

Abstract
Delandistrogene moxeparvovec is an rAAVrh74 vector-based gene transfer therapy that delivers a transgene encoding delandistrogene moxeparvovec micro-dystrophin, an engineered, functional form of dystrophin shown to stabilize or slow disease progression in DMD. It is approved in the US and in other select countries. Two serious adverse event cases of immune-mediated myositis (IMM) were reported in the phase Ib ENDEAVOR trial (NCT04626674). We hypothesized that immune responses to the micro-dystrophin transgene product may have mediated these IMM events. An interferon-gamma ELISpot assay was used to detect T cell responses to delandistrogene moxeparvovec micro-dystrophin peptide pools. ELISpot analysis suggested that IMM resulted from T cell-mediated responses directed against specific micro-dystrophin peptides corresponding to exons 8 and 9 (Case 1) and exon 8 (Case 2) of the DMD gene. In silico epitope mapping based on the patients’ HLA-I alleles indicated greater probability for peptides derived from exons 8 and/or 9 to bind HLA-I, providing further evidence that peptides derived from corresponding micro-dystrophin regions may have higher immunogenic potential. Collectively, these data suggest that patients with DMD gene deletions involving exons 8 and/or 9 may be at increased risk of IMM following delandistrogene moxeparvovec micro-dystrophin gene therapy infusion.
Keywords: AAV vector, Delandistrogene moxeparvovec, Duchenne muscular dystrophy, Dystrophin, Gene transfer therapy, Immune-mediated myositis
Subject terms: Neuromuscular disease, Gene therapy
Introduction
Duchenne muscular dystrophy (DMD) is a rare, X-linked, progressive neuromuscular disease caused by mutations in the DMD gene that result in the absence of functional dystrophin and continuous muscle damage beginning from birth1–4. Impaired motor function can be observed as early as 18–36 months of age when individuals with DMD do not achieve the developmental milestone of walking, have a waddling gait, or experience frequent falls and difficulty climbing stairs; these symptoms typically progress to loss of ambulation during early adolescence5,6. Delandistrogene moxeparvovec is a single-administration recombinant adeno-associated virus rhesus isolate serotype 74 vector-based gene transfer therapy that delivers a transgene encoding delandistrogene moxeparvovec micro-dystrophin, an engineered, functional form of dystrophin shown to stabilize or slow disease progression in DMD7–9. It is approved in the US and in other select countries10.
The adverse drug reactions identified in clinical trials of delandistrogene moxeparvovec to date include infusion-related reaction, vomiting, nausea, thrombocytopenia, pyrexia, liver injury, and immune-mediated myositis (IMM)9,11–15. With the exception of liver injury and IMM, these events are believed to be caused by an innate immune response to the adeno-associated virus vector capsid component of the construct16,17; liver injury is hypothesized to be caused by a T cell response to the adeno-associated virus capsid16. Two cases of IMM were observed in patients with DMD gene deletion mutations involving exons 3–43 and exons 8–9 approximately 1 month after infusion with delandistrogene moxeparvovec in ENDEAVOR (SRP-9001-103; NCT04626674), an ongoing, open-label, phase Ib, multi-cohort study to evaluate delandistrogene moxeparvovec micro-dystrophin expression and safety in patients with DMD. It was hypothesized that these IMM events may have resulted from a T cell-based response due to lack of self-tolerance to specific region(s) encoded by the transgene10. Similarly, rare instances of IMM have also been observed post-treatment in clinical trials of other investigational DMD gene therapies, which are believed to be caused by immune system reactions to shortened dystrophin in conjunction with the patient’s specific genetic mutation18,19. Understanding the intricacies of the immune response underlying these cases of IMM is paramount in elucidating potential complications associated with gene therapy interventions for DMD. Therefore, to better understand these IMM events, we conducted a clinical and immunologic investigation of the two patients who experienced IMM following delandistrogene moxeparvovec administration in ENDEAVOR.
Results
Overview of the IMM cases: clinical investigation
IMM Case 1 (ENDEAVOR Cohort 2)
The clinical course of IMM experienced by this patient after treatment with delandistrogene moxeparvovec has been described20. Briefly, the patient, a 9-year-old ambulatory male with a deletion of exons 3–43 of the DMD gene, developed IMM 4–5 weeks post-infusion with delandistrogene moxeparvovec. He presented with dysphonia (day 31 post-infusion), upper and lower limb weakness, and dysphagia (day 32). Investigations revealed a creatine kinase level of 16,000 U/L (day 30) with a peak of ~ 28,000 U/L on the day of admission. Other serologic tests (renal function, liver function, complete blood counts, erythrocyte sedimentation rate, B-type natriuretic peptide, troponins, viral studies, and coagulation studies) showed no abnormalities. Cardiac evaluation with electrocardiogram, echocardiogram, and troponin testing was normal. Liver ultrasound showed no abnormalities. The patient had a positive enzyme-linked immunosorbent spot (ELISpot) result against the transgene protein product on day 30. The patient was admitted to the hospital on day 35. A magnetic resonance imaging (MRI) analysis was performed on day 38, for which the patient was intubated and extubated post-procedure to bilevel positive airway pressure. Bilevel positive airway pressure was continued on days 39–52 following insertion of a nasogastric tube for an incident of aspiration. Management included prednisone/methylprednisolone 1.0 mg/kg as part of the gene therapy treatment (days −1 to 112; tapered to 0.5 mg/kg starting on day 112) and six rounds of plasmapheresis (~ days 39–51) were initiated. Immunosuppressive treatment with tacrolimus was started after plasmapheresis on day 52 when muscle biopsy results suggested IMM. The muscle biopsy showed an abundance of CD8- and CD4-positive T cells and CD68-positive macrophages in the endomysium that surrounded and invaded individual myofibers. These inflammatory cells appeared to selectively attack myofibers expressing the rod domain (dystrophin 1) and transgene-derived N-terminus (dystrophin 3) but not those expressing the native C-terminus (dystrophin 2) of the DMD gene20. At discharge (day 55), the patient did not require respiratory support and could walk with slight assistance by day 64 (Fig. 1A). The patient was weaned off tacrolimus by day ~ 980 without re-emergence of any symptoms of IMM.
Fig. 1.

Outcome of the IMM cases. (A) Outcome of IMM Case 1, (B) Outcome of IMM Case 2. *Steroids were administered as part of gene therapy treatment. BID twice daily, BiPAP bilevel positive airway pressure, BNP brain natriuretic peptide, BUN blood urea nitrogen, CK creatine kinase, cMRI cardiac magnetic resonance imaging, CXR chest X-ray, ECG electrocardiogram, ECHO echocardiogram, EDVi end-diastolic volume, ELISpot enzyme-linked immunosorbent spot, ESR erythrocyte sedimentation rate, Hct hematocrit, Hg hemoglobin, IMM immune-mediated myositis, INR international normalized ratio, IVIG intravenous immunoglobulin, LFT liver function test, MRI magnetic resonance imaging, NG nasogastric tube, NSAA North Star Ambulatory Assessment, PTT partial thromboplastin time, Strep Streptococcus pyogenes, WBC white blood cell.
IMM Case 2 (ENDEAVOR Cohort 5a)
The patient, a 7-year-old ambulatory male with a deletion of exons 8–9 of the DMD gene, was enrolled and dosed in Cohort 5a, a cohort specifically designed to better understand the risk of IMM in patients with deletion mutations in exons 1–17, a region overlapping with sequences present in the delandistrogene moxeparvovec micro-dystrophin transgene. Prior to screening, he had a series of multiple common childhood infections requiring antibiotic therapy. At baseline, he had mildly elevated troponin-I levels. Post-infusion, his family indicated that he was well for 2 weeks, apart from nausea and vomiting. Three weeks post-infusion, the family reported instances of falling, increased use of an adaptive chair, and less activity. The patient subsequently developed a fever and sore throat, was diagnosed with a Streptococcus pyogenes infection, and started on cefalexin. At week 4, his family reported unusual fatigue and weakness despite antibiotic treatment. He was admitted to the hospital on day 29 post-infusion due to concerns regarding strength testing on neurologic examination. Investigations performed revealed a positive viral panel, normal chest radiography, normal echocardiogram, and troponin-I levels ~ 3 × the baseline levels. A swallow study performed was normal, overnight oximetry was not concerning, and echocardiogram/electrocardiogram remained reassuring. He was started on high-dose intravenous corticosteroids for 5 days. In addition, he was started on tacrolimus and intravenous immunoglobulin. Muscle biopsy with immunohistochemistry was not performed for this patient. The IMM was assessed as resolved with sequelae (asthenia); at discharge (day 35), strength had improved significantly, with North Star Ambulatory Assessment total score returning to baseline levels at week 24 post-gene therapy infusion (Fig. 1B). Approximately one year (day 362) post-infusion, tacrolimus had been weaned from 1.5 mg to 1 mg twice daily in response to hyperkalemia as well as elevation of cystatin C and blood urea nitrogen. On day 397, the patient exhibited a recurrence of IMM symptoms (pain, weakness, frequent falls) and was hospitalized. The dosage of tacrolimus was increased to 1.5 mg (day 398) and then to 2 mg (day 401) twice daily. On day 400, the patient experienced chest pain, and troponin-I was elevated to 7.77 ng/mL; an echocardiogram revealed mild decline of left ventricular ejection fraction to 41%. The patient was administered with intravenous methylprednisolone for 5 days and was started on rituximab on day 402. Symptoms stabilized and the patient remained hemodynamically stable and was discharged 10 days post-admission, on day 407. Cardiac MRI post-discharge on day 437 showed slightly decreased subepicardial late gadolinium enhancement along the lateral wall of the left ventricle, with normal left ventricular size and ejection fraction of 56%; spironolactone was added to the treatment regimen (Fig. 1B). To date, the patient remains on corticosteroids, monthly intravenous immunoglobulin, and rituximab, and has transitioned from tacrolimus to sirolimus to prevent recurrent hyperkalemia and cystatin C elevations.
Ex vivo identification of micro-dystrophin T cell epitopes
Cellular immune response to micro-dystrophin and use of micro-dystrophin (MDys) peptide pools to determine targets of T cell response
The interferon-gamma (IFN-γ) ELISpot assay was used to detect T cells directed at specific delandistrogene moxeparvovec micro-dystrophin peptides (Fig. 2). ELISpot analysis of leukocytes taken from the two patients with IMM suggested the presence of T cells directed against specific delandistrogene moxeparvovec micro-dystrophin peptides with elevated responses to peptides from MDys pool 1 in Case 1 (at weeks 4, 10, 12, 24, 52, and 104 post-gene therapy infusion; Fig. 3A) and Case 2 (at weeks 2, 4, 5, 6, 7, 8, 9, 10, 11, 12, 15, 20, and 24 post-gene therapy infusion; Fig. 3B), which contains peptides corresponding to DMD gene exons 1–10. No clinically relevant elevated responses to peptides from MDys pools 2 and 3 were observed at time of assessments performed in association with the IMM event (Fig. 3). To identify the specific regions of micro-dystrophin potentially responsible for the T cell immune response, T cell epitope mapping was performed using 15 matrix pools designed from the peptides from pool 1. ELISpot analysis revealed that the patient samples contained T cells directed against three peptide pools in Case 1 (of which the common peptides were 38 and 39; Fig. 4A) and two peptide pools in Case 2 (of which the common peptide was 32; Fig. 4B). These peptides (32, 38, and 39) map to exons 8 and 9 of the DMD gene (Fig. 4).
Fig. 2.

ELISpot assay pools. (A) Overview of the full-length dystrophin domains and exons alongside delandistrogene moxeparvovec micro-dystrophin. Combinations of peptides from regions of delandistrogene moxeparvovec micro-dystrophin were selected to form the MDys peptide pools—MDys pools 1, 2, and 3—used for the IFN-γ ELISpot assay, (B) Genomic deletions of the two patients who had an IMM event.
Adapted from Iannaccone ST, et al. J Neurol. 2024;271(8):5659–5664. Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/). ABD actin binding domain, CR cysteine-rich domain, cont. continued, CT C-terminal domain, ELISpot enzyme-linked immunosorbent spot, H hinge domain, IFN-γ interferon-gamma, MDys micro-dystrophin, R spectrin‑like repeat domain.
Fig. 3.

Cellular immune response to micro-dystrophin. (A) IMM Case 1, (B) IMM Case 2. BL baseline, D day, IFN-γ interferon-gamma, IMM immune-mediated myositis, MDys micro-dystrophin, PBMC peripheral blood mononuclear cell, SFC spot-forming cell, W week.
Fig. 4.

ELISpot analysis of micro-dystrophin peptide pool MDys1 to identify potential T cell targets. (A): IMM Case 1, (B): IMM Case 2. *MDys pool, peptide numbers, and IFN-γ response omitted for brevity. ABD actin binding domain, ELISpot enzyme-linked immunosorbent spot, H hinge domain, IFN-γ interferon-gamma, IMM immune-mediated myositis, MDys micro-dystrophin, N N-terminal domain, PHA phytohemagglutinin, SD standard deviation.
Genotypes of patients in ENDEAVOR with mutations in exons 1–17 or 59–71
Patients enrolled in ENDEAVOR were required to have a definitive diagnosis of DMD based on documentation of clinical findings and prior confirmatory clinical diagnostic genetic testing. Twenty-one patients who were enrolled in ENDEAVOR had genetic mutations that overlapped the transgene region, including 17 patients with mutations in exons 1–17 and four patients with mutations in exons 59–71 (Table 1). Only two of the 17 patients with mutations involving exons 1–17 described in the present study experienced IMM. Notably, four patients with deletions involving exons 8 and/or 9 (including a patient with a nonsense single nucleotide deletion) had no clinical evidence of IMM and no clinically relevant elevations in ELISpot analysis. The human leukocyte antigen class I (HLA-I) genotypes of the six patients (two with IMM and four without IMM) are shown in Table 2.
Table 1.
Genotypes of patients in ENDEAVOR with genetic mutations in exons 1–17 or 59–71.
| Patient genotypes with mutations in exons 1–17 (n = 17) | |||
|---|---|---|---|
| Mutation type | Exon location | IMM event | |
| Deletions | 3–7 | N | |
| 3–43 | Y | ||
| 8–9 | Y | ||
| 8–9 | N | ||
| 8–12 | N | ||
| 10–11 | N | ||
| 12–16 | N | ||
| 12–30 | N | ||
| Intron 7–exon 8 (40 bp) | N | ||
| Duplications | 2 | N | |
| 2–9 | N | ||
| 8–11 | N | ||
| 12–16 | N | ||
| 14–19 | N | ||
|
Nonsense (premature stop) |
Single nucleotide deletion (c.907del) | 9 | N |
| Single nucleotide substitution | 11 | N | |
| Single nucleotide substitution | 14 | N | |
| Patient genotypes with mutations in exons 59–71 (n = 4) | |||
|---|---|---|---|
| Mutation type | Exon location | IMM event | |
| Splice site | N/A: intron 62 | N | |
| N/A: intron 69 | N | ||
|
Nonsense (premature stop) |
Single nucleotide deletion | 66 | N |
| Single nucleotide deletion | 67 | N | |
Bold rows indicate patients bearing deletions involving exons 8 and/or 9 of the DMD gene.
IMM immune-mediated myositis, N/A not applicable.
Table 2.
HLA-I genotypes of six patients with mutations in exons 8 and/or 9 of the DMD gene.
| IMM event | Dystrophin mutation | HLA-I genotype | ||
|---|---|---|---|---|
| Locus | Allele 1 | Allele 2 | ||
| Yes | Deletion exons 3–43 | A | A*24:02:01 | A*68:01:02 |
| B | B*39:01:01 | B*40:08 | ||
| C | C*02:02:02 | C*07:02:01 | ||
| Yes | Deletion exons 8–9 | A | A*11:01:01 | A*32:01:01 |
| B | B*35:01:01 | B*35:01:01 | ||
| C | C*04:01:01 | C*04:01:01 | ||
| No | Intron 7-exon 8 (40 bp) | A | A*02:01:01 | A*24:20:01 |
| B | B*40:01:02 | B*40:01:02 | ||
| C | C*04:01:01 | C*07:02:01 | ||
| No | Single nucleotide deletion, exon 9 | A | A*02:01:01 | A*02:05:01 |
| B | B*50:01:01 | B*51:01:01 | ||
| C | C*06:02:01 | C*16:01:01 | ||
| No | Deletion exons 8–9 | A | A*02:01:01 | A*02:03:01 |
| B | B*39:09:01 | B*52:01:01 | ||
| C | C*07:02:01 | C*12:02:02 | ||
| No | Deletion exons 8–12 | A | A*03:01:01 | A*34:02:01 |
| B | B*14:01:01 | B*15:01:01 | ||
| C | C*03:04:01 | C*08:02:01 | ||
In silico epitope mapping to determine potential immunogenicity of micro-dystrophin peptides
One prerequisite for immune detection is the presentation of peptide fragments derived from the transgene product, facilitated by HLA-I or HLA-II21. An in silico tool (NetMHCpan-4.1) was used to determine the propensity of each 9-mer peptide encoded by dystrophin exons 1–17 to bind each HLA-I molecule allele expressed by the patients22,23. We focused on HLA-I-mediated peptide presentation because the T cell responses of the two IMM cases appeared to be of cytotoxic nature and therefore dependent on CD8positive T cell responses. The highest epitope scores were attributed to sequences encoded by exon 8 (Case 1, 66.72; Case 2, 60.44) and exon 9 (Case 1, 20.02; Case 2, 41.56) of the DMD gene. These results indicated a greater probability for peptides derived from exons 8 and 9 to bind HLA-I, and consequently, a higher potential for these peptides to be presented at the cell surface and activate a response from specific T cells compared with peptides derived from other exons among 1–17 (Fig. 5). This suggests that these epitopes have a higher potential to be presented at the cell surface to CD8positive T cells and in turn activate a cytotoxic response, as long as the patients are not immunologically tolerant to this region of the micro-dystrophin sequence due to their DMD gene mutations.
Fig. 5.

In silico HLA-I epitope mapping and scoring. (A) IMM Case 1, (B) IMM Case 2. The sums of transformed scores (epitope scores) are shown for the protein sequences derived from each exon 1 to 17. HLA human leukocyte antigen, IMM immune-mediated myositis.
Patients who do not naturally produce a portion of the dystrophin protein may be at risk of developing an immune response to that portion of the micro-dystrophin transgene product following gene transfer therapy. This was the case in the two patients who developed IMM, whose DMD gene deletions comprised exons 8 and 9. We aimed to investigate whether the risk of IMM might be reflected by the epitope scores in the six patients with deletions involving exons 8 and/or 9. To calculate the epitope scores, it was important to account for the impact of the mutation on natural dystrophin expression in order to consider the epitope scores only for the non-expressed regions of the DMD gene (where no tolerance is expected). Table 3 displays the epitope scores calculated for exons 8 and 9, considering each patient’s mutation. For the patient with a 40 bp deletion between intron 7 and exon 8, one question was whether the alternative translation start site in exon 8 is used24,25. Both possibilities were considered. Results showed that, while the epitope scores calculated for exon 8 were relatively high in the two patients who developed IMM, one out of the four patients without IMM who carried a deletion of exons 8 and 9 had a score in the same range. For exon 9, the epitope scores were higher in the two patients who developed IMM compared with the patients who did not, reflecting the occurrence of the clinical event.
Table 3.
Epitope scores for exons 8 and 9 in patients with mutations in these exons who did or did not develop IMM.
| Mutation | Deletion exons 3–43 | Deletion exons 8–9 | Intron 7–exon 8 (40 bp) | Single nucleotide deletion, exon 9 (c.907del) | Deletion exons 8–9 | Deletion exons 8–12 |
|---|---|---|---|---|---|---|
| IMM | Yes | Yes | No | No | No | No |
| Exon 8 | 66.7 | 60.4 | 25.4 (31.9)* | 0 | 77.6 | 50.2 |
| Exon 9 | 20.0 | 41.6 | 0 (10.6)* | 18.3 | 19.3 | 16.9 |
*For each patient, only the epitopes in the dystrophin protein sequence that cannot be naturally expressed due to his particular mutation were considered. In the patient with a splice site deletion between intron 7 and exon 8, the alternative translation start site present in exon 8 may or may not be used; the numbers in parentheses correspond to the latter situation.
This epitope scoring approach was also applied to peptides 38–39 (overlapping between exons 8 and 9 and driving the T cell response in the first IMM case) and peptide 32 (exon 8, identified in the second IMM case) (Supplementary Table 1). For peptides 38–39, the patient with a deletion of exons 8–9 who did not have IMM showed the highest score, while the first IMM case displayed the next highest score. The second IMM case showed a low score for peptides 38–39. For peptide 32, the two IMM cases showed the highest scores among the six patients with mutations in exons 8 and/or 9.
Discussion
This study aimed to investigate the intricacies of the IMM events observed in two patients during the ENDEAVOR clinical trial of delandistrogene moxeparvovec in patients with DMD. Delandistrogene moxeparvovec delivers a transgene encoding a micro-dystrophin that retains the key functional domains of dystrophin, including domains corresponding to exons 1–17 and exons 59–71 of the DMD gene. Through a comprehensive clinical and immunologic investigation, we identified key insights into the underlying mechanisms and potential risk factors associated with IMM in the context of gene therapy for DMD.
Given the temporal relationship of T cell activation concurrent with the onset of IMM, results of this study suggest that T cell-mediated muscle inflammation is a likely immunologic driver of IMM. Three lines of evidence converge to strongly suggest that T cell immune responses directed against micro-dystrophin peptides corresponding to exons 8 and/or 9 of the DMD gene were associated with the IMM events in these two patients. Firstly, ELISpot analysis of leukocytes taken from the two patients with IMM identified T cells directed against three peptides that mapped to exons 8 and 9 of the DMD gene (Figs. 3, 4; summarized in Fig. 6). These peptides are also included in delandistrogene moxeparvovec micro-dystrophin (Fig. 2). Secondly, in silico epitope mapping suggests greater probability for peptides derived from exons 8 and 9 to bind HLA-I molecules of the two patients with IMM, increasing the potential for these specific peptide sequences to drive a cytotoxic immune response (Fig. 5; summarized in Fig. 6). Despite deletions in both MDys pools 1 and 2 in Case 1, the patient did not have T cell-mediated responses against epitopes from pool 2. One explanation for this finding is that the peptides included in MDys pool 2 cover the sequence encoded by a part of exons 10–17. Our in silico epitope scoring approach suggests that the risk of T cell response is lower for any of these exons compared with exons 8 and 9 in both IMM cases (Fig. 5). This higher risk results from both the numbers and the HLA-I binding properties of the peptides encoded by each exon. Lastly, both patients who developed IMM had mutations that deleted exons 8 and/or 9, raising the potential for their immune systems to recognize the corresponding protein sequences as foreign. In people with normal dystrophin, or those with dystrophin mutations, such as duplications, that could potentially allow for some level of expression of these sequences, T cells recognizing these self-peptides would be expected to be negatively selected, preventing them from entering circulation26. However, when these sequences are fully or partially deleted, as in the six cases described in this article, T cells recognizing the corresponding peptides may escape negative selection and create potential to attack muscles expressing these sequences. The two patients who developed IMM showed high scores for exon 8 and, remarkably, higher epitope scores for exon 9 than the four other patients who did not have IMM (Table 3). More data are needed to determine whether the in silico epitope scoring approach described in this article may help assess the risk of IMM in patients with DMD treated with delandistrogene moxeparvovec. In addition to HLA alleles and T cell epitopes, other factors such as the underlying inflammatory state of patients with DMD or immunosuppressive treatments may influence the risk of IMM, while HLA-II alleles may also contribute to modulating immune responses.
Fig. 6.

Summary of ELISpot and in silico HLA-epitope mapping findings. ELISpot enzyme-linked immunosorbent spot, HLA human leukocyte antigen, IMM immune-mediated myositis, MHC major histocompatibility complex, TCR T cell receptor.
The occurrences of IMM in ENDEAVOR are consistent with similar cases reported in other clinical trials evaluating gene therapies for DMD (ClinicalTrials.gov number, NCT04281485 and Eudra-CT number, 2020-002093-27)19,27,28. The overlapping presentations, potentially including severe muscle weakness, loss of ambulation, respiratory muscle involvement, muscle edema on MRI, and T cell infiltration on muscle biopsy, highlight a shared pattern of myositis associated with these investigational treatments19. In response to these observations, a collaborative effort among sponsors of ongoing DMD gene therapy trials was initiated to minimize risks for future participants19,29. This study is part of these ongoing efforts. Clinical trial protocols were adjusted to exclude patients with genetic mutations overlapping transgene sequences, thereby mitigating potential risks. It is important to note that at the time of publication of the findings of this collaborative effort, the patients who experienced IMM all had relatively large deletions in their DMD gene, and the present study identifying the specific sequences driving these responses had yet to be performed. Accordingly, the first recorded case of IMM in ENDEAVOR led to a protocol update for the pivotal, phase III EMBARK study (SRP-9001-301; NCT05096221) excluding individuals with genetic mutations between or including exons 1–17 for participant safety, as research into these risks was ongoing30.
The later emergence of the second case of IMM in ENDEAVOR in a patient with a deletion involving only exons 8 and 9 of the DMD gene, combined with the work described here that also implicated only exons 8 and 9 even in the first case of IMM in ENDEAVOR reported in a patient with a larger DMD gene deletion of exons 3–43, enabled the narrowing of the delandistrogene moxeparvovec contraindication specifically to deletions in exons 8 and/or 910. The risk of IMM in patients with other mutations who potentially receive delandistrogene moxeparvovec is presently undetermined. Findings from this study suggest that the size of the DMD gene mutation may not be of particular significance, but rather it is the deletion of specific sequences with elevated immunogenic potential that poses a higher risk of IMM.
In an earlier DMD gene therapy trial investigating a different vector, promoter, and transgene construct, immunogenic T cell responses were detected in a patient with a deletion in exons 3–17 that were directed against non-self epitopes encoded by the transgene; however, no clinical manifestations of IMM were reported31. It should be noted that, aside from the two patients described in ENDEAVOR, IMM has not been observed in any other patients with genetic mutations contained in exons 1–17 or 59–71 overlapping the transgene. In ENDEAVOR, four of six patients who had a genetic deletion (including nonsense single nucleotide deletions) involving exons 8 and/or 9 of the DMD gene have not shown any signs of IMM to date (Table 1). Therefore, risk factors other than the specific DMD gene deletion, like the patient’s HLA genotype (as suggested by our in silico epitope scoring approach), may contribute to the development and severity of IMM.
Limited data are available from patients with mutations in exons 1–17 and exons 59–71 who received delandistrogene moxeparvovec treatment in clinical trials. Study 101 (SRP-9001-101; NCT03375164) and Study 102 (SRP-9001-102; NCT03769116) excluded patients with mutations in exons 1–17 and exons 59–79. In ENDEAVOR, patients with any mutation in exons 1–79 were initially included to allow for evaluation of more patients. However, the development of the first case of IMM in Cohort 2 resulted in a change to the genetic criteria for all cohorts of ENDEAVOR that excluded patients with mutations in exons 1–17 (except for Cohort 5, which was designed to better understand the risk associated with these mutations). This informed the inclusion criteria for subsequent studies, including those for EMBARK, which also excluded patients with mutations in exons 1–17; patients with mutations in exons 59–71 were not excluded. The US FDA Prescribing Information (June 2023) includes a warning that patients with deletions in the DMD gene in exons 1–17 and/or exons 59–71 may be at risk of a severe IMM reaction10. As of May 31, 2024, 21 patients with a DMD gene mutation in the exon 1–17 region, five of which were deletions in the exon 1–17 region (no patients with a deletion in exons 8 and/or 9 were dosed), and 15 patients with a DMD gene mutation in the exon 59–71 region have received the FDA-approved delandistrogene moxeparvovec-rokl gene therapy product; there have been no IMM events reported in these 36 patients.
The broader implications of our results indicate that peptides in the micro-dystrophin sequence derived from exons 8 and/or 9 of the DMD gene have an elevated potential for T cell activation. As the field of precision genetic medicine advances to address the underlying cause of other diseases, similar events of immune responses to transgenes may occur. Insights derived from the interplay between transgenes and the immune system in DMD may contribute to the safety and success of these future gene therapies.
Conclusion
Our immunologic investigation sheds light on IMM in patients with DMD treated with delandistrogene moxeparvovec, highlighting the potentially heightened immunogenicity of exons 8 and/or 9 of the DMD gene for patients bearing deletions in this region. Notably, not all patients with genetic mutations involving these exons developed IMM, suggesting a complex interplay of factors. These findings underscore the need for ongoing research to better understand the risk factors associated with IMM and to develop strategies for the safe administration of DMD gene therapies, particularly in patients with potentially higher-risk mutations in the DMD gene.
Methods
ENDEAVOR study design and participants
ENDEAVOR was approved by a central institutional review board through Advarra and was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines. Informed consent was obtained from all legal guardians of minor participants prior to performing any procedures required for this study in compliance with all applicable guidelines. ENDEAVOR is an ongoing, open-label, phase Ib, multi-cohort study to assess delandistrogene moxeparvovec micro-dystrophin expression as well as safety and functional outcomes following a single administration of commercial process delandistrogene moxeparvovec material in patients with DMD. Part 1 of ENDEAVOR (baseline through week 12) assesses delandistrogene moxeparvovec micro-dystrophin expression and safety, while Part 2 (baseline up to week 156 [3 years]) will assess long-term safety outcomes before participants enter EXPEDITION (SRP-9001-305; NCT05967351) to complete 5 years of follow-up. ENDEAVOR methodology has been previously published13. Eligibility required a definitive DMD diagnosis based on documentation of clinical findings and prior confirmatory clinical diagnostic genetic testing. Of relevance to this study, ENDEAVOR Cohort 2 (ambulatory patients aged ≥ 8 to < 18 years) included patients with definitive DMD gene mutations, and Cohort 5 included patients with genetic mutations partially or fully contained in exons 1–17 of the DMD gene but not those with a full deletion mutation of exons 9–13, as similar mutations have previously been associated with IMM13. Cohort 5 consists of two subgroups: 5a (ambulatory patients aged ≥ 4 to < 9 years) and 5b (non-ambulatory patients of all ages).
Clinical and immunologic investigations
Ex vivo identification of micro-dystrophin T cell epitopes
To determine the region(s) of the micro-dystrophin most effective at stimulating T cell responses, we synthesized 18-mer long peptides with 11 amino acid overlaps that covered the entirety of the protein expressed by the micro-dystrophin transgene. The individual peptides were divided into three pools (i.e., MDys pools 1, 2, and 3) based on the region of the protein from which they were derived. We exposed T cells to each pool of peptides and assessed their activation by measuring the amount of IFN-γ produced using an IFN-γ ELISpot assay. The assay was designed to detect the specific peptide pool(s) that elicited a T cell response in the patients (Fig. 2). An analysis was performed at the following timepoints: Case 1–baseline, day 2, and weeks 1, 2, 4, 10, 12, 24, 52, and 104; Case 2–baseline, day 2, and weeks 2, 4, 5, 6, 7, 8, 9, 10, 11, 12, 15, 20, and 24. To identify a specific T cell epitope in pool 1, 51 individual peptides of pool 1 (exons 1–10) were distributed into 15 peptide pools. The matrix pooling strategy was adapted to ensure that each peptide was present in two separate pools (Fig. 4).
In silico epitope mapping
Based on the patients’ HLA-I genotypes, individual ELrank values displayed by NetMHCpan-4.1 were used to calculate epitope scores for each dystrophin exon from 1–17. As low ELrank numbers correspond to higher affinities, ELranks of each 9-mer peptide/HLA allele combination were transformed according to the below formula and summed for each exon.
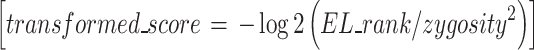 |
The zygosity of the allele, whether homozygous or heterozygous, was accounted for in this transformation. Any negative transformed scores were set to 0, establishing an upper ELrank threshold of 1.0 for heterozygous alleles and 4.0 for homozygous alleles. To calculate epitope scores adjusted to each patient’s mutation, only the ELrank values of the peptides not naturally expressed by the patient due to his mutation were considered.
Statistical analysis
Data analyses were primarily descriptive in nature. For continuous variables, means, medians, standard deviations, and ranges were calculated. For categorical variables, frequency counts and percentages were calculated.
Supplementary Information
Acknowledgements
The authors would like to thank the patients and their families for participating in ENDEAVOR, as well as the investigators and trial staff involved in ENDEAVOR. This study is part of work being conducted by the Cross Company Collaborative Group, a joint working group consisting of four sponsors currently running gene therapy trials in Duchenne muscular dystrophy and two academic investigators. This study was sponsored by Sarepta Therapeutics, Inc., Cambridge, MA, USA and funded by Sarepta Therapeutics, Inc., Cambridge, MA, USA and F. Hoffmann-La Roche Ltd, Basel, Switzerland. Medical writing and editorial support were provided by David Kabanda, MBChB, and Leo Mahachi, PhD, of Nucleus Global, in accordance with Good Publication Practice (GPP) 2022 guidelines (https://www.ismpp.org/gpp-2022) and were funded by Sarepta Therapeutics, Inc., Cambridge, MA, USA and F. Hoffmann-La Roche Ltd, Basel, Switzerland.
Author contributions
R.A.P., I.H.M., S.K., H.H., A.H., D.R.A., L.R.R.K.: substantial contributions to the conception or design of the work; and the acquisition, analysis, or interpretation of data; drafted the work or substantively revised it; approved the submitted version (and any substantially modified version that involves the author’s contribution to the study); agreed both to be personally accountable for the author’s own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature. G.S., C.W., A.P.M., E.P., D.A.G., S.M., S.T.I, C.M.Z.: substantial contributions to the interpretation of data; drafted the work or substantively revised it; approved the submitted version (and any substantially modified version that involves the author’s contribution to the study); agreed both to be personally accountable for the author’s own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature.
Data availability
The datasets presented in this article are not readily available. Qualified researchers may request access to the data that support the findings of this study from Sarepta Therapeutics, Inc., Cambridge, MA, USA, by contacting the Sarepta Medical Information Team at medinfo@sarepta.com.
Declarations
Competing interests
R.A.P., I.H.M., S.K., D.R.A., E.P., D.A.G., and S.M. are employees of Sarepta Therapeutics and may have stock options. H.H., A.H., G.S., and C.W. are employees of F. Hoffmann-La Roche Ltd and may have stock options. A.P.M. is an employee of Roche Products Ltd and may have stock options. S.T.I. receives research support from industry (Elpida, Novartis, Biogen, Sarepta Therapeutics, PTC Therapeutics, Scholar Rock, FibroGen, RegenxBio, and ReveraGen) and the Department of Defense W81XWH2010293, Parent Project Muscular Dystrophy, and Cure SMA. She has served on medical advisory boards for Novartis, Biogen, Genentech, and Sarepta Therapeutics. She receives partial salary support from the following grants: National Institutes of Health Wellstone Muscular Dystrophy Center P50HD087351, NeuroNEXT U24NS107176, and the Muscular Dystrophy Association. C.M.Z. receives research support from Biogen and Novartis and has served on an advisory board for Sarepta Therapeutics. L.R.R.K. is an employee of Sarepta Therapeutics and may have stock options and is a co-inventor of AAVrh74.MHCK7.SRP-9001-dys technology.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-84077-w.
References
- 1.Bushby, K. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol.9, 77–93. 10.1016/S1474-4422(09)70271-6 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Crisafulli, S. et al. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J. Rare Dis.15, 141. 10.1186/s13023-020-01430-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendell, J. R. et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol.74, 637–647. 10.1002/ana.23982 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Emery, A. E. Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul. Disord.1, 19–29. 10.1016/0960-8966(91)90039-u (1991). [DOI] [PubMed] [Google Scholar]
- 5.Norcia, G. et al. Early gross motor milestones in Duchenne muscular dystrophy. J. Neuromuscul. Dis.8, 453–456. 10.3233/jnd-210640 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duan, D., Goemans, N., Takeda, S., Mercuri, E. & Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers7, 13. 10.1038/s41572-021-00248-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asher, D. et al. Clinical development on the frontier: gene therapy for duchenne muscular dystrophy. Expert Opin. Biol. Ther.20, 263–274. 10.1080/14712598.2020.1725469 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Zheng, C. & Baum, B. J. Evaluation of promoters for use in tissue-specific gene delivery. Methods Mol. Biol.434, 205–219. 10.1007/978-1-60327-248-3_13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendell, J. et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol.77, 1121–1131. 10.1001/jamaneurol.2020.1484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.US Food and Drug Administration. ELEVIDYS™ (delandistrogene moxeparvovec-rokl) Highlights of prescribing information. https://www.fda.gov/media/169679/download (2023). Accessed Dec 2024.
- 11.Mendell, J. R. et al. Long-term safety and functional outcomes of delandistrogene moxeparvovec gene therapy in patients with Duchenne muscular dystrophy: a phase 1/2a nonrandomized trial. Muscle Nerve69, 93–98. 10.1002/mus.27955 (2024). [DOI] [PubMed] [Google Scholar]
- 12.Mendell, J. R. et al. Practical considerations for delandistrogene moxeparvovec gene therapy in patients with Duchenne muscular dystrophy. Pediatr. Neurol.153, 11–18. 10.1016/j.pediatrneurol.2024.01.003 (2024). [DOI] [PubMed] [Google Scholar]
- 13.Zaidman, C. M. et al. Delandistrogene moxeparvovec gene therapy in ambulatory patients (aged ≥4 to <8 years) with Duchenne muscular dystrophy: 1-year interim results from Study SRP-9001-103 (ENDEAVOR). Ann. Neurol.10.1002/ana.26755 (2023). [DOI] [PubMed] [Google Scholar]
- 14.Mendell, J. R. et al. Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy. Front. Cell. Dev. Biol.11, 1167762. 10.3389/fcell.2023.1167762 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goedeker NL et al. Management of patients following investigational delandistrogene moxeparvovec gene therapy for Duchenne muscular dystrophy: Delphi panel consensus considerations based on clinical trial experience. Presented at the 2023 American Society of Gene and Cell Therapy Annual Meeting, Los Angeles, CA, USA. https://investorrelations.sarepta.com/static-files/ad325995-7c11-4504-b37f-1fbbba67f383. (2023). Accessed Dec 2024.
- 16.Ertl, H. C. J. Immunogenicity and toxicity of AAV gene therapy. Front Immunol.13, 975803. 10.3389/fimmu.2022.975803 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salmon, F., Grosios, K. & Petry, H. Safety profile of recombinant adeno-associated viral vectors: focus on alipogene tiparvovec (Glybera®). Expert Rev. Clin. Pharmacol.7, 53–65. 10.1586/17512433.2014.852065 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Khan, S. et al. T-cell response to micro-dystrophin in a patient treated with delandistrogene moxeparvovec gene therapy: a case of immune-mediated myositis. Presented at the 28th International Annual Congress of the World Muscle Society (WMS 2023). Charleston, SC, USA, 2023. https://investorrelations.sarepta.com/static-files/51d79e1a-7ef7-46c6-93e3-c63b7be11238. (2023). Accessed Dec 2024.
- 19.Bonnemann, C. G. et al. Dystrophin immunity after gene therapy for Duchenne’s muscular dystrophy. N. Engl. J. Med.388, 2294–2296. 10.1056/NEJMc2212912 (2023). [DOI] [PubMed] [Google Scholar]
- 20.Iannaccone, S. T. et al. Immune-mediated myositis following gene therapy for Duchenne muscular dystrophy: a case report. J. Neurol.271, 5659–5664. 10.1007/s00415-024-12431-z (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Elsen, P. J. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front. Immunol.2, 48. 10.3389/fimmu.2011.00048 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nilsson, J. B. et al. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning. Sci. Adv.9, eadj6367. 10.1126/sciadv.adj6367 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454. 10.1093/nar/gkaa379 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winnard, A. V., Mendell, J. R., Prior, T. W., Florence, J. & Burghes, A. H. Frameshift deletions of exons 3–7 and revertant fibers in Duchenne muscular dystrophy: mechanisms of dystrophin production. Am. J. Hum. Genet.56, 158–166 (1995). [PMC free article] [PubMed] [Google Scholar]
- 25.Gangopadhyay, S. B. et al. Dystrophin in frameshift deletion patients with Becker muscular dystrophy. Am. J. Hum. Genet.51, 562–570 (1992). [PMC free article] [PubMed] [Google Scholar]
- 26.Wu, Y. X., Jin, S. H. & Cui, J. Autophagy and immune tolerance. Adv. Exp. Med. Biol.1206, 635–665. 10.1007/978-981-15-0602-4_28 (2019). [DOI] [PubMed] [Google Scholar]
- 27.ClinicalTrials.gov. NCT04281485: a phase 3 study to evaluate the safety and efficacy of PF-06939926 for the treatment of Duchenne muscular dystrophy. https://clinicaltrials.gov/study/NCT04281485 (2024). Accessed Dec 2024.
- 28.Clinicaltrialsregister.eu. 2020-002093-27: microdystrophin (GNT0004) gene therapy clinical trial in Duchenne muscular dystrophy. https://www.clinicaltrialsregister.eu/ctr-search/trial/2020-002093-27/FR (2020). Accessed Dec 2024.
- 29.Bonnemann, C. et al. A collaborative analysis by clinical trial sponsors and academic experts of anti-transgene SAEs in studies of gene therapy for DMD; Poster 44. Presented at the Muscular Dystrophy Association (MDA) Clinical & Scientific Conference, Dallas, USA. https://www.mdaconference.org/abstract-library/a-collaborative-analysis-by-clinical-trial-sponsors-and-academic-experts-of-anti-transgene-saes-in-studies-of-gene-therapy-for-dmd/ (2023). Accessed Dec 2024.
- 30.ClinicalTrials.gov. NCT05096221: a gene transfer therapy study to evaluate the safety and efficacy of delandistrogene moxeparvovec (SRP-9001) in participants with Duchenne muscular dystrophy (DMD) (EMBARK). https://clinicaltrials.gov/study/NCT05096221 (2022). Accessed Dec 2024.
- 31.Mendell, J. R. et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med.363, 1429–1437. 10.1056/NEJMoa1000228 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this article are not readily available. Qualified researchers may request access to the data that support the findings of this study from Sarepta Therapeutics, Inc., Cambridge, MA, USA, by contacting the Sarepta Medical Information Team at medinfo@sarepta.com.