

Abstract
BACKGROUND
CD8+ T regulatory (Treg) cells that recognize the nonclassical class 1b molecule Qa-1/human leukocyte antigen E (Q/E CD8+ Treg cells) are important in maintaining self-tolerance. We sought to investigate the role that these T cells play in type 1 diabetes (T1D) pathogenesis and whether an intervention targeting this mechanism may delay T1D progression.
METHODS
We conducted a phase 1/2, randomized, double-blind, placebo-controlled trial of the autologous dendritic cell therapy AVT001 that included participants at least 16 years of age, within 1 year of T1D diagnosis, and with ex vivo evidence of a defect in Q/E CD8+ Treg function. Patients were randomly assigned in a 2:1 ratio to AVT001 or placebo, which was administered in three monthly intravenous infusions. The primary end point was safety; efficacy end points included changes from baseline in C-peptide area under the curve (AUC) during a 4-hour mixed meal, hemoglobin A1c (HbA1c), and insulin dose.
RESULTS
Sixteen patients received AVT001, and nine received placebo. Similar rates and severity of adverse events were observed in both groups. None of the patients in the AVT001 group had serious adverse events through visit day 360. Compared with placebo, treatment with ATV001 was associated with less decline from baseline log-transformed C-peptide AUC (nmol/l), with the treatment effect between AVT001 and placebo at day 150 of 0.09 (95% confidence interval [CI], 0.03 to 0.15) and at day 360 of 0.10 (95% CI, 0.04 to 0.15). No clear differences in change in HbA1c and insulin dose from baseline were observed between groups. Estimated treatment effects of AVT001 versus placebo at day 360 were −0.17% (95% CI, −0.60 to 0.26%) for HbA1c and −0.06 U/kg/day (95% CI, −0.14 to 0.02) for daily insulin dose.
CONCLUSIONS
In this phase 1/2 trial, AVT001 did not result in dose-limiting adverse events. Potential signals of efficacy observed here warrant further evaluation in a fully powered trial. (Funded by Avotres Inc. and the Division of Diabetes, Endocrinology, and Metabolic Diseases; ClinicalTrials.gov number, NCT03895996.)
Introduction
Type 1 diabetes (T1D) is a complex autoimmune disease characterized by destruction of the insulin-producing beta cells in the pancreas. Disease progression is marked by loss of beta-cell function, resulting in hyperglycemia and increased risk of long-term complications.1 Despite decades of research, the pathogenesis of T1D is still not fully understood, hampering the development of effective therapies.2–4
Preservation of residual beta-cell function after clinical diagnosis, as measured by stimulated C-peptide, likely contributes to improved glycemic control and is associated with lower risk of hypoglycemia and diabetes-related complications.1,5 Several drugs have been shown to slow disease progression after diagnosis of stage 3 T1D,6–9 which is typically defined as the onset of symptoms and signs of T1D. Despite evidence of some beneficial effects on beta-cell function, none of these drugs have been approved for treatment of stage 3 disease. Autologous cell therapies, such as polyclonal T regulatory (Treg) cells and tolerogenic dendritic cells, are in the early stages of development, with insufficient proof of clinical efficacy to date.
Among the diverse T cell–mediated regulatory pathways (Treg pathways) explored in T1D, a potentially promising one is the Qa-1 (mouse homologue of human leukocyte antigen E [HLA-E])/HLA-E–restricted CD8+ (Q/E CD8+) Treg pathway. Our preclinical studies identified and validated this pathway’s biologic role in maintaining peripheral self-tolerance through self–nonself discrimination.10–17 The role of this pathway in immune regulation has been independently identified and corroborated by several other groups.18–20
The distinguishing feature of the Q/E CD8+ Treg pathway is the recognition of a “common target structure,” a complex of the Qa-1/HLA-E molecule coupled with a peptide derived from the leader sequence of heat shock protein 60 (Hsp60sp).12–15 This complex, termed HLA-E/Hsp60sp, is preferentially expressed on intermediate-avidity T cells. This intermediate-avidity T-cell pool includes activated self-reactive T cells.12–15 The T-cell receptor of Q/E CD8+ Treg cells specifically recognizes this “common target structure” expressed on the self-reactive T cells. This T–T-cell interaction then leads to a selective down-regulation of the self-reactive T cells. Failure of such down-regulation may contribute to a variety of autoimmune diseases.14,15,21 On the other hand, infection and tumor immunity largely depend upon high-avidity T cells,21,22 and thus, they are unaffected.
In our preclinical studies, we found that the Q/E CD8+ Treg pathway was defective in 9 of 10 (90.0%) patients with clinically diagnosed T1D tested.23 Importantly, in approximately 90% of such patients, triggering their CD8+ Treg cells with immature autologous dendritic cells primed with the oligopeptide Hsp60sp, which is AVT001, corrected this defect ex vivo.23 AVT001 is an autologous dendritic cell vaccine engineered to reactivate the defective Q/E CD8+ Treg pathway.
The primary aim of this first-in-human study was to assess the safety and tolerability of AVT001; the secondary objectives were to assess potential correction of the functional defect of the Q/E CD8+ Treg pathway in vivo by AVT001 and to assess the effect on preserving beta-cell function within 1 year of diagnosis of clinical (stage 3) T1D.
Methods
TRIAL DESIGN AND OVERSIGHT
We conducted this phase 1/2, randomized, placebo-controlled, double-blind, parallel-group trial at Joslin Diabetes Center in Boston, Massachusetts. The trial included a screening and cell collection period of up to 3 months, a treatment period of three monthly infusions of AVT001 or placebo, and a posttreatment follow-up period of 21 months. The results through day 360 (D360) are reported in this article.
The participants or the parents or guardians for minor participants provided written informed consent, and additionally, minor participants provided assent before trial entry. Ethical oversight was provided by the Joslin Committee on Human Studies. An independent data and safety monitoring board (DSMB) met after the first 3 participants, after 6 participants, and after 15 participants had received at least one dose and had completed an in-clinic visit at least 1 month postdose. The DSMB reviewed unblinded data to assess the safety and tolerability of treatment. After each meeting, the DSMB recommended continuation of the trial as planned. The trial was sponsored, designed, and conducted by Avotres Inc. through collaboration with Joslin Diabetes Center, the Connell and O’Reilly Families Cell Manipulation Core Facility at Dana-Farber Cancer Institute, and IQVIA. The sponsor and investigator designed the trial. The study team from Joslin Diabetes Center and the sponsor collected the data. The sponsor and IQVIA analyzed the data.
PARTICIPANTS AND RANDOMIZATION
Participants were enrolled from June 2019 to September 2021. Initially, eligible participants were 18 years of age or older. After DSMB safety review of the first 15 participants, the protocol, which is provided with the full text of this article at evidence.nejm.org, was amended in February 2021 to extend the lower age limit to 16 years of age. All participants had a clinical diagnosis of T1D within 1 year before first dosing, and diagnosis was confirmed by at least one positive autoantibody of glutamic acid decarboxylase (GADA), insulinoma-associated protein 2 (IA-2A), or zinc transporter 8 (ZnT8A). Eligibility also required a demonstrated defect in the Q/E CD8+ Treg pathway that was correctable by AVT001 ex vivo as assessed by a sponsor-developed CD8+ T-cell inhibition assay.23 This assay was developed to detect whether one’s Q/E CD8+ Treg cells have the normal function to selectively down-regulate self-reactive T cells.
Eligible participants were randomly assigned in a 2:1 ratio to receive AVT001 or placebo. A block size of three was used for the first six participants to facilitate DSMB safety and tolerability review. Thereafter, block sizes were randomly assigned. Treatment assignments were concealed from the investigator, participants, and sponsor. Only the manufacturer of AVT001 and placebo and the independent statistician from IQVIA had access to the randomization codes.
TREATMENT
Participants, regardless of treatment assignment, underwent leukapheresis to obtain primary monocytes. Leukapheresis was performed at the Kraft Family Blood Donor Center or Joslin Diabetes Center. AVT001 and matching placebo were produced at the Connell and O’Reilly Families Cell Manipulation Core Facility at Dana-Farber Cancer Institute.
AVT001 was an individualized preparation of autologous immature dendritic cells derived by culture from the participant’s adherent primary monocytes with granulocyte-macrophage colony-stimulating factor and interleukin-4 for 6 days loaded passively with the synthetic oligopeptide (QMRPVSRVL) of Hsp60sp. Dendritic cells were fixed using 2% paraformaldehyde for 10 minutes at 4°C24 and washed three times. The final drug product was a cryopreserved preparation of 20 ml AVT001 suspension in 10% dimethyl sulfoxide and 12.5% human serum albumin, which contained approximately 10 million synthetic oligopeptide-loaded, CD11c+ immature dendritic cells per dose. Placebo volume was also 20 ml, containing only 12.5% human serum albumin with 10% dimethyl sulfoxide. Treatment was administered monthly by intravenous infusion for a total of three doses, with an opaque sleeve to maintain masking.
END POINTS AND ASSESSMENTS
The primary objective was to determine the safety profile and infusion-related tolerability of three monthly doses of AVT001 in persons with T1D. Safety end points included rate of treatment-emergent adverse events (TEAEs), rate and severity of local intravenous-site reactions, rate of severe hypoglycemic events, clinically significant changes from baseline in clinical laboratory parameters, and changes from baseline in vital signs and electrocardiograms.
The secondary efficacy assessments focused on the Q/E CD8+ Treg pathway and diabetes-related metabolic and clinical measures. The efficacy end points included assessment of the functional activity of the Q/E CD8+ Treg pathway; changes from baseline in the area under the curve (AUC) of the stimulated C-peptide levels during a 4-hour mixed-meal tolerance test (MMTT; 360 ml of Boost High Protein; Nestlé Health Science); and changes from baseline in hemoglobin A1c (HbA1c), daily insulin dose (U/kg/day), and diabetes-related autoantibody levels: GADA, IA-2A, ZnT8A, and insulin autoantibodies (IAAs). Cell collection for the baseline CD8+ T-cell inhibition assay was performed during screening. Baseline MMTT was performed in the morning on the same day but before the first infusion of AVT001 or placebo. The functional activity of the Q/E CD8+ Treg pathway was assessed using a modification of the previously published CD8+ T-cell inhibition assay23 as described in Section 3 of the Supplementary Appendix. Details of the C-peptide and autoantibody assays also appear in Section 3 of the Supplementary Appendix.
STATISTICAL ANALYSIS
The primary objective of this trial was to assess the safety and infusion-related tolerability of AVT001 compared with placebo in patients with T1D. A sample size of 14 participants treated with AVT001 would provide at least 80% power to observe at least one occurrence of a TEAE with a true underlying incidence of 12%. The sample size provided the same power to detect at least one occurrence of local tolerability events relating to the intravenous administration of the study drug. To account for an estimated 10% potential dropout before the completion of the month 5 safety and efficacy assessment postfirst dose, a total sample size of approximately 24 participants was planned to be randomly assigned in a 2:1 ratio of AVT001 to placebo.
For the primary end points, in the safety population, TEAEs, and posttreatment adverse events (PTAEs) were summarized by system organ class and preferred term. Local site reactions and severe hypoglycemic events were reported. Changes from baseline were summarized for hematology, chemistry, and urine microalbumin. Corresponding shift tables based on Common Terminology Criteria for Adverse Events (CTCAE), version 4.03 toxicity grade and laboratory normal range were also reported. Abnormal postbaseline changes in vital signs and electrocardiograms were summarized.
For the secondary end points, changes from baseline in ln[C-peptide AUC+1], C-peptide AUC, HbA1c, daily insulin dose (U/kg/day), and autoantibody levels were analyzed by the mixed-effect model for repeated measurements (MMRM) with a random intercept using a backward stepwise variable selection process. The pharmacodynamic population for the MMRM was a priori defined as all participants in the safety population with at least one postbaseline assessment for the functional activity of the Q/E CD8+ Treg pathway by the CD8+ T-cell inhibition assay; thus, all participants were included. The difference of least-squares means between treatment groups (AVT001 vs. placebo), the associated standard error, and the two-sided 95% confidence interval (CI) for each visit were reported at D150 and D360. Values for functional activity of the Q/E CD8+ Treg pathway measured by percentage of inhibition at D150 and D360 were compared with screening values (baseline) using a two-sampled t-test The difference of means and the corresponding two-sided 95% CIs were reported. The reported CIs have not been adjusted for multiple comparisons and should not be used in place of hypothesis testing. Statistical analyses were performed with SAS software, version 9.4 (SAS Institute).
Results
PARTICIPANTS
Of 32 patients screened, 27 met all eligibility criteria and were randomly assigned. Two participants withdrew from the study before leukapheresis, and 25 underwent leukapheresis and received AVT001 (n=16) or placebo (n=9) (Fig. 1). The representativeness of the study population compared with the population of individuals with T1D is described in Table S1.
Figure 1.
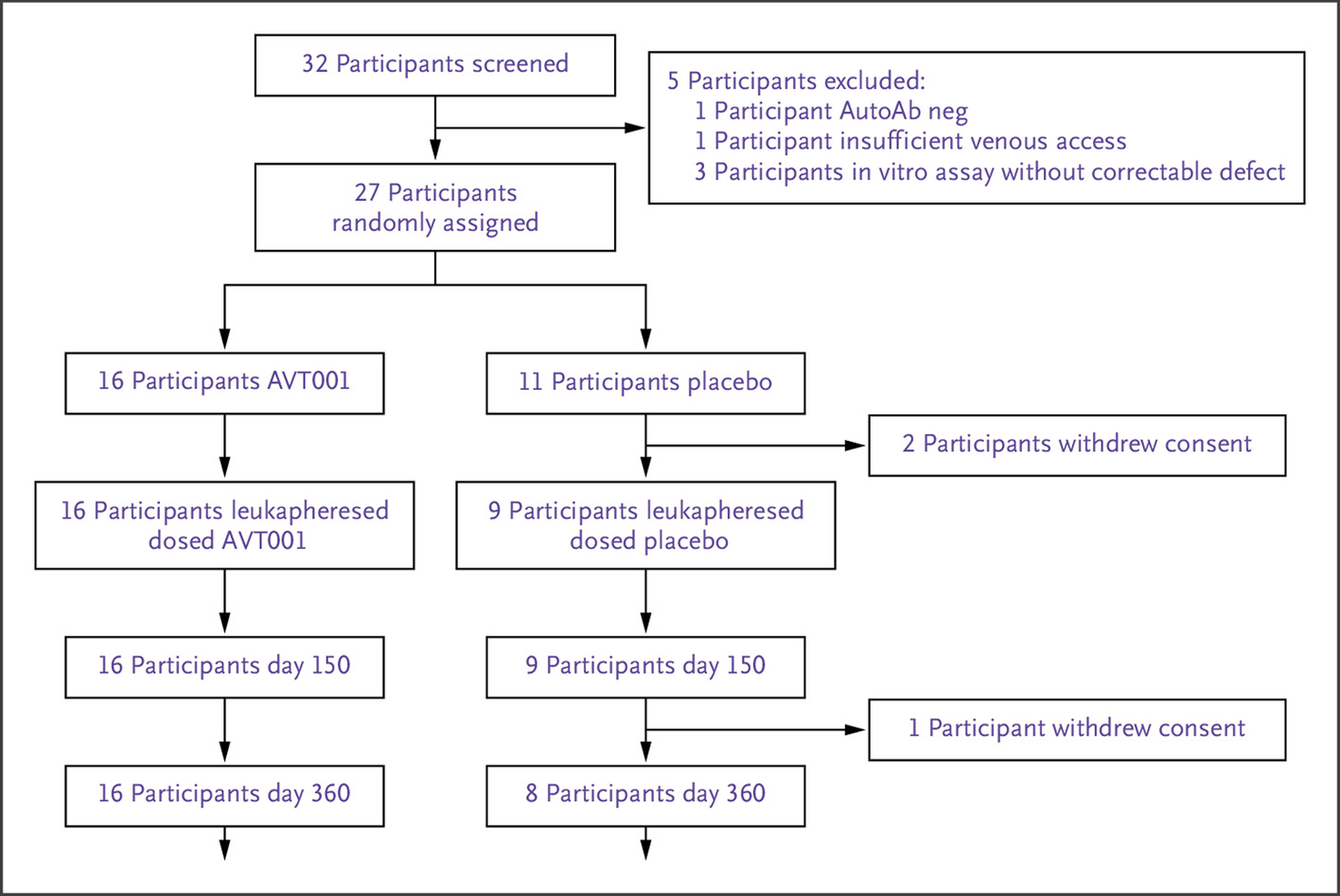
CONSORT Diagram.
Of 32 participants screened, 5 did not meet the screening criteria, and the remaining 27 patients were randomly assigned. Before leukapheresis, two participants randomly assigned to placebo withdrew consent. Sixteen participants in the AVT001 group and nine participants in the placebo group received study treatment as planned and completed the follow-up visits through day 150. One participant withdrew from the study before the visit on day 360. AutoAb neg denotes autoantibody negative.
Participants’ insulin delivery modality and continuous glucose monitoring use were not adjusted as part of this trial. One participant in the AVT001 group did not use continuous glucose monitoring, whereas all other participants used continuous glucose monitoring at some point during the course of the trial. The treatment groups were generally well balanced with respect to baseline characteristics, including age, time from diagnosis, body-mass index (BMI), unstimulated C-peptide, daily insulin dose, and autoantibodies GADA and IAA (Table 1). Compared with those in the placebo group, at baseline, participants in the AVT001 group had lower mean C-peptide AUC (0.53 vs. 0.61 nmol/l), lower peak C-peptide (0.73 vs. 0.84 nmol/l), lower percentage of inhibition evaluated by the CD8+ T-cell inhibition assay (−11.2 vs. 3.5%), higher HbA1c (6.0 vs. 5.7), higher percentage of positive IA-2A (81.3 vs. 55.6%), and higher ZnT8A (68.8 vs. 22.2%) (Table 1).
Table 1.
Baseline Characteristics.*
| Baseline Characteristics | AVT001 (N=16) | Placebo (N=9) |
|---|---|---|
| Age — yr | 26.5 (9.0) | 26.2 (6.2) |
| Time since diagnosis — months | 7.9 (2.7) | 8.8 (2.0) |
| Body-mass index (kg/m2) | 23.0 (3.3) | 25.2 (3.0) |
| C-peptide area under the curve — nmol/l | 0.53 (0.09) | 0.61 (0.06) |
| Peak C-peptide — nmol/l | 0.73 (0.50) | 0.84 (0.25) |
| Unstimulated C-peptide — nmol/l | 0.23 (0.20) | 0.22 (0.12) |
| HbA1c — % | 6.0 (0.7) | 5.7 (0.7) |
| Daily insulin dose — U/kg per day | 0.4 (0.2) | 0.4 (0.2) |
| Age category — n (%) | ||
| <18 | 2 (12.5) | 1 (11.1) |
| 18 to <25 | 7 (43.8) | 4 (44.4) |
| 25 to <40 | 6 (37.5) | 4 (44.4) |
| ≥40 | 1 (6.3) | 0 (0.0) |
| Gender — n (%) | ||
| Male | 10 (62.5) | 7 (77.8) |
| Female | 6 (37.5) | 2 (22.2) |
| Race — n (%) | ||
| White | 13 (81.3) | 9 (100.0) |
| African American | 1 (6.3) | 0 (0.0) |
| Native American | 1 (6.3) | 0 (0.0) |
| Multiracial | 1 (6.3) | 0 (0.0) |
| Positive autoantibodies — n (%) | ||
| Autoantibody of glutamic acid decarboxylase — >20 DK units/ml | 15 (93.8) | 8 (88.9) |
| Autoantibody of insulinoma-associated protein 2 — >5 DK units/ml | 13 (81.3) | 5 (55.6) |
| Autoantibody of zinc transporter 8 — >0.020 index | 11 (68.8) | 2 (22.2) |
| Insulin autoantibodies — >0.010 index | 15 (93.8) | 9 (100) |
| Functional activity of Q/E CD8+ T regulatory pathway evaluated by CD8+ T-cell inhibition assay as a percentage of inhibition† | −11.2 (9.9) | 3.5 (7.1) |
Numbers are shown as mean (standard deviation) unless otherwise noted. DK denotes digestive and kidney (units/ml); HbA1c, hemoglobin A1c; and Q/E CD8+, Qa-1 (mouse homologue of human leukocyte antigen E)/human leukocyte antigen E–restricted CD8+.
The CD8+ T-cell inhibition assay was developed by Avotres Inc. Technical details can be found in Section 3 of the Supplementary Appendix.
All 25 participants received the full course of three doses and were followed through D150 as planned. One participant in the placebo group withdrew from the trial before D360 (Fig. 1).
SAFETY
The safety population included all 25 participants treated with AVT001 or placebo. There were no deaths, no serious adverse events, no local site reactions, and no severe hypoglycemic events reported during 360 days of follow-up. No differences between AVT001 and placebo groups were observed for clinically significant changes from baseline in laboratory parameters, vital signs, and electrocardiograms (Tables S2, S3, S4, and S5).
TEAEs and PTAEs are summarized in Table 2. Rates of decreased blood bicarbonate and anemia were higher in AVT001; all such events were grade 1, except for one grade 2 anemia (Table 2 and Table S6). All adverse events were CTCAE grade 1 or grade 2, except for two instances of grade 3 events of neutrophil count decrease (one each in the AVT001 and placebo groups).
Table 2.
Treatment-Emergent Adverse Events and Posttreatment Adverse Events by Number of Participants When There Were Two or More Reported in Either Group by Preferred Term.*
| Adverse Events | AVT001 (N=16) | Placebo (N=9) |
|---|---|---|
| TEAEs by preferred term | ||
| Participants with any TEAE | 9 (56.3) | 6 (66.7) |
| Neutrophil count decreased | 4 (25.0) | 1 (11.1) |
| Hyponatremia | 3 (18.8) | 1 (11.1) |
| Cough | 3 (18.8) | 0 (0.0) |
| White blood-cell count decreased | 2 (12.5) | 1 (11.1) |
| Ear pain | 2 (12.5) | 0 (0.0) |
| Lymphocyte count decreased | 2 (12.5) | 0 (0.0) |
| Dizziness | 1 (6.3) | 2 (22.2) |
| Headache | 0 (0.0) | 2 (22.2) |
| PTAEs by preferred term | ||
| Participants with any PTAE | 16 (100.0) | 8 (88.9) |
| White blood-cell count decreased | 5 (31.3) | 4 (44.4) |
| Neutrophil count decreased | 4 (25.0) | 1 (11.1) |
| Blood alkaline phosphatase increased | 3 (18.8) | 1 (11.1) |
| Blood bicarbonate decreased | 3 (18.8) | 0 (0.0) |
| Lymphocyte count decreased | 3 (18.8) | 0 (0.0) |
| Corona virus infection | 2 (12.5) | 1 (11.1) |
| Anemia | 2 (12.5) | 0 (0.0) |
| Hypocalcemia | 2 (12.5) | 0 (0.0) |
| Blood bilirubin increased | 1 (6.3) | 2 (22.2) |
Treatment-emergent adverse events (TEAEs) are defined as adverse events that started on or after the first dose of study medication through 30 days following the last dose. Posttreatment adverse events (PTAEs) are defined as adverse events that started more than 30 days following the last dose through day 360.
EFFICACY
The efficacy analyses included all 25 treated participants. For changes from baseline in ln[C-peptide AUC+1], after the backward stepwise variable selection process, the final model was change from baseline of ln[C-peptide AUC+1] (nmol/L) as the dependent variable; its baseline value (nmol/L) as the covariate; and treatment group, visit, BMI (kg/m2), the CD8+ T-cell inhibition assay, and the treatment group-by-visit interaction as fixed effects. The treatment effects assessed by differences of least-squares means of AVT001 versus placebo estimated from the model were 0.09 nmol/l (95% CI, 0.03 to 0.15) at D150 and 0.10 nmol/l (95% CI, 0.04 to 0.15) at D360, respectively, suggesting an association between AVT001 treatment and the preservation of endogenous insulin secretion at D150 and through D360 (Fig. 2A).
Figure 2.
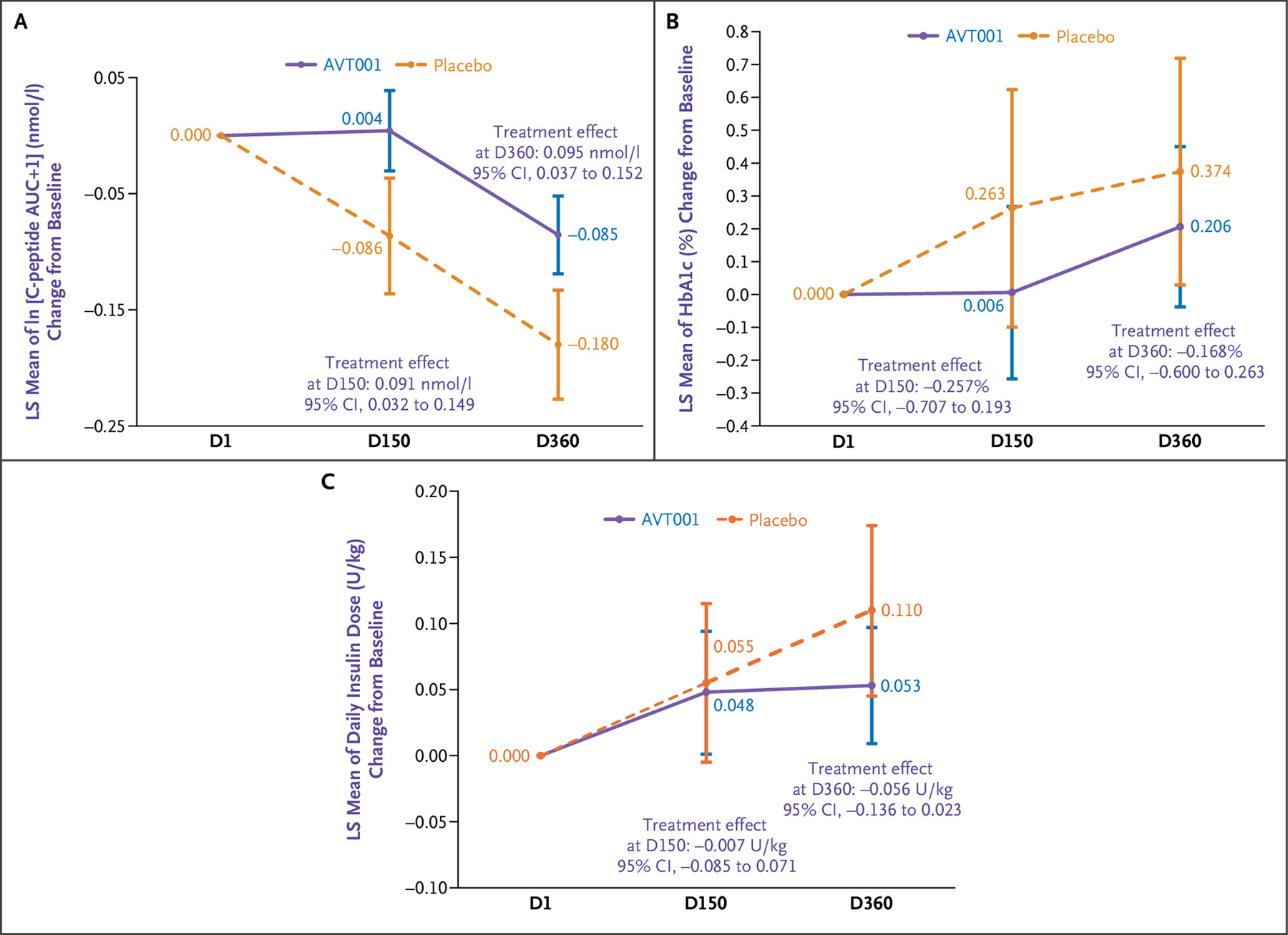
LS Mean of ln[C-Peptide AUC+1] (nmol/l) Change from Baseline, Its 95% CI, and Treatment Effects of AVT001 versus Placebo at Visit D150 and Visit D360 Estimated by MMRM.
The change from baseline (D1) for ln[C-peptide AUC+1] is set as zero per definition (Panel A). LS mean of hemoglobin A1c (HbA1c; percentage) change from baseline, its 95% CI, and treatment effects of AVT001 versus placebo at visit D150 and visit D360 estimated by MMRM. Change from baseline for HbA1c is set as zero per definition (Panel B). LS mean of insulin use (U/kg/day) change from baseline, its 95% CI, and treatment effects of AVT001 versus placebo at visit D150 and visit D360 estimated by MMRM. Change from baseline insulin use is set as zero per definition (Panel C). The widths of all the CIs have not been adjusted for multiple comparisons and should not be used in place of hypothesis testing. AUC denotes area under the curve; CI, confidence interval; D, day; LS, least squares; and MMRM, mixed-effect model for repeated measurements.
For changes from baseline in C-peptide AUC, a similar MMRM was used, and the treatment effects assessed by the differences of least-squares means of AVT001 versus placebo were 0.15 nmol/l (95% CI, 0.06 to 0.25) at D150 and 0.15 nmol/l (95% CI, 0.05 to 0.24) at D360, respectively, consistent with the results for ln[C-peptide AUC+1] (Fig. S1). Similar results were also observed in post hoc exploratory analyses using a simpler MMRM or two-sample t-tests (Tables S7 and S8).
In post hoc subgroup analyses, preservation of C-peptide AUC at D150 was associated with younger age, lower baseline insulin use, higher fasting C-peptide, and higher C-peptide AUC (Fig. S2).
Similarly, MMRMs were applied to HbA1c; daily insulin dose; and autoantibodies GADA, IA-2A, ZnT8A, and IAA (Table S9). There were no observed associations between AVT001 treatment and these outcomes at either D150 or D360. Estimated treatment effects of AVT001 versus placebo at D360 were −0.168% (95% CI, −0.60 to 0.26) for HbA1c and −0.06 U/kg per day (95% CI, −0.14 to 0.02) for daily insulin dose (Fig. 2B and 2C).
Functional activity of the Q/E CD8+ Treg pathway was assessed by the sponsor’s CD8+ T-cell inhibition assay. Comparing the percentages of inhibition on D150 and D360 with the baseline, the differences of means, estimated by the two-sample t-test, were 20.5% (95% CI, 6.5 to 34.5) and 20.5% (95% CI, 5.4 to 35.6), respectively, in the AVT001 group (Fig. 3A), and they were −1.7% (95% CI, −14.8 to 11.3) versus −4.1% (95% CI, −16.9 to 8.8), respectively, in the placebo group (Fig. 3B). These results are consistent with an association between AVT001 treatment and correction of the defect as measured by the percentage of inhibition.
Figure 3.
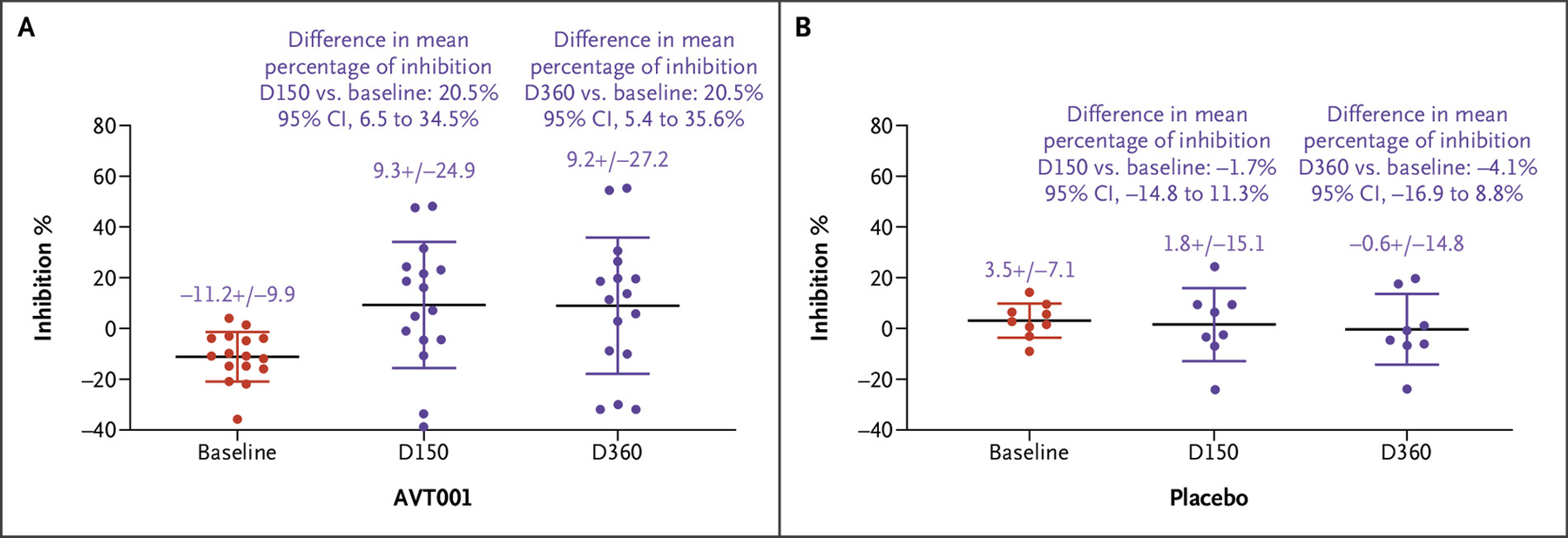
Q/E CD8+ T Regulatory Cell–Based Inhibition Evaluated by the CD8+ T-Cell Inhibition Assay in Participants Who Received AVT001 Treatment.
CD8+ T cells, isolated from participants with type 1 diabetes (T1D) who were treated with AVT001 at screening (baseline), D150, and D360, were assessed for their specific recognition of the “common target structure” of the HLA-E/heat shock protein 60sp complex expressed on the TH1 target cells by the CD8+ T-cell inhibition assay. More details can be found in Section 3 of the Supplementary Appendix. Data show the mean ± standard deviation of 16 participants with T1D receiving AVT001 treatment. The difference of means at D150 versus at baseline in AVT001 was 20.5% (95% CI, 6.5 to 34.5%). The difference of means at D360 versus at baseline was 20.5% (95% CI, 5.4 to 35.6%) (Panel A). Q/E CD8+ T regulatory cell function evaluated by the CD8+ T-cell inhibition assay in participants who received placebo. The difference of means at D150 versus at baseline in placebo was −1.7% (95% CI, −14.8 to 11.3%), and the difference of means at D360 versus at baseline was −4.1% (95% CI, −16.9 to 8.8%), which showed no statistically significant difference between the two groups (Panel B). The widths of the CIs have not been adjusted for multiple comparisons and should not be used in place of hypothesis testing. CI denotes confidence interval; D, day; and Q/E CD8+, Qa-1 (mouse homologue of human leukocyte antigen E)/human leukocyte antigen E–restricted CD8+.
Discussion
This phase 1/2 trial of treatment with three monthly infusions of AVT001 did not identify dose-limiting side effects at follow-up of 150 and 360 days. The only adverse event above grade 2 observed was neutrophil count decrease, which occurred in both AVT001 and placebo groups. This may potentially be related to the T1D disease state because low neutrophil counts have been described in T1D.25 As shown in Figure 2A and Figure S1, treatment with AVT001 was associated with differences in ln[C-peptide AUC+1] and C-peptide AUC from baseline between AVT001 compared with placebo on D150 and D360. No similar associations were observed for other secondary outcomes, including HbA1c and insulin dose. These efficacy outcomes could be further investigated in a future trial.
Participants were 16 years of age and older, within 1 year of clinical diagnosis of T1D, and with an ex vivo correctable defect in the function of the Q/E CD8+ Treg pathway as determined by the sponsor’s CD8+ T-cell inhibition assay. Limitations of this trial include being a relatively small, single-center experience with limited duration of follow-up. Durability of efficacy was unknown before trial initiation, and alternative dosing regimens may result in different effects, which will be important aspects to consider in a future trial.
Because this trial was designed as a first-in-human safety and tolerability trial, it is limited in size and age distribution of the study participants. The mean ages in the AVT001 and placebo groups were 26.5 and 26.2 years of age, respectively. In general, the decline in C-peptide in adults after the clinical diagnosis of T1D is slower than that in children and adolescents.26 This slower rate of decline in C-peptide may make it more difficult to demonstrate a treatment effect compared with placebo. We speculate that treatment efficacy could be more apparent and impactful in younger individuals.
The CD8+ T-cell inhibition assay, designed to assess the specific function of the Q/E CD8+ Treg pathway, was used to evaluate whether AVT001 could correct the defect of the function of the Q/E CD8+ Treg cells in vivo. The results suggest that AVT001 treatment was associated with apparent correction of the defect of the function of the Q/E CD8+ Treg cells in vivo (Fig. 3A) as measured by the percentage of inhibition, but no similar correction was observed in the placebo group (Fig. 3B). If the apparent correction of the defect of the Q/E CD8+ Treg cells observed in this trial is confirmed in future larger efficacy trials powered to evaluate this outcome, it would provide mechanistic evidence for the effect of AVT001 on clinical efficacy. Although there could be potential alternative explanations as to how infusion of AVT001 could affect the dysfunction of the Q/E CD8+ Treg pathway assessed by the CD8+ T-cell inhibition assay that was associated with preservation of C-peptide, the Hsp60sp peptide-loaded dendritic cells being fixed before cryopreservation excludes several of these possibilities.
The defect of the Q/E CD8+ Treg pathway was observed in 29 of 32 (90.6%) screened participants in this trial (Fig. 1), similar to the 90% reported previously,23 providing additional evidence that this defect is common in persons with T1D.
In summary, in this first-in-human phase 1/2 trial, treatment with AVT001 did not demonstrate dose-limiting adverse events. Compared with placebo, treatment with AVT001 was associated with evidence of preservation of endogenous insulin secretion but not with change in HbA1c and insulin dose at D360. The apparent correction of the defect in the Q/E CD8+ Treg pathway by AVT001 provides preliminary mechanistic evidence supporting this approach. These early-phase results suggesting that an autologous dendritic cell–based vaccine targeting correction of the defect of the Q/E CD8+ Treg pathway may delay progression of T1D warrant further evaluation in a larger trial.
Supplementary Material
Disclosures
Sponsored by Avotres Inc. The Joslin Clinical Research Center is supported by the Joslin Diabetes Research Center through the Division of Diabetes, Endocrinology, and Metabolic Diseases (grant number P30DK036836).
We thank Mikayla Mackey and David Y. I. Kim of the Joslin Diabetes Center and Dan Zhao and Yifan Liu of Avotres Inc. for their contributions to the conduct of the clinical trial and the preparation of the manuscript. In addition, we acknowledge the late Drs. Leonard Chess and Benvenuto Pernis for their extraordinary contributions to the discovery and identification of the Qa-1/human leukocyte antigen-E–restricted CD8+ T cell–mediated regulatory pathway and its role in immune regulation, without which the AVT001 product could not have advanced to its current clinical development status. We also thank the patients, their families, and all investigational site members involved in this trial. Medical writing and editorial support, under the direction of the authors, were provided by Karen Segal, Ph.D., and were funded by Avotres Inc. according to Good Publication Practice guidelines.
Footnotes
Author disclosures and other supplementary materials are available at evidence.nejm.org.
References
- 1.Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care 2003;26: 832–836. DOI: 10.2337/diacare.26.3.832. [DOI] [PubMed] [Google Scholar]
- 2.Green EA, Cooke AC, Piganelli JD, Richardson SJ, Wen L, Wong FS. Editorial: immunopathology of type 1 diabetes. Front Immunol 2022;13:852963. DOI: 10.3389/fimmu.2022.852963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ni Q, Pham NB, Meng WS, Zhu G, Chen X. Advances in immunotherapy of type I diabetes. Adv Drug Deliv Rev 2019;139:83–91. DOI: 10.1016/j.addr.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Skyler JS. New insights into halting type 1 diabetes. Lancet Diabetes Endocrinol 2021;9:475–476. DOI: 10.1016/S2213-8587(21)00169-8. [DOI] [PubMed] [Google Scholar]
- 5.Rickels MR, Evans-Molina C, Bahnson HT, et al. High residual C-peptide likely contributes to glycemic control in type 1 diabetes. J Clin Invest 2020;130:1850–1862. DOI: 10.1172/JCI134057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobsen LM, Bundy BN, Greco MN, et al. Comparing beta cell preservation across clinical trials in recent-onset type 1 diabetes. Diabetes Technol Ther 2020;22:948–953. DOI: 10.1089/dia.2020.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quattrin T, Haller MJ, Steck AK, et al. Golimumab and beta-cell function in youth with new-onset type 1 diabetes. N Engl J Med 2020;383:2007–2017. DOI: 10.1056/NEJMoa2006136. [DOI] [PubMed] [Google Scholar]
- 8.Gitelman SE, Bundy BN, Ferrannini E, et al. Imatinib therapy for patients with recent-onset type 1 diabetes: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol 2021;9:502–514. DOI: 10.1016/S2213-8587(21)00139-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forlenza GP, McVean J, Beck RW, et al. Effect of verapamil on pancreatic beta cell function in newly diagnosed pediatric type 1 diabetes: a randomized clinical trial. JAMA 2023;329:990–999. DOI: 10.1001/jama.2023.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang H, Zhang SI, Pernis B. Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science 1992;256:1213–1215. DOI: 10.1126/science.256.5060.1213. [DOI] [PubMed] [Google Scholar]
- 11.Koh D-R, Fung-Leung W-P, Ho A, Gray D, Acha-Orbea H, Mak T-W. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8-/- mice. Science 1992;256:1210–1213. DOI: 10.1126/science.256.5060.1210. [DOI] [PubMed] [Google Scholar]
- 12.Jiang H, Ware R, Stall A, Flaherty L, Chess L, Pernis B. Murine CD8+ T cells that specifically delete autologous CD4+ T cells expressing V beta 8 TCR: a role of the Qa-1 molecule. Immunity 1995;2:185–194. DOI: 10.1016/S1074-7613(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 13.Ware R, Jiang H, Braunstein N, et al. Human CD8+ T lymphocyte clones specific for T cell receptor V beta families expressed on autologous CD4+ T cells. Immunity 1995;2:177–184. DOI: 10.1016/S1074-7613(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 14.Jiang H, Wu Y, Liang B, et al. An affinity/avidity model of peripheral T cell regulation. J Clin Invest 2005;115:302–312. DOI: 10.1172/JCI23879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, Zhang L, Liang B, et al. Perceiving the avidity of T cell activation can be translated into peripheral T cell regulation. Proc Natl Acad Sci USA 2007;104:20472–20477. DOI: 10.1073/pnas.0709878104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Y, Zheng Z, Jiang Y, Chess L, Jiang H. The specificity of T cell regulation that enables self-nonself discrimination in the periphery. Proc Natl Acad Sci USA 2009;106:534–539. DOI: 10.1073/pnas.0811843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang H, Chess L. The specific regulation of immune responses by CD8+ T cells restricted by the MHC class Ib molecule, Qa-1. Annu Rev Immunol 2000;18:185–216. DOI: 10.1146/annurev.immunol.18.1.185. [DOI] [PubMed] [Google Scholar]
- 18.Hu D, Ikizawa K, Lu L, Sanchirico ME, Shinohara ML, Cantor H. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol 2004;5:516–523. DOI: 10.1038/ni1063. [DOI] [PubMed] [Google Scholar]
- 19.Leavenworth JW, Tang X, Kim HJ, Wang X, Cantor H. Amelioration of arthritis through mobilization of peptide-specific CD8+ regulatory T cells. J Clin Invest 2013;123:1382–1389. DOI: 10.1172/JCI66938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Zaslavsky M, Su Y, et al. KIR+CD8+ T cells suppress pathogenic T cells and are active in autoimmune diseases and COVID-19. Science 2022;376:eabi9591. DOI: 10.1126/science.abi9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang H, Chess L. How the immune system achieves self-nonself discrimination during adaptive immunity. Adv Immunol 2009;102: 95–133. DOI: 10.1016/S0065-2776(09)01202-4. [DOI] [PubMed] [Google Scholar]
- 22.Jiang H, Chess L. Qa-1/HLA-E-restricted regulatory CD8+ T cells and self-nonself discrimination: an essay on peripheral T-cell regulation. Hum Immunol 2008;69:721–727. DOI: 10.1016/j.humimm.2008.08.279. [DOI] [PubMed] [Google Scholar]
- 23.Jiang H, Canfield MS, Gallagher M, et al. HLA-E-restricted regulatory CD8(+) T cells are involved in the development and control of human autoimmune type 1 diabetes. J Clin Invest 2010;120:3641–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu F, Zheng H, Qi Y, et al. PFA-fixed Hsp60sp-loaded dendritic cells as a vaccine for the control of mouse experimental allergic encephalomyelitis. Cell Mol Immunol 2014;11:169–174. DOI: 10.1038/cmi.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valle A, Giamporcaro GM, Scavini M, et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013;62:2072–2077. DOI: 10.2337/db12-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao W, Gitelman S, DiMeglio LA, Boulware D, Greenbaum CJ. Fall in C-peptide during first 4 years from diagnosis of type 1 diabetes: variable relation to age, HbA1c, and insulin dose. Diabetes Care 2016;39:1664–1670. DOI: 10.2337/dc16-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.