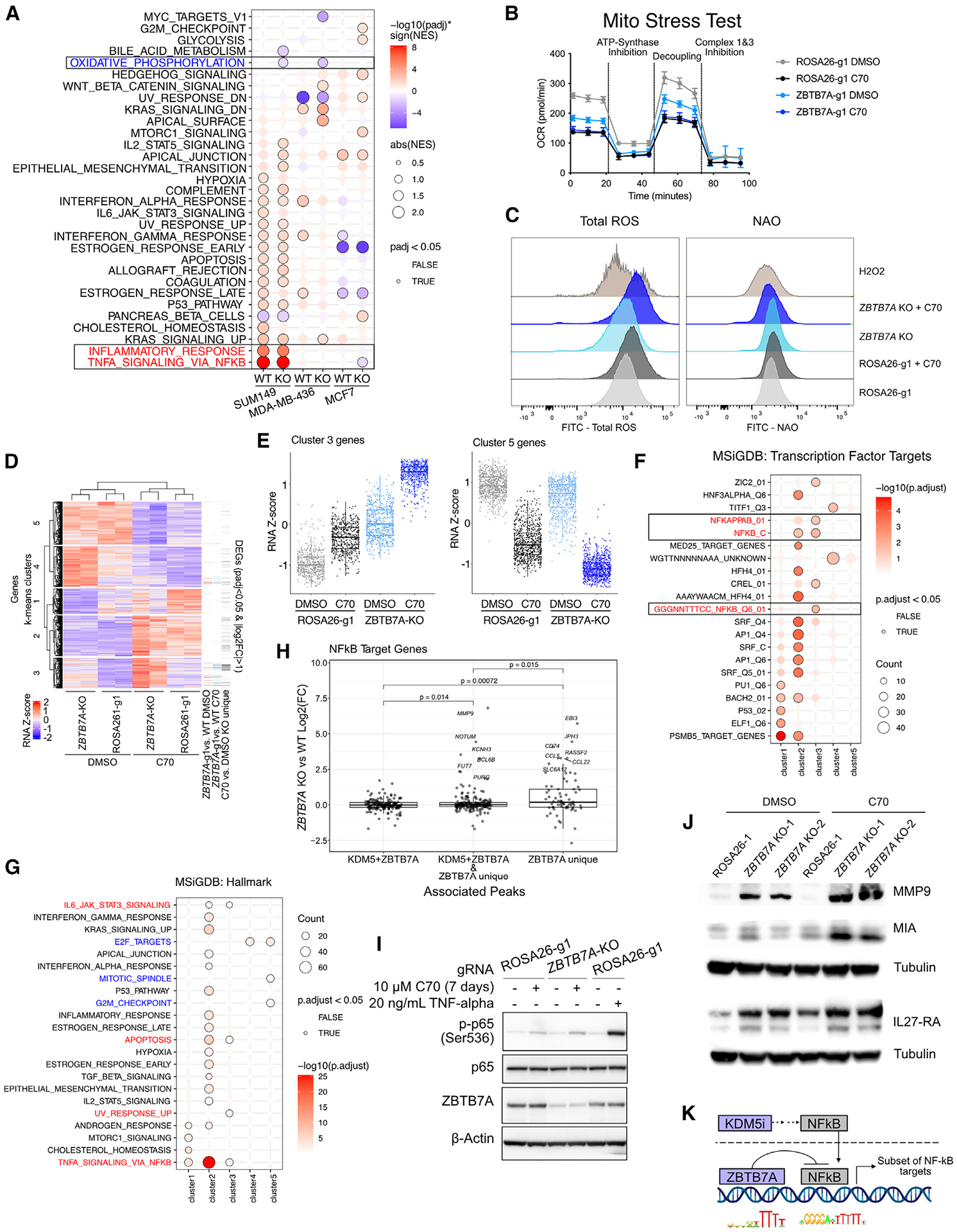
(A) GSEA on genes ranked by log2(FC) for ±10 μM C70 for 7 days. The analysis was performed in all three cell lines with wild-type (i.e., ROSA26-g1) or ZBTB7A KO.
(B) Oxygen consumption rate (OCR) in ROSA26-g1 and ZBTB7A KO SUM149 cells ± pre-treatment with 10 μM C70 for 6 days. Values are the mean ± standard deviation. N = 5 for all conditions except ZBTB7A KO + C70, which had one outlier well removed (N = 4).
(C) Ridge plot depicting flow cytometry for total ROS detection with the Total Reactive Oxygen Species (ROS) Assay Kit and for mitochondrial cardiolipins with nonyl acridine orange (NAO). SUM149 cells were treated with or without 10 μM C70 for 5 days. One millimolar H2O2 (7 h for ROS and 2 h for NAO) was used as a positive control.
(D) Heatmap of DEGs upon C70 treatment in either SUM149 ROSA26-g1 or ZBTB7A-g1 (padj < 0.05). Genes are ordered based on k-means clustering (k = 5) and samples are ordered based on hierarchical clustering.
(E) RNA Z scores of cluster 3 and 5 genes from (D). Box plots represent mean, first and third quantile, and min and max values.
(F and G) Overrepresentation analysis for (F) MSigDB transcription factor targets and (G) MSigDB Hallmark pathways within each gene cluster specified by (D).
(H) Plot of NF-κB target genes associated with KDM5 + ZBTB7A peaks, ZBTB7A unique peaks, or both (KDM5 + ZBTB7A and ZBTB7A unique). Target genes were defined by the union of MSigDB transcription factor target gene sets (GGGNNTTTCC_NFKB_Q6_01, NFKAPPAB_01, NFKAPPAB65_01, NFKB_C, NFKB_Q6_01, and NFKB_Q6). The p value was determined by the t test. Box plots represent mean, first and third quantile, and min and max values.
(I) Immunoblot for phospho-NF-κB p65 (Ser536) in SUM149 ROSA26-g1 and ZBTB7A KO cells ± 10 μM C70 for 7 days. Cells treated with 20 ng/mL TNF-α for 5 min were used as positive control. Image is the left side part of a larger blot with additional lanes.
(J) Immunoblot for NF-κB targets MMP9, MIA, and IL-27-RA in SUM149 ROSA26-g1 or ZBTB7A KO cell lines ± 10 μM C70 for 6 days. Tubulin was used as loading control.
(K) Diagram of proposed interaction between ZBTB7A and KDM5 inhibition on NF-κB signaling.
See also Figures S4 and S5.
To more globally analyze how ZBTB7A KO alters transcriptional response to C70, we re-clustered all DEGs upon C70 treatment in WT or ZBTB7A KO cells (padj < 0.05) (Figure 5D). We then categorized the genes into five clusters, each with unique responses to ZBTB7A and/or C70 (Figures 5D, 5E, and S5B). Cluster 1 genes had a muted upregulation following C70 treatment in ZBTB7A KO cells compared to WT (Figure 5D), and they showed enrichment in mitochondrion-encoded genes (Figures S5C and S5D). We also assessed if upregulated genes are clustered to particular genomic regions potentially reflecting co-regulation due to shared enhancer activity. Only one cluster, cluster 2, displayed cytogenetic enrichment to a specific genomic region, 1q32 (Figure S5C). Among all clusters detected, cluster 3 was particularly prominent because it was enriched in genes differentially expressed due to either C70 treatment or ZBTB7A deletion (Figure 5D). These genes increased upon loss of ZBTB7A and had a further increase with C70 treatment (Figure 5E). When we analyzed each cluster for overlap with MSigDB transcription factor targets, cluster 3 was significantly enriched for NF-κB target genes (Figure 5F), which was also the transcription factor motif enriched among the predicted targets of ZBTB7A unique peaks (Figure 4F). To explore this in more detail, we analyzed changes in the expression of NF-κB targets in ZBTB7A KO cells compared to WT and found significantly higher expression of NF-κB targets associated with ZBTB7A unique compared to ZBTB7A and KDM5 overlapping peaks (Figure 5G).
When comparing each cluster to Hallmark gene sets, we found significant enrichment of TNF-α signaling via NF-κB, apoptosis, and IL-6/JAK/STAT3 signaling in both clusters 2 and 3, both of which show highest gene expression levels in the C70-treated ZBTB7A KO cells (Figures 5E, 5H, and S5B). Cluster 2 showed the most significant enrichment for TNF-α signaling via NF-κB even though the NF-κB transcription factor targets were specifically enriched in cluster 3 (Figures 5F and 5H).
To examine NF-κB activation after KDM5i and ZBTB7A KO, we assessed phospho(S536)-p65 and nuclear p65 levels and the activity of an NF-κB-driven GFP reporter. C70 increased phospho(S536)-p65 as well as marginally increasing nuclear p65 levels, with ZBTB7A KO having no observable effect (Figures 5I and S5E). Similarly, C70 induced GFP expression driven by a minimal NF-κB promoter, with no additive effect observed for the ZBTB7A KO (Figure S5F). However, immunoblot analysis of NF-κB targets selected from the overlapping topmost enriched genes in cluster 3 (Figure 5E) and NF-κB targets (Figure 5F) revealed that the expression of several proteins, including MMP9, MIA, and IL-27-RA, was significantly higher in ZBTB7A KO cells and upregulated by C70 (Figure 5J). Taken together, our data imply that KDM5i acts upstream and leads to the activation and nuclear localization of NF-κB. ZBTB7A, however, acts at the DNA level, in which ZBTB7A unique peaks repress a subset of NF-κB direct-target genes (Figure 5K).
In contrast to cluster 3 genes that were the most upregulated by C70 in ZBTB7A KO, cluster 5 genes had the largest decrease in expression in C70-treated ZBTB7A KO cells (Figures 5D and 5E). This cluster was specifically enriched for proliferative pathways, such as E2F targets, mitotic spindle, and G2M checkpoint (Figure 5G), which were the same pathways specifically enriched among KDM5A sites with diminished binding in the ZBTB7A KO (Figure 4G). Since these peaks were associated with activator function (Figure 4E), we hypothesize that ZBTB7A and KDM5A co-bound sites function as transcriptional activators of pro-proliferation pathways and that combined ZBTB7A KO and KDM5i leads to a stronger downregulation of these target genes.
Last, we reanalyzed our RNA-seq data for differential expression of endogenous retroelements with ERVmap because KDM5B was previously reported to derepress retroelements.40 We found that KDM5i led to increased expression of endogenous retroelements (mostly LINEs [long interspersed nuclear elements] and SINEs [short interspersed nuclear elements]) in both the ROSA26-g1 control cells and all three KO derivates (Figure S5G; Table S6).41 However, while 4,732 endogenous retroelements were significantly differentially expressed between DMSO- and C70-treated parental SUM149 cells (mostly upregulated), this number was much higher (12,097) in ZBTB7A KO and lower in KDM5A and KDM5B KO lines (4,072 and 2,474, respectively). In addition, we also noted that, in contrast to the limited changes seen in the expression of protein-coding genes in KDM5A KO cells, the expression of 2,143 endogenous retroelements was significantly different between KO and WT cells, whereas this was not seen in KDM5B KO (Table S6). Thus, some of the phenotypic consequences of C70 treatment and deletion of KDM5 genes might be via their regulation of endogenous retroelement expression.
ZBTB7A expression alters KDM5-associated phenotypes in patient samples
We next tested if ZBTB7A expression altered KDM5-associated phenotypes in patient samples using the TCGA and METABRIC cohorts. Corroborating our findings in cell-line models, KDM5A/B expression was significantly positively correlated with proliferation (E2F targets, G2M checkpoint, and mitotic spindle) and negatively correlated with NF-κB and other inflammation-related pathways (Figure 6A). In addition, these correlations were stronger among samples with low ZBTB7A expression. Analysis of TNBC samples from the METABRIC cohort also demonstrated that KDM5A/B expression is more strongly associated with many of the select pathways in ZBTB7A-low samples, but the differences were less prominent (Figure 6B).
Figure 6. Associations between ZBTB7A and KDM5 expression and tumor features in patient samples.
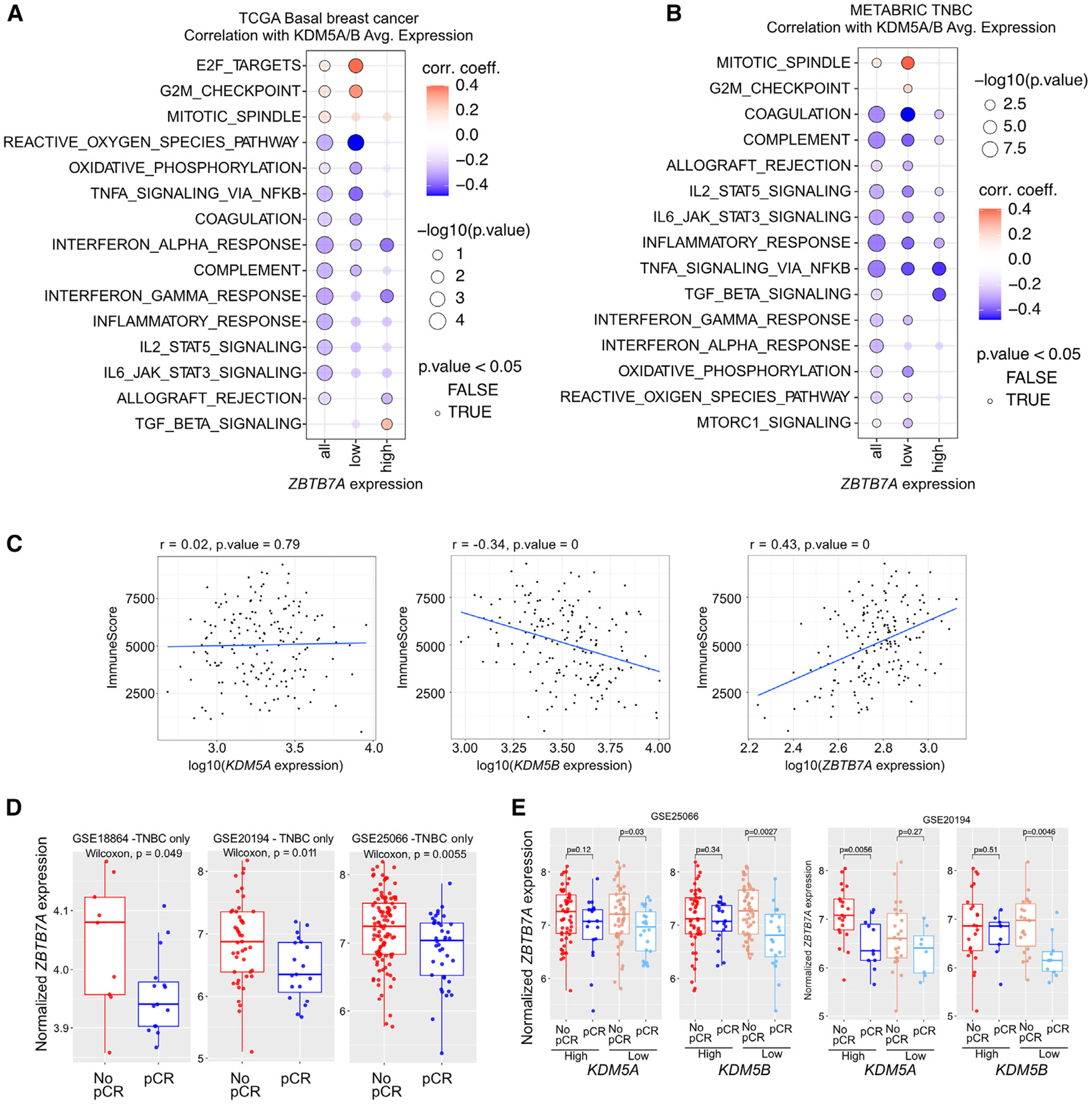
(A and B) Correlation between KDM5A/B expression and select pathways across basal tumors from TCGA (A) and TNBC samples from METABRIC (B). Correlation coefficients and −log10(p) are plotted. The samples were subset into high and low ZBTB7A expression based on upper and lower tertiles.
(C) Correlation between KDM5A, KDM5B, and ZBTB7A expression with estimated immune infiltration scores from bulk RNA-seq data. Data are from basal tumors in TCGA. Immune scores were calculated from bulk RNA-seq via “Estimation of Stromal and Immune cells in Malignant Tumors Using Expression Data” (ESTIMATE).
(D) Boxplots depicting the expression of ZBTB7A in TNBC from patients with pCR or no pCR from the indicated cohorts. Box plots represent mean, first and third quantile, and min and max values. Mann-Whitney U test was used.
(E) Boxplots depicting the expression of ZBTB7A in breast tumors from patients with pCR or no pCR from the indicated cohorts and divided based on the expression levels of KDM5A or KDM5B. Box plots represent mean, first and third quantile, and min and max values. Mann-Whitney U test was used.
Due to the significant inverse association between KDM5A/B expression and inflammation-related pathways, we also assessed the correlation between KDM5A, KDM5B, or ZBTB7A expression with estimated immune infiltration scores among basal breast cancer samples in the TCGA cohort. We found that ZBTB7A was significantly positively correlated with immune infiltration, KDM5B was negatively correlated, and KDM5A had no association (Figure 6C).
Finally, we wanted to determine if ZBTB7A expression modified response to neoadjuvant chemotherapy by itself or associated with KDM5 expression. We found that the expression of ZBTB7A in TNBC was significantly higher in tumors from patients who did not achieve pathologic complete response (pCR) in three independent cohorts (Figure 6D).42–44 The association of higher ZBTB7A expression with lack of pCR was significant only in tumors with lower expression of KDM5B, whereas for KDM5A this was less consistent (Figure 6E).
These analyses of clinical samples validate our findings in the cell-line models and confirm the role for ZBTB7A as a transcriptional modulator of NF-κB and inflammation-related pathways.
DISCUSSION
Through CRISPR KO viability screens ± C70 KDM5 inhibitor, we identified several factors that modulate sensitivity to KDM5i in basal breast cancer, including ZBTB7A. We demonstrated that ZBTB7A interacts and co-localizes with KDM5A/B at genomic regions with high H3K4me3 levels and that a subset of KDM5A binding might be ZBTB7A dependent.
Functional differences between the KDM5 paralogs have been described previously, but the underlying mechanism remains unresolved.45 For example, KDM5A and not KDM5B was found to promote MYC-driven transcription in multiple myeloma by altering RNA polymerase II (RNAPII) promoter-proximal pausing,22 while KDM5B was specifically found to suppress endogenous retroelement expression in melanoma by recruiting the H3K9 methyltransferase SETDB1.40 Our work provides some mechanistic insight into these distinctions, in which we found significant differences in genomic binding between KDM5A and KDM5B. Specifically, KDM5A was primarily restricted to promoters, whereas KDM5B exhibited extensive binding across both promoter and non-promoter regions. This indicates that KDM5A could be a promoter-specific regulator of transcription in agreement with prior findings that KDM5A is a key modulator of RNAPII pausing at MYC-driven promoters.22 In addition, the intergenic binding of KDM5B could explain why this paralog was found to regulate endogenous retroelement expression in melanoma,40 supporting our data in SUM149 cells. Further investigation into the intergenic KDM5B peaks in our study unveiled an association with the cohesin/CTCF complex, indicating a potential role of KDM5B in the regulation of chromatin topology. This finding is particularly intriguing in the context of previous research from our lab, which demonstrated that KDM5B physically associates with CTCF.7 Overall, our results indicate that distinct genomic binding patterns may contribute, at least in part, to paralog-specific KDM5 activity, but further work is needed to elucidate the underlying mechanisms.
ZBTB7A has previously been described as a key regulator of metabolism in leukemias, where loss of ZBTB7A increased glycolysis and sensitized to glycolytic inhibition.46–48 Here, we identified ZBTB7A as a regulator of oxidative phosphorylation in TNBC and propose that this may contribute to increased KDM5 inhibitor sensitivity upon loss of ZBTB7A. Initially, we found that peaks with decreased KDM5A binding in the ZBTB7A KO were significantly enriched for downstream targets of the NRF1 transcription factor, a regulator of nuclear genes essential for both respiration and mitochondrial DNA transcription.37 Several observations support the hypothesis that reduced mitochondrial respiration can sensitize cells to KDM5i. First, our C70-resistant derivative cell line significantly upregulated oxidative phosphorylation genes, especially members of the electron transport chain. Second, prior studies found that melanoma cells with high KDM5B expression had an increased reliance on mitochondrial respiration.49 Inhibition of ATP synthase decreased the emergence of cells with high KDM5B expression after treatment with a panel of anti-cancer drugs, and KDM5B overexpression led to increased oxygen consumption and mitochondrial ATP production.49 This potential link between mitochondrial activity and KDM5 inhibitor sensitivity is interesting given that α-ketoglutarate, a key product of the TCA cycle, is a required co-factor for KDM5 demethylase activity. In addition, since the KDM5 inhibitor C70 is an α-ketoglutarate competitor,16 altering α-ketoglutarate levels could alter the effective concentration of C70.
Mitochondrial dysfunction can also contribute to ROS and oxidative stress. We found that C70 treatment induced ROS production in both WT and ZBTB7A KO cells. However, ROS levels were higher within the ZBTB7A KO treated with C70. This is in line with previous observations in which ZBTB7A was found to protect against oxidative stress to promote cell survival during viral infection.50 It also agrees with previous work where KDM5A was found to mitigate the accumulation of ROS and protect against defects in mitochondrial membrane potential in response to cisplatin treatment.51 Interestingly, regulation of mitochondrial activity via KDM5s may be evolutionarily conserved, in that Drosophila KDM5/Lid was identified as a direct activator of genes required for mitochondrial structure/function, and mutant KDM5/Lid led to elevated ROS production.52 Taken together, these results indicate that both ZBTB7A and KDM5 activity may protect against oxidative damage and that loss of both could have a compounding effect on cell viability.
Last, we found that both KDM5i and loss of ZBTB7A led to an induction of NF-κB target genes and that the NF-κB pathway was depleted in the C70-resistant SUM149CR cell line. We hypothesize that KDM5 activity and ZBTB7A may influence NF-κB signaling through distinct mechanisms. First, KDM5i may act upstream, since C70 increased phosphorylated p65 (Ser536), led to slightly elevated levels of p65 in the nucleus, and induced GFP expression driven by a minimal NF-κB binding motif. It is unclear how inhibition of KDM5 activity activates NF-κB, but previous research implicates the DNA-sensing cGAS-STING pathway, an upstream activator of NF-κB signaling.40,53 KDM5B, but not KDM5A KO, was found to derepress STING expression and induce interferon signaling in breast cancer cell lines with high cytosolic DNA levels.53 Similarly, KDM5B KO was found to increase tumor immunogenicity through the derepression of retroelements and subsequent activation of cGAS-STING.40 Aligned with these findings, our RNA-seq data revealed a significant increase in retroelement expression after C70 treatment. In contrast, we hypothesize that ZBTB7A regulates NF-κB at the DNA level, whereby ZBTB7A binding represses nearby NF-κB target genes. Supporting this hypothesis, we found that the targets of ZBTB7A unique binding sites were significantly enriched for the NFKB1 transcription factor motif.
Given that NF-κB is generally a pro-survival pathway that activates anti-apoptotic factors and cell-cycle-promoting genes, it was unexpected to see this pathway diminished and less responsive to C70 in the SUM149CR C70-resistant cell line. However, there are several reports indicating that NF-κB could both inhibit and promote cell death depending on the context and type of stressor placed on the cell.54,55 Most relevantly, NF-κB signaling enhanced oxidative-stress-induced killing with H2O2.54 Therefore, increased NF-κB activity could render the cells more sensitive to increased ROS observed with ZBTB7A KO and C70 treatment. Cells adapt to long-term ROS by upregulating antioxidant defense mechanisms.56 Thus, the upregulation of MMP9, an NF-κB target that has been shown to protect from ROS in some cellular contexts,57 in ZBTB7A KO cells could reflect such a coping mechanism.
Limitations of the study
Our study was performed in human breast cancer cell lines with validation of findings in clinical samples. However, we do not know to what degree cell lines and cell culture reflect the physiological conditions in human breast cancer, which is especially a concern for changes in metabolic pathways. A limitation of the experiments using KO cell lines is that the consequences of short-term and long-term deletion of a gene can be different, as we have seen previously for KDM5B.10 Thus, while KDM5A deletion in the CRISPR screen (relatively short-term assay and assessing more acute loss of KMD5A) may have decreased response to C70, this effect is lost in the stable KDM5A KO cell line used for validation.
The KDM5 inhibitor we used blocks the activity of all KDM5 family members. Further studies would be required to assess the effects of the specific inhibition of KDM5A or KDM5B in human breast tumors.
RESOURCE AVAILABILITY
Lead contact
Requests for further information and resources and reagents should be directed to and will be fulfilled by the lead contact, Kornelia Polyak, Dana-Farber Cancer Institute, 450 Brookline Avenue, SM1070B, Boston, MA 02215, USA (kornelia_polyak@dfci.harvard.edu).
Materials availability
Breast cancer cell line derivatives will be made available upon request and following the execution of an MTA.
Data and code availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. All raw genomic data were deposited with GEO under accession no. GSE259252. This study did not generate custom code. The raw mass spectrometry data have been deposited in the public proteomics repository MassIVE (http://massive.ucsd.edu) using the identifier MSV000094452. The data should be accessible at ftp://massive.ucsd.edu/v07/MSV000094452. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
STAR★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Breast cancer cohort data
TCGA data was downloaded from cBioPortal (https://www.cbioportal.org/) for the Breast Invasive Carcinoma (TCGA, PanCancer Atlas) dataset (ID = brca_tcga_pan_can_atlas_2018).
Breast cancer cell lines and derivation of the C70-resistant model
Breast cancer cell lines were obtained from ATCC, or generously provided by Dr. Steve Ethier (U. Michigan) and cultured following the provider’s recommendations. The identity of the cell lines was confirmed based on STR and exome-seq analyses. Cells were regularly tested for mycoplasma. SUM149 and SUM159 were cultured in DMEM/F12 supplemented with 5% FBS, 5 μg/mL insulin, and 1 μg/mL hydrocortisone. MCF7 was cultured in DMEM/F12 supplemented with 10% FBS, and 10 μg/mL insulin. FCIBC02 was cultured in DMEM/F12 supplemented with 10% FBS. MDA-MB-436 was cultured in McCoy’s supplemented with 10% FBS, and 10 μg/mL insulin. SKBR3 was cultured in McCoy’s supplemented with 10% FBS. MDA-MB-361 was cultured in McCoy’s supplemented with 20% FBS. T-47D was cultured in RPMI supplemented with 10% FBS and 10 μg/mL insulin. BT-474 and HCC1419 were cultured in RPMI supplemented with 10% FBS. All cell lines were supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin. All cells were cultured at 37°C with 5% CO2. C70 resistant SUM149CR cells were derived by growing the cells in the presence of 10 μM C70 and retested IC50 in every 5th passage. It took ~45 days to achieve resistance determined based on a significant shift in IC50. Subsequently the cells were maintained in 10 μM C70-containing culture media.
CRISPR libraries
Human CRISPR knockout library (H3) was generated by Drs. Xiaole Shirley Liu and Myles Brown (Addgene #133914).
METHOD DETAILS
Cellular viability and growth assays
Viability and growth assays were performed in 96-well plates (N = 6 well per condition). Cells were treated with inhibitors and cultured at 37 °C with 5% CO2. The medium was replaced with fresh medium (with or without inhibitors) every 2–3 days. Plates were fixed in an ice-cold 3:1 mixture of methanol and glacial acetic acid for at least 10 minutes. Fixed cells were washed twice with 1X PBS and stained with 1μg/mL DAPI at 37 °C for 20 minutes. The cells were washed twice with 1X PBS and the number of DAPI stained cells were acquired using the automated Celigo Image Cytometer from Nexcelom.
CRISPR screen and data analyses
Human CRISPR knockout library (H3) was generated by Drs. Xiaole Shirley Liu and Myles Brown (Addgene #133914). We followed the screen protocol for adherent cells provided by Addgene (Addgene #133914). Briefly, 200 million SUM149 or SUM149CR cells were infected with the pooled lentiviral CRISPR knock-out H3 library at a multiplicity of infection of 0.3 to ensure most cells received only one viral construct. This resulted in ~60 million infected cells and 500X library coverage. After 5 days of puromycin selection, more than 60 million cells were pelleted and stored as the day 0 control. The remaining cells were split and cultured for 10 doublings with 0.01% DMSO or 10 μM C70. To limit changes in gRNA distribution due to sampling, 60 million cells per condition (500X library coverage) were seeded each passage or pelleted at the treatment endpoint. Genomic DNA was isolated from the pelleted samples via phenol/chloroform extraction and PCR was performed to construct each sequencing library (see key resources table for primer information). The libraries were sequenced at 30–40 million reads per sample to ensure at least 300X library coverage. The libraries were sequenced on an Illumina NS500 Single End 75bp with a 10% PhiX spike in.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti IL27-RA | Cell Signaling Technology | Cat# 41220 |
| Rabbit monoclonal anti MMP-9 | Cell Signaling Technology | Cat# 13667; RRID:AB_2798289 |
| Rabbit polyclonal anti MIA | Peprotech | Cat# 500-P243; RRID:AB_1268631 |
| Rabbit polyclonal anti p44/42 MAPK | Cell Signaling Technology | Cat# 9102; RRID:AB_330744 |
| Rabbit monoclonal anti NRF1 | abcam | Cat# ab175932; RRID:AB_2629496 |
| Rabbit polyclonal anti CTCF | abcam | Cat# ab70303; RRID:AB_1209546 |
| Rabbit Gamma Globulin | Life Technologies | Cat# 31887; RRID:AB_2532980 |
| Mouse IgG Isotype Control | Life Technologies | Cat# 10400C; RRID:AB_2532980 |
| Armenian Hamster IgG Isotype Control (eBio299Arm), eBioscience | Life Technologies | Cat# 14-4888-81; RRID:AB_470128 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Life Technologies | Cat# 65–6120; RRID:AB_2533967 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Life Technologies | Cat# 62–6520; RRID:AB_2533947 |
| Goat anti-Armenian Hamster IgG (H+L) Secondary Antibody, HRP | Life Technologies | Cat# PA1–32045; RRID:AB_10985178 |
| Mouse monoclonal anti-beta-Actin (AC-74) | Sigma | Cat# A2228; RRID:AB_476697 |
| Monoclonal Anti-α-Tubulin antibody produced in mouse | Sigma | Cat# T5168; RRID:AB_477579 |
| Armenian Hamster monoclonal anti-ZBTB7A (13E9) | Life Technologies | Cat# 14-3309-82; RRID:AB_2043856 |
| Armenian Hamster monoclonal anti-ZBTB7A (13E9) | SantaCruz | Cat# sc-33683; RRID:AB_668999 |
| Rabbit monoclonal anti-ZBTB7A (EPR13178(B)) | Abcam | Cat# ab175918 |
| Recombinant monoclonal anti-KDM5A | Active Motif | Cat# 91211; RRID:AB_2793805 |
| Anti-KDM5A / Jarid1A / RBBP2 antibody (ab70892) | abcam | ab70892; RRID:AB_2280628 |
| Rabbit polyclonal anti-KDM5B | Sigma | Cat# HPA027179; RRID:AB_1851987 |
| Rabbit monoclonal anti-KDM5B (E2X6N) | Cell Signaling Technology | Cat# 15327; RRID:AB_2798737 |
| Rabbit polyclonal anti-KDM5B | Novus Biologicals | Cat# 22260002; RRID:AB_10004656 |
| Rabbit polyclonal anti-KDM5C | Abcam | Cat# ab34718; RRID:AB_881090 |
| Rabbit polyclonal anti-H3K4me3 | Diagenode | Cat# C15410003–50 |
| Anti-Histone H3 (tri methyl K4) antibody - ChIP Grade (ab8580) | Abcam | Cat# ab8580; RRID:AB_306649 |
| Mouse monoclonal anti-H3 | Active Motif | Cat# 39763; RRID:AB_265052 |
| RhoA (67B9) Rabbit mAb #2117 | Cell Signaling Technology | Cat# 2117: RRID:AB_10693922 |
| Anti-PKN2 antibody (ab87812) | Abcam | Cat# ab87812; RRID:AB_2042690 |
| Rabbit monoclonal anti-MTA1 (D17G10) | Cell Signaling Technology | Cat# 5647; RRID:AB_10705601 |
| Rabbit polyclonal anti-MTA2 | Cell Signaling Technology | Cat# 15793 |
| Rabbit monoclonal anti-MBD2 (EPR18361) | Abcam | Cat# ab188474 |
| Rabbit monoclonal anti-MBD3 (N87) | Cell Signaling Technology | Cat# 14540; RRID:AB_2798504 |
| Rabbit polyclonal anti-CHD3 | Cell Signaling Technology | Cat# 4241; RRID:AB_10557102 |
| Rabbit monoclonal anti-CHD4 (D8B12) | Cell Signaling Technology | Cat# 11912; RRID:AB_2751014 |
| Phospho-NF-κB p65 (Ser536) (93H1) Rabbit mAb #3033 | Cell Signaling Technology | Cat# 3033; RRID:AB_331284 |
| Recombinant Anti-NF-κB p65 antibody [E379] (ab32536) | Abcam | Cat# ab32536; RRID:AB_776751 |
| Phospho-NFkB p50 (Ser337) Polyclonal Antibody | Life Technologies | Cat# PA5-37658; RRID:AB_2554266 |
| NFkB p50 Monoclonal Antibody (5D10D11) | Life Technologies | C# MA5-15870; RRID:AB_11153885 |
| Bacterial and virus strains | ||
| Endura ElectroCompetent Cells | Lucigen | Cat# 60242-2 |
| One Shot Stbl3 Chemically Competent E. coli | Life Technologies | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| KDM5 inhibitor C70 | This paper; Johansson et al.16; Tumber et al.58; Hinohara et al.10 | N/A |
| Nonyl Acridine Orange (Acridine Orange 10-Nonyl Bromide) | Life Technologies | Cat# A1372 |
| Recombinant Human TNF-alpha Protein | R&D Systems | Cat# 210-TA |
| Critical commercial assays | ||
| Thermo Scientific | Active Rho Pull-Down and Detection Kit | Cat# 16116 |
| Thermo Scientific | Active Rac1 Pull-Down and Detection Kit | Cat# 16118 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| Seahorse FluxPaks | Agilent | Cat# 102340-100 |
| Seahorse XF DMEM assay medium pack, pH 7.4 | Agilent | Cat# 103680-100 |
| Total Reactive Oxygen Species (ROS) Assay Kit 520 nm | Invitrogen | Cat# 88-5930 |
| Deposited data | ||
| All raw genomic data | GEO | GSE259252 |
| Processed CRISPR screen data | This paper | Table S2 |
| Processed mass spectrometry data (qPLEX-RIME) | This paper | Table S3 |
| Raw histone mass spectrometry data | MassIVE | MSV000094452 |
| Experimental models: Cell lines | ||
| SUM149 cell line | Steve Ethier (University of Michigan) | N/A |
| SUM149 cell line, ROSA26-g1 | This paper | N/A |
| SUM149 cell line, ROSA26-g2 | This paper | N/A |
| SUM149 cell line, ZBTB7A-g1 | This paper | N/A |
| SUM149 cell line, ZBTB7A-g2 | This paper | N/A |
| SUM149 cell line, KDM5A-g1 | This paper | N/A |
| SUM149 cell line, KDM5A-g2 | This paper | N/A |
| SUM149 cell line, KDM5B-g1 | This paper | N/A |
| SUM149 cell line, KDM5B-g2 | This paper | N/A |
| SUM149 cell line, RHOA-g1 | This paper | N/A |
| SUM149 cell line,RHOA-g2 | This paper | N/A |
| SUM149 cell line,PKN2-g1 | This paper | N/A |
| SUM149 cell line, PKN2-g2 | This paper | N/A |
| MDA-MB-436 cell line | ATCC | HTB-130; RRID:CVCL_0623 |
| MDA-MB-436 cell line, ROSA26-g1 | This paper | N/A |
| MDA-MB-436 cell line, ZBTB7A-g2 | This paper | N/A |
| MCF7 cell line | Marc Lippman (University of Michigan) | N/A |
| MCF7 cell line, ROSA26-g1 | This paper | N/A |
| MCF7 cell line, ZBTB7A-g1 | This paper | N/A |
| SUM159 cell line | Steve Ethier (University of Michigan) | |
| FCIBC02 cell line | Massimo Christofanelli (Fox Chase Cancer Center) | |
| T-47D cell line | ATCC | HTB-133; RRID:CVCL_0553 |
| BT-474 cell line | ATCC | HTB-20 |
| MDA-MB-361 cell line | ATCC | HTB-27; RRID:CVCL_0620 |
| HCC1419 cell line | ATCC | CRL-2326; RRID:CVCL_1251 |
| SKBR3 cell line | ATCC | HTB-30; RRID:CVCL_0033 |
| Oligonucleotides | ||
| ROSA26-g1: GGTGATCTAGTATTTCTTG | This paper | N/A |
| ROSA26-g2: TGCGGTCAGGTCACGCCGC | This paper | N/A |
| NonTargeting-g1: ACCGGAACGATCTCGCGTA | This paper | N/A |
| ZBTB7A-g1: CCGTCAGCACAGCCAACGT | This paper | N/A |
| ZBTB7A-g2: TTGAAGTACTGGCTGCAGG | This paper | N/A |
| KDM5A-g1: TCCAGAATGCTTAGATGTG | This paper | N/A |
| KDM5A-g2: TGTCCTAAATGTGTCGCCG | This paper | N/A |
| KDM5B-g1: CACCTTCGCCTAGTCACAC | This paper | N/A |
| KDM5B-g2: GACTGGCATCTGTAAGGTG | This paper | N/A |
| RHOA-g1: AAACACATCAGTATAACAT | This paper | N/A |
| RHOA-g2: CCACTCACCTAAACTATCA | This paper | N/A |
| PKN2-g1: TAATGGAATATGCTGCCGG | This paper | N/A |
| PKN2-g2: TAGATATCATACTTTGACG | This paper | N/A |
| CRISPR PCR round 1 Forward: AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG | This paper | N/A |
| CRISPR PCR round 1 Reverse: TCTACTATTCTTTCCCCTGCACTGTGACTGTGGGCGATGTGCGCTCTG | This paper | N/A |
| CRISPR PCR round 2 Staggered 1 Forward: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTtCAAGGTCAtcttgtggaaaggacgaaacaccg | This paper | N/A |
| CRISPR PCR round 2 Staggered 1 Reverse: CAAGCAGAAGACGGCATACGAGATCAAGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTacgatcgatTCTACTATTCTTTCCCCTGCACTGT | This paper | N/A |
| CRISPR PCR round 2 Staggered 2 Forward: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTatGCATAACTtcttgtggaaaggacgaaacaccg | This paper | N/A |
| CRISPR PCR round 2 Staggered 2 Reverse: CAAGCAGAAGACGGCATACGAGATGCATAACTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTcgatcgatTCTACTATTCTTTCCCCTGCACTGT | This paper | N/A |
| CRISPR PCR round 2 Staggered 3 Forward: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTgatCTCTGATTtcttgtggaaaggacgaaacaccg | This paper | N/A |
| CRISPR PCR round 2 Staggered 3 Reverse: CAAGCAGAAGACGGCATACGAGATCTCTGATTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTgatcgatTCTACTATTCTTTCCCCTGCACTGT | This paper | N/A |
| CRISPR PCR round 3 Forward: AATGATACGGCGACCACCGA | This paper | N/A |
| CRISPR PCR round 3 Reverse: CAAGCAGAAGACGGCATACGA | This paper | N/A |
| Recombinant DNA | ||
| Human CRISPR Knockout Library (H3) | Addgene | Cat# 133914 |
| lentiCRISPR v2 | Addgene | Cat# #52961 |
| pLV-5xNFkBminiP-d1EGFP | This paper | Vector builder |
| Software and algorithms | ||
| Visualization Pipeline for RNA-seq analysis (VIPER) | Bitbucket; Cornwell et al. (2018)59 | https://bitbucket.org/cfce/viper/src/master/ |
| Containerized Bioinformatics workflow for Reproducible ChIP/ATAC-seq Analysis (CoBRA) | Bitbucket; Qiu et al.60 | https://bitbucket.org/cfce/cobra/src/master/ |
| Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) (version: 0.5.9) | SourceForge; Li et al.61 | https://sourceforge.net/p/mageck/wiki/Home/ |
| MAGeCKFlute (version 1.14.0) | Bioconductor; Wang et al.62 | https://www.bioconductor.org/packages/release/bioc/html/MAGeCKFlute.html |
| Cell Ranger (version: 5.0.1) | 10X Genomics | https://www.10xgenomics.com/support/software/cell-ranger/latest |
| Seurat (version 4.3.0) | CRAN | https://cran.r-project.org/web/packages/Seurat/index.html |
| ERVmap (version 1.1) | Github; Tokuyama etal. (2018)41 | https://github.com/mtokuyama/ERVmap |
| ClusterProfiler (version: 4.2.2) | Bioconductor; Wu et al.; Yu et al.63,64 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Enrichr | Ma’ayan Lab (Icahn School of Medicine, Mount Sinai)65–67 | https://maayanlab.cloud/Enrichr/ |
| Molecular Signatures Database (MSigDB) Compute Overlaps Function | Broad Institute, Inc., Massachusetts Institute of Technology, and Regents of the University of California68,69 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| RStudio (version: 4.2.0) | Posit | https://posit.co |
| Affinity Designer (version: 1.10.4) | Affinity | https://affinity.serif.com/en-us/ |
| GraphPadPrism (version: 9) | GraphPad Software Inc. | https://www.graphpad.com/updates/prism-900-release-notes |
| FlowJo (version: 10.8.2) | FlowJo, LLC | https://www.flowjo.com/ |
Generation of single CRISPR/Cas9 knock-out cells
Construction of lenti-CRISPR/Cas9 vectors targeting ZBTB7A, KDM5A, KDM5B, RHOA, and PKN2 was performed following the protocol associated with the backbone vector lentiCRISPR V2 (Addgene #52961)70 The sgRNA sequences used are listed in key resources table. Knockouts were verified by western blot analysis after puromycin selection.
Generation of NFkB reporter cells
A lentiviral vector containing the NF-kB reporter (pLV-5xNFkBminiP-d1EGFP) was designed based on the backbone of the HypoxCR vector (Addgene #59946) and obtained via fee service from VectorBuilder (https://en.vectorbuilder.com/). The vector contained DNA sequences encoding d1EGFP and mCherry downstream of the 5xNF-kB response element-minimal promoter and CMV promoter, respectively. Cells were transduced with the vector and selected for with Blasticidin.
ChIP-seq
SUM149 expressing ROSA26-g1 −/+ 10 μM C70 for 7 days, ZBTB7A-g1 (ZBTB7A-KO), KDM5A-g1 (KDM5A-KO), and KDM5B-g1(KDM5B-KO) from the lentiCRISPR v2 backbone (see key resources table) were cultured in biological duplicates in 15 cm dishes to about 80% confluence. For ChIP-seq of histone modifications (H3K4me3), each dish was washed once with PBS then crosslinked in fixing buffer (50 mM HEPES-NaOH pH 7.5, 100 mM NaCl, 1 mM EDTA) containing 1% PFA (Electron Microscopy Sciences, 15714) for 10 minutes at room temperature. For ChIP-seq of chromatin binding proteins (KDM5A, KDM5B, ZBTB7A), each dish was washed once with PBS then crosslinked in PBS containing 2 mM DSG (Fisher Scientific 20593) for 30 minutes at room temperature. DSG was then removed, and the samples were further crosslinked with fixing buffer (50 mM HEPES-NaOH pH 7.5, 100 mM NaCl, 1 mM EDTA) containing 1% PFA for 10 minutes at 37 °C. After PFA fixation, all samples were quenched with glycine at a final concentration of 0.125 M for 5 minutes at room temperature. Cells were washed and harvested in ice cold PBS. Nuclei were extracted by first resuspending the cells in lysis buffer (1 mL per 5 million cells) for 10 minutes at 4C (50 mM HEPES-NaOH pH 8, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% IGEPAL CA-630, 0.25% Triton X-100). Nuclei were pelleted and washed once in wash buffer (10 mM Tris-HCl pH 8, 200 mM NaCl, 1 mM EDTA), 1 mL of wash buffer per 10 million cells (histone modification ChIP) or 25 million cells (chromatin modification ChIP). The nuclei were then pelleted and resuspended in 1 mL of shearing buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.1% SDS), containing 10 million (histone modification ChIP) or 25 million cells (chromatin modification ChIP). Samples were transferred to 1 mL AFA Fiber tubes (Covaris 520130), and sonicated in a Covaris E220 sonicator with the following settings: Peak Incident Power 150, Duty Cycles 5%, cycles per Burst 200. Histone modification ChIP samples were sonicated for 900 seconds. Chromatin binding protein ChIP samples were sonicated for 1,200 seconds. After sonication, Triton X-100 and NaCl were added to a final concentration of 1% Triton X-100 and 150 mM NaCl. The samples were then pre-cleared with 50 μL of Dynabeads Protein G (Fisher Scientific, 10004D) for 1 hour at 4 °C. For histone modification ChIP, primary antibodies were added to 1 mL of pre-cleared chromatin from ~10 million cells (H3K4me3 = Diagenode C154100003 at 5 μg/mL) and incubated at 4 °C overnight. For chromatin binding protein ChIP, pre-cleared samples were aliquoted, and volumes adjusted using shearing buffer containing 1% Triton X-100 and 150 mM NaCl before adding the appropriate primary antibodies for overnight incubation at 4 °C. Chromatin binding protein ChIP samples contained 500 μL of pre-cleared chromatin from ~5 million cells (ZBTB7A = SantaCruz sc-33683X at 5 μg/mL, KDM5A = Abcam ab70892 at 4 μg/mL, KDM5B = Novus/sdix 22260002 at 5 μg/mL). After overnight immunoprecipitation, crosslinked complexes were precipitated with Dynabeads G (Fisher Scientific, 10004D) for 2 hours at 4°C. The beads were then washed once with low salt wash buffer (20 mM Tris-HCl pH 8, 150 mM NaCl, 0.1% SDS, 1% Triton-X 100, 2 mM EDTA) for 5 minutes at 4°C, once with high salt wash buffer (20 mM Tris-HCl pH 8, 500 mM Nacl, 0.1% SDS, 1% Triton-X 100, 2 mM EDTA) for 5 minutes at 4 °C, once with wash buffer (10 mM Tris-HCl pH 8, 250 mM LiCl, 1% IGEPAL CA-630, 1% Sodium Deoxycholate, 1 mM EDTA) for 5 minutes at 4 °C, and once with TE buffer (10 mM Tris-HCl pH8, 1 mM EDTA) quickly at room temperature. DNA was eluted from the beads in 1% SDS, 100 mM NaHCO3 for 30 minutes at room temperature. Crosslinks were reversed at 65°C overnight. RNA and protein were digested with 0.2 mg/mL RNase A for 30 minutes at 37°C followed by 0.2 mg/mL Proteinase K for 1 hour at 55 °C. DNA was purified with phenol chloroform extraction and ethanol precipitation. Libraries were prepared and sequenced at the Molecular Biology Core Facilities (MBCF) at the Dana-Farber Cancer Institute (DFCI). Libraries were prepared with the automated Swift 2S ligation chemistry and sequenced to 40M 50 bp reads pairs.
RNA-seq
SUM149 and SUM149CR were incubated in biological triplicates for 2 and 7 days in 0.01% DMSO or 10 μM. SUM149 KOs (ROSA26-g1, KDM5A-g1, KDM5B-g1, and ZBTB7A-g1), MCF7 KOs (ROSA26-g1 and ZBTB7A-g1), and MDA-MB-436 KOs (ROSA26-g1 and ZBTB7A-g2) were incubated in biological duplicates for 7 days in 0.01% DMSO or 10 μM C70. Cells were cultured to ~80% confluence in 10 cm dishes and RNA was collected using the RNeasy Mini Kit (Qiagen 74104 or 74106) and submitted to the Molecular Biology Core Facilities (MBCF) at the Dana-Farber Cancer Institute for library prep and sequencing. RNA underwent polyA enrichment before library prep and was sequenced on an Illumina NovaSeq to generate 40M 150bp read pairs (80M reads total) per sample.
qPLEX-RIME
SUM149, SUM159, MCF7, and T-47D were cultured to ~80% confluence in 4–5 biological replicates in 15 cm dishes. Samples were washed once with PBS then crosslinked in PBS containing 2 mM DSG (Fisher Scientific 20593) for 30 minutes at room temperature. DSG was then removed, and the samples were further crosslinked with fixing buffer (50 mM HEPES-NaOH pH 7.5, 100 mM NaCl, 1 mM EDTA) containing 1% PFA for 10 minutes at 37°C. After PFA fixation, all samples were quenched with glycine at a final concentration of 0.125 M for 5 minutes at room temperature. Cells were washed and harvested in ice cold PBS, pelleted, and snap frozen.
The nuclear fraction was extracted by first resuspending the pellet in 10ml of LB1 buffer (50 mM Hepes–KOH, pH 7.5; 140 mM NaCl; 1 mM EDTA;10% Glycerol; 0.5% NP-40 or Igepal CA-630; 0.25% Triton X-100) for 10 min at 4oC. Cells were pelleted, resuspended in 10 ml of LB2 buffer (10 mM Tris–HCL, pH8.0; 200 mM NaCl; 1 mM EDTA; 0.5 mM EGTA) and mixed at 4oC for 5 minutes. Cells were pelleted and resuspended in 300 ul of LB3 buffer (10 mM Tris–HCl, pH 8; 100 mM NaCl; 1 mM EDTA; 0.5 mM EGTA; 0.1% Na–Deoxycholate; 0.5% N-lauroylsarcosine) and sonicated in a waterbath sonicator (Diagenode bioruptor). 30μl of 10% Triton-X was added and the lysate centrifuged for 10 minutes at 20,000 rcf to separate debris. Samples were incubated with primary antibody at 4 °C overnight [anti-KDM5B (Novus Biologics 22260002)]. Samples were then precipitated with Dynabeads Protein G for 2 hr. The beads were washed 10 times in 1ml of RIPA buffer and twice in 100mM ammonium hydrogen carbonate (AMBIC) solution. For the second AMBIC wash, the beads were transferred to new tubes.
Samples were digested and purified with the Ultra-Micro C18 Spin Columns (Harvard Apparatus) as previously described31 After purification, each sample was dried and reconstituted in 100ul 0.1M TEAB (triethylammonium bicarbonate) and labelled with the TMT-10plex reagents (Thermo Fisher). The peptide mixture was fractionated with Reversed-Phase spin columns at high pH (Pierce, #84868) and each fraction was analyzed on a Dionex Ultimate 3000 UHPLC system coupled with the LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). Mobile phase A was composed of 2% acetonitrile, 0.1% formic acid, 5% dimethyl sulfoxide (DMSO) and mobile phase B was composed of 80% acetonitrile, 0.1% formic acid, 5% DMSO. The precursor scans were performed in the Orbitrap in the range of 380–1500 m/z at 60K resolution. The MS2 scans were performed in the ion trap with CID collision energy 30% and in the Orbitrap with HCD collision energy 40% back-to-back for each precursor. The raw data were processed on Proteome Discoverer 2.1 using the SequestHT search engine. The node for SequestHT included the following settings: Precursor Mass Tolerance 20ppm, Fragment Mass Tolerance 0.5Da for the CID spectra and 0.05Da for the HCD spectra, Dynamic Modifications were Oxidation of M (+15.995Da), Deamidation of N/Q (+0.984Da) and Static Modifications were TMT6plex at any N-Terminus/K (+229.163Da).
Antibodies and inhibitors
Full list with catalog numbers available in key resources table. Antibodies used for Immunoblotting were anti-beta-Actin (Sigma, A2228), anti-alpha Tubulin (Sigma, T5168), anti-ZBTB7A (Invitrogen, 14-3309-82), anti-KDM5A (Abcam, ab70892), anti-KDM5B (Sigma, HPA027179), anti-KDM5C (Abcam, ab34718), anti-H3K4me3 (Abcam, ab8580), anti-H3 (Active Motif, 39763), anti-RHOA (Cell Signaling Technology, 2117), anti-PKN2 (Abcam, ab87812), anti-MTA1 (Cell Signaling Technology, 5647), anti-MTA2 (Cell Signaling Technology, 15793), anti-MBD2 (Abcam, ab188474), anti-MBD3 (Cell Signaling Technology, 14540), anti-CHD3 (Cell Signaling Technology, 4241), anti-CHD4 (Cell Signaling Technology, 11912), anti-phospho-p65 (Ser536) (Cell Signaling Technology, 3033), anti-p65 (Abcam, ab32536), anti-phospho-p50 (Ser337) (Invitrogen, PA5–37658), anti-p50 (Invitrogen, MA5–15870), Goat anti-Mouse IgG Secondary HRP (Invitrogen, 62–6520), Goat anti-Rabbit IgG Secondary HRP (Invitrogen, 65–6120), and Goat anti-Armenian Hamster IgG Secondary HRP (Invitrogen, PA1–32045). Antibodies used for Immunoprecipitation during Co-IP were anti-ZBTB7A (SantaCruz, sc-33683X), anti-ZBTB7A C-terminus (Abcam, ab175918), anti-KDM5A (Active Motif, 91211), and anti-KDM5B (Cell Signaling Technology, 15327). Additionally, isotype controls used were Rabbit IgG (Invitrogen, 31887), Mouse IgG (Invitrogen, 10400C), and Armenian Hamster IgG (Invitrogen, 14-4888-81). Antibodies used for Immunoprecipitation during ChIP-seq were anti-ZBTB7A (SantaCruz, sc-33683X), anti-KDM5A (Abcam, ab70892), anti-KDM5B (Novus Biologicals, 22260002), and anti-H3K4me3 (Diagenode, C15410003–50). Antibodies used for qPLEX-RIME were anti-KDM5B (Novus Biologicals, 22260002). The KDM5-C70 inhibitor was provided by the National Center for Advancing Translational Sciences (NCATS) (NCGC ID = NCGC00371443). Previous reports on the synthesis of KDM5-C70 are described in Tumber et al. (2017)58 and Hinohara et al. (2018)10.
Immunoblotting and immunoprecipitation experiments
For whole-cell lysates, cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 5 mM EDTA). To determine the levels of GTP-bound RhoA and Rac1, we used the Active Rho Pull-Down and Active Rac1 Pull-Down and detection kits from Thermo Scientific (Thermo Scientific, 16116 and 16118). SUM149 cells were treated with 0.01% DMSO or 10 μM C70 for 3 days before processing with the pull-down kits. The samples were collected at two different confluence levels to assess the effect of confluence on active GTPase levels: 50% and 100% confluence. For the Active Rho Pull-Down kit, an anti-RhoA specific antibody from Cell Signaling Technology (Cell Signaling Technology, 2117) was used for immunoblotting instead of the pan-RhO (RhoA, RhoB, RhoC) antibody provided in the kit. For cell fractionation experiments, cells were first incubated in hypotonic buffer (10 mM HEPES pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 0.5% NP-40, and 0.2 mM EDTA) for 10 minutes at 4°C. The samples were then centrifuged at 1,800xg for 5 minutes and the supernatant stored as the cytoplasmic fraction. The pelleted nuclei were resuspended in nuclear extraction buffer (20 mM HEPES pH 7.4, 420 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 0.2 mM EDTA), incubated for 10 minutes at 4°C, and sonicated in a cup horn sonicator (Qsonica Q500, 5 minutes net sonication time, 75% amplitude, 20 seconds On/10 seconds Off cycle). The lysate was then centrifuged at 13,000 rpm for 10 minutes and the supernatant stored as the nuclear fraction. For NRF1, cellular fractionation was performed with the Cell Fractionation Kit (CST, #9038) according to the manufacturers protocol. Briefly, different fractions were isolated from cell suspensions via specific isolation buffer and separately collected after centrifugation. For experiments only cytoplasmic fraction and cytoskeletal/nuclear fraction was used. For co-immunoprecipitation experiments, cells were incubated in hypotonic buffer (1 mL per 10 million cells) (10 mM HEPES pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 0.5% NP-40) for 10 minutes at 4°C, then centrifuged at 1,800xg for 5 minutes and supernatant discarded. The nuclear pellet was then resuspended in nuclear extraction buffer (300 μl per 10 million cells) (20 mM HEPES pH 7.4, 300 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 0.2 mM EDTA), incubated for 10 minutes at 4 °C and sonicated in a cup horn sonicator (Qsonica Q500, 5 minutes net sonication time, 75% amplitude, 20 seconds On/10 seconds Off cycle). The nuclear lysate was then cleared via centrifugation at 13,000 rpm for 10 minutes. Part of the nuclear lysate was stored as input, and to the rest 1 volume of CoIP dilution buffer was added (20 mM Tris-HCl pH 8.0, 4.5 mM MgCl2, 1.0 mM CaCl2, 0.5% NP-40, 0.2 mM EDTA). Then 1 mg of diluted nuclear lysate was moved to a 1.5 ml tube (Eppendorf LoBind Microcentrifuge Tubes: Protein, Cat# 13-698-794) and volume adjusted to 1 ml. The sample was pre-cleared with 25 μl of Protein A/G Magnetic Beads blocked with 0.5% BSA (Thermo Scientific, 8803) for 1 hour at 4°C. Pre-cleared samples were then incubated with primary antibodies or IgG controls (4 μg/ml) overnight at 4°C. After overnight incubation, protein complexes were precipitated with 25 μl of Protein A/G beads blocked with 0.5% BSA for 2 hours at 4°C. The protein-bead complexes were washed twice with 1 mL of wash buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 0.2 mM EDTA) and once with LoTE (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA). Protein complexes were then eluted with 100 μL of sample buffer containing 1X NuPAGE sample reducing agent (Invitrogen NP0009) and 1X NuPAGE LDS Sample Buffer (Invitrogen NP0007) in nuclear extraction buffer for 10 minutes at room temperature. 20 μL of the eluted sample was used for western blotting. For all immunoblots, proteins were denatured with heating at 95°C for 10 minutes in 1X NuPAGE sample reducing agent (Invitrogen NP0009) and 1X NuPAGE LDS Sample Buffer (Invitrogen NP0007). The proteins were then resolved on a 4–12% Bis-Tris polyacrylamide gel (Invitrogen WG1402BOX) in MOPS SDS running buffer and transferred to PVDF membranes using a wet NuPAGE transfer buffer system. The membranes were blocked with 2.5% milk powder in 0.1% Tween20 in TBS (TBS-T) for 1 hour at room temperature followed by incubation with primary antibodies in 2.5% milk TBS-T overnight. The membranes were washed and incubated for 1 hour at room temperature with the appropriate secondary antibodies, then washed and developed with Clarity Western ECL substrate (Bio-Rad 1705061) or Clarity Max Western ECL substrate (Bio-Rad 1705062).
Mass spectrometry analysis of histone modifications
Cells were treated for 48 hours with 0.01% DMSO or 10 μM C70 for 48 hours. They were then collected via scraping in PBS, pelleted, and snap-frozen in liquid nitrogen. Histones were isolated from cell nuclei using acid extraction, biochemically prepared, and analyzed by mass spectrometry against a reference of stable isotope-labeled synthetic peptide standards exactly as described (Creech et al., 2015).71
scRNA-seq library prep
SUM149 and SUM149CR were seeded in 10 cm dishes and treated with 0.01% DMSO or 10 μM C70 for 7 days. The cells were then trypsinized and counted. One million cells were pelleted, washed 1 time, and resuspended in 1 mL of ice cold 0.04% BSA in PBS. Library construction was performed using the 10X Genomics 3’ v3.1 library kit as described and sequenced on an Illumina NovaSeq.
Flow cytometry experiments
SUM149 cells expressing ROSA26-g1 or ZBTB7A-g1 from the lentiCRISPR v2 vector (see key resources table) were grown in 0.01% DMSO or 10 μM C70 for 5 days. Cells were passaged once during treatment and seeded to be 50% confluent by the 5-day endpoint. For detecting reactive oxygen species, 7 hours of 1 mM H2O2 treatment was used as a positive control. Cells were trypsinized, counted, and resuspended in 100 μL per million cells with 1X ROS assay reagent in assay buffer from the Total Reactive Oxygen Species (ROS) Assay Kit 520 nm (Thermo Fisher, 88–5930). The samples were then incubated for 1 hour at 37 °C, centrifuged at 200xg for 5 minutes, and resuspended in 500 μL of ice cold FACS buffer (1% BSA and 2 mM EDTA in PBS). For detecting mitochondrial cardiolipins, 2 hours of 1 mM H2O2 treatment was used as a positive control. Cells were trypsinized, counted, and resuspended in 1 mL per million cells with 0.4 μM Nonyl Acridine Orange (NAO) (Fisher Scientific, A1372) in PBS. The samples were then incubated for 20 minutes at 37 °C, centrifuged at 200xg for 5 minutes, and resuspended in 500 μL of ice cold FACS buffer. All samples were kept on ice, and fluorescence intensities were acquired on an LSRFortessa cytometer (BD Biosciences) through the FITC channel and analyzed via FlowJo.
Mito-Stress Test
To measure the effect of ZBTB7A-KO and C70 treatment on mitochondrial respiration, we used Agilent’s Seahorse XF Cell Mito Stress Test Kit (Agilent, 103015–100). SUM149 cells expressing ROSA26-g1 or ZBTB7A-g1 from the lentiCRISPR v2 vector (see key resources table) were pre-treated with 0.01% DMSO or 10 μM C70 for a total of 6 days. The cells were first seeded in 10 cm dishes and treated with −/+ 10 μM C70 for 4 days. Afterwards, cells were trypsinized and re-seeded in Agilent Seahorse XF24 cell culture microplates (100777–004) for an additional 2 days of pre-treatment: 40,000 cells per well in 100 μL of media −/+ 10 μM C70. Cells were seeded such that they form a 100% confluent monolayer by the time of the assay. We then followed the protocol as described for the Seahorse XF Cell Mito Stress Test Kit (Agilent, 103015–100). The following final compound concentrations were used during the assay: 1.5 μM Oligomycin, 0.75 μM FCCP, and 0.5 μM Rotenone/Antimycin A.
QUANTIFICATION AND STATISTICAL ANALYSIS
Software used in this study
See key resources table. Visualization Pipeline for RNA-seq analysis (VIPER), Containerized Bioinformatics workflow for Reproducible ChIP/ATAC-seq Analysis (CoBRA), Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) (v 0.5.9), Cell Ranger (v5.0.1), Seurat (v4.3.0), ERVmap (v1.1), deepTools2.0, RStudio (v 4.2.0), MAGeCKFlute (v1.14.0), ClusterProfiler (v4.2.2), ChIPseeker (v1.30.3), GenomicRanges (v1.46.1), Affinity Designer (v 1.10.4), GraphPadPrism (v 9), FlowJo (v 10.8.2).
ChIP-seq data analysis
Peak calling and data analysis
All samples were processed through the computational pipeline developed at the Dana-Farber Cancer Institute Center for Functional Cancer Epigenetics (CFCE) using primarily open-source programs.60,72 Samples were analyzed in duplicates, and each sample was normalized to a 1% input. Sequence tags were aligned with Burrows-Wheeler Aligner (BWA)73 to build hg19 and uniquely mapped, non-redundant reads were retained. These reads were used to generate binding sites with Model-Based Analysis of ChIP-Seq 2 (MACS v2.1.1.20160309), with a q-value (FDR) threshold of 0.01.74 We evaluated multiple quality control criteria based on alignment information and peak quality: (i) sequence quality score; (ii) uniquely mappable reads (reads that can only map to one location in the genome); (iii) uniquely mappable locations (locations that can only be mapped by at least one read); (iv) peak overlap with Velcro regions, a comprehensive set of locations – also called consensus signal artifact regions – in the genome that have anomalous, unstructured high signal or read counts in next-generation sequencing experiments independent of cell line and of type of experiment; (v) number of total peaks (the minimum required was 10,000); (vi) high-confidence peaks (the number of peaks that are tenfold enriched over background); (vii) percentage overlap with known DHS sites derived from the ENCODE Project (the minimum required to meet the threshold was 80%); and (viii) peak conservation (a measure of sequence similarity across species based on the hypothesis that conserved sequences are more likely to be functional).
Peak cluster definitions
In Figures 3B and S3G, peak clusters were defined based on whether there was overlap in peak calls for ZBTB7A, KDM5A, and/or KDM5B in the untreated ROSA26-g1 samples. Peaks were considered present for a given factor if they were called in both replicates and absent if there was no peak call in either replicate. Heatmaps were created with deepTools2.0 using the computeMatrix and plotHeatmap functions on the Glaxy server (https://usegalaxy.org/) using bigwig files from CoBRA60 and self-defined peak regions as input. Feature annotations and nearest genes for each peak were obtained with the annotatePeak function from ChIPseeker (v1.30.3) in R (v4.1.3).
Differential binding analyses
Peaks from all samples were merged to create a union set of sites for each transcription factor and histone mark using bedops.75 Sample-sample correlation and differential peaks analysis were performed by the CoBRA pipeline.60 Read densities were calculated for each peak for each sample and used for the comparison of cistromes across samples. Sample similarity was determined by hierarchical clustering using the spearman correlation between samples. Differential peaks were identified by DEseq2 with adjusted P ≤ 0.05. A total number of reads in each sample was applied to the size factor in Deseq2, which can normalize the sequencing depth between samples. KDM5A unchanged peaks (Figure 3) were defined as regions with KDM5A peak calls across all untreated ROSA26-g1 replicates and padj > 0.05 for deseq between ZBTB7A-KO and ROSA26-g1.
Target gene identification, GSEA, and motif analysis: To identify target genes using only peak information, we used Cistrome-GO (http://go.cistrome.org/), which calculates regulatory potential scores for each gene based on both the number and proximity of peaks.32 In Figures 3H–3J and S3J, we used the top 500 predicted target genes based on regulatory potential score to look for enrichment of certain pathways.
Overlap with Hallmark pathways was calculated using the compute overlap function from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/). Overlap with consensus transcription factor target genes from ENCODE/ChEA and overlap with transcription factor motifs from TRANSFAC/JASPAR were calculated using enrichr (https://maayanlab.cloud/Enrichr/). To assess similarity of our peak sets with published ChIP-seq tracks, we used the Cistrome DB toolkit (http://dbtoolkit.cistrome.org/) “What factors have significant overlap with your peak set?” with peak number to use set to “All peaks in each sample”. The Giggle scores from this analysis (-log1 (padj)*odds ratio from Fisher’s Exact Test, see Layer et al. 2018 for more information) are plotted in Figures 3K and S3I.33,34 In Figure S3I, only the maximum Giggle scores identified for each factor are plotted. To determine the regulatory potential of each peak set, we performed binding and expression target analysis (BETA), in which we integrated the peak information with RNA-seq comparing the ZBTB7A KO and wild-type SUM149 cells.32 For promoter-enriched peak sets (e.g., KDM5A down and ZBTB7A+KDM5A/B), we used a window of 3 kb from each gene’s TSS to select peaks, and for non-promoter enriched peak sets (i.e., ZBTB7A unique), we used the default parameter of 100 kb. To annotate the genes in the volcano plots with their nearest peaks, we used the output from the annotatePeak function in the ChIPseeker (v1.30.3) package. The p values in these graphs are from the previously described BETA analyses. Finally, for enrichment analyses, we selected high probability target genes for each peak set based on a rank product score of less than 0.001 from the BETA output. Enrichment of transcription factor motifs from published ChIP-seq across Cistrome was calculated using the epigenetic Landscape In Silico deletion Analysis (Lisa) tool (http://lisa.cistrome.org/). Overlap with Hallmark pathways was calculated using the compute overlap function from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/). Motif enrichment scores were calculated using simple enrichment analysis from MEME-suite (https://meme-suite.org/meme/doc/sea.html) for ZBTB7A consensus sequence GACCC. Default shuffled input sequences were used as control.
RNA-seq data analysis
RNA-seq data was analyzed using the Visualization Pipeline for RNA-seq analysis (VIPER) and aligned to version hg19 of the human genome59 The following config.yaml parameters were adjusted: RPKM_threshold = 1.0 and min_num_samples_expressing_at_threshold = 2. Samples were analyzed in duplicates. GSEA was performed by ranking genes based on log2(FC) and using the GSEA function from the ClusterProfiler (v4.2.2) package in R. Public gene sets tested in the manuscript include MSigDB’s Hallmark (H1) and positional gene sets (C1), KEGG, and Reactome. TNBC subtypes used for GSEA in Figure S4K were from our prior publication.39 The heatmaps in Figures S2D and 4A are the default output of VIPER, which used the top 1,000 variable genes for plotting. PCA plots were computed using log2 transformed count data of all genes within each sample with the prcomp function form the stats (v4.1.3) package in R. The heatmap in Figure 5D was created using all C70-responsive DEGs, which were defined as padj < 0.05 when comparing −/+ C70 in either SUM149 ROSA26-g1 or SUM149 ZBTB7A-g1. The heatmap was generated using row normalized Z-scores of FPKM values with the ComplexHeatmap (v2.10.0) function in R. Rows were clustered via k-means with k = 5. Enrichment of gene sets within the heatmap clusters was determined using the enricher function from the clusterProfiler (v4.2.2) package in R. Transcription factor target gene sets were taken from “TFT: transcription factor targets” in MSigDB.
TCGA and METABRIC data analyses: Because these cohorts are bulk RNA-seq, we used the ISOpure algorithm to estimate gene expression levels specifically coming from the tumor fraction and used these deconvoluted matrices76 for the analyses. First, we assessed if KDM5 activity correlated with select Hallmark pathways we found to be influenced by KDM5 inhibition in our study. We used average expression of KDM5A and KDM5B as a surrogate for KDM5 activity and tested for correlation with pathway enrichment scores (gene set variation analysis, GSVA) across basal TCGA samples.77 Immune scores were calculated from bulk RNA-seq via “Estimation of STromal and Immune cells in MAlignant Tumours using Expression data” (ESTIMATE).78
Endogenous retroelement expression analysis
Differential expression of endogenous retroelements was assessed from bulk RNA-seq data using the ERVmap pipeline (v1.1).41
CRISPR screen data analysis
CRISPR data were analyzed by MAGeCK (v0.5.9) essentially as described79–81 Briefly, read counts for each sgRNA were obtained from fastq files with mageck count.61 To obtain significantly enriched and depleted sgRNAs in the C70 treated versus untreated samples, we used the MAGeCK TEST algorithm, in which read counts were normalized to a set of 3,842 control guides targeting the AAVS1 (3,540), CCR5 (99), and ROSA26 (203) loci. Genes with p value less than 0.001 were defined as candidate hits and colored in the rank plots in Figure 2. The MageckFlute package (v1.14.0)61 was used to visualize the data.
scRNA-seq data analysis
Raw bcl2 files from the NovaSeq run were converted to fastq using cellranger mkfastq (Cell Ranger v5.0.1). Count information was obtained using cellranger count and aligned to the GRCh38 reference transcriptome provided by 10X: refdata-gex-GRCh38–2020-A.tar.gz. The filtered h5 files output from cellranger count were then loaded and analyzed with Seurat (v4.3.0). To remove poor quality cells, cells with less than 1,000 total RNA counts or percent mitochondrial counts greater than 3 absolute deviations from the median were filtered out. To remove doublets, cells with total RNA greater than 3 absolute deviations from the median were also filtered out. The filtered data was then normalized with Seurat’s NormalizeData function, which normalized the gene expression for each cell by total expression, and then multiplied by a scaling factor of 10,000 and log-transformed the results. We identified the top 2,000 most variable features using FindVariableFeatures with selection.method = “vst”. The data was then scaled using ScaleData, which makes the mean expression across cells 0 and variance 1. The scaled data was then used for clustering and visualization via UMAP using the first 30 PC dimensions. Hexagonal plots were created as we previously described.10,82 Code to generate the plots was generated by Dr. Hua-Jun Wu and can be found on Bitbucket (https://bitbucket.org/mthjwu/hexplot/src/master/). To create the hexagonal plots, we first defined cell identity signatures for SUM149, C70 (i.e., SUM149 + 10 μM C70 for 7 days), and SUM149CR. For each cell type, we compared its bulk RNA-seq (3 replicates) with the other two cell types combined (3 replicates each) and obtained DEGs via deseq. We chose the top 66 up- and down-regulated genes based on padj values as the up- and down-signatures of each cell type. The top 66 genes were chosen since this was the smallest number of DEGs (padj<0.05) identified for a given cell type, and we sought to use the same number of genes to define each gene signature. Using these gene signatures, we then calculated cell identity scores for each cell within our filtered and scaled scRNA-seq data set. The cell identity score was the average scaled expression of the upgene signature minus the average scaled expression of the down- gene signature. We randomly selected 1,000 sets of up and down signatures, each matching the size of the original true signatures, which allowed us to generate a bootstrap distribution for the cell identity score. From this distribution, we calculated the bootstrap p-value. Single cells were then classified based on this p-value, using a cutoff of 5%. Cells that didn’t meet the threshold for any signature were labeled as unclassified. Hexagonal plots were utilized to visually represent the bootstrap classification of single cells within SUM149 and SUM149CR −/+ C70. Cells that clearly exhibited identity (i.e., passed the 5% threshold for only 1 of the 3 identities) were positioned along the edge of the plot. Cell-to-cell distance were calculated in each group using “Embeddings” function of the Seurat package with PCA dimension reduction. Gini indexes of all the genes within each group were calculated using “ineq” package.
qPLEX RIME data analysis
Peptide intensities were normalized using median scaling and protein level quantification was obtained by the summation of the normalized peptide intensities. A statistical analysis of differentially-regulated proteins was carried out using qPLEXanalyzer a Bioconductor R-package.31, which internally uses limma R-package from Bioconductor.83 Multiple testing correction of p-values was applied using the Benjamini-Hochberg method.84 to control the false discovery rate.
Supplementary Material
Highlights.
Deletion of ZBTB7A transcription factor and core SAGA complex synergizes with KDM5 inhibitors
KDM5A chromatin binding is altered by ZBTB7A deletion
Loss of ZBTB7A potentiates KDM5 inhibition-induced signaling changes by modulating NF-κB
High ZBTB7A in triple-negative breast cancer with no response to neoadjuvant chemotherapy
ACKNOWLEDGMENTS
We thank members of our laboratories for their critical reading of the manuscript and useful discussions. We thank the Dana-Farber Cancer Institute Molecular Biology Core Facility for their outstanding sequencing service. This research was supported by National Cancer Institute R35 CA197623 (K.P.) and P01 CA250959 (K.P., H.W.L., and M.B.), by the Ludwig Center at Harvard (K.P.), and in part by the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (A.S., S.C.K., G.R., M.L.-N., and A.J). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DECLARATION OF INTERESTS
K.P. serves on the scientific advisory boards of Ideaya Biosciences and Scorpion Therapeutics, holds equity options in Scorpion Therapeutics and Ideaya Biosciences, and receives sponsored research funding from Novartis, where she also consults. H.W.L. receives research funding from Novartis.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114991.
REFERENCES
- 1.Feinberg AP, Koldobskiy MA, and Göndör A (2016). Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet 17, 284–299. 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380. 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen C, and Vakoc CR (2015). Gain-of-function mutation of chromatin regulators as a tumorigenic mechanism and an opportunity for therapeutic intervention. Curr. Opin. Oncol 27, 57–63. 10.1097/CCO.0000000000000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinohara K, and Polyak K (2019). Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 29, 569–579. 10.1016/j.tcb.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan S, Norgard RJ, and Stanger BZ (2019). Cellular Plasticity in Cancer. Cancer Discov. 9, 837–851. 10.1158/2159-8290.CD-19-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747–752. 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto S, Wu Z, Russnes HG, Takagi S, Peluffo G, Vaske C, Zhao X, Moen Vollan HK, Maruyama R, Ekram MB, et al. (2014). JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer Cell 25, 762–777. 10.1016/j.ccr.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou MR, Cao J, Liu Z, Huh SJ, Polyak K, and Yan Q (2014). His-tone demethylase jumonji AT-rich interactive domain 1B (JARID1B) controls mammary gland development by regulating key developmental and lineage specification genes. J. Biol. Chem 289, 17620–17633. 10.1074/jbc.M114.570853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi HJ, Joo HS, Won HY, Min KW, Kim HY, Son T, Oh YH, Lee JY, and Kong G (2018). Role of RBP2-Induced ER and IGF1R-ErbB Signaling in Tamoxifen Resistance in Breast Cancer. J. Natl. Cancer Inst 110, 400. 10.1093/jnci/djx207. [DOI] [PubMed] [Google Scholar]
- 10.Hinohara K, Wu HJ, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, Madsen T, Fassl A, Egri SB, Papanastasiou M, et al. (2018). KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell 34, 939–953.e9. 10.1016/j.ccell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao J, Liu Z, Cheung WKC, Zhao M, Chen SY, Chan SW, Booth CJ, Nguyen DX, and Yan Q (2014). Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 6, 868–877. 10.1016/j.celrep.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein BJ, Piao L, Xi Y, Rincon-Arano H, Rothbart SB, Peng D, Wen H, Larson C, Zhang X, Zheng X, et al. (2014). The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 6, 325–335. 10.1016/j.celrep.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Shi L, Gui B, Yu W, Wang J, Zhang D, Han X, Yao Z, and Shang Y (2011). Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 71, 6899–6908. 10.1158/0008-5472.CAN-11-1523. [DOI] [PubMed] [Google Scholar]
- 14.Hou J, Wu J, Dombkowski A, Zhang K, Holowatyj A, Boerner JL, and Yang ZQ (2012). Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am. J. Transl. Res 4, 247–256. [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Liu L, Holowatyj A, Jiang Y, and Yang ZQ (2016). Integrated genomic and functional analyses of histone demethylases identify oncogenic KDM2A isoform in breast cancer. Mol. Carcinog 55, 977–990. 10.1002/mc.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson C, Velupillai S, Tumber A, Szykowska A, Hookway ES, Nowak RP, Strain-Damerell C, Gileadi C, Philpott M, Burgess-Brown N, et al. (2016). Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol 12, 539–545. 10.1038/nchembio.2087. [DOI] [PubMed] [Google Scholar]
- 17.Paroni G, Bolis M, Zanetti A, Ubezio P, Helin K, Staller P, Gerlach LO, Fratelli M, Neve RM, Terao M, and Garattini E (2019). HER2-positive breast-cancer cell lines are sensitive to KDM5 inhibition: definition of a gene-expression model for the selection of sensitive cases. Oncogene 38, 2675–2689. 10.1038/s41388-018-0620-6. [DOI] [PubMed] [Google Scholar]
- 18.Maeda T, Hobbs RM, and Pandolfi PP (2005). The transcription factor Pokemon: a new key player in cancer pathogenesis. Cancer Res. 65, 8575–8578. 10.1158/0008-5472.CAN-05-1055. [DOI] [PubMed] [Google Scholar]
- 19.Lunardi A, Guarnerio J, Wang G, Maeda T, and Pandolfi PP (2013). Role of LRF/Pokemon in lineage fate decisions. Blood 121, 2845–2853. 10.1182/blood-2012-11-292037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Constantinou C, Spella M, Chondrou V, Patrinos GP, Papachatzopoulou A, and Sgourou A (2019). The multi-faceted functioning portrait of LRF/ZBTB7A. Hum. Genomics 13, 66. 10.1186/s40246-019-0252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blumbach K, Zweers MC, Brunner G, Peters AS, Schmitz M, Schulz JN, Schild A, Denton CP, Sakai T, Fässler R, et al. (2010). Defective granulation tissue formation in mice with specific ablation of integrin-linked kinase in fibroblasts - role of TGFbeta1 levels and RhoA activity. J. Cell Sci 123, 3872–3883. 10.1242/jcs.063024. [DOI] [PubMed] [Google Scholar]
- 22.Ohguchi H, Park PMC, Wang T, Gryder BE, Ogiya D, Kurata K, Zhang X, Li D, Pei C, Masuda T, et al. (2021). Lysine Demethylase 5A is Required for MYC Driven Transcription in Multiple Myeloma. Blood Cancer Discov. 2, 370–387. 10.1158/2643-3230.BCD-20-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmlinger D, and Tora L (2017). Sharing the SAGA. Trends Biochem. Sci 42, 850–861. 10.1016/j.tibs.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu CJ, Prazak L, Fajardo M, Yu S, Tyagi N, and Di Cesare PE (2004). Leukemia/lymphoma-related factor, a POZ domain-containing transcriptional repressor, interacts with histone deacetylase-1 and inhibits cartilage oligomeric matrix protein gene expression and chondrogenesis. J. Biol. Chem 279, 47081–47091. 10.1074/jbc.M405288200. [DOI] [PubMed] [Google Scholar]
- 25.Pavlenko E, Ruengeler T, Engel P, and Poepsel S (2022). Functions and Interactions of Mammalian KDM5 Demethylases. Front. Genet 13, 906662. 10.3389/fgene.2022.906662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, and Tyers M (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F, et al. (2021). The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 30, 187–200. 10.1002/pro.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368. 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishibuchi G, Shibata Y, Hayakawa T, Hayakawa N, Ohtani Y, Sinmyozu K, Tagami H, and Nakayama J-I (2014). Physical and Functional Interactions between the Histone H3K4 Demethylase KDM5A and the Nucleosome Remodeling and Deacetylase (NuRD) Complex. J. Biol. Chem 289, 28956–28970. 10.1074/jbc.m114.573725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi WI, Jeon BN, Yoon JH, Koh DI, Kim MH, Yu MY, Lee KM, Kim Y, Kim K, Hur SS, et al. (2013). The proto-oncoprotein FBI-1 interacts with MBD3 to recruit the Mi-2/NuRD-HDAC complex and BCoR and to silence p21WAF/CDKN1A by DNA methylation. Nucleic Acids Res. 41, 6403–6420. 10.1093/nar/gkt359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papachristou EK, Kishore K, Holding AN, Harvey K, Roumeliotis TI, Chilamakuri CSR, Omarjee S, Chia KM, Swarbrick A, Lim E, et al. (2018). A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat. Commun 9, 2311. 10.1038/s41467-018-04619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, and Liu XS (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protoc 8, 2502–2515. 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. (2011). Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12, R83. 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Layer RM, Pedersen BS, DiSera T, Marth GT, Gertz J, and Quinlan AR (2018). GIGGLE: a search engine for large-scale integrated genome analysis. Nat. Methods 15, 123–126. 10.1038/nmeth.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu H, Pardoll D, and Jove R (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809. 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren R, Horton JR, Chen Q, Yang J, Liu B, Huang Y, Blumenthal RM, Zhang X, and Cheng X (2023). Structural basis for transcription factor ZBTB7A recognition of DNA and effects of ZBTB7A somatic mutations that occur in human acute myeloid leukemia. J. Biol. Chem 299, 102885. 10.1016/j.jbc.2023.102885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scarpulla RC (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev 88, 611–638. 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 38.Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen CH, Brown M, Zhang X, Meyer CA, and Liu XS (2019). Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735. 10.1093/nar/gky1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jovanovic B, Temko D, Stevens LE, Seehawer M, Fassl A, Murphy K, Anand J, Garza K, Gulvady A, Qiu X, et al. (2023). Heterogeneity and transcriptional drivers of triple-negative breast cancer. Cell Rep. 42, 113564. 10.1016/j.celrep.2023.113564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang SM, Cai WL, Liu X, Thakral D, Luo J, Chan LH, McGeary MK, Song E, Blenman KRM, Micevic G, et al. (2021). KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 598, 682–687. 10.1038/s41586-021-03994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tokuyama M, Kong Y, Song E, Jayewickreme T, Kang I, and Iwasaki A (2018). ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc. Natl. Acad. Sci. USA 115, 12565–12572. 10.1073/pnas.1814589115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, Vidaurre T, Holmes F, Souchon E, Wang H, et al. (2011). A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 305, 1873–1881. 10.1001/jama.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Popovici V, Chen W, Gallas BG, Hatzis C, Shi W, Samuelson FW, Nikolsky Y, Tsyganova M, Ishkin A, Nikolskaya T, et al. (2010). Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 12, R5. 10.1186/bcr2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, et al. (2010). Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol 28, 1145–1153. 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoo J, Kim GW, Jeon YH, Kim JY, Lee SW, and Kwon SH (2022). Drawing a line between histone demethylase KDM5A and KDM5B: their roles in development and tumorigenesis. Exp. Mol. Med 54, 2107–2117. 10.1038/s12276-022-00902-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Redondo Monte E, Kerbs P, and Greif PA (2020). ZBTB7A links tumor metabolism to myeloid differentiation. Exp. Hematol 87, 20–24.e1. 10.1016/j.exphem.2020.05.010. [DOI] [PubMed] [Google Scholar]
- 47.Redondo Monte E, Wilding A, Leubolt G, Kerbs P, Bagnoli J, Hiddemann W, Enard W, Theurich S, and Greif PA (2019). Loss of ZBTB7A Enhances Glycolysis and Beta Oxidation in Myeloid Leukemia. Blood 134, 1453. 10.1182/blood-2019-128378. [DOI] [Google Scholar]
- 48.Redondo Monte E, Wilding A, Leubolt G, Kerbs P, Bagnoli JW, Hartmann L, Hiddemann W, Chen-Wichmann L, Krebs S, Blum H, et al. (2020). ZBTB7A prevents RUNX1-RUNX1T1-dependent clonal expansion of human hematopoietic stem and progenitor cells. Oncogene 39, 3195–3205. 10.1038/s41388-020-1209-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, et al. (2013). Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1B-high Cells. Cancer Cell 23, 811–825. 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu X, Trimarco JD, Williams CA, Barrera A, Reddy TE, and Heaton NS (2022). ZBTB7A promotes virus-host homeostasis during human coronavirus 229E infection. Cell Rep. 41, 111540. 10.1016/j.celrep.2022.111540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu C, Zheng Z, Li W, Tang D, Zhao L, He Y, and Li H (2022). Inhibition of KDM5A attenuates cisplatin-induced hearing loss via regulation of the MAPK/AKT pathway. Cell. Mol. Life Sci 79, 596. 10.1007/s00018-022-04565-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu X, and Secombe J (2015). The Histone Demethylase KDM5 Activates Gene Expression by Recognizing Chromatin Context through Its PHD Reader Motif. Cell Rep. 13, 2219–2231. 10.1016/j.celrep.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu L, Cao J, Cai WL, Lang SM, Horton JR, Jansen DJ, Liu ZZ, Chen JF, Zhang M, Mott BT, et al. (2018). KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 16, e2006134. 10.1371/journal.pbio.2006134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaltschmidt B, Kaltschmidt C, Hofmann TG, Hehner SP, Dröge W, and Schmitz ML (2000). The pro- or anti-apoptotic function of NF-κB is determined by the nature of the apoptotic stimulus. Eur. J. Biochem 267, 3828–3835. 10.1046/j.1432-1327.2000.01421.x. [DOI] [PubMed] [Google Scholar]
- 55.Baichwal VR, and Baeuerle PA (1997). Apoptosis: Activate NF-κB or die? Curr. Biol 7, R94–R96. 10.1016/s0960-9822(06)00046-7. [DOI] [PubMed] [Google Scholar]
- 56.Pickering AM, Vojtovich L, Tower J, and A Davies KJ (2013). Oxidative stress adaptation with acute, chronic, and repeated stress. Free Radic. Biol. Med 55, 109–118. 10.1016/j.freeradbiomed.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walter L, Canup B, Pujada A, Bui TA, Arbasi B, Laroui H, Merlin D, and Garg P (2020). Matrix metalloproteinase 9 (MMP9) limits reactive oxygen species (ROS) accumulation and DNA damage in colitis-associated cancer. Cell Death Dis. 11, 767. 10.1038/s41419-020-02959-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tumber A, Nuzzi A, Hookway ES, Hatch SB, Velupillai S, Johansson C, Kawamura A, Savitsky P, Yapp C, Szykowska A, et al. (2017). Potent and Selective KDM5 Inhibitor Stops Cellular Demethylation of H3K4me3 at Transcription Start Sites and Proliferation of MM1S Myeloma Cells. Cell Chem. Biol 24, 371–380. 10.1016/j.chembiol.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cornwell M, Vangala M, Taing L, Herbert Z, Köster J, Li B, Sun H, Li T, Zhang J, Qiu X, et al. (2018). VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinform. 19, 135. 10.1186/s12859-018-2139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qiu X, Feit AS, Feiglin A, Xie Y, Kesten N, Taing L, Perkins J, Gu S, Li Y, Cejas P, et al. (2021). CoBRA: Containerized Bioinformatics Workflow for Reproducible ChIP/ATAC-seq Analysis. Genom. Proteom. Bioinform 19, 652–661. 10.1016/j.gpb.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang B, Wang M, Zhang W, Xiao T, Chen CH, Wu A, Wu F, Traugh N, Wang X, Li Z, et al. (2019). Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc 14, 756–780. 10.1038/s41596-018-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene Set Knowledge Discovery with Enrichr. Curr. Protoc 1, e90. 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Creech AL, Taylor JE, Maier VK, Wu X, Feeney CM, Udeshi ND, Peach SE, Boehm JS, Lee JT, Carr SA, and Jaffe JD (2015). Building the Connectivity Map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods 72, 57–64. 10.1016/j.ymeth.2014.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taing L, Bai G, Cousins C, Cejas P, Qiu X, Herbert ZT, Brown M, Meyer CA, Liu XS, Long HW, and Tang M (2021). CHIPS: A Snakemake pipeline for quality control and reproducible processing of chromatin profiling data. F1000Res. 10, 517. 10.12688/f1000research.52878.1. [DOI] [Google Scholar]
- 73.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. (2012). BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920. 10.1093/bioinformatics/bts277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quon G, Haider S, Deshwar AG, Cui A, Boutros PC, and Morris Q (2013). Computational purification of individual tumor gene expression profiles leads to significant improvements in prognostic prediction. Genome Med. 5, 29. 10.1186/gm433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 1. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612. 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao T, Li W, Wang X, Xu H, Yang J, Wu Q, Huang Y, Geradts J, Jiang P, Fei T, et al. (2018). Estrogen-regulated feedback loop limits the efficacy of estrogen receptor-targeted breast cancer therapy. Proc. Natl. Acad. Sci. USA 115, 7869–7878. 10.1073/pnas.1722617115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li W, Köster J, Xu H, Chen CH, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281. 10.1186/s13059-015-0843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen CH, Xiao T, Xu H, Jiang P, Meyer CA, Li W, Brown M, and Liu XS (2018). Improved design and analysis of CRISPR knockout screens. Bioinformatics 34, 4095–4101. 10.1093/bioinformatics/bty450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shu S, Wu H-J, Ge JY, Zeid R, Harris IS, Jovanović B, Murphy K, Wang B, Qiu X, Endress JE, et al. (2020). Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol. Cell 78, 1096–1113.e8. 10.1016/j.molcel.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benjamini Y, and Yosef H (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Royal Statis. Soc. Series B (Methodol.) 57, 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. All raw genomic data were deposited with GEO under accession no. GSE259252. This study did not generate custom code. The raw mass spectrometry data have been deposited in the public proteomics repository MassIVE (http://massive.ucsd.edu) using the identifier MSV000094452. The data should be accessible at ftp://massive.ucsd.edu/v07/MSV000094452. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.