

Abstract
The FucO protein, a member of the group III “iron-activated” dehydrogenases, catalyzes the interconversion between l-lactaldehyde and l-1,2-propanediol in Escherichia coli. The three-dimensional structure of FucO in a complex with NAD+ was solved, and the presence of iron in the crystals was confirmed by X-ray fluorescence. The FucO structure presented here is the first structure for a member of the group III bacterial dehydrogenases shown experimentally to contain iron. FucO forms a dimer, in which each monomer folds into an α/β dinucleotide-binding N-terminal domain and an all-α-helix C-terminal domain that are separated by a deep cleft. The dimer is formed by the swapping (between monomers) of the first chain of the β-sheet. The binding site for Fe2+ is located at the face of the cleft formed by the C-terminal domain, where the metal ion is tetrahedrally coordinated by three histidine residues (His200, His263, and His277) and an aspartate residue (Asp196). The glycine-rich turn formed by residues 96 to 98 and the following α-helix is part of the NAD+ recognition locus common in dehydrogenases. Site-directed mutagenesis and enzyme kinetic assays were performed to assess the role of different residues in metal, cofactor, and substrate binding. In contrast to previous assumptions, the essential His267 residue does not interact with the metal ion. Asp39 appears to be the key residue for discriminating against NADP+. Modeling l-1,2-propanediol in the active center resulted in a close approach of the C-1 hydroxyl of the substrate to C-4 of the nicotinamide ring, implying that there is a typical metal-dependent dehydrogenation catalytic mechanism.
In Escherichia coli and other enterobacteria the anaerobic metabolism of l-fucose and l-rhamnose requires the enzyme lactaldehyde:propanediol oxidoreductase (FucO), which is encoded by the fucO gene of the fucose regulon (6, 14, 16, 24, 32). The breakdown of these methylpentoses generates the intermediate metabolite l-lactaldehyde, which under anaerobic conditions, with NADH as a cofactor, is reduced by FucO to l-1,2-propanediol, which is excreted as a fermentation product (14). In mutant strains of E. coli adapted to grow on l-1,2-propanediol, FucO catalyzes the oxidation of the polyol to l-lactaldehyde, which is subsequently oxidized to l-lactate by a specific aldehyde dehydrogenase (41) and introduced into the general metabolism. FucO, which is induced regardless of the respiratory conditions of the culture, remains fully active in the absence of oxygen (11). In the presence of oxygen, this enzyme becomes oxidatively inactivated by a metal-catalyzed oxidation mechanism (10).
FucO is an iron-dependent metalloenzyme that is inactivated by other metals, such as zinc, copper, or cadmium (40), and has been reported to be a homodimer formed by monomers consisting of 383 amino acids and having a molecular mass of 40,644 Da. The iron in the active center accounts for the oxidative inactivation of FucO mentioned above (10). A putative iron-binding motif encompassing a 15-amino-acid stretch which includes three strictly conserved His residues was proposed by Bairoch (1) and Cabiscol et al. (10). Two of the histidine residues are in an HXXXH motif, situated in an α helix, correctly spaced to chelate the metal ion with their imidazole rings. This motif has also been described in other enzymes containing iron (43) and zinc (3, 5). The FucO enzyme reduces l-lactaldehyde and, with similar efficiency, glycol aldehyde (9), using NADH as a cofactor. However, FucO is unable to utilize NADPH. The reaction catalyzed by this enzyme is reversible with a catalytic efficiency that is 2 orders of magnitude higher in the direction of reduction (6, 7). The enzyme is able to oxidize l-1,2-propanediol and utilizes propanol, ethylene glycol, ethanol, and glycerol as alternative substrates (6).
According to Reid and Fewson (31), the oxidoreductases that catalyze the interconversion of alcohols, aldehydes, and ketones can be divided into three major categories: (i) NAD(P)-dependent alcohol dehydrogenases, (ii) NAD(P)-independent alcohol dehydrogenases, and (iii) flavin adenine dinucleotide-dependent alcohol oxidases. The first category can, in turn, be divided into the following three groups on the basis of the coenzyme binding site: group I, medium-chain zinc-dependent dehydrogenases; group II, short-chain zinc-independent dehydrogenases; and group III, “iron-activated” dehydrogenases. However, the finding that glycerol dehydrogenase of Bacillus stearothermophilus, an enzyme with high levels of sequence similarity to the group III enzymes, is zinc dependent has led some authors (35) to propose that this group should be renamed the family III metal-dependent polyol dehydrogenases. The available information about the three-dimensional structure of members of this family of metalloproteins includes information for two zinc-dependent enzymes, glycerol dehydrogenase from B. stearothermophilus (35) and dehydroquinate synthase from Aspergillus nidulans (13), and an enzyme with an unknown function from Thermotoga maritima (TM0920) (37) whose sequence is very similar to the sequence of Klebsiella pneumoniae 1,3-propanediol dehydrogenase (18); for this reason, TM0920 has been proposed, but not experimentally proved, to be an iron-dependent enzyme. Recently, the crystal structure of the E. coli alcohol dehydrogenase YqhD has been solved (42). Both T. maritima TM0920 and E. coli YqhD have been shown to bind NADP+, although they use distinct metal ions for catalysis (Fe2+ and Zn2+, respectively). In this report we describe the three-dimensional structure of an iron-dependent type III dehydrogenase from E. coli, and in this study we confirmed experimentally the presence of an Fe2+ metal ion at the active site.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All the strains used were E. coli K-12 derivatives. Strain ECL1 (HfrC phoA8 relA1 tonA22 T2r) was the wild type in this study (23). Strains M15(pREP4) (QIAGEN) and BL21 (Pharmacia Biotech) were used to express the His6-tagged FucO and glutathione S-transferase (GST)-FucO proteins, respectively.
Plasmid pQE32 (QIAGEN) was used for expression of N-terminal His6-tagged proteins. For expression of GST fusion proteins, the polylinker of plasmid pGEX-3X (Pharmacia Biotech) was appropriately modified by introduction of a NotI restriction site upstream from the BamHI site of the polylinker in order to obtain a native protein having an N-terminal end without extra residues after factor Xa cleavage.
Growth of cells and preparation of cell extracts.
Cells were routinely grown aerobically on Luria-Bertani (LB) medium, and cell extracts were prepared as described elsewhere (6). Where indicated below, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM to induce expression of the cloned genes. Ampicillin and kanamycin were used at concentrations of 100 μg/ml and 50 μg/ml, respectively.
Cloning, expression, and purification.
The full-length fucO gene from wild-type E. coli strain ECL1 was cloned into plasmid pQE32, generating plasmid pCM1. For this construction, the fragment containing the fucO gene was obtained by PCR using primers FucO-pQE fw and FucO-pQE rev (Table 1) bearing BamHI and KpnI restriction sites, respectively. Overproduction of His6-tagged FucO was achieved in transformed cells of strain M15(pREP4) bearing the recombinant plasmid pCM1 after IPTG induction in LB medium containing ampicillin and kanamycin for 4 h at 37°C. Cells were harvested, and the pellets were resuspended in 50 mM sodium phosphate buffer, pH 8.0, containing 0.3 M NaCl and 5 mM 2-mercaptoethanol (buffer A) and sonically disrupted. The cell extract was centrifuged at 15,000 × g for 30 min, and the supernatant was incubated with a nickel-nitrilotriacetic acid resin (QIAGEN) for 1 h with gentle shaking. After the mixture was loaded into a column, the resin was first washed with 50 mM sodium phosphate buffer, pH 6.0, containing 0.3 M NaCl, 5 mM 2-mercaptoethanol, and 10% glycerol (buffer B). Stepwise elutions were performed with buffer B containing 40 mM, 100 mM, and 200 mM imidazole to eliminate contaminant protein. His-tagged FucO was eluted with 400 mM imidazole in buffer B. The purity of the FucO preparation (>98%) and the molecular mass were verified by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and matrix-assisted laser desorption ionization—time of flight mass spectrometry, respectively. The FucO preparations used for crystallization were dialyzed against 5 mM sodium phosphate buffer and then concentrated to 10 mg/ml.
TABLE 1.
Oligonucleotides used in this study
| Primer | Sequence (5′-3′) | Expt | Sitea |
|---|---|---|---|
| FucO-pQE fw | CGCGGATCCTTATGATGGCTAACAGAATG | fucO cloning in pQE32 | BamHI |
| FucO-pQE rev | CGGGGTACCGCGCATTTACCAGGCGGTATG | fucO cloning in pQE32 | KpnI |
| FucO-pGEX fw | ATGATGGCTAACAGAATGATTCTG | fucO cloning in modified pGEX-3X | Start codon |
| FucO-pGEX rev | CGCGGATCCGCGCATTTACCAGGCGGTATG | fucO cloning in modified pGEX-3X | BamHI |
| H200A fw | GGTGTCGATGCGCTCACTGCTGCTATTGAGGGGTA | Mutagenesis, His200 → Ala | Ala |
| H200A rev | TACCCCTCAATAGCAGCAGTGAGCGCATCGACACC | Mutagenesis, His200 → Ala | Ala |
| H200F fw | GGTGTCGATGCGCTCACTTTTGCTATTGAGGGGTA | Mutagenesis, His200 → Phe | Phe |
| H200F rev | TACCCCTCAATAGCAAAAGTGAGCGCATCGACACC | Mutagenesis, His200 → Phe | Phe |
| D196L fw | TAGCATGAGTGAGCGCAAGGACACCCGTCGCAGCT | Mutagenesis, Asp196 → Leu | Leu |
| D196L rev | AGCTGCGACGGGTGTCCTTGCGCTCACTCATGCTA | Mutagenesis, Asp196 → Leu | Leu |
| D39G fw | ATTGCACCAGCGTTTTACCGGTGACGATCAGCGCC | Mutagenesis, Asp39 → Gly | Gly |
| D39G rev | GGCGCTGATCGTCACCGGTAAAACGCTGGTGCAAT | Mutagenesis, Asp39 → Gly | Gly |
| GXDXXG fw | TTAAAGCCCCAACAGCATCCCGACCAAACCATGCC | Mutagenesis, Gly17 → Asp | Asp |
| GXDXXG rev | GGCATGGTTTGGTCGGGATGCTGTTGGGGCTTTAA | Mutagenesis, Gly17 → Asp | Asp |
| GEG fw | TATCCTGTGGAGAACCCTCACCAATAGCGATCAGG | Mutagenesis, Gly97 → Glu | Glu |
| GEG rev | CCTGATCGCTATTGGTGAGGGTTCTCCACAGGATA | Mutagenesis, Gly97 → Glu | Glu |
| M252L fw | CAACATTCGAGAAGCCCTTACCCGCAACATACTGC | Mutagenesis, Met252 → Lys | Lys |
| M252L rev | GCAGTATGTTGCGGGTAAGGGCTTCTCGAATGTTG | Mutagenesis, Met252 → Lys | Lys |
| ΔN10 fw | GAAACGGCATGGTTTGGTCGGGGTGCTGTTGG | 10-amino-acid (β1) deletion | |
| ΔN10 rev | GGGAATTCATCAGCGCATTTACCAG | 10-amino-acid (β1) deletion | EcoRI |
Underlined site in the sequence.
To avoid the His6 tag in experiments in site-directed mutagenesis studies, wild-type and mutant FucO preparations were purified using the GST gene fusion system with recognition sites for factor Xa cleavage that yielded precisely the wild-type N-terminal end. The fucO gene was amplified by PCR with the FucO-pGEX fw and FucO-pGEX rev primers (Table 1). The PCR fragment was digested and cloned into the NotI (blunted by nuclease S1 treatment) and BamHI restriction sites of modified plasmid pGEX-3X, yielding plasmid pCM2. In this construction the reported ATG start codon of fucO was in frame with GST. The GST-FucO fusion protein was overexpressed in strain BL21(DE3) carrying the recombinant plasmid pGEX-pCM2 after IPTG (0.5 mM) induction in LB medium containing ampicillin for 3 to 4 h at 37°C. Cells were collected by centrifugation at 4°C, and the cell pellet was suspended in phosphate-buffered saline (PBS) (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.3) and sonicated on ice. The cell extract, diluted 10-fold, was applied to an affinity chromatography column containing glutathione-Sepharose 4B resin (Pharmacia) at room temperature. After the column was washed with PBS, factor Xa cleavage buffer was passed through to equilibrate the matrix before cleavage of the bound GST fusion protein. Digestion was performed by applying to the column a factor Xa solution (50 U) and subsequent overnight incubation at room temperature. The cleaved FucO protein was eluted with PBS and dialyzed against 10 mM Tris-HCl, pH 7.3.
SDS-PAGE was performed as described by Laemmli (21). The protein concentration was determined by using the method of Lowry et al. (26) with bovine serum albumin as the standard.
Size exclusion chromatography.
Fast protein liquid chromatography was carried out on a Superdex 200 column. The purified protein (0.5 mg) in 10 mM Tris-HCl (pH 7.3) and 10% (vol/vol) glycerol was loaded onto a column previously equilibrated with the same buffer. The protein was eluted using the same buffer at a flow rate of 0.5 ml/min.
Cross-linking reactions.
All cross-linking reactions were carried out at 25°C. Samples (usually 50 μl [total volume]) contained 50 mM HEPES—NaOH, pH 8.0, and 1 mM bis(sulfosuccinimidyl)suberate (DSS). The reactions were started by addition of protein (25 μg), and the reaction mixtures were incubated for 10 and 20 min. The reactions were stopped by addition of gel-loading buffer and immediate incubation in boiling water for 5 min. Samples were analyzed by SDS-PAGE (usually a 10% polyacrylamide resolving gel and a 5% polyacrylamide stacking gel), and the cross-linked and uncross-linked proteins were identified by staining with Coomassie brilliant blue R-250.
Crystallization, X-ray data collection, and fluorescence measurement.
For crystallization, the FucO abortive ternary complex was formed by incubation of the purified protein with 4 mM NAD+ and 4 mM glycol aldehyde. One microliter of the protein ternary complex (10 mg/ml) in the appropriate buffer was mixed with 1 μl of reservoir solution (0.8 M ammonium sulfate, 0.1 M morpholineethanesulfonic acid [MES], pH 6.0), and crystals were grown by the sitting-drop vapor diffusion method at 20°C. Needle-like crystals that were 0.2 by 0.01 by 0.01 mm were cryoprotected with glycerol and flash frozen in liquid nitrogen for data collection at 100 K. The crystals are trigonal and belong to space group P3121, with the following unit cell parameters: a = b = 109 Å and c = 184.4 Å. There are two monomers in the asymmetric unit. An X-ray diffraction data set to 2.75 Å was collected at microfocus beamline ID13 of the ESFR synchrotron light source in Grenoble, France. Due to radiation damage, data were obtained by irradiating the crystal in six different places along the needle. The data were processed with XDS (19) and were merged with XSCALE (19) (Table 2). A fluorescence XANES spectrum was recorded around the Fe-K absorption edge (7.122 keV) at beamline ID29, which demonstrated the presence of the metal ion in the crystals.
TABLE 2.
Data collection and refinement statistics
| Parameter | Value |
|---|---|
| Data collection | |
| Space group | P 31 2 1 |
| Cell constants (Å) | a = b = 109.42, c = 184.46 |
| Wavelength (Å) | 0.978 |
| No. of measurements | 88,939 |
| No. of unique reflections | 26,635 |
| Resolution range (Å) | ∞-2.85 |
| Completeness (%)a | 88.2 (67.2) |
| Rmerge (%)a,b | 0.124 (0.45) |
| <I/σ(I)>a | 8.16 (2.62) |
| Refinement | |
| Resolution range used for refinement (Å) | 30.0-2.85 |
| No. of reflections used | 25,260 |
| Crystallographic Rfactor (free Rfactor)c | 0.258 (0.287) |
| No. of solvent molecules | 46 |
| R.m.s. deviation from target values | |
| Bonds (Å) | 0.010 |
| Angles (°) | 1.49 |
| Avg temp factor, protein atoms (Å2) | 45.3 |
| Avg temp factor, Fe atoms (Å2) | 45.4 |
The values in parenthesis indicate the outermost 3.07 to 2.85-Å resolution shell.
Rmerge = ΣhklΣi|Ii(hkl) − <I(hkl)>|/ΣhklΣiIi(hkl), where Ii is the ith measurement of reflection I (hkl).
Rfactor = Σhkl||Fobs| − k |Fcalc||/Σhkl |Fobs|. The free Rfactor is the same for a test set of 5% reflections not used during refinement.
Structure solution and refinement.
The structure of FucO was solved by the molecular replacement method using the program AMoRe (28). The starting model was the structure of the T. maritima 1,3-propanediol dehydrogenase monomer (Protein Data Bank code 1O2D) (37). Two monomers were clearly identified from the rotation and translation functions in the asymmetric unit. Cycles of model building using TURBO-FRODO (34) were alternated with refinement using the program CNS (8). Noncrystallographic symmetry restraints were used initially. Bulk solvent correction was applied, and atomic temperature factors were refined. Refinement was completed with a final round performed with REFMAC (27), including TLS (translation, liberation, and screw rotation). The final monomer included 382 of the 383 amino acids of the chemical sequence, 1 NAD+ molecule, 1 Fe2+ ion, and 46 water molecules. The final model has good stereochemical parameters, as analyzed with PROCHECK (22), with no residues in disallowed regions of the Ramachandran plot (Table 2).
DNA manipulation and site-directed mutagenesis.
Bacterial genomic DNA was obtained as described by Silhavy et al. (39). For large-scale preparation, a crude DNA sample was purified on a column (QIAGEN). DNA manipulations were performed essentially as described by Sambrook and Russell (36). DNA sequencing was done by using an automated ABI 377 DNA sequencer and fluorescent dye termination methods. DNA fragments were amplified by PCR with E. coli chromosomal DNA as the template. When necessary, specific restriction sites were introduced at the 5′ end of the primers to facilitate cloning of the fragments in the appropriate vector. PCRs were performed with Pfu DNA polymerase under standard conditions. Site-directed mutagenesis was performed using the QuikChange PCR-based mutagenesis procedure (Stratagene) with pCM2 as the template. The primers used to construct each of the mutants are listed in Table 1. The appropriate substitutions and the absence of unwanted mutations were confirmed by sequencing the inserts in both directions.
Metal analysis.
The Fe and Zn contents of the homogeneous proteins were determined by inductively coupled plasma mass spectrometry with a Perkin-Elmer Elan-6000. In all cases, prior to metal analysis samples were applied to a Sephadex G-25 PD-10 column (Pharmacia) equilibrated with MilliQ water (resistivity, >18 MΩ) in order to eliminate reagents and metals not bound to the enzyme. Fractions were collected in metal-free polypropylene tubes. Blank water samples, representing the flowthrough collected in the void volume of the column, were also analyzed, and they contained no significant detectable metal. In all cases, to avoid adsorption of the metalloproteins to the tube walls, samples were sonicated for 5 min before they were digested with 1% nitric acid. Solutions to be tested were adjusted to metal concentrations ranging from 20 to 200 ppb and carried an internal standard (500 ppb of rhodium).
Enzyme and NAD+ binding assays.
FucO activity was measured by determining NADH-dependent l-lactaldehyde reduction to l-1,2-propanediol using a molar absorption coefficient of 6.22 × 10−3 M−1 cm−1 at 340 nm and glycol aldehyde as an alternative substrate (9). The oxidation reaction was measured with l-1,2-propanediol and NAD+ as described previously (40). Kinetic determinations were performed for each condition with eight different concentrations of cofactor bracketing an interval from 37.5 to 250 μM. The initial velocities obtained during the first 30 s of the reaction were determined. The Km and Vmax were determined by linear regression analysis of the data plotted according to the method of Lineweaver and Burk (25). Parameters were expressed as means for at least three different measurements. For the NAD+ loading assays the increase in absorbance at 360 nm was monitored upon stepwise addition of the cofactor to an enzyme suspension (1 mg/ml) in 10 mM Tris-HCl (pH 7.3) that had previously been treated with charcoal to release all bound cofactor. Measurements were performed 10 min after each addition (2).
Protein structure accession number.
The atomic coordinates of FucO have been deposited in the Protein Data Bank under accession code 2bl4.
RESULTS AND DISCUSSION
Structure of FucO monomer.
Each FucO subunit consists of a single polypeptide chain of 383 residues composed of 10 β strands (β1 to β10), 16 α helices (α1 to α16), and one 310 helix (Fig. 1). The molecule, like other proteins in the family, folds into two structural domains that are separated by a deep cleft (Fig. 2A). The N-terminal domain (residues 1 to 182) is formed by an α/β region containing the dinucleotide-binding fold, with β1 to β6 and β10 forming the central β-sheet, where strand β1 is swapped between monomers (see below). Strands β2 to β6 are connected by helices α1 to α5, whereas the connection between β6 and β10 is a long loop that includes an antiparallel β-ribbon formed by β8 and β9. The C-terminal part (residues 183 to 383), in contrast, is an all-helical domain formed by 10 α helices (α7 to α16) with a 310 helix (residues 361 to 365) between α15 and α16. The enzyme forms a dimer in the crystal (Fig. 2B).
FIG. 1.

Sequence alignment of several group III iron-dependent alcohol dehydrogenases. Amino acid sequences are shown for lactaldehyde:1,2-propanediol oxidoreductase from E. coli (FucO) (accession no. P11549), 1,2-propanediol dehydrogenase from S. enterica serovar Typhimurium (1,2-Prd) (accession no. Q8ZMC8), alcohol dehydrogenase II from Z. mobilis (ADH2) (accession no. P06758), 1,3-propanediol oxidoreductase from K. pneumoniae (1,3-Prd) (accession no. Q59477), and alcohol dehydrogenase IV from S. cerevisiae (ADH4) (accession no. P10127). Amino acid residues that are identical in all sequences are indicated by red shading. Residues that are conserved are indicated by red type and enclosed in blue boxes. The secondary structure of FucO is shown above the sequences. Green circles indicate residues that interact with the NAD+ cofactor. Blue circles indicate residues that coordinate the Fe2+ ion. The black triangle indicates an essential histidine, which putatively interacts with the substrate.
FIG. 2.

Crystal structure of FucO. (A) Ribbon diagram of the FucO subunit in a complex with iron and the cofactor NAD+. The structure consists of two domains, an α/β domain and an all-α domain. The rainbow color ramp for the model ranges from blue for the N-terminal segment to red for the C-terminal segment. Iron is represented by a purple ball, and the bound NAD+ is represented by a ball-and-stick model. (B) Ribbon diagram of the quaternary structure of FucO. In the dimer one subunit is yellow and orange, and the other subunit is light blue and dark blue. Note the swapping of strand β1 between monomers.
Active site and metal coordination.
The two domains define between them a deep cleft where the active center of the enzyme is found. Helix α5 together with strands β8 and β9 of the β-ribbon and the intervening loop between β7 and β8 of the N-terminal domain and helices α7, α11, and α12 of the bundle of the C-terminal domain constitute the opposing faces of the cleft. The location of the binding site for the Fe2+ ion is defined in the face formed by the C-terminal domain. Fe2+ is tetrahedrally coordinated through an ion dipole interaction with residues Asp196, His200, His263, and His277 (Fig. 3). In a previous study (30) His267 was also thought to be involved in Fe2+ binding, but mutation of this residue did not significantly affect the metal ion binding. In this previous study, participation of His263 and His277 was also proposed and demonstrated by site-directed mutagenesis. In the present structure His267 is not coordinated with the Fe2+ ion. As discussed below, this residue seems to be a good candidate to interact with the substrate, which would be consistent with the inactivation observed when it is mutated. Participation of His263 and His277 in the metal coordination was also proposed in the previous study and was demonstrated by site-directed mutagenesis. In the present work we mutated the two other Fe2+ binding residues, Asp196 and His200. Mutant FucO proteins bearing the mutation D196L, H200A, or H200F were purified, and their metal contents were determined. The three mutants were not able to bind Fe2+ and had iron contents of less than 0.01 atom/subunit, whereas wild-type FucO protein, which was examined in parallel as a control, had 0.53 atom/subunit.
FIG. 3.

Close-up of the FucO active site: stereo representation showing the binding sites of the iron metal (purple), the cofactor NAD+ (grey sticks), and the modeled substrate l-1,2-propanediol (green sticks). Carbon atoms are represented by grey balls, oxygen atoms are represented by red balls, and nitrogen atoms are represented by blue balls. For clarity, only the residues that coordinate the iron, the substrate, and the essential His267 residue are shown. The diagram was drawn with MOLSCRIPT (20).
l-1,2-Propanediol oxidoreductase has been reported to be activated by Fe2+ and Mn2+ and to be inhibited by Zn2+, Cu2+, and Cd2+, among other divalent cations (40). Incubation of the iron enzyme in the presence of concentrations of Zn2+ as low as 5 or 10 μM inactivated the enzyme and seemed to displace Fe2+, suggesting that the two metals compete for a single site and are mutually exclusive. Furthermore, inhibition of FucO by Zn2+ was blunted by the presence of Fe2+, but the protective effect required concentrations of Fe2+ that were more than 1 order of magnitude higher than those of Zn2+, indicating that although FucO is active only with Fe2+, the enzyme has, in fact, a higher affinity for Zn2+ (Fig. 4).
FIG. 4.
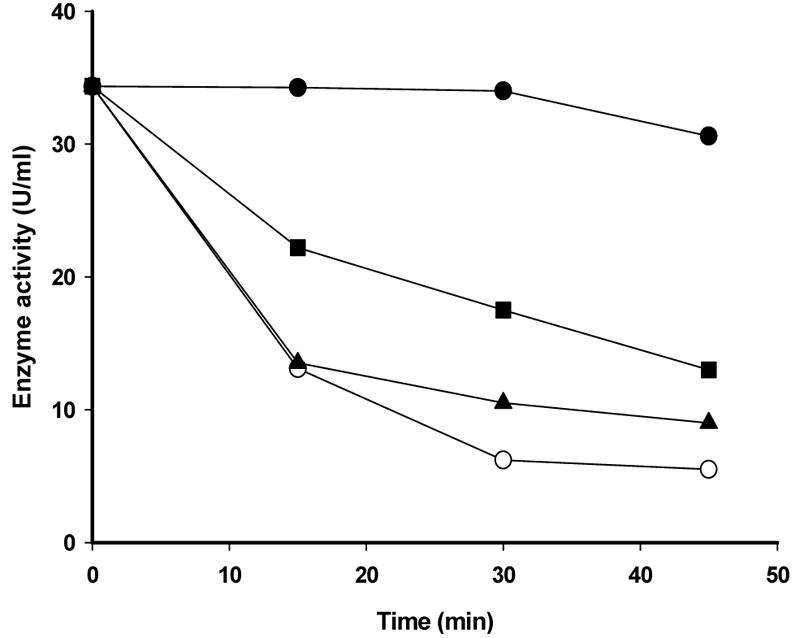
Time course of inhibition of lactaldehyde:propanediol oxidoreductase activity by Zn2+. His6-FucO was incubated with 0.1 mM ZnCl2 in the absence of FeSO4 (○) or in the presence of 0.1 mM FeSO4 (▴) or 1 mM FeSO4 (▪). Control incubations in the absence of any metal were performed in parallel (•). At different times, aliquots were taken to measure the enzymatic activity following NADH oxidation using glycol aldehyde as the substrate.
Location of the cofactor binding site and NAD+ specificity.
The crystals grew in the presence of 2 mM NAD+, and both the 2Fo-Fc and Fo-Fc Fourier maps clearly showed the presence of the cofactor molecule (Fig. 5A). The cofactor binds in the cleft defined by the two domains interacting mainly with residues of the N-terminal domain (Fig. 5B). The adenine ring interacts with Thr140 of the N-terminal domain and Met181 of α6 at the intervening sequences between domains, located at the upper end of the cleft. The exocyclic nitrogen at C-6 of the six-member ring of the purine interacts with the carbonyl oxygens of Thr140 and Met181, while N-7 of the five-member ring of the purine interacts with the side chain oxygen of Thr140 (Fig. 6). The hydroxyl group at position 2 of the adenine ribose is hydrogen bonded with the carboxyl of the side chain of Asp39 at the end of strand β3. Interactions with the pyrophosphate group include the following hydrogen bonds: the adenine-proximal phosphate interacts with Ser99 of α5, and the adenine-distal phosphate interacts with Thr141 and with the nitrogen amide atom of Gly98 in the glycine-rich turn formed by residues 96 to 98 (Fig. 6).
FIG. 5.

NAD+ binding site. (A) Stereo view of the Fo-Fc OMIT map of NAD+ in the FucO subunit. The density is well defined for the adenine moiety, whereas the electron density of the nicotinamide ring is weaker. The diagrams were drawn with BOBSCRIPT (15). (B) Comparison of the orientation of the nicotinamide ring in the FucO (blue cofactor) and TM0920 (red cofactor) active centers.
FIG. 6.

Diagram of the interactions of NAD+ with FucO. Carbon atoms are represented by black circles, oxygen atoms are represented by red circles, and nitrogen atoms are represented by blue circles. Hydrogen bonds are indicated by dashed green lines. The spiked atoms are the atoms involved in hydrophobic contacts, and the spiked circle segments surround hydrophobic van der Waals partners. The diagram was drawn with LIGPLOT (44).
This glycine-rich motif and a following α helix are part of the NAD+ recognition locus in the dehydrogenases, which have a typical dinucleotide-binding fold. However, in the FucO sequence there is another glycine-rich motif (GXGXXG) between residues 15 and 20, which has been reported to be part of the recognition site for many dehydrogenases (31, 38, 45). To assess the participation of this glycine-rich motif in the recognition of NAD+ by FucO, we obtained by site-directed mutagenesis two mutant FucO clones, one containing the mutation G97E and the other containing the mutation G17D. The G97E mutation completely abolished FucO activity, whereas G17D did not affect it significantly. Furthermore, the NAD+ binding capacity was measured for the two mutant enzymes and compared to that of wild-type FucO. Both the wild-type enzyme and the G17D mutant enzyme bound NAD+ up to saturation (2 mol of NAD+ per mol of protein) upon addition of the cofactor to an NAD+ unloaded preparation (Fig. 7). In contrast, mutant G97E did not bind a significant amount of NAD+. Thus, we concluded that of the two glycine-rich motifs that might interact with the cofactor, only the one between positions 96 and 98 is relevant to NAD+ binding. In addition, this is consistent with the location of NAD+ in the structure of the enzyme-cofactor complex, as determined in this study.
FIG. 7.
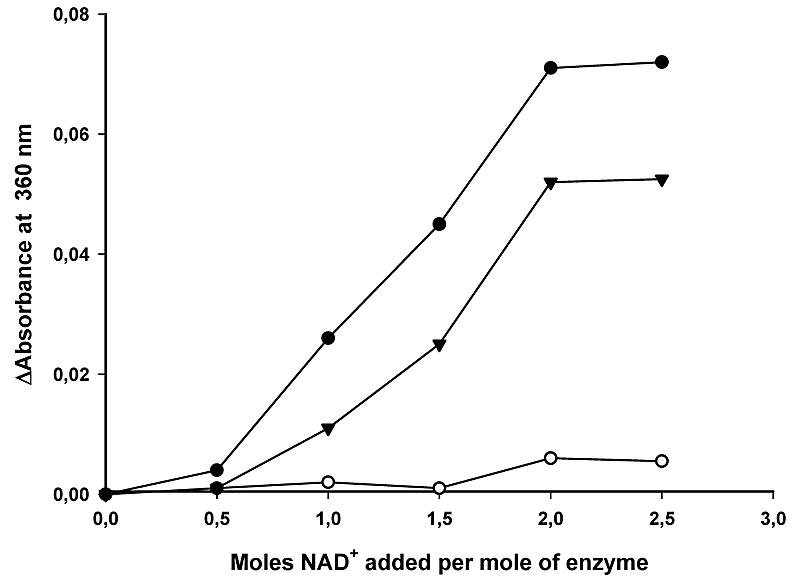
Titration of E. coli wild-type FucO (•), mutant G17D FucO (▴), or mutant G97E FucO (○) with NAD+. The enzyme suspensions were titrated at 23°C with stepwise additions of NAD+, and the absorbance at 360 nm was determined 10 min after each addition.
The nicotinamide ribose O-2 hydroxyl group forms a hydrogen bond with the nitrogen atom of the side chain of the conserved Lys162 residue. The nicotinamide ring lies in a hydrophobic pocket formed by Val164, Phe254, Val153, Leu189, Met185, Pro186, and Leu42, among others (Fig. 6), with the C-4 and C-5 ring atoms in close proximity to the Fe2+. The nicotinamide ring orientation in the FucO active center facilitates hydride transfer to the A face of the ring during the enzyme's catalytic cycle. It should be emphasized that when the locations of the nicotinamide ring in FucO and in the 1,3-propanediol dehydrogenase of T. maritima (37) are compared, a significant difference in orientation is detected as the ring is closer to the N terminal domain in the T. maritima enzyme (Fig. 5B). The flexibility of the nicotinamide nucleotide moiety in the NAD+ molecule permits a certain mobility in this part of the cofactor molecule in a space large enough to allow the movement required for the correct location of the substrate and may explain the weakness of the electron density observed in the nicotinamide area.
FucO is an NAD+ or NADH enzyme, depending on the oxidation or reduction in its catalytic cycle. Analysis of the structure clearly indicates that its inability to use NADP(H) is due to steric hindrance of the 2′ phosphate of the adenosine moiety with Asp39. Consistently, one of the structural differences between FucO and the NADP(H)-dependent enzyme from T. maritima is that in the latter, residue 39 corresponds to a glycine. We mutated the Asp39 of FucO to glycine. The D39G mutant protein allowed utilization of NADP(H), and, as expected, this mutant enzyme also had the capacity to use NAD(H). To check the catalytic efficiency of the mutant enzyme with either of the two cofactors, we examined the concentration kinetics of the reduction reaction for the wild-type and mutant enzymes. As shown in Table 3, the affinity of the mutant FucO for NADP(H) was similar to the affinity either of the mutant or of the wild-type enzyme for NAD(H). Nevertheless, the Kcat of the mutant enzyme was lower for NADP(H). These results indicated that the D39G mutation allows FucO to use NADP(H) as a cofactor, although the catalytic efficiency is slightly lower. Rosell et al. (33), working with ADH8, an amphibian NADP(H)-dependent dehydrogenase, showed by site-directed mutagenesis that several amino acids (namely, Gly223-Thr224-His225 and a conserved Lys228) define a binding pocket for the terminal phosphate of NADP(H). Close inspection of the residues in the vicinity of Asp39 of our enzyme allowed us to identify a group of residues (Lys40, Thr41, and Gln44) that seem to mimic this binding pocket for the terminal phosphate. This observation may explain the similar affinity of the cofactor center for the spurious NADP(H) or the physiological NAD(H), provided that the steric and electrostatic hindrance with Asp39 is eliminated by mutating this amino acid to glycine. At present, we cannot explain why the mutation lowers the Vmax of the reaction.
TABLE 3.
Coenzyme specificity and kinetic parameters of wild-type FucO and the D39G mutant
| FucO enzyme | Coenzyme | Km (M) | Vmax (U/mg) | Kcat (s−1) | Kcat/Km (s−1 M−1) |
|---|---|---|---|---|---|
| Wild type | NADH | 0.35 × 10−4 | 148 | 96.3 | 2.7 × 106 |
| D39G mutant | NADH | 0.66 × 10−4 | 160 | 104 | 1.5 × 106 |
| NADPH | 0.36 × 10−4 | 48 | 31.2 | 0.86 × 106 |
Location of the substrate binding site.
Although we grew crystals of FucO in the presence of up to 2 mM glycol aldehyde as a substrate to form the abortive ternary complex with NAD+, in no case could an electron density corresponding to the glycol aldehyde be found on the Fourier maps. Docking of the substrate molecule l-1,2-propanediol (Fig. 3) in the active center resulted in a model in which the O-1 hydroxyl of the substrate interacts with the Fe2+ ion. In this model His267 does not interact directly with the substrate (both the O-1 and O-2 hydroxyl oxygens of the substrate are 4.2 Å from the Nɛ2 nitrogen of His267), but a water-mediated hydrogen bond is possible. The loss of the latter interaction may explain the inactivation of enzyme activity when this residue is mutated to alanine (30).
As indicated above, the space available at the area around the location of the nicotinamide moiety of NAD+ allows a certain mobility of the ring, opening several possibilities for modeling entry of the substrate. The position of the l-1,2-propanediol substrate was determined by examining the position of the substrate glycerol in the very similar active center of the glycerol dehydrogenase of B. stearothermophilus (35). In this model the nicotinamide ring of the cofactor was slightly pulled toward the C-terminal domain face of the intermediate cleft of the protein structure. The docking of the substrate caused close approach of the C-1 hydroxyl of the substrate to C-4 of the nicotinamide ring (3.1 Å) and to the Fe2+ ion (3.3 Å between the l-1,2-propanediol C-1 and Fe2+ or 2.6 Å between the corresponding hydroxyl O-1 and Fe2+), an appropriate arrangement for a typical metal-dependent dehydrogenation catalytic mechanism. In such a mechanism the Fe2+ ion lowers the pKa of the C-1 hydroxyl, and the hydride is thus transferred to C-4 of the nicotinamide ring.
Quaternary structure of FucO.
Since the FucO protein molecular mass, as estimated by gel filtration, has been reported to be close to 80,000 Da, it has been generally accepted that native FucO in E. coli is a dimeric protein (6, 24). The presence of homodimers in FucO purified preparations is consistent with the dimeric arrangement observed in the crystal structure (Fig. 2B). The two monomers of the dimer are related by a twofold noncrystallographic symmetry axis. The interface area between the two monomers (1,094 Å2) is indicated in Fig. 8A, and there are several possible interactions, which are mainly hydrophobic, with some hydrogen bonds and salt bridges. The dimer is maintained mainly by the swapping of strand β1 between monomers, leading on each subunit to a 10-strand β-sheet in which the first strand belongs to the partner monomer (Fig. 2B). Besides the β-sheet-type H-bonding interactions between strands β1 and β2 (residues 4 to 14) of the N-terminal domain, additional contacts between the α8 and α10 helices (residues 212 to 226 and 237 to 254, respectively) of the C-terminal domain are observed.
FIG. 8.

Interface analysis of the FucO dimer structure. (A) The interface area of the wild-type FucO subunit interactions is red. (B) The residual interface area of the ΔN10 FucO mutant is red. The diagrams in panels A and B were generated with GRASP (29). (C) Analysis by cross-linking of the β1 deletion effect in FucO dimerization. The electrophoretic mobility of the cross-linked species obtained after reaction of DSS with wild-type or ΔN10 mutant FucO was determined by SDS-PAGE. Lane 1, control wild-type FucO; lanes 2 and 3, cross-linked wild-type FucO with 1 mM DSS after 10 and 20 min, respectively; lane 4, control ΔN10 FucO mutant; lanes 5 and 6, cross-linked ΔN10 FucO mutant with 1 mM DSS after 10 and 20 min, respectively; lane M, molecular mass markers (BENCHMARK prestained protein ladder; Gibco BRL).
The key role of the N-terminal β strands was confirmed by studying the dimerization of a mutant enzyme with residues 1 to 10 of the FucO sequence deleted. The sequence modification in this mutant, which exhibited no oxidoreductase activity, greatly diminished the estimated interface area according to the GRASP program (29) (Fig. 8B). The differences in oligomeric states between the wild-type FucO and the ΔN10 mutant FucO were analyzed by size exclusion chromatography. The apparent molecular mass derived from the elution volume of the wild-type FucO corresponded to a dimer, whereas the elution volume of the mutant FucO corresponded to a monomer (not shown). The oligomeric states were also established in cross-linking experiments with DSS. These experiments revealed no band with the dimer mobility in the PAGE lane corresponding to the mutant, which contrasted with the band observed in the lane corresponding to the wild-type enzyme, thus indicating that there was a significant decrease in the dimerization capacity of the mutant enzyme (Fig. 8C). Note that the ΔN10 mutant FucO is more sensitive to degradation by the cross-linker than the wild-type enzyme.
Structural comparison with other proteins.
Group III “iron-activated” alcohol dehydrogenases from E. coli and its orthologs form a conserved family of microbial enzymes. A BLAST search analysis of the amino acid sequence of the E. coli FucO showed similarity with various iron-dependent group III dehydrogenases, such as 1,2-propanediol dehydrogenase from Salmonella enterica serovar Typhimurium (accession no. Q8ZMC8), alcohol dehydrogenase II from Zymomonas mobilis (accession no. P06758), 1,3-propanediol oxidoreductase from K. pneumoniae (accession no. Q59477), and alcohol dehydrogenase IV from Saccharomyces cerevisiae (accession no. P10127). Analysis of these sequences showed that the largest number of strictly conserved residues is at the active site involved in metal coordination or in cofactor binding (Fig. 1).
A structural comparison using the DALI server (17) of E. coli FucO and other protein structures revealed that the most structurally similar proteins were (i) the putative 1,3-propanediol dehydrogenase TM0920 from T. maritima (PDB accession code 1O2D) with a root mean square deviation of 2.0 Å for the superimposition of 344 Cα atoms and 28% sequence identity and (ii) the alcohol dehydrogenase YqhD from E. coli (PDB accession code 1OJ7) with an rmsd of 2.5 Å for the superimposition of 372 Cα atoms and 24% sequence identity. Both of these structures have a highly analogous N-terminal domain consisting of an α/β region containing the dinucleotide-binding fold and a C-terminal all-helix domain. The fold of the second domain is characteristic of the group III metal-dependent alcohol dehydrogenases and has not been reported in any other type of protein. The interactions of two monomers through N-terminal β-strand swapping (β1 and β2) together with the α-helical contacts mentioned above are also found in the T. maritima TM0920 and E. coli YqhD enzymes. Analysis of the structure of the glycerol dehydrogenase of B. stearothermophilus also showed this form of monomer-monomer interaction. We concluded that this model of dimerization constitutes a specific feature of most of the proteins reported to belong to the group III metal-dependent dehydrogenases. On the other hand, as noticed by Sulzenbacher et al. (42), A. nidulans dehydroquinate synthase (13) (PDB accession code 1DQS), which has a similar monomer fold (rmsd, 3.0 Å; 14% sequence identity), does not show the N-terminal β-strand swapping. Therefore, the 10-strand β-sheet is formed by elements that all belong to the same monomer. Strand 1 is thus not extended out of the sheet but is folded over strand 2. Superposition of the 1DQS structure with our structure indicated that in the former, the swapped conformation is not possible because β1 would clash with a loop of the all-α-helix C terminus at residues 255 and 256. Among the members of this group, dehydroquinate synthase shows the lowest sequence similarity and a distinct catalytic activity and is therefore the most distantly related.
Domain or secondary structure element swapping has been proposed as a rapid mechanism for the evolution from monomeric to oligomeric proteins (4, 12). Because it is based on the interchange of identical structural elements, the resulting interactions are guaranteed, as occurs with the β-sheet hydrogen bonds in the present structure. Additional interactions can be gained from mutations that reinforce the union, leading finally to a new interface between subunits. It is clear, however, that evolution to dimeric forms can follow completely different paths in closely related proteins, as shown for the 1DQS protein. An open question is whether there are any subtle structure determinants that drive molecular evolution one way or the other.
Acknowledgments
We thank Robin Rycroft for editorial assistance.
This work was supported by grants from the Ministerio de Educación y Ciencia of Spain (grant BFU 2004-03586/BMC to J.A. and grant BIO2002-00379 to M.C.) and by grants from the Generalitat de Catalunya (grant SGR 2000-0042 to J.A. and grant SGR2001-00346 to M.C.). C.M. was a recipient of a predoctoral fellowship from the Ministerio de Educación y Ciencia of Spain, and L.B. received a contract from the Ministerio de Educación y Ciencia of Spain.
Footnotes
This work is dedicated to E. C. C. Lin, who used this and other enzymes as the basis for a school of molecular evolution, a school in which J.A. “grew up” as a scientist.
REFERENCES
- 1.Bairoch, A. 1991. PROSITE: a dictionary of sites and patterns in proteins. Nucleic Acids Res. 19:2241-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baldoma, L., and J. Aguilar. 1987. Involvement of lactaldehyde dehydrogenase in several metabolic pathways of Escherichia coli K12. J. Biol. Chem. 262:13991-13996. [PubMed] [Google Scholar]
- 3.Becker, A. B., and R. A. Roth. 1993. Identification of glutamate-169 as the third zinc-binding residue in proteinase III, a member of the family of insulin-degrading enzymes. Biochem. J. 292:137-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennet, M. J., S. Choe, and D. S. Eisenberg. 1994. Domain swapping: entangling alliances between proteins. Proc. Natl. Acad. Sci. USA 91:3127-3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bode, W., F. X. Gomis-Ruth, and W. Stockler. 1993. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. 331:134-140. [DOI] [PubMed] [Google Scholar]
- 6.Boronat, A., and J. Aguilar. 1979. Rhamnose-induced propanediol oxidoreductase in Escherichia coli: purification, properties, and comparison with the fucose-induced enzyme. J. Bacteriol. 140:320-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boronat, A., and J. Aguilar. 1981. Experimental evolution of propanediol oxido-reductase in Escherichia coli: comparative analysis of the wild type and mutant enzymes. Biochim. Biophys. Acta 672:98-107. [DOI] [PubMed] [Google Scholar]
- 8.Brünger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D 54:905-921. [DOI] [PubMed] [Google Scholar]
- 9.Caballero, E., L. Baldoma, J. Ros, A. Boronat, and J. Aguilar. 1983. Identification of lactaldehyde dehydrogenase and glycolaldehyde dehydrogenase as functions of the same protein in Escherichia coli. J. Biol. Chem. 258:7788-7792. [PubMed] [Google Scholar]
- 10.Cabiscol, E., J. Aguilar, and J. Ros. 1994. Metal-catalyzed oxidation of Fe2+ dehydrogenases. J. Biol. Chem. 269:6592-6597. [PubMed] [Google Scholar]
- 11.Cabiscol, E., J. Badia, L. Baldoma, E. Hidalgo, J. Aguilar, and J. Ros. 1992. Inactivation of propanediol oxidoreductase of Escherichia coli by metal-catalyzed oxidation. Biochim. Biophys. Acta 1118:155-160. [DOI] [PubMed] [Google Scholar]
- 12.Canals, A., J. Pous, A. Guasch, A. Benito, M. Ribó, M. Vilanova, and M. Coll. 2001. The structure of engineered domain-swapped ribonuclease dimer and its implications for the evolution of proteins toward oligomerization. Structure 9:967-976. [DOI] [PubMed] [Google Scholar]
- 13.Carpenter, E. P., A. R. Hawkins, J. W. Frost, and K. A. Brown. 1998. Structure of dehydroquinate synthase reveals an active site capable of multistep catalysis. Nature 394:299-302. [DOI] [PubMed] [Google Scholar]
- 14.Cocks, G. T., J. Aguilar, and E. C. C. Lin. 1974. Evolution of l-1,2-propanediol metabolism in E. coli by recruitment of enzymes for l-fucose and l-lactate metabolism. J. Bacteriol. 118:83-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esnouf, R. M. 1999. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. Sect. D 55:938-940. [DOI] [PubMed] [Google Scholar]
- 16.Hacking, A. J., and E. C. C. Lin. 1976. Disruption of the fucose pathway as a consequence of a genetic adaptation to propanediol as a carbon source in Escherichia coli. J. Bacteriol. 126:1166-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holm, L., and C. Sander. 1995. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20:478-480. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, E. A., and E. C. C. Lin. 1987. Klebsiella pneumoniae 1,3-propanediol: NAD+-oxidoreductase. J. Bacteriol. 169:2050-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kabsch, W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26:795-800. [Google Scholar]
- 20.Kraulis, P. J. 1991. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946-950. [Google Scholar]
- 21.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283-291. [Google Scholar]
- 23.Lin, E. C. C. 1976. Glycerol dissimilation and its regulation in bacteria. Annu. Rev. Microbiol. 30:535-578. [DOI] [PubMed] [Google Scholar]
- 24.Lin, E. C. C., and T. T. Wu. 1984. Functional divergence of the l-fucose system in mutants of Escherichia coli, p. 135-163. In R. P. Mortlock (ed.), Microorganisms as model systems for studying evolution. Plenum Publishing Co., New York, N.Y.
- 25.Lineweaver, H., and D. Burk. 1984. Determination of the enzyme dissociation constants. J. Am. Chem. Soc. 56:658-666. [Google Scholar]
- 26.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-273. [PubMed] [Google Scholar]
- 27.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D 53:240-255. [DOI] [PubMed] [Google Scholar]
- 28.Navaza, J. 1994. AMoRe: an automated package for molecular replacement. Acta Crystallogr. Sect. A 50:157-163. [Google Scholar]
- 29.Nicholls, A., R. Bharadwaj, and B. Honig. 1993. GRASP: graphical representation and analysis of surface properties. Biophys. J. 64:A166-A166. [Google Scholar]
- 30.Obradors, N., E. Cabiscol, J. Aguilar, and J. Ros. 1998. Site-directed mutagenesis studies of the metal-binding center of the iron-dependent propanediol oxidoreductase from Escherichia coli. Eur. J. Biochem. 258:207-213. [DOI] [PubMed] [Google Scholar]
- 31.Reid, M. F., and C. A. Fewson. 1994. Molecular characterization of microbial alcohol dehydrogenases. Crit. Rev. Microbiol. 20:13-56. [DOI] [PubMed] [Google Scholar]
- 32.Ros, J., and J. Aguilar. 1985. Propanediol oxidoreductases of Escherichia coli, Klebsiella pneumoniae and Salmonella typhimurium. Aspects of interspecies structural and regulatory differentiation. Biochem. J. 231:145-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosell, A., E. Valencia, X. Pares, I. Fita, J. Farres, and W. F. Ochoa. 2003. Crystal structure of the vertebrate NADP(H)-dependent alcohol dehydrogenase (ADH8). J. Mol. Biol. 330:75-85. [DOI] [PubMed] [Google Scholar]
- 34.Roussel, A., and C. Cambilleau. 1989. TURBO-FRODO, p. 77-79. In Silicon Graphics geometry partners directory. Silicon Graphics, Mountain View, CA.
- 35.Ruzheinikov, S. N., J. Burke, S. Sedelnikova, P. J. Baker, R. Taylor, P. A. Bullough, N. M. Muir, M. G. Gore, and D. W. Rice. 2001. Glycerol dehydrogenase: structure, specificity and mechanism of a family III polyol dehydrogenase. Structure 9:789-802. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 37.Schwarzenbacher, R., F. von Delft, J. M. Canaves, L. S. Brinen, X. Dai, A. M. Deacon, M. A. Elsliger, S. Eshaghi, R. Floyd, A. Godzik, C. Grittini, S. K. Grzechnik, C. Guda, L. Jaroszewski, C. Karlak, H. E. Klock, E. Koesenema, J. S. Kovarik, A. Kreusch, P. Kuhn, S. A. Lesley, D. McMullan, T. M. McPhillips, M. A. Miller, M. D. Miller, A. Morse, K. Moy, J. Ouyang, R. Page, A. Robb, K. Rodrigues, T. L. Selby, G. Spraggon, R. C. Stevens, H. Van den Bedem, J. Velasquez, J. Vincent, X. Wang, B. West, G. Wolf, K. O. Hodgson, J. Wooley, and I. A. Wilson. 2004. Crystal structure of an iron-containing 1,3-propanediol dehydrogenase (TM0920) from Thermotoga maritima at 1.3 Å resolution. Proteins 54:174-177. [DOI] [PubMed] [Google Scholar]
- 38.Scrutton, N. S., A. Berry, and R. N. Perham. 1990. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343:38-43. [DOI] [PubMed] [Google Scholar]
- 39.Silhavy, T. J., M. L. Berman, and L. Enquist. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 40.Sridhara, S., T. T. Wu, T. M. Chused, and E. C. C. Lin. 1969. Ferrous activated nicotinamide adenine dinucleotide-linked dehydrogenase from a mutant of Escherichia coli capable of growth on 1,2-propanediol. J. Bacteriol. 93:87-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sridhara, S., and T. T. Wu. 1969. Purification and properties of lactaldehyde dehydrogenase from Escherichia coli. J. Biol. Chem. 244:5233-5238. [PubMed] [Google Scholar]
- 42.Sulzenbacher, G., K. Alvarez, R. H. H. van den Heuvel, C. Versluis, S. Spinelli, V. Campanacci, C. Valencia, C. Cambillau, H. Eklund, and M. Tegoni. 2004. Crystal structure of E. coli alcohol dehydrogenase YqhD: evidence of a covalently modified NADP coenzyme. J. Mol. Biol. 342:489-502. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki, H., and K. Kishimoto. 1994. Site-directed mutagenesis studies on the iron-binding domain and the determinant for the substrate oxygenation site of porcine leukocyte arachidonate 12-lipoxygenase. Biochim. Biophys. Acta 1210:308-316. [DOI] [PubMed] [Google Scholar]
- 44.Wallace, A. C., R. A. Laskowski, and J. M. Thornton. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8:127-134. [DOI] [PubMed] [Google Scholar]
- 45.Wierenga, R. K., P. Terpstra, and W. G. Hol. 1986. Prediction of the occurrence of the ADP-binding βαβ-fold in proteins, using an amino acid sequence fingerprint. J. Mol. Biol. 187:101-107. [DOI] [PubMed] [Google Scholar]