

ABSTRACT
Aims
To analyze the effect of APOE ε4 on fluid biomarkers and the correlations between blood molecules and CSF biomarkers in AD patients.
Methods
This study enrolled 575 AD patients, 131 patients with non‐AD dementia, and 112 cognitively normal (CN) participants, and AD patients were divided into APOE ε4 carriers and non‐carriers. Cerebrospinal fluid (CSF) biomarkers and blood‐derived biomolecules were compared between AD and CN groups, between non‐AD dementia and CN groups, as well as within APOE ε4 subgroups of AD patients. Utilizing Spearman's correlation analysis and quantile regression analysis, the relationships between blood‐derived biomolecules and CSF biomarkers were analyzed in APOE ε4 carriers and non‐carriers.
Results
The levels of CSF biomarkers and blood molecules exhibited significant differences between the AD and CN groups, including Aβ42, t‐tau, p‐tau 181, high‐density lipoprotein, low‐density lipoprotein (LDL), and uric acid. In AD patients, APOE ε4 carriers had increased levels of CSF t‐tau, p‐tau 181, and plasma LDL. In the correlation and regression analyses, the negative relationships between plasma TG and t‐tau, between plasma TG and p‐tau 181 levels, as well as the positive relationship between serum IgA and CSF Aβ42, were observed significantly in APOE ε4+ AD groups, but not in APOE ε4− AD group.
Conclusion
APOE ε4 is associated with accelerated progression of AD pathology. The blood‐derived biomolecules correlated with CSF biomarkers in APOE ε4 carriers are related to neuroinflammation and lipid metabolism, which may indicate the role of APOE ε4 in AD pathophysiology and offer insights for diagnostic and therapeutic strategies for AD.
Trial Registration
ClinicalTrials.gov identifier: NCT03653156
Keywords: Alzheimer's disease, apolipoprotein E, biomarker, high‐density lipoprotein, immunoglobulin, low‐density lipoprotein, uric acid
APOE ε4 is associated with accelerated progression of AD pathology. The blood‐derived biomolecules correlated with CSF biomarkers in APOE ε4 carriers are related to neuro‐inflammation and lipid metabolism, which may indicate the role of APOE ε4 in AD pathophysiology and offer insights for diagnostic and therapeutic strategies for AD.

1. Introduction
Alzheimer's disease (AD) stands as one of the most challenging and complex neurodegenerative disorders nowadays, affecting millions of individuals worldwide [1]. The apolipoprotein E (APOE) gene has garnered considerable attention for its association with risk of AD [2, 3]. The APOE gene is characterized by three common alleles, known as ε2, ε3, and ε4. Each individual inherits one allele from each parent, leading to the formation of six distinct genotypic combinations. Notably, the ε4 allele has been consistently associated with an increased risk of developing sporadic AD [4, 5], while the precise mechanisms through which APOE ε4 contributes to AD pathology are complex and not fully understood.
Recent research has turned its focus toward discovering the influence of APOE ε4 on AD biomarkers, which include amyloid‐beta (Aβ) and tau proteins in cerebrospinal fluid (CSF), as well as neuroimaging markers [4, 6]. The relationships between APOE ε4, CSF, and blood biomarkers have been discussed in several studies, which indicate that APOE genotypes may have effects on several CSF biomarkers and blood‐derived biomolecules, including cholesterol, immunoglobulin, complement factor, uric acid (UA), etc [7, 8, 9, 10, 11, 12, 13]. However, the relationships between APOE ε4 and blood‐derived biomolecules, as well as the correlations between CSF biomarkers and blood‐derived biomolecules, are still in need of further investigation.
Moreover, blood‐based biomarkers are less medically invasive and less expensive, when compared to CSF and PET biomarker assessments. Thus, blood‐based biomarker measurements have been recognized as a valuable method for early diagnosis and monitoring of AD progression [14]. The identification of novel blood‐based biomarkers for AD is imperative, and exploring the correlations between blood‐derived biomolecules and CSF biomarkers may facilitate the discovery of potential blood biomarkers for AD.
Based on cross‐sectional data from an ongoing cohort study, we analyzed the effects of APOE ε4 on AD biomarkers in CSF and blood‐derived biomolecules. Additionally, we investigated the correlations between CSF biomarkers and blood‐derived biomolecules in AD patients stratified according to APOE ε4 carrying status, aiming to provide a comprehensive understanding of how APOE genotypes might influence AD pathophysiology through the blood‐derived biomolecules. This study could illuminate the associations between APOE genotypes, AD biomarkers, and blood‐derived biomolecules, thus providing valuable insights that may pave the way for novel diagnostic and therapeutic strategies for AD.
2. Methods
2.1. Study Design and Participants
The China Cognition and Aging Study (COAST) is a nationwide prospective cohort study on dementia in China (Study ID Number: SYXWJ001). In this cross‐sectional study based on the COAST, we recruited AD patients who met the enrollment criteria from January 2021 to December 2023. Patients diagnosed with AD, according to the National Institute of Aging and Alzheimer's Association (NIA‐AA) criteria [15], were consecutively enrolled from the Innovation Center for Neurological Disorders, Department of Neurology, Xuanwu Hospital, Capital Medical University. A total of 112 cognitively normal (CN) individuals were identified from subjects who had undergone routine medical assessment in the hospital. Another 131 patients with non‐AD dementia (including frontotemporal dementia, Parkinson's disease dementia, and Lewy Body dementia) were also enrolled from the inpatients of Xuanwu Hospital, Capital Medical University. The CN participants and the patients with non‐AD dementia (non‐ADD) were age‐matched with the AD patients (Figure 1).
FIGURE 1.
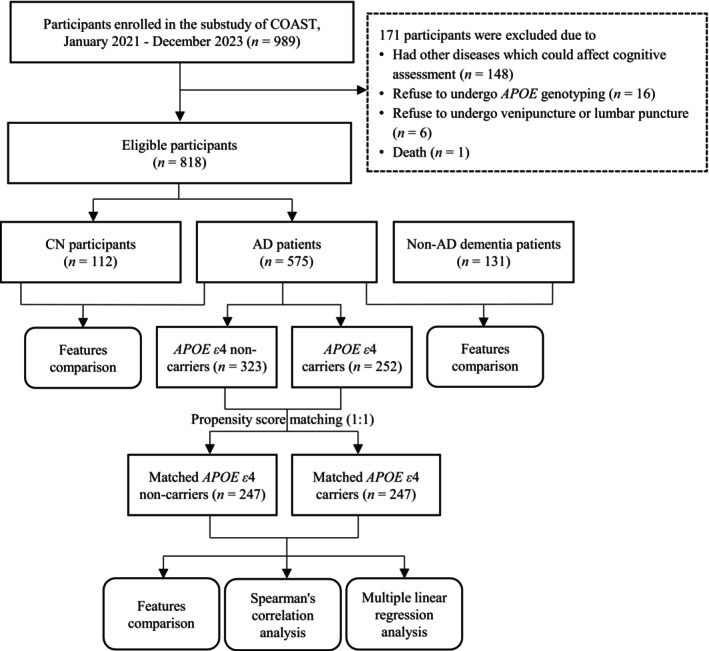
Study design. The patients with non‐AD dementia included patients with frontotemporal dementia, Parkinson's Disease dementia, and Lewy Body dementia. The other diseases which could affect cognitive assessment included corticobasal degeneration, multiple sclerosis, substance abuse, severe systematic diseases, active infections, chronic wasting disease, autoimmune diseases, hematological system diseases, newly onset cardiovascular diseases, stroke, and traumatic brain injury. In AD patients, the APOE ε4 carriers were matched with the non‐carriers by age, sex, and educational level. AD, Alzheimer's disease; APOE, apolipoprotein E; CN, cognitively normal; COAST, China Cognition and Aging Study.
The protocol of this study was approved by the Ethics Committee of Xuanwu Hospital, Capital Medical University, and followed the tenets of the Helsinki Declaration. Written or oral informed consent was obtained from the participants or their legal guardians prior to the study procedures.
In this study, we defined the inclusion criteria as participants who (1) had complete basic demographic data (including age and sex); (2) underwent both one blood sample assessment and one CSF sample assessment from January 2021 to December 2023; (3) underwent APOE genotyping at baseline. The exclusion criteria of this study were defined as participants who (1) had corticobasal degeneration, multiple sclerosis, or substance abuse; (2) had severe systematic diseases, active infections, chronic wasting disease, autoimmune diseases, hematological system diseases, or cancer; (3) had newly onset cardiovascular diseases or stroke; (4) had traumatic brain injury recently; (5) refuse to undergo venipuncture for blood sample assessment or lumbar puncture for CSF collection; (6) refuse to sign the informed consent.
2.2. Collection of Demographic Information
All of the eligible participants underwent collection of demographic information, including age, sex, education level, body mass index (BMI), history of drinking, smoking, hypertension, hyperlipidemia, and diabetes mellitus. The following information was self‐reported by the participants or the legal guardians of the patients, including education level, history of drinking, smoking, hypertension, hyperlipidemia, and diabetes mellitus.
2.3. Assessments of Cognitive Function
The global cognitive function of the participants was assessed by the Mini‐Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MoCA) scales [16, 17]. Besides meeting other inclusion and exclusion criteria, the MMSE scores of the cognitively normal group should also fall within the normal range: MMSE > 22 points for illiteracy, > 23 points for those with primary school education, > 24 points for those with middle school education, > 26 points for those with college education or above [16, 18, 19]. The neurology physician who performed the cognitive test and gave AD diagnosis to the patients was blinded to the results of blood, CSF sample, and APOE alleles testing.
2.4. Blood Sample Assessment
All individuals had undergone routine blood testing. Fasting plasma samples were collected before 8 a.m. by venipuncture in heparin‐lithium anticoagulant tubes. After centrifugation, serum was then separated and stored at −80°C until assayed. Utilizing standard assays, the plasma samples were divided into aliquots and placed in polypropylene tubes for biochemical analyses. In this study, we focused on four variables in the biochemical blood test, including UA, high‐density lipoprotein (HDL), low‐density lipoprotein (LDL), and triglycerides (TG). Nonfasting blood samples were collected with 5 mL serum‐separating vacutainer tubes and centrifuged at 1300 g for 10 min to separate the serum for immunological test. The serum‐derived molecules which underwent further analysis included Immunoglobulin A (IgA) and complement 3 (C3).
2.5. CSF Sample Assessment
Samples of CSF from all the eligible participants were obtained by lumbar puncture in the L3/4 or L4/5 interspace without reported serious adverse effects, collected in polypropylene tubes, centrifuged, and stored frozen at −80°C until analysis, according to standard operating procedures. The concentrations of the biomarkers in CSF were measured with the use of enzyme‐linked immunosorbent assay kits according to the manufacturers' instructions. The kits used for the measurement of AD biomarkers in CSF were INNOTEST β‐AMYLOID (1–40) for Aβ40, INNOTEST β‐AMYLOID (1–42) for Aβ42, INNOTEST hTAU Ag for total tau protein (t‐tau), and INNOTEST PHOSPHO‐TAU (181P) for phosphorylated tau 181 (p‐tau 181), all FUJIREBIO. All biomarker results were required to adhere to the quality‐control standards, which included ensuring that biomarker concentrations fell within the specified assay ranges of the respective kits and maintaining consistent measurement uniformity across all plates by using a validation control included on each plate. Furthermore, to protect the confidentiality of the participants and to minimize the selection bias, the laboratory technicians who conducted the CSF sample analyses were blinded to the diagnosis and other clinical information related to this study of the participants.
2.6. APOE Alleles Testing
Genotyping for APOE (gene map locus 19q13.2) was conducted utilizing allelic discrimination technology (TaqMan; Applied Biosystems) or equivalent methods. Genotypes for the two single‐nucleotide polymorphisms (rs429358 C/T and rs7412 C/T), which define APOE ε2, ε3, and ε4, were determined using real‐time fluorescence quantitative polymerase chain reaction with nucleic acid detection reagents. The APOE ε4 carriers (APOE ε4+ group) were defined as participants who carried the APOE ε4/ ε4, APOE ε3/ ε4, or APOE ε2/ ε4, while the APOE ε4 non‐carriers (APOE ε4− group) were defined as the ones who carried the APOE ε2/ ε2, APOE ε2/ ε3 or APOE ε3/ ε3.
2.7. Statistical Analysis
For the continuous numerical variables, outliers of each variable were defined as with z‐scores over three and were excluded from the following analysis [20]. In AD patients, propensity score matching (PSM) was conducted to match the APOE ε4 carriers with the non‐carriers by age, sex, and education level. Demographic variables, cognitive function, the levels of AD biomarkers in CSF, and the blood‐derived biomolecules between the AD patients and the CN participants, between the patients with non‐AD dementia and the CN participants, as well as between the matched APOE ε4 carriers and non‐carriers of AD patients were compared. Kolmogorov–Smirnov test was used to test for normal distribution. Continuous variables conforming to normal distribution were presented as means (SD) and compared by a two‐tailed t‐test, while non‐normal distributed continuous variables were presented as median (interquartile range) and compared by nonparametric test. Categorical variables were presented as number (percentage) and compared by Chi‐Squared test (χ 2 test). Statistical significance was defined as a two‐sided p < 0.05. The correlations of blood‐derived biomolecules and AD biomarkers in CSF were assessed using Spearman's correlation analysis and visually represented in heat maps. Quantile regression analyses were further conducted to validate the differences of correlations between blood‐derived biomolecules and CSF biomarkers in APOE ε4 subgroups. The models were all constructed at 0.50 quantile and adjusted by confounding factors including age, sex, education level, and disease duration. To account for multiple testing, two‐sided p‐values were adjusted according to the method of Benjamini/Hochberg (B/H) to control the false discovery rate. An association was considered to be statistically significant, if its corresponding B/H‐adjusted p‐value was below 0.05, corresponding to an FDR of 5%. Statistical analyses were conducted using the R software, version 4.3.2 (R Foundation for Statistical Computing, Vienna, Austria). We used the following R packages: “MatchIt” for PSM; “corrplot” and “ggplot2” for plotting of correlation heatmaps; “quantreg” for quantile regression analysis.
3. Results
In total, 989 individuals were screened for eligibility, during which 148 patients with other diseases which could affect cognitive assessment and one dead patient were excluded. Participants who refused to undergo APOE genotyping (n = 16) or venipuncture and lumbar puncture (n = 6) were also excluded. A total of 818 individuals (575 AD patients, 131 patients with non‐AD dementia, and 112 CN participants) aged between 50 and 87 years old were eligible for this study (Figure 1). The median age was 64 years old, and 457 (55.9%) participants were females. Of all the participants included in this study, 342 (41.8%) cases carried APOE ε4 allele. Among the 575 AD patients enrolled in this study, 252 cases (43.8%) were APOE ε4 carriers (Table 1). The number and the proportion of missing data and outliers (with z‐scores over three) for each variable used for further analysis are shown in Table S1.
TABLE 1.
Demographic and clinical features of CN participants, AD and non‐ADD patients.
| Characteristics | CN | AD | Non‐ADD | p (AD vs. CN) | p (non‐AD vs. CN) |
|---|---|---|---|---|---|
| Participants, n | 112 | 575 | 131 | ||
| Female, n (%) | 58 (51.8) | 344 (59.8) | 76 (58.0) | 0.162 | 0.162 |
| Age (years), median (IQR) | 64.50 (60.75, 68.25) | 64.00 (58.00, 70.00) | 64.00 (59.00, 69.00) | 0.916 | 0.916 |
| APOE ε4 carriers, n (%) | 41 (36.6) | 252 (43.8) | 49 (37.4) | 0.382 | 0.999 |
| Education level, n (%) | |||||
| Illiteracy | 0 (0.0) | 20 (3.7) | 2 (1.8) | 0.029* | 0.098 |
| Primary school | 9 (8.5) | 75 (13.9) | 19 (17.0) | ||
| Junior high school | 26 (24.5) | 133 (24.7) | 24 (21.4) | ||
| Senior high school | 30 (28.3) | 152 (28.3) | 40 (35.7) | ||
| Bachelor's degree | 39 (36.8) | 146 (27.1) | 27 (24.1) | ||
| Master's degree and above | 2 (1.9) | 12 (2.3) | 0 (0.0) | ||
| BMI (kg/m2), median (IQR) | 24.22 (22.49, 27.09) | 23.44 (21.39, 25.35) | 23.81 (22.03, 25.77) | 0.002** | 0.254 |
| Smoking, n (%) | 24 (21.4) | 116 (20.2) | 36 (27.5) | 0.862 | 0.694 |
| Drinking, n (%) | 24 (21.4) | 99 (17.2) | 36 (27.5) | 0.353 | 0.353 |
| History | |||||
| Hypertension, n (%) | 47 (42.0) | 197 (34.4) | 51 (38.9) | 0.308 | 0.727 |
| Diabetes mellitus, n (%) | 21 (18.8) | 100 (17.5) | 20 (15.3) | 0.852 | 0.852 |
| Hyperlipidemia, n (%) | 15 (13.4) | 60 (10.4) | 14 (16.5) | 0.689 | 0.689 |
| Cognitive function | |||||
| MMSE, median (IQR) | 27 (25, 28) | 18 (12, 23) | 19 (13, 23) | < 0.001*** | < 0.001*** |
| MoCA, median (IQR) | 22 (20, 25) | 13 (8, 18) | 12 (7, 17) | < 0.001*** | < 0.001*** |
Note: Data were presented as number (percentage), mean ± SD, or median (IQR). All p values were adjusted by Benjamini/Hochberg (B/H) method.
Abbreviations: AD, Alzheimer's disease; BMI, body mass index; CN, cognitively normal; IQR, interquartile range; MMSE, mini‐mental State Examination; MoCA, Montreal Cognitive Assessment; non‐ADD, non‐AD dementia; SD, standard deviation.
B/H adjusted p < 0.05.
B/H adjusted p < 0.01.
B/H adjusted p < 0.001.
3.1. Differences of Clinical Features in CN, AD, and Non‐ADD Groups
The demographic characteristics and levels of CSF biomarkers and blood molecules from the groups of CN, AD, and non‐ADD are exhibited in Table 1 and Figure 2. AD patients were more likely to have a lower BMI, when compared with CN participants (p = 0.001). AD patients demonstrated significantly lower scores in cognitive assessments (MMSE and MoCA) (p < 0.001) in comparison to the CN group (Table 1). The levels of CSF Aβ42 and plasma UA from the AD patients were significantly lower than that from the CN group (p < 0.001; p = 0.016). The levels of CSF t‐tau and p‐tau 181 levels in CSF, as well as plasma HDL and LDL from the AD patients were significantly higher than that from the CN group (p < 0.001; p < 0.001; p < 0.001; p = 0.004) (Table 1, Figure 2). However, patients with non‐AD dementia only demonstrated significantly lower scores in cognitive assessments (MMSE and MoCA) (p < 0.001) in comparison to the CN group. The levels of CSF biomarkers and blood‐derived molecules did not exhibit differences with statistical significance between CN and non‐ADD groups (Table 1).
FIGURE 2.
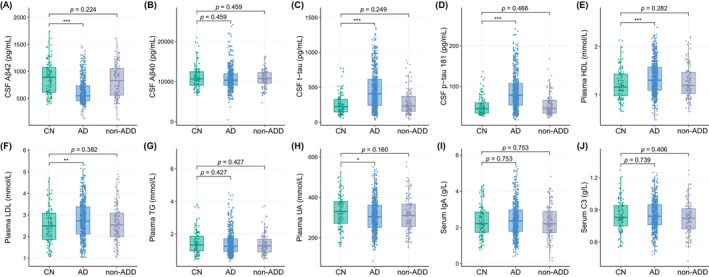
Differences of CSF biomarkers and blood biomolecules between the groups of CN, AD, and non‐ADD. All p values were adjusted by Benjamini/Hochberg (B/H) method. Aβ, amyloid‐beta; AD, Alzheimer's disease; CN, cognitively normal; CSF, cerebrospinal fluid; C3, complement C3; HDL, high‐density lipoprotein; IgA, Immunoglobulin A; LDL, low‐density lipoprotein; non‐ADD, non‐AD dementia; p‐tau, phosphorylated tau; TG, triglycerides; t‐tau, total tau; UA, uric acid. *B/H adjusted p < 0.05, **B/H adjusted p < 0.01, ***B/H adjusted p < 0.01.
3.2. Differences of Clinical Features in APOE ε4+ and APOE ε4− AD Patients
In AD patients, 247 APOE ε4 non‐carriers were successfully matched with APOE ε4 carriers in a ratio of 1:1, by PSM adjusted for age, sex, and education level. In Table 2, we summarized the demographic characteristics and levels of fluid biomarkers from the matched APOE ε4 carriers and non‐carriers. Demographic information, including age, sex, AOO, disease duration, educational level, smoking, drinking, BMI, etc., demonstrated no significant difference between the two APOE subgroups. The results of cognitive function assessments (including MMSE and MoCA), and levels of some blood‐derived biomolecules (including HDL, TG, UA, IgA, and C3) indicated no significant difference between the two groups (Table 2). The levels of plasma LDL, CSF t‐tau, and p‐tau 181 from the APOE ε4 carrier group were significantly higher than that from the non‐carrier group in AD patients (p = 0.015, p = 0.001, p = 0.001) (Table 2, Figure 3).
TABLE 2.
Demographic and clinical features of matched APOE ε4− and APOE ε4+ AD patients.
| Characteristics | All matched AD patients | Matched APOE ε4− AD patients | Matched APOE ε4+ AD patients | p |
|---|---|---|---|---|
| Patients, n | 494 | 247 | 247 | |
| Female, n (%) | 307 (62.1) | 151 (61.1) | 156 (63.2) | 0.711 |
| Age (years), median (IQR) | 65 (58, 70) | 65 (58, 70) | 66 (59, 71) | 0.490 |
| AOO (years), median (IQR) | 62 (55, 68) | 62 (55, 67) | 63 (55, 68) | 0.511 |
| Disease duration (years), median (IQR) | 2 (1, 4) | 2 (1, 3) | 2 (1, 4) | 0.341 |
| Education level, n (%) | ||||
| Illiteracy | 17 (3.7) | 9 (3.9) | 8 (3.5) | 0.267 |
| Primary school | 67 (14.5) | 32 (13.7) | 35 (15.2) | |
| Junior high school | 118 (25.5) | 51 (21.9) | 67 (29.1) | |
| Senior high school | 125 (27.0) | 70 (30.0) | 55 (23.9) | |
| Bachelor's degree | 125 (27.0) | 63 (27.0) | 62 (27.0) | |
| Master's degree and above | 11 (2.3) | 8 (3.5) | 3 (1.3) | |
| BMI (kg/m2), mean ± SD | 23.52 ± 3.14 | 23.46 ± 2.96 | 23.57 ± 3.31 | 0.692 |
| Smoking, n (%) | 95 (19.2) | 48 (19.4) | 47 (19.0) | 0.999 |
| Drinking, n (%) | 81 (16.4) | 43 (17.4) | 38 (15.4) | 0.627 |
| History | ||||
| Hypertension, n (%) | 167 (33.9) | 78 (31.6) | 89 (36.3) | 0.309 |
| Diabetes mellitus, n (%) | 88 (17.9) | 50 (20.2) | 38 (15.6) | 0.218 |
| Hyperlipidemia, n (%) | 54 (10.9) | 33 (13.4) | 21 (8.5) | 0.113 |
| Cognitive function | ||||
| MMSE, median (IQR) | 18 (12, 23) | 18 (12, 24) | 19 (13, 23) | 0.698 |
| MoCA, median (IQR) | 13 (8, 18) | 13 (8, 19) | 13 (8, 18) | 0.904 |
Note: Data were presented as number (percentage), mean ± SD, or median (IQR). APOE ε4−, APOE ε4 non ‐ carriers; APOE ε4+, APOE ε4 carriers.
Abbreviations: AD, Alzheimer's disease; AOO, age of onset; APOE, apolipoprotein E; BMI, body mass index; IQR, interquartile range; MMSE, Mini‐mental State Examination; MoCA, Montreal Cognitive Assessment; SD, standard deviation.
FIGURE 3.
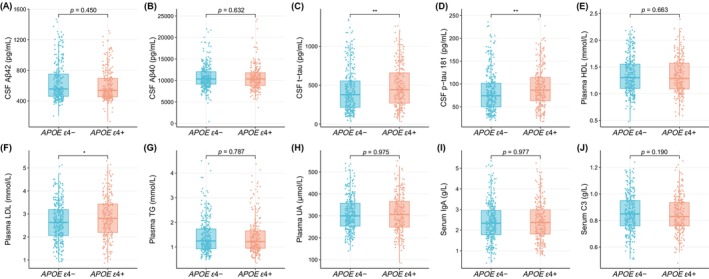
Differences of CSF biomarkers and blood biomolecules between the APOE ε4‐ and APOE ε4+ groups in AD patients. Aβ, amyloid‐beta; AD, Alzheimer's disease; APOE, apolipoprotein E; CSF, cerebrospinal fluid; C3, complement C3; HDL, high‐density lipoprotein; IgA, Immunoglobulin A; LDL, low‐density lipoprotein; p‐tau, phosphorylated tau; TG, triglycerides; t‐tau, total tau; UA, uric acid. *p < 0.05, **p < 0.01.
3.3. Correlation Analysis
Among the 494 AD patients selected by PSM, correlations between the blood‐derived biomolecules with AD biomarkers in CSF are presented in Figure 4 and Table S2, as are the results from the APOE ε4 subgroups (Figure 4; Table S3). The negative correlations between plasma LDL and CSF Aβ42 levels (r range: −0.163 to −0.109), between plasma TG and CSF t‐tau levels (r range: −0.235 to −0.184), between plasma UA and CSF t‐tau levels (r range: −0.190 to −0.152), between serum C3 and CSF t‐tau levels (r range: −0.182 to −0.116), and between plasma TG and CSF p‐tau 181 levels (r range: −0.195 to −0.172), as well as the positive correlation between plasma HDL and CSF t‐tau levels (r range: 0.169 to 0.193) were observed significant (B/H adjusted p < 0.05) in total AD and APOE ε4+ AD groups, but not in APOE ε4− AD group (Figure 4; Table S2, 3). The level of serum IgA was positively and significantly correlated with CSF Aβ42 level (r = 0.211, B/H adjusted p = 0.030) only in APOE ε4+ AD group (Figure 4; Table S3). The level of plasma HDL was positively and significantly correlated with CSF p‐tau 181 level in total AD and APOE ε4− AD groups (r range: 0.169 to 0.231, B/H adjusted p < 0.05), but not in APOE ε4+ AD group (Figure 4; Table S2, 3). The positive correlation between plasma LDL and CSF p‐tau 181 levels (r = 0.143), as well as the negative correlation between serum C3 and CSF p‐tau 181 levels (r = −0.130) were observed significant (B/H adjusted p < 0.05) only in total AD group (Figure 4; Table S2).
FIGURE 4.
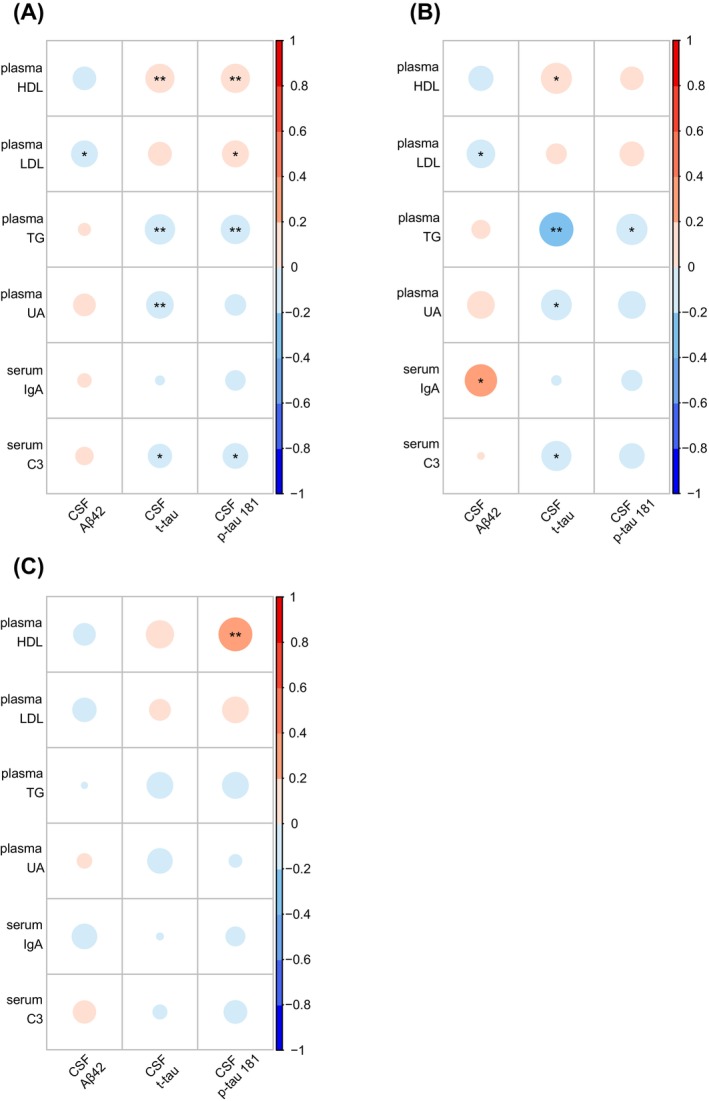
Heat maps of correlations between blood‐derived biomolecules and CSF biomarkers. Shown are the correlations between blood‐derived biomolecules and CSF biomarkers in all matched AD patients (A), APOE ε4+ AD patients (B), and APOE ε4− AD patients (C). All p values were adjusted by Benjamini/Hochberg (B/H) method. Aβ, amyloid‐beta; AD, Alzheimer's disease; APOE, apolipoprotein E; CSF, cerebrospinal fluid; C3, complement C3; HDL, high‐density lipoprotein; IgA, Immunoglobulin A; LDL, low‐density lipoprotein; p‐tau, phosphorylated tau; TG, triglycerides; t‐tau, total tau; UA, uric acid. *B/H adjusted p < 0.05, **B/H adjusted p < 0.01, ***B/H adjusted p < 0.001.
3.4. Quantile Regression Analysis
Differences of associations between the blood‐derived biomolecules with CSF biomarkers in matched APOE subgroups were further validated by quantile regression analysis (at 50 quantile) and adjusted for possible confounding factors, including age, sex, education level, and disease duration (Figures 5 and S1). In the APOE ε4+ AD group, the level of serum IgA significantly and positively predicted CSF Aβ42 level (β = 43.75, B/H adjusted p = 0.011), the level of plasma TG significantly and negatively predicted CSF t‐tau level (β = −101.83, B/H adjusted p < 0.001) and CSF p‐tau 181 level (β = −14.76, B/H adjusted p < 0.001) (Figure 5A–C). In the APOE ε4− AD group, the level of plasma HDL significantly and positively predicted CSF p‐tau 181 level (β = 26.95, B/H adjusted p = 0.049), and the levels of plasma TG significantly and negatively predicted CSF p‐tau 181 level (β = −9.68, B/H adjusted p = 0.026) (Figure 5D,E). In total AD patients, the levels of plasma TG significantly and negatively predicted CSF t‐tau level (β = −62.68, B/H adjusted p = 0.006) and CSF p‐tau 181 level (β = −9.06, B/H adjusted p = 0.001). The level of plasma HDL significantly and negatively predicted CSF p‐tau 181 level (β = 21.94, B/H adjusted p = 0.008) (Figure S1).
FIGURE 5.
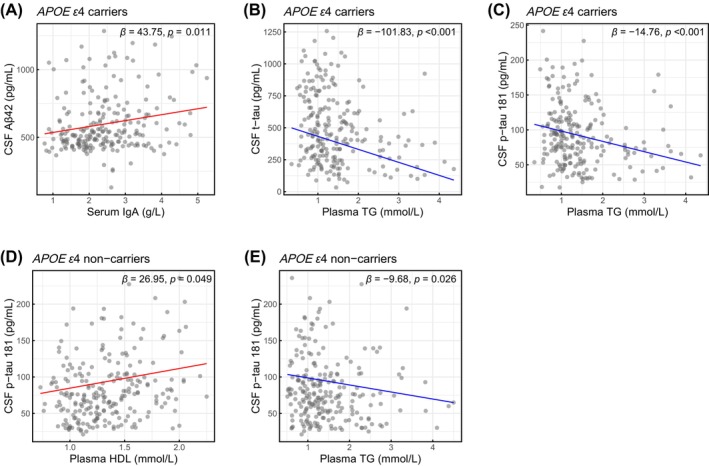
Associations between blood‐derived molecules and CSF biomarkers in APOE subgroups of AD patients. Utilizing the quantile regression method (at 0.50 quantile), shown are the relationships between serum IgA and CSF Aβ42 levels in APOE ε4 carriers (A), between plasma TG and CSF t‐tau levels in APOE ε4 carriers (B), between plasma TG and CSF p‐tau 181 levels in APOE ε4 carriers (C), between plasma HDL and CSF p‐tau 181 levels in APOE ε4 non‐carriers (D), and between plasma TG and CSF p‐tau 181 levels in APOE ε4 non‐carriers (E). All models were adjusted for age, sex, education level, and disease duration. All p values were adjusted by Benjamini/Hochberg (B/H) method. Aβ, amyloid‐beta; AD, Alzheimer's disease; APOE, apolipoprotein E; CSF, cerebrospinal fluid; HDL, high‐density lipoprotein; IgA, Immunoglobulin A; p‐tau, phosphorylated tau; TG, triglycerides; t‐tau, total tau.
4. Discussion
Polymorphism in the APOE gene is recognized as a major genetic risk factor for late‐onset AD (LOAD), with the APOE ε4 allele conferring an increased risk [4, 5]. However, the exact mechanisms by which APOE ε4 contributes to AD pathology are intricate and not completely understood. Moreover, further investigation is required to elucidate the relationships between APOE ε4, CSF biomarkers, and blood‐derived molecules. In this study, APOE ε4 carriers in AD patients exhibited higher levels of CSF t‐tau and p‐tau 181, indicating a more progressive type of disease than APOE ε4 non‐carriers. In the correlation analysis, the blood‐derived biomolecules associated with CSF biomarkers differed between APOE ε4 carriers and non‐carriers among AD patients, suggesting distinct roles of APOE genotypes in AD pathology.
Our findings revealed variations in fluid biomarker levels from the two APOE subgroups, which indicated the different roles of APOE genotypes in the progression of Alzheimer's disease. In AD participants, the increased levels of t‐tau and p‐tau 181 in CSF from APOE ε4 carriers aligned with previous studies, which suggested that APOE ε4 may be associated with tau pathology and thus leading to more progressive type of AD [21, 22]. We further conducted correlation and regression analyses to reveal the potential mechanisms by which APOE ε4 may influence AD pathology.
Cholesterol plays an indispensable role in the central nervous system, serving as a crucial component essential for axonal growth, synaptogenesis, and synaptic plasticity, which are fundamental to learning, memory formation, and neuronal repair [23]. Up until now, the evidence for the involvement of lipids and lipoproteins in the development of dementia remains inconclusive. Studies have reported that dyslipidemia is supposed to be one of the risk factors of AD [24, 25]. However, the relationships between plasma LDL, HDL, and AD remain controversial [26, 27, 28, 29, 30, 31, 32, 33]. In our study, the elevated plasma LDL and HDL levels in AD patients were consistent with previous findings [26, 27, 30, 33], but not supported by some other studies [28, 29, 31, 32]. Aligned with the previous study which found that a higher level of TG may be a protective factor of dementia development [34], the negative correlation between TG and CSF t‐tau and p‐tau 181 in APOE ε4+ AD subgroup indicated that TG may delay the AD progression by protecting neurons from damage and degeneration. According to previous studies, APOE may influence the risk of AD by disrupting the homeostasis of lipid metabolism [35, 36]. Our results showed that the correlations of HDL, LDL, and TG with the CSF biomarkers were differed between the APOE ε4 subgroups, which suggest that APOE ε4 may play a role in the lipid metabolism by interfering with different lipids in AD patients, thus further influence the AD pathology.
Neuro‐inflammation has been proved to be one of the vital mechanisms involved in the progression of AD [37]. As the most abundant complement protein, complement C3 is a reactant during the acute phase and may serve as an indicator of inflammation [38]. A plethora of research data showed that decreased levels of complement C3 may result in impaired immunological responses, consequently leading to reduced clearance of plaques in the AD‐prone brain, thus offering a plausible explanation of the phenomenon [11, 39, 40]. In the correlation analyses, serum complement C3 level was negatively related to CSF t‐tau protein in APOE ε4+ AD group, which may be supported by a proposed theory suggesting that APOE ε4 may have effects on the involvement of complement C3 in the amplification of pathological processes in the AD brain [11]. Bonham et al. also reported a conceptual model of the AD pathogenic cascade where a synergistic relationship between complement C3 and APOE ε4 results in advanced Alzheimer's associated pathology [41]. IgA is the most prominent immunoglobulin isotype found on mucosal surfaces, such as saliva, tears, and respiratory secretions [42]. According to the results reported by Pocevičiūtė et al. [10], the normal IgA response to AD‐related inflammatory events may be disturbed in APOE ε4 carriers. In the correlation and regression analyses, the positive relationship between IgA and CSF Aβ42 was significant only in APOE ε4+ AD group, which indicates that APOE ε4 may play a role in the inflammatory response related to IgA in AD patients. Further investigations are warranted to elucidate the precise mechanism underlying the influence of APOE ε4 on neuro‐inflammation in AD pathology.
Recognized as a systemic antioxidant, UA is believed to exert protective effects against neurodegeneration by scavenging reactive oxygen species [43]. In our study, AD patients exhibited a lower level of plasma UA than CN participants, which was consistent with previous studies [13, 44]. In our study, plasma UA was positively and significantly correlated with CSF t‐tau among APOE ε4+ AD patients in Spearman's correlation analysis, while this relationship was not validated by quantile regression analysis. These findings indicate that the effect of APOE ε4 on oxidative stress in AD pathology may be interfered by factors like age and sex, which still warrant further examination and validation.
Up to now, the diagnosis of AD primarily relies on the quantitative assessment of Aβ and tau protein levels in the CSF, or the utilization of neuroimaging modalities such as amyloid and tau protein positron emission tomography (PET), both of which are medically invasive and costly [45, 46]. However, recent investigations have underscored the relevance of early alterations in peripheral blood signatures in the onset of AD, and the role of blood biomarkers in the diagnosis of AD has been receiving more attention [47, 48]. In our study, the levels of plasma HDL, LDL, TG, and serum complement C3 were associated with levels of CSF biomarkers of AD. These assumptions suggested that blood‐derived biomolecules obtain the potential to be novel biomarkers to indicate the progression of AD. Our findings may illuminate future progress in AD pathology and the discovery of candidate blood biomarkers of AD, thus providing a foundation for early detection and therapeutic interventions of AD.
The limitations of this study need to be acknowledged. This study was conducted at a single study center of the hospital and had a limited sample size. This circumstance may introduce inherent selection biases, potentially affecting the reliability and generalizability of the results. Moreover, some of the data used for analysis are self‐reported by the patients (including education level, history of drinking, smoking, hypertension, statin therapy, and diabetes mellitus), which may introduce recall bias into the study. Another limitation of our study is its observational nature, which prevents us from determining a causative relationship. Further longitudinal studies with larger sample size will be necessary to confirm the associations between APOE ε4 and fluid biomarkers of AD, as well as to explore the underlying mechanism of APOE ε4 in AD pathology.
5. Conclusions
Our study revealed that APOE ε4 was associated with accelerated progression of AD pathology, manifesting as increased t‐tau and p‐tau 181 levels in CSF of AD patients. According to the correlation analysis between blood‐derived molecules and CSF biomarkers, APOE ε4 may contribute to the pathogenesis of AD by disrupting lipid metabolism and neuroinflammation. These findings suggested that APOE ε4 may play a crucial role in the pathogenesis of AD, and provided valuable insights for the exploration of novel biomarkers and the development of individualized therapeutic approaches.
Author Contributions
W.W. designed and supervised the study. P.Z., Q.W., and D.G. recruited patients. J.J. collected samples for COAST dataset in this study. B.Z. and W.W. performed the statistical analysis. M.Q. wrote the first draft of the manuscript. All authors contributed to the interpretation of data and revision of the manuscript. W.W. and M.Q. obtained funding for the study.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Appendix S1.
Acknowledgments
We acknowledge the dedication of the participants and their families, without whom the study would not be possible to be conducted. We additionally thank all of the participating researchers in the COAST. We acknowledge the altruism of the participants and their families and contributions of the COAST and support staff at Xuanwu Hospital for their contributions to this study.
Bote Zhao and Peixi Zang contributed equally to the work.
Funding: This study is supported by grants from National Key R&D Plan of the Ministry of Science and Technology (2022YFC3501404), National Natural Science Foundation of China (82271452), the Youth Program of National Natural Science Foundation of China (82101503), and Beijing Natural Science Foundation (L232136).
Contributor Information
Jianping Jia, Email: jjp@ccmu.edu.cn.
Wei Wang, Email: wangwei76@xwhosp.org.
Data Availability Statement
Both raw and processed data of the COAST dataset that support the findings of the current study will be made available upon request to the corresponding author and the COAST committee to ensure that the privacy of the participants is protected. All analyses were conducted in R, version 4.3.2. Code used to generate the results presented in the manuscript are available from the corresponding author upon request.
References
- 1. Scheltens P., De Strooper B., Kivipelto M., et al., “Alzheimer's Disease,” Lancet 397, no. 10284 (2021): 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lim Y. Y. and Mormino E. C., “APOE Genotype and Early β‐Amyloid Accumulation in Older Adults Without Dementia,” Neurology 89, no. 10 (2017): 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. For the Alzheimer's Disease Neuroimaging I , Toledo J. B., Habes M., et al., “APOE Effect on Amyloid‐β PET Spatial Distribution, Deposition Rate, and Cut‐Points,” Journal of Alzheimer's Disease 69, no. 3 (2019): 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamazaki Y., Zhao N., Caulfield T. R., Liu C.‐C., and Bu G., “Apolipoprotein E and Alzheimer Disease: Pathobiology and Targeting Strategies,” Nature Reviews Neurology 15, no. 9 (2019): 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saunders A. M., Strittmatter W. J., Schmechel D., et al., “Association of Apolipoprotein E Allele ϵ4 With Late‐Onset Familial and Sporadic Alzheimer's Disease,” Neurology 43, no. 8 (1993): 1467–1472. [DOI] [PubMed] [Google Scholar]
- 6. Quan M., Wang Q., Qin W., et al., “Shared and Unique Effects of ApoEε4 and Pathogenic Gene Mutation on Cognition and Imaging in Preclinical Familial Alzheimer's Disease,” Alzheimer's Research & Therapy 15, no. 1 (2023): 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dong L., Mao C., Liu C., et al., “Association Between Common Variants of APOE, ABCA7, A2M, BACE1, and Cerebrospinal Fluid Biomarkers in Alzheimer's Disease: Data From the PUMCH Dementia Cohort,” Journal of Alzheimer's Disease 85, no. 4 (2022): 1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giannisis A., Al‐Grety A., Carlsson H., et al., “Plasma Apolipoprotein E Levels, Isoform Composition, and Dimer Profile in Relation to Plasma Lipids in Racially Diverse Patients With Alzheimer's Disease and Mild Cognitive Impairment,” Alzheimer's Research & Therapy 15, no. 1 (2023): 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koch M., DeKosky S. T., Goodman M., et al., “Association of Apolipoprotein E in Lipoprotein Subspecies With Risk of Dementia,” JAMA Network Open 3, no. 7 (2020): e209250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pocevičiūtė D., Nuñez‐Diaz C., Roth B., et al., “Increased Plasma and Brain Immunoglobulin A in Alzheimer's Disease Is Lost in Apolipoprotein E ε4 Carriers,” Alzheimer's Research & Therapy 14, no. 1 (2022): 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rasmussen K. L., Nordestgaard B. G., Frikke‐Schmidt R., and Nielsen S. F., “An Updated Alzheimer Hypothesis: Complement C3 and Risk of Alzheimer's Disease‐A Cohort Study of 95,442 Individuals,” Alzheimers Dement 14, no. 12 (2018): 1589–1601. [DOI] [PubMed] [Google Scholar]
- 12. Royall D. R., Al‐Rubaye S., Bishnoi R., and Palmer R. F., “Few Serum Proteins Mediate APOE's Association With Dementia,” PLoS One 12, no. 3 (2017): e0172268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu N., Dubreuil M., Zhang Y., et al., “Gout and the Risk of Alzheimer's Disease: A Population‐Based BMI‐Matched Cohort Study,” Annals of the Rheumatic Diseases 75, no. 3 (2016): 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Teunissen C. E., Verberk I. M. W., Thijssen E. H., et al., “Blood‐Based Biomarkers for Alzheimer's Disease: Towards Clinical Implementation,” Lancet Neurology 21, no. 1 (2022): 66–77. [DOI] [PubMed] [Google Scholar]
- 15. McKhann G. M., Knopman D. S., Chertkow H., et al., “The Diagnosis of Dementia due to Alzheimer's Disease: Recommendations From the National Institute on Aging‐Alzheimer's Association Workgroups on Diagnostic Guidelines for Alzheimer's Disease,” Alzheimer's & Dementia 7, no. 3 (2011): 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Folstein M. F., Folstein S. E., and McHugh P. R., “"Mini‐Mental State" A Practical Method for Grading the Cognitive state of Patients for the Clinician,” Journal of Psychiatric Research 12, no. 3 (1975): 189–198. [DOI] [PubMed] [Google Scholar]
- 17. Nasreddine Z. S., Phillips N. A., Bédirian V., et al., “The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool for Mild Cognitive Impairment,” Journal of the American Geriatrics Society 53, no. 4 (2005): 695–699. [DOI] [PubMed] [Google Scholar]
- 18. Spering C. C., Hobson V., Lucas J. A., Menon C. V., Hall J. R., and O'Bryant S. E., “Diagnostic Accuracy of the MMSE in Detecting Probable and Possible Alzheimer's Disease in Ethnically Diverse Highly Educated Individuals: An Analysis of the NACC Database,” Journals of Gerontology. Series A, Biological Sciences and Medical Sciences 67, no. 8 (2012): 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Bryant S. E., Humphreys J. D., Smith G. E., et al., “Detecting Dementia With the Mini‐Mental State Examination in Highly Educated Individuals,” Archives of Neurology 65, no. 7 (2008): 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hodge V. and Austin J., “A Survey of Outlier Detection Methodologies,” Artificial Intelligence Review 22, no. 2 (2004): 85–126. [Google Scholar]
- 21. Farfel J. M., Yu L., De Jager P. L., Schneider J. A., and Bennett D. A., “Association of APOE With Tau‐Tangle Pathology With and Without β‐Amyloid,” Neurobiology of Aging 37 (2016): 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bejanin A., Iulita M. F., Vilaplana E., et al., “Association of Apolipoprotein E ɛ4 Allele With Clinical and Multimodal Biomarker Changes of Alzheimer Disease in Adults With Down Syndrome,” JAMA Neurology 78, no. 8 (2021): 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mauch D. H., Nägler K., Schumacher S., et al., “CNS Synaptogenesis Promoted by Glia‐Derived Cholesterol,” Science 294, no. 5545 (2001): 1354–1357. [DOI] [PubMed] [Google Scholar]
- 24. Silva M. V. F., Loures C. D. M. G., Alves L. C. V., De Souza L. C., Borges K. B. G., and Carvalho M. D. G., “Alzheimer's Disease: Risk Factors and Potentially Protective Measures,” Journal of Biomedical Science 26, no. 1 (2019): 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. European As, Dementia Biobank Mendelian Randomization C , Luo J., et al., “Genetic Associations Between Modifiable Risk Factors and Alzheimer Disease,” JAMA Network Open 6, no. 5 (2023): e2313734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wingo T. S., Cutler D. J., Wingo A. P., et al., “Association of Early‐Onset Alzheimer Disease With Elevated Low‐Density Lipoprotein Cholesterol Levels and Rare Genetic Coding Variants of APOB,” JAMA Neurology 76, no. 7 (2019): 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee H., Kim K., Lee Y. C., et al., “Associations Between Vascular Risk Factors and Subsequent Alzheimer's Disease in Older Adults,” Alzheimer's Research & Therapy 12, no. 1 (2020): 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ferguson E. L., Zimmerman S. C., Jiang C., et al., “Low‐ and High‐Density Lipoprotein Cholesterol and Dementia Risk Over 17 Years of Follow‐Up Among Members of a Large Health Care Plan,” Neurology 101, no. 21 (2023): e2172–e2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reitz C., Tang M. X., Luchsinger J., and Mayeux R., “Relation of Plasma Lipids to Alzheimer Disease and Vascular Dementia,” Archives of Neurology 61, no. 5 (2004): 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kjeldsen E. W., Thomassen J. Q., Juul Rasmussen I., Nordestgaard B. G., Tybjærg‐Hansen A., and Frikke‐Schmidt R., “Plasma High‐Density Lipoprotein Cholesterol and Risk of Dementia: Observational and Genetic Studies,” Cardiovascular Research 118, no. 5 (2022): 1330–1343. [DOI] [PubMed] [Google Scholar]
- 31. van Velsen E. F., Vernooij M. W., Vrooman H. A., et al., “Brain Cortical Thickness in the General Elderly Population: The Rotterdam Scan Study,” Neuroscience Letters 550 (2013): 189–194. [DOI] [PubMed] [Google Scholar]
- 32. den Heijer T., Hofman A., Koudstaal P. J., and Breteler M. M., “Serum Lipids and Hippocampal Volume: The Link to Alzheimer's Disease?,” Annals of Neurology 57, no. 5 (2005): 779–780. [DOI] [PubMed] [Google Scholar]
- 33. Pedrini S., Doecke J. D., Hone E., et al., “Plasma High‐Density Lipoprotein Cargo Is Altered in Alzheimer's Disease and Is Associated With Regional Brain Volume,” Journal of Neurochemistry 163, no. 1 (2022): 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou Z., Ryan J., Tonkin A. M., et al., “Association Between Triglycerides and Risk of Dementia in Community‐Dwelling Older Adults: A Prospective Cohort Study,” Neurology 101, no. 22 (2023): e2288–e2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sienski G., Narayan P., Bonner J. M., et al., “ APOE4 Disrupts Intracellular Lipid Homeostasis in Human iPSC‐Derived Glia,” Science Translational Medicine 13, no. 583 (2021): eaaz4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haney M. S., Pálovics R., Munson C. N., et al., “ APOE4/4 Is Linked to Damaging Lipid Droplets in Alzheimer's Disease Microglia,” Nature 628, no. 8006 (2024): 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Akiyama H., Barger S., Barnum S., et al., “Inflammation and Alzheimer's Disease,” Neurobiology of Aging 21, no. 3 (2000): 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Veteleanu A., Pape S., Davies K., et al., “Complement Dysregulation and Alzheimer's Disease in Down Syndrome,” Alzheimers Dement 19, no. 4 (2023): 1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maier M., Peng Y., Jiang L., Seabrook T. J., Carroll M. C., and Lemere C. A., “Complement C3 Deficiency Leads to Accelerated Amyloid Beta Plaque Deposition and Neurodegeneration and Modulation of the Microglia/Macrophage Phenotype in Amyloid Precursor Protein Transgenic Mice,” Journal of Neuroscience 28, no. 25 (2008): 6333–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wyss‐Coray T., Yan F., Lin A. H., et al., “Prominent Neurodegeneration and Increased Plaque Formation in Complement‐Inhibited Alzheimer's Mice,” Proceedings of the National Academy of Sciences of the United States of America 99, no. 16 (2002): 10837–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bonham L. W., Desikan R. S., Yokoyama J. S., and For the Alzheimer's Disease Neuroimaging Initiative , “The Relationship Between Complement Factor C3, APOE ε4, Amyloid and Tau in Alzheimer's Disease,” Acta Neuropathologica Communications 4, no. 1 (2016): 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Corthésy B., “Multi‐Faceted Functions of Secretory IgA at Mucosal Surfaces,” Frontiers in Immunology 4 (2013): 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu H., Reynolds G. P., and Wei X., “Uric Acid and High‐Density Lipoprotein Cholesterol Are Differently Associated With Alzheimer's Disease and Vascular Dementia,” Journal of Alzheimer's Disease 73, no. 3 (2020): 1125–1131. [DOI] [PubMed] [Google Scholar]
- 44. Du N., Xu D., Hou X., et al., “Inverse Association Between Serum Uric Acid Levels and Alzheimer's Disease Risk,” Molecular Neurobiology 53, no. 4 (2016): 2594–2599. [DOI] [PubMed] [Google Scholar]
- 45. Abubakar M. B., Sanusi K. O., Ugusman A., et al., “Alzheimer's Disease: An Update and Insights Into Pathophysiology,” Frontiers in Aging Neuroscience 14 (2022): 742408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greenberg B. D., Pettigrew C., Soldan A., et al., “CSF Alzheimer Disease Biomarkers: Time‐Varying Relationships With MCI Symptom Onset and Associations With Age, Sex, and ApoE4,” Neurology 99, no. 15 (2022): e1640–e1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Di Costanzo A., Paris D., Melck D., et al., “Blood Biomarkers Indicate That the Preclinical Stages of Alzheimer's Disease Present Overlapping Molecular Features,” Scientific Reports 10, no. 1 (2020): 15612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Padala S. P. and Newhouse P. A., “Blood‐Based Biomarkers in Alzheimer's Disease: A Mini‐Review,” Metabolic Brain Disease 38, no. 1 (2023): 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Data Availability Statement
Both raw and processed data of the COAST dataset that support the findings of the current study will be made available upon request to the corresponding author and the COAST committee to ensure that the privacy of the participants is protected. All analyses were conducted in R, version 4.3.2. Code used to generate the results presented in the manuscript are available from the corresponding author upon request.