

Abstract
Splicing factors are affected by recurrent somatic mutations and copy number variations in several types of haematologic and solid malignancies, which is often seen as prima facie evidence that splicing aberrations can drive cancer initiation and progression. However, numerous spliceosome components also ‘moonlight’ in DNA repair and other cellular processes, making their precise role in cancer difficult to pinpoint. Still, few would deny that dysregulated mRNA splicing is a pervasive feature of most cancers. Correctly interpreting these molecular fingerprints can reveal novel tumour vulnerabilities and untapped therapeutic opportunities. Yet multiple technological challenges, lingering misconceptions, and outstanding questions hinder clinical translation. To start with, the general landscape of splicing aberrations in cancer is not well defined, due to limitations of short-read RNA sequencing not adept at resolving complete mRNA isoforms, as well as the shallow read depth inherent in long-read RNA-sequencing, especially at single-cell level. Although individual cancer-associated isoforms are known to contribute to cancer progression, widespread splicing alterations could be an equally important and, perhaps, more readily actionable feature of human cancers. This is to say that in addition to ‘repairing’ mis-spliced transcripts, possible therapeutic avenues include exacerbating splicing aberration with small-molecule spliceosome inhibitors, targeting recurrent splicing aberrations with synthetic lethal approaches, and training the immune system to recognize splicing-derived neoantigens..
Introduction
In 1977, two groups led by Richard Roberts and Phillip Sharp simultaneously observed the presence of “amazing [loop-like] sequence arrangements” in adenoviral DNA–RNA heteroduplexes, suggesting that the mature messenger RNA (mRNA) molecule is produced by intramolecular joining of short segments of “split genes”1,2. In doing so, they discovered mRNA splicing, a key step in transcript processing, wherein non-contiguous exons are transcribed to form a long pre-mRNA precursor followed by the removal of intervening intronic sequences. Shortly thereafter, introns were identified in many eukaryotic organisms, with the number of spliced genes increasing on average with organismal complexity.
Of note, exons are not always spliced together precisely or sequentially: common alterations include alternative 5’ and 3’ exon boundaries, exon skipping, and intron retention. In humans, it is estimated that over 95% of multi-exon genes undergo alternative splicing3,4, a subset of which can encode proteins with distinct, and sometimes even opposing functions5. In fact, alternative splicing plays a critical role in many cellular processes. They include cell differentiation, development, and proliferation, intracellular signaling and metabolism, and intrinsic and extrinsic apoptosis6–11. It is therefore not surprising that alterations in RNA splicing have been linked to human diseases, including cancer.
Over the past twenty years, the discovery of highly recurrent mutations in genes encoding splicing factors across a variety of tumor types has provided compelling genetic evidence for a direct causal relationship between splicing dysfunction and cancer12. Even in tumors without splicing factor mutations, mRNA splicing patterns are patently aberrant, resulting in the profound re-shaping of cancer transcriptomes and proteomes13,14. This allows aberrant mRNA splicing to enable virtually all recognized ‘Hallmarks of Cancer’15, impacting not only tumor initiation, but also progression, metastatic spread, drug responses, etc12. These far-reaching effects informed the development of first-in-class approaches to target cancer-promoting spliced isoforms or regulators thereof, raising early hopes of translating mRNA splicing research to the clinic12.
10+ years later, our still incomplete understanding of how dysregulated splicing contributes to cancer pathogenesis remains the chief obstacle to achieving this goal. At first glance, the havoc that mutant splicing factors wreak upon pre-cancerous cells appears indiscriminate, with thousands of exons per tumor assembled out of order and thousands of introns retained in otherwise mature transcripts. There might be a method to this madness, i.e., a small number of aberrantly spliced mRNAs that are key to neoplastic transformation. However, currently there is no clear consensus on how these driver events might be identified and whether they could and should be reversed, either genetically or pharmacologically. Building such a consensus would require insights from experts in computational biology, RNA biology, structural/molecular biology, medicinal chemistry, immunology, and hematology/oncology.
Most recent effort in that direction was expended at the Forbeck Forum on Therapeutic Targeting of mRNA Splicing in Cancer held September 21–24, 2023, at the Asilomar Conference Grounds, the site of the 1975 scientific summit that had yielded the influential “Summary Statement of the Asilomar Conference on Recombinant DNA Molecules”16. Not having to worry any longer about hefty recombinant DNA biosafety issues allowed the 2023 Forum participants to focus on “recombinant” (i.e., mis-spliced) RNA molecules and their role in cancer. The resultant Roadmap article is not meant to serve as a comprehensive catalogue of all recent developments in this dynamic field, which have been summarized previously12. Rather, our goal was to highlight basic science advancements, ranging from long-read RNA-sequencing to single-cell proteomics, that could facilitate clinical use of compounds targeting - or taking advantage of - aberrant splicing patterns in cancer.
Dysfunction of splicing regulators in cancer
RNA splicing is a highly regulated process performed by the spliceosome - a very large complex consisting of both small nuclear RNA (snRNA) [G] and protein components - along with additional regulatory splicing factor proteins that fine-tune its activity. The spliceosome recognizes core regulatory sequences in the pre-mRNA, which include the 5’ and 3’ splice sites (5’ and 3’ SS) that mark intron-exon boundaries and flank the branch point site [G] (BPS) and the polypyrimidine tract [G] (Py-tract) (Figure 1a). Splicing reactions are carried out by either of the two spliceosomal complexes, major U2-type and minor U12-type, which differ in a subset of snRNA components and in the splice site sequences they recognize. The major U2-type spliceosome preferentially recognizes GT-AG splice sites and is responsible for the removal of ~99% of introns. It contains over 300 components – including snRNAs that interact with Sm proteins [G] and additional proteins to form small nuclear ribonucleoprotein [G] (snRNP) particles. The minor U12-type spliceosome recognizes both GT-AG and AT-AC sites and is involved in the removal of less than 1% of introns. It utilizes distinct spliceosomal snRNA and protein components, including ZRSR2. Remarkably, as detailed below, both RNA and core protein components of the spliceosome are altered in human tumors (Figure 1b).
Figure 1. Splicing factor alterations in human tumors.
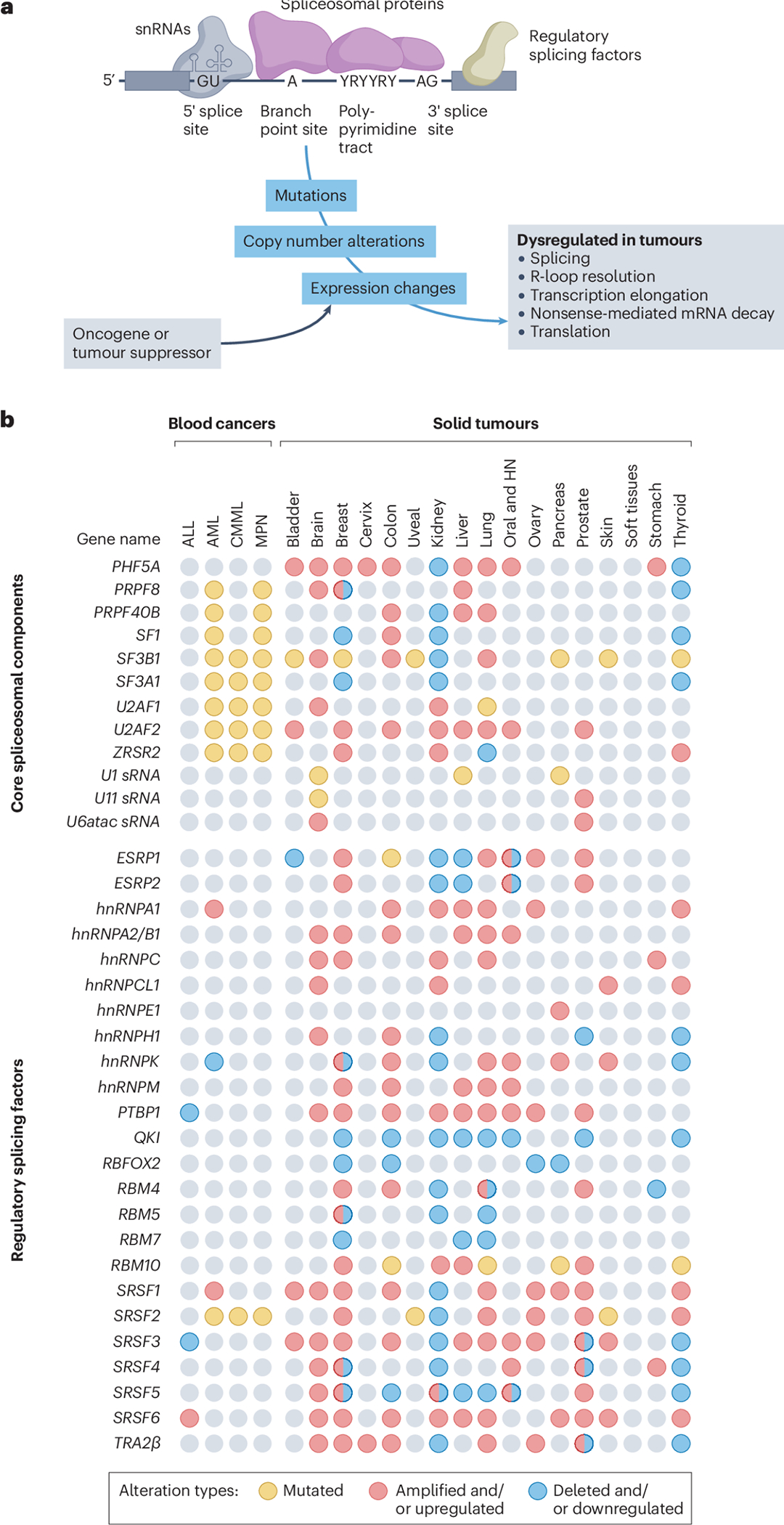
a, Core spliceosomal proteins (purple) and small nuclear RNAs (snRNAs) (grey) are critical components of constitutive RNA splicing, whereas regulatory splicing factors (tan) have a role in alternative splicing. These are depicted assembled on a pre-mRNA molecule composed of exons (grey rectangles) and introns (black lines). The 5′ and 3′ splice sites are depicted along with highly conserved dinucleotides GU and AG, which define intron boundaries, the adenosine residue serving as the branch point site, and the polypyrimidine tract. Tumours exhibit splicing factor mutations, copy number alterations, or expression changes. In some tumours, these splicing factor expression changes are induced directly or indirectly by oncogenes or tumour suppressors. Splicing factor alterations lead to dysregulation of alternative splicing but also of other processes listed on the right.
b, Genomic events that affect expression and function of selected splicing factors in blood (left) and solid (right) cancers. They affect both select core (top) and regulatory (bottom) spliceosome components. The data presented have been curated from the literature and represent key splicing factors that are frequently altered in primary or metastatic human tumours compared with normal tissues (see references in Supplementary Table 1). Splicing factors can be mutated (single nucleotide changes), upregulated with or without gene amplification, or downregulated with or without gene deletion. A limitation of these data is that given their non-focal nature, amplification and shallow deletions are more difficult to functionally interpret than deep deletions. Note that for some splicing factors, individual studies have reported them to be either upregulated or downregulated in the same tumour type. These discrepancies can often be explained by differences in tumour subtypes, tumour stages, and even spatial localization within the tumour tissues (for example, carcinoma in situ versus the invasive tumour front). ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; CMML, chronic myelomonocytic leukaemia; HN, head and neck cancers; MPN, myeloproliferative neoplasms.
Pleiotropic mutations in core spliceosomal proteins
Landmark studies performed concurrently and published almost simultaneously in 2011 described recurrent driver mutations in human genes encoding core components of the spliceosome [G], namely splicing factor 3b subunit 1 (SF3B1), serine and arginine-rich splicing factor 2 (SRSF2, previously known as SC35), zinc finger CCCH-type RNA binding motif and serine/arginine rich 2 (ZRSR2), and U2 small nuclear RNA auxiliary factor 1 (U2AF1)17–20. These mutations, found in both hematologic malignancies and solid tumors, for the first time implicated splicing factors as bona fide cancer drivers21 (Figure 1a,b). All four proteins play key roles during the splicing reaction and are critical for 3’SS recognition: SF3B1 is a component of the U2 snRNP that is involved in BPS recognition and assembly of the pre-spliceosomal complex A, SRSF2 interacts with U1 and U2 snRNPs and mediates recognition of the 5’ and 3’SS, U2AF1 binds to the AG dinucleotide at the 3’SS and is critical for U2 snRNP binding, and ZRSR2 has been implicated in 3’SS recognition for both U2-and U12-type introns. Follow-up molecular studies have revealed that hotspot mutations at specific amino acid residues in SF3B1, SRSF2, and U2AF1 either lead to a gain of function or alter the spliceosome specificity, resulting most frequently in the selection of alternative 3’ splice sites22–26. In contrast, mutations in ZRSR2, which are distributed across the whole gene, lead to a loss of function and improper retention of U12-type introns27,28.
Up until recently, it was assumed that mutations in these genes impact tumor formation and progression mostly by affecting RNA splicing. However, it is now well-understood that mutations in spliceosome components can have additional tumorigenic properties, since their targets are involved in numerous cellular processes beyond splicing. Since splicing occurs on nascent pre-mRNA molecules, it is intimately linked to transcript elongation, with both processes affecting each other29. One important aspect of this crosstalk is that splicing factors U2AF1 and SRSF2 regulate both transcription elongation and nucleocytoplasmic export30–32. SRSF2 additionally has been implicated as a reader of the 5-methylcytosine RNA modification, which in turn affects RNA stability and nuclear export33. Of note, imbalanced levels of nuclear mRNAs (spliced or un-spliced) result in the formation of R-loops [G], triple-stranded structures consisting of a DNA:RNA hybrid and a displaced strand of DNA34. These structures need to be resolved as they are causally connected to double-stranded DNA damage, chromosomal instability, and aneuploidy35. Independently, SRSF2 is involved in the formation of the exon junction complex (EJC)36 [G], a key step required for nonsense-mediated mRNA decay (NMD) [G], a co-translational process that eliminates transcripts containing premature stop codons37. Therefore, cells with mutations in core splicing factors might be expected to have multiple potentially tumorigenic features, namely defects in transcription elongation, R-loop resolution, DNA damage repair, NMD, and translational regulation.
This indeed turned out to be the case. For example, a recent study found that the SF3B1K700E mutation reduces the elongation rate of RNA polymerase II along gene bodies and its density at promoters in leukemia cell models38. This elongation defect results from disrupted pre-spliceosome assembly due to impaired protein-protein interactions of mutant SF3B1. This SF3B1K700E-mediated transcriptional elongation defects also reduces chromatin accessibility and H3K4me3 marks at promoters38, suggesting that modulation of histone tail modifications could counteract the effects of mutant SF3B1K700E. Furthermore, mutations in splicing factors SF3B1, SRSF2 and U2AF1 were all found to augment the formation of R-loops, most likely due to prolonged transcriptional pausing and the availability of free RNA ends generated during splicing-related RNA cleavage events39–43. Importantly, suppression of R-loops has been shown to partially correct the proliferation defect of hematopoietic progenitor cells with mutations in splicing factors, suggesting that these mutations, at least in myelodysplastic neoplasms, lead to chronic insult to the genome and ensuing cell cycle arrest. In turn, there is evidence that DNA damage-induced splicing aberrations preferentially affect hundreds of genes involved in DNA repair, cell cycle progression, and programmed cell death44.
Cancer cells with mutations in key splicing factors also exhibit significant perturbations in NMD. SRSF2-mutants cancer cell lines were found to exhibit enhanced mRNA decay45. Mechanistically, binding of mutant SRSF2 to RNA facilitates the deposition of the EJC downstream from premature stop codons to a greater extent than that of wild-type SRSF2. This in turn boosts the association of key NMD factors with the transcript and augments mRNA decay45. Similarly, cells with mutations in SF3B1 or U2AF1 not only exhibited elevated DNA replication stalling, DNA damage, and chromosomal instability but were more sensitive to NMD inhibition compared to their wild-type counterparts46. Finally, U2AF1 bearing the S34F mutation was shown to directly affect translation of hundreds of transcripts via direct mRNA binding in the cytoplasm47.
Collectively, these findings highlight the complex functions of splicing regulators. Especially when mutated, they can have pleotropic effects, both indirectly (via mRNA splicing and altered proteomes) and directly, by “moonlighting” in a variety of cellular processes. This routine repurposing of RNA-binding proteins48 make seemingly distinct steps in gene expression, from transcriptional regulation to post-translational control, functionally interconnected49,50.
Wide-reaching mutations in snRNAs
Beyond its protein constituents, critical components of the spliceosome are the snRNAs, whose deregulation can profoundly affect splicing patterns. U1 and U2 snRNAs are primarily responsible for the recognition of 5′ splice sites and branch points, respectively, which are critical steps for defining the beginning and the end of introns that will be excised by the major spliceosome (Figure 1a). As snRNAs are short in length and lack poly(A) tails [G], they are not detected by many routinely used RNA-sequencing protocols. However, more targeted approaches designed to capture short noncoding RNAs have revealed that snRNA genes exhibit variable expression levels in human cell lines and adult and fetal tissues51. This suggested that by impacting the efficiency of snRNPs assembly, they play important roles in controlling tissue- and cell-type–specific splicing patterns. Comparable variations in snRNA levels have been detected also across tumors. For example, U6atac snRNA, which regulates the activity of the minor U12 spliceosome, is elevated in primary and metastatic prostate cancer samples, where its levels correlate with metastatic spread52. Mechanistically, U6atac has been shown to control a cluster of genes involved in cell proliferation53, potentially explaining its association with disease progression. And just like core splicing factors, snRNAs influence many steps of RNA processing, not just splicing. For example, U1 precludes selection of proximal polyadenylation signals in introns and last exons (an activity dubbed telescripting54,55), and in doing so, promotes cancer cell migration and invasion in vitro56.
Thus, it comes as no surprise that recurrent mutations in U1 and U2 genes have been detected across several liquid and solid tumor types, namely in blood, liver, prostate, and pancreatic cancers, as well as in pediatric brain tumors57–59. While the impact of common U2 mutations (e.g., U2 c.28) hasn’t been extensively studied, U1 mutations typically change the preferred A-U base-pairing between this snRNA and the 5’SS to the C-G base-pairing, redirecting splicing towards cryptic 5’SS events. In addition, mutations in U11 snRNA, which is responsible for 5′SS recognition of minor introns, have been reported in the Sonic Hedgehog subtype of medulloblastomas57. Collectively, tumors with U1 or U11 snRNA mutations exhibit widespread splicing alterations, with a bias towards cryptic 5’SS, including those mapping to known oncogenes and tumor suppressors57,58.
Aberrant expression of regulatory splicing factors
While recurrent mutations in core spliceosome components of the splicing machinery are common in hematological malignancies (30–80% of patients), these are much less common in solid tumors (1–4% of patients), with the exception of SF3B1 mutations in uveal melanoma12. Solid tumors, on the other hand, exhibit frequent changes in expression levels in regulatory splicing factors that fine-tune the activity of the spliceosome (Figure 1b), most notably RNA-binding proteins of serine/arginine-rich (SR) [G] and heterogeneous nuclear ribonucleoprotein (HNRNP) [G] families. These proteins participate in the splicing of canonical exons, but are particularly adept at regulating inclusion or skipping of non-constitutive “cassette” exons60. To perform these functions, they recognize and bind cis-acting exonic and/or intronic sequences and strengthen or weaken the spliceosome’s recognition of the corresponding splice sites.
In tumors, changes in levels of regulatory splicing factors are sometimes due to focal gene amplifications or “deep” bi-allelic deletions61–65 (which could credential them as cancer drivers), but more commonly due to more elusive “soft-wired” gene expression changes at the transcriptional, post-transcriptional, or post-translational levels63,66–70 (Figure 1b). For example, in B- and T-cell acute lymphoblastic leukemias (B-/T-ALL), where mutations in the core spliceosome components are not observed, many members of the SR protein family are dysregulated at the post-transcriptional level whereby the inclusion of a stop codon-containing poison exon [G] regulates their protein levels71,72. Furthermore, in T-ALL SRSF6 protein levels can be sustained via the activity of ubiquitin-specific peptidase 7 (USP7)72. Similarly, the levels of multiple splicing factors, including SR proteins, are upregulated in breast, lung, kidney, melanoma and others tumors, as a result of increased poison exon skipping in their respective transcripts73,74 (Figure 1b).
Many of these splicing factor up- and down-modulation events could be traced back to bona fide cancer genes dysregulated in the majority of all cancers, such as MYC75 and TP5376. Indeed, evidence shows that tumors with alterations in MYC and TP53 have widespread splicing aberrations63,66–68,77. They can occur by a diverse range of direct and indirect mechanisms (Figure 1a). For example, the MYC oncoprotein is known to bind directly to the promoter regions and regulate the expression of assorted splicing factors, including members of the SR and HNRNP protein families63,68,78–80. Most notably, it transcriptionally upregulates both snRNP assembly genes and their upstream regulators67 such as PRMT5, an arginine methyltransferase that methylates Sm and other RNA binding proteins. These coordinated regulatory effects are key for the biogenesis of snRNPs, effective pre-mRNA splicing, cell survival and proliferation67. Consequently, the MYC–PRMT5 signaling axis is critical to splicing fidelity of exons with weak 5’SS. Therefore, it is a targetable vulnerability of SF3B1 and SRSF2-mutant cancers81. Moreover, as MYC selectively amplifies pre-mRNA synthesis82,83, it increases the pressure on the spliceosome to process more intron-containing transcripts66,67. Thus, MYC-dependent tumors are particularly sensitive to inhibition of the splicing machinery, providing opportunities for synthetic lethal approaches for these aggressive cancers. Although MYC is notable for its ability to profoundly dysregulate tumor cell transcriptomes84, many other cancer drivers have outsized impacts on mRNA splicing. For example, the PI3K–AKT pathway, which is frequently hyperactivated in multiple cancers, regulates multiple SR proteins, both directly85 and indirectly, through the SR protein kinases SRPKs86. As a result, PI3K hotspot mutations drive not only widespread transcriptomic changes but also aberrant splicing that has been shown to support tumor growth in breast cancer models87.
More tissue-restricted oncogenes are also known to influence the landscape of their respective cancers. For instance, in pancreatic cancer, TP53 hotspot missense mutations giving rise to the oncogenic form of p53 are associated with aberrant splicing, when compared to truncating, tumor-suppressive TP53 mutations77. Specifically, TP53 mutant cells upregulate the expression of splicing factor HNRNPK, which preferentially binds to C-rich exonic sequences. As a result, cytosine-rich exons corresponding to KRAS-suppressing GTPase-activating proteins are more frequently included in mutant tumors, leading to heightened KRAS activity and more robust tumor growth77. Similarly, adult gliomas driven by mutant isocitrate dehydrogenase (IDH) dysregulate splicing factors PTBP1, SRSF3, and RBFOX1 compared to their IDH wild-type counterparts, with ensuing changes to splicing patterns88,89.
The effects of splicing factor up- and down-modulation events on tumor phenotypes vary significantly, depending on histology and unique properties of individual family members. In breast tumors, upregulation of specific SR protein family members promotes tumor formation, while others impact cell migration and metastasis63,79. Very recently, deep deletions of SRSF3 and HNRNPU were observed in B-ALL with acquired resistance to chemotherapy, but not in paired diagnostic samples61. There are many other examples of dysregulated splicing factors promoting distinct tumor phenotypes90–93. While such observations could be instrumental in elucidating novel oncogenic and tumor suppressive pathways, the task of identifying key affected transcripts remains challenging, due primarily to the multitude of downstream splicing events.
Mapping splicing alterations in tumors
Unlike mutations in many classical cancer genes (e.g., Ras family members or the PI3 kinases), mutations affecting the spliceosome function have unusually broad, pleiotropic effects on cancer transcriptomes. This puts an emphasis on the precise resolution of RNA isoforms, which is a much more challenging task than merely analyzing difference in gene expression at the level of whole transcripts.
Isoform detection with short-read RNA-sequencing
Over the past twenty years, high-throughput approaches to profile RNA isoforms, from exon-level microarrays to deep short-read RNA-sequencing, have enabled researchers to catalog differences in alternative splicing patterns in cancer cell lines and patients samples with some degree of confidence12. Most cancer studies to date, particularly those based on large-cohorts like The Cancer Genome Atlas (TCGA) and Therapeutically Applicable Research to Generate Effective Treatments (TARGET), have used short-read RNA-sequencing to map and measure splicing in tumor samples14,94. These approaches rely on mapping millions of short reads onto reference or de novo-reconstructed transcriptomes, identifying sequences that are differentially included or skipped, and then quantifying splicing differences between cancer cohorts (Figure 2). Yet, accurate detection and quantification of splicing remains challenging, especially in clinical samples. While most researchers acknowledge that shallow RNA-sequencing (<30 million reads per sample) is not sufficient for accurate splicing detection and isoform mapping, there is growing evidence that even very deep short-read RNA-sequencing (>100 million reads) is not sufficient to adequately define the splicing landscape in certain tissues95.
Figure 2. Four key steps towards clinical translation of mRNA splicing research.
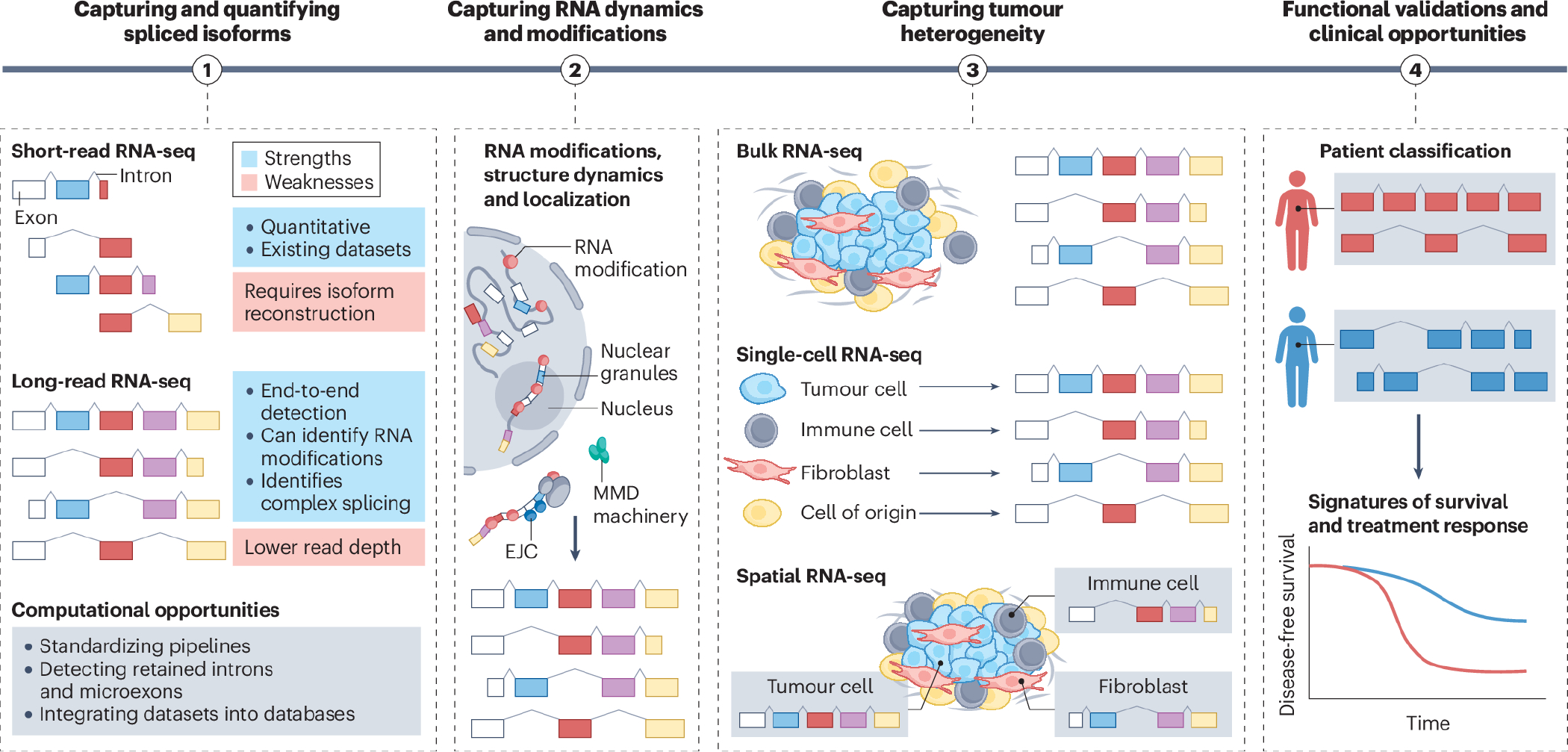
The first step towards translating mRNA splicing research is to capture and quantify isoforms. The strengths and weaknesses are listed for current RNA-sequencing (RNA-seq) approaches, along with computational challenges that remain be addressed. Second, understanding the role of RNA processing in cancer requires profiling of complex repertoires of RNA isoforms, their structure and folding, any chemical modifications (including m7G, N6-methyladenosine (m6A) and m5C ψ), A-I editing (represented by red bubbles), the cellular localization (to specific nuclear comportments or phase-separated granules), the stability and turnover (including cytoplasmic degradation by the nonsense mediated mRNA decay (NMD) pathway of transcripts containing an exon junction complex (EJC) downstream of their stop codon), and the RNA dynamics. All of these factors can impact splicing, stability, nuclear export and translation. Third, capturing spliced isoform heterogeneity and their spatial localization will enable isoforms expressed in tumour cells to be distinguished from those originating in the tumour microenvironment. This is critical for splicing-directed therapies that are currently being developed, often with the assumption that all cells within the tumour express the same isoform. Lastly, identifying spliced isoforms in tumours offers multiple clinical opportunities. For example, classifying patients based on their isoform profile might reveal novel tumour subtypes, some of which might be associated with distinct survival patterns, prognosis, and drug responses.
On the biological side, the problem is that the highly dynamic nature of RNA processing often results in the abundance of rare splicing events, which are increasingly being detected and reported as more samples are sequenced96,97. On the technical side, the heavy reliance on complementary DNA (cDNA) sequencing opens the door for reverse transcription artifacts. For example, the so-called ‘falsitrons’ [G] are artifactual alternative splicing events characterized by missing exonic fragments, which are only detected by reverse transcription-based protocols and, unlike true ‘exitrons’98,99 [G], cannot be experimentally validated using direct long-read RNA-sequencing of native RNA molecules100 (see the next section). Finally, on the computational side, there are almost as many methods to identify spliced isoforms as there are laboratories studying splicing. The most commonly used ones are replicate multivariate analysis of transcript splicing (rMATS), mixture-of-isoforms (MISO), LeafCutter, Modeling Alternative Junction Inclusion Quantification (MAJIQ), SUPPA, and Vertebrate Alternative Splicing and Transcription Tools (VAST-TOOLS)101–108. They rely on either ‘event-centric’ approaches, which focus on identifying and characterizing individual alternative splicing events within genes, or more challenging ‘transcript-centric’ calculations, which consider entire transcript isoforms resulting from alternative splicing as units of analysis. However, even the event-centric approaches often disagree with one another, which is not to say that some are always right or always wrong109. Comparative analyses have shown that each method has its strengths and weaknesses with regard to specific splicing events, and those called by one method but missed by another can be validated using orthogonal approaches104.
Nonetheless, short-read RNA-sequencing approaches are clearly sub-optimal for the reconstruction of full-length isoforms and are even less adept at profiling intron retention109,110. This is because accurate quantification of intron retention events would require reads that span the full length of introns. Such coverage is usually not feasible, given that the mean length of human introns is >3kb111; and reads that partially overlap with introns may not provide sufficient information to accurately estimate the abundance of retained introns. It is also challenging to determine whether any given set of reads maps to translatable protein-coding mRNAs, to long non-coding RNAs, or in fact to NMD substrates. Finally, most computational tools for RNA splicing analysis lack batch correction methods, thus limiting our ability to compare data across studies and models. To the best of our knowledge, the recently developed MOCCASIN is the only tool that addresses this challenge by accounting for both known and unknown confounders in mRNA splicing analysis112.
Isoform detection with long-read RNA-sequencing
Advances in long-read approaches have enabled the detection of end-to-end, full-length RNA molecules using the Pacific Biosciences (PacBio) or Oxford Nanopore Technologies (ONT) RNA-sequencing platforms. These approaches have two important advantages: they do not rely on transcriptome reconstruction required for short-read data and they facilitate the discovery of novel isoforms that are not present in reference transcriptomes (Figure 2). Additionally, by enabling direct long-read RNA-sequencing, ONT can detect chemical (epitranscriptomic) modifications in the RNA (described further below). Unsurprisingly, recent long-read profiling experiments have revealed the increased complexity of the splicing repertoire in tumor samples compared to normal tissues and uncovered novel tumor-specific isoforms missed by current short-read sequencing113–116.
While long-read RNA-sequencing approaches hold the promise of a harmonized view of splicing aberrations in cancer, there is no single universally appropriate method for isoform calling. Detection of de novo events, absent from reference transcriptomes, remains particularly challenging96. This is partly due to technical issues, such as persistent errors in base calling encountered with ONT sequencing. Consequently, accurate mapping and alignment of reads in the context of tumors, which often exhibit novel isoforms absent in reference transcriptomes and tumor-specific gene fusions and rearrangements, can be problematic. To some extent, this challenge can be addressed computationally with algorithms like Full-Length Alternative Isoform analysis of RNA (FLAIR) and Error Statistics PRomoted Evaluator of Splice Site Options (ESPRESSO), which account for splice site motifs and correct identified base calling errors accordingly117,118. An additional limitation of long-read RNA-sequencing is the bias against very long transcripts (those greater than 6 kilobases), therefore making it more challenging to capture retained introns96,119. On the opposite side of the length spectrum a commonly encountered problem is the mapping of microexons [G], a class of exons shorter than 30 nucleotides in size that play key roles in brain development and neuropathologies120,121. Recent studies suggest that microexons are also expressed in non-neuronal tissues and tumors, can encode short peptides and putative neoantigens, and are associated with survival in patients with cancer122–126. It is anticipated that dedicated local alignment methods can improve the mapping of these frequently misaligned exons127,128.
However, the most basic limitation of current long-read approaches is that, until recently, they yielded far fewer reads per sample, thus limiting their utility for isoform quantification across transcriptionally diverse cancer tissues96. To address this limitation, studies have combined long-read RNA-sequencing for isoform identification with short-read RNA-sequencing for isoform quantification, an effective but complex and expensive approach for characterizing the splicing repertoire in tumors113–115,119. Additionally, targeted long-read RNA-sequencing, which uses probe captures to enrich for isoforms of interest, can help to increase read coverage for the targets of interest and enable isoform quantification using only long-read data. A recent study focusing on 468 clinically actionable cancer genes (including frequently mutated genes as well as approved and pre-clinically druggable targets) discovered novel isoforms of tumor suppressor genes (e.g., TP53) that are targeted by NMD in breast cancer cell lines, revealing a common RNA-associated mechanism for tumor suppressor gene inactivation129. Finally, recent advances in ONT technologies as well as concatenation methods for PacBio workflows are increasing the throughput and one day might reach more than 100 million reads per sample, replacing short-read technologies.
To sum up, for this relatively new field of long-read RNA-sequencing there are not only two distinct sequencing platforms (i.e., PacBio vs. ONT) but also a variety of library generation protocols with their inherent limitations and biases (e.g., direct RNA vs. cDNA sequencing) and a rapidly growing number of computational tools. Recently, the Long-read RNA-Seq Genome Annotation Assessment Project (LRGASP) consortium evaluated the use of these technologies and computational approaches for isoform analysis96. In brief, the LRGASP study revealed that libraries with longer, more accurate reads produce better annotated catalogues than those with increased read depth, whereas greater read depth improved transcript quantification. Moreover, when aiming to detect rare and novel transcripts or when using reference-free approaches, incorporating additional orthogonal data and replicate samples is advisable. The key message is that no one method is a clear winner, and the choice of experimental and computational approaches should be driven by the project’s aims96. The LRGASP consortium also experimentally validated many novel transcripts detected by long- and short-read RNA-sequencing methods in individual samples, confirming the existence of an important pool of rare RNA isoforms96. This effort provides a benchmark for current practices, opens the door for improved and more standardized pipelines, and suggests future directions for method development96.
Profiling RNA modifications and dynamics
Our understanding of the role of RNA processing in cancer relies on our ability to define mRNA repertoires in tumors, profiling their overall structure, stability, dynamics, and last but not least, post-transcriptional modifications (Figure 2). The latter is now known to modulate the expression of oncogenes and tumor suppressor genes by affecting nuclear export, stability, and translation130. For example, high levels of the m6A RNA modification destabilize mRNAs encoding metabolic enzymes in the citric acid (TCA) cycle, setting up a decrease in oxidative-phosphorylation metabolism in hematopoietic stem and progenitor cells and favoring the development of leukemia 131. Perhaps not surprisingly, deposition of the m6A mark onto mRNA (usually by METTL3–METTL14 methyltransferases) also affects splicing patterns. One known mechanism is based on the m6A reader YTHDC1 promoting exon inclusion via recruitment of the pre-mRNA splicing factor SRSF3132. In addition to these ‘hyperlocal’ cis effects, m6A can affect splicing transcriptome-wide by virtue of being deposited onto the U6 snRNA by METTL16133. Of note, METTL3, 14 and 16 are all essential genes in human acute myeloid leukemia cell lines134, suggesting that these enzymes could be potential therapeutic targets in AML and leading to the clinical development of STM2457, a highly selective first-in-class inhibitor of METTL3135.
Additional RNA modifications are likely to play their own roles in splicing of cancer-related genes, pseudouridylation of alternatively spliced regions being one recent example136. The growing spectrum of such modifications can be captured by ONT-based platforms, which enable direct sequencing of the native RNA molecules, rather than the resulting cDNA137. While very promising, these technologies still require large amounts of RNA, and may not be generally applicable to clinical samples for which RNA material may be limited. Therefore, high-throughput profiling of RNA modifications and associated splicing events in larger cohorts of cancer patients remains a largely unmet need, especially with respect to disease-specific signatures and tumor evolution.
Capturing splicing heterogeneity at the single cell level
Most splicing studies published to date have relied on bulk RNA-sequencing, where a small (usually frozen) piece of the tumor is ground in a homogenizer, and pools of mRNAs from different compartments are extracted and sequenced. However, bulk approaches are unable to resolve which cells within a given tissue express the detected isoforms. Thus, we have a very limited understanding of isoform heterogeneity and clonality within the neoplastic compartment or within the other cell types present in the tumor microenvironment. Also largely unknown is how intratumoral heterogeneity of alternative splicing impacts disease progression and drug responses. For this, a transition to single-cell and spatial approaches is urgently needed, since only these approaches can fully unravel the role of RNA splicing in tumor evolution and therapeutic resistance (Figure 2). One frequently encountered problem is that the commonly used single-cell RNA-sequencing or spatial transcriptomics 10x Genomics platforms, at least as standardly implemented, only yield sequences less than 100 nucleotides from either 5’ or more commonly 3’ ends of mRNA. Therefore, they don’t capture information about full transcripts and their internal exons. Another key problem is obtaining sufficient read coverage for the quantification of isoforms in the limited material obtained from a single cell or a spatial spot, often requiring amplification steps that can introduce major biases138–141. Finally, some of the current spatial transcriptomic approaches rely on pre-designed transcript probes that are unable to distinguish between spliced isoforms.
While original dedicated single-cell approaches, such as Smart-seq and Smart-seq2, were able to generate read coverage along the whole transcript, their throughput was limited to only several hundred cells142,143. This lower cell number (as compared to the 10X Genomics platform, which interrogates 5,000–10,000 cells per run) sharply reduced the number of cell types and states that can be captured in one experiment. Several newer approaches are being developed to map spliced isoforms in single cells at scale144–148, but these have not yet been extensively applied to tumor tissues. For example, Smart-seq3 combines full-length transcriptome coverage with a 5′ unique molecular identifier for duplication correction, and has greatly increased sensitivity compared to Smart-seq2. It typically allows to detect and reconstruct in silico thousands more transcripts per cell144. Other workflows implement two-step approaches. Examples include single-cell isoform RNA-Seq (ScISOr-Seq) which combines short-read 3′ sequencing with long-read RNA-sequencing (PacBio or ONT)145; single-cell COrrected Long-Read sequencing (scCOLOR-seq), which is a single-cell corrected long-read RNA-sequencing approach which overcomes the high error rate inherent in ONT reads146; and single cell Nanopore sequencing analysis of Genotypes and Phenotypes Simultaneously (scNanoGPS), which performs independent deconvolution of error-prone long-reads into single-cells and single-molecules and calculates both genotypes and phenotypes in individual cells147. Finally, Genotyping of Transcriptomes (GoT)-Splice enables genotyping of transcriptomes with long-read single-cell RNA-sequencing and proteogenomics for single-cell profiling of transcriptomes, surface proteins, somatic mutations, and RNA splicing148.
These single-cell and long-read approaches are being increasingly used to characterize the splicing landscape of human tumors across cell types. Recent studies revealed cell type-specific isoforms and alternative polyadenylation site usages in tumor, immune and/or stromal cells in dissociated single cells from ovarian and kidney tumor samples147,149. In addition, at the tissue level, a handful of studies have combined spatial transcriptomics with long-read RNA-sequencing to understand how splicing contributes to tissue development and disease in situ; yet these have mostly been used in the context of brain development and neurodegeneration150,151, and are yet to be applied to tumors.
Both single-cell and spatial transcriptomic approaches suffer from gene drop-out even at the whole transcript level, which is likely exacerbated at the isoform level. Future technological advances that decrease cost, increase read depth, and improve accuracy and throughput would aid our ability to map splicing in tumors. For example, Multiplexed Arrays Sequencing (MAS-seq) library preparation concatenates transcripts to optimally utilize PacBio long-read RNA-sequencing and increase transcript yield by 5–10-fold152. Thus, more tumor studies benefiting from these advantages are expected in the near future, assisting with the development of splicing-based prognostic and predictive biomarkers.
Functional relevance of splicing aberrations
The rapid advances in sequencing technologies will continue to improve our ability to profile splicing aberrations with great resolution and accuracy. However, as the catalog of recurrent splicing events grows, so will the list of questions about any functional roles that these isoforms might (or might not) play in cancer. There are excellent examples of alternatively spliced mRNA isoforms that have been functionally validated153,154, but the extent to which splicing-derived proteoforms [G] are actually expressed and display distinct functions has been the subject of debate155–157. Therefore, considerable efforts have been expanded to develop tools to assess the functional impact of cancer-associated splicing events, including their utility as prognostic or predictive biomarkers.
Annotation of splicing-derived proteoforms
While splicing events have been suggested to play functional roles in tumor progression67,77,80,158–164, it remains challenging to determine which isoforms are clinically relevant. Improvements are needed for both computational predictions and high-throughput functional testing in pre-clinical model systems. On the computational side, novel methods for prioritizing and assessing the functional relevance of recurrent spliced isoforms found in large-scale transcriptomic datasets are rapidly emerging. These include methods for functional annotation and profiling at the isoform level165, for inferring interaction networks that connect alternative splicing events to functional annotations and to co-regulated genes or pathways166, for predicting the functional impact of the altered splicing patterns13,14, and for integrating with large-scale proteomics datasets167,168. Still needed are databases that integrate splicing signatures from large sets of independent studies. A first example of such a database is MAJIQlopedia, a MAJIQ-based encyclopedia of splicing variations that encompasses 86 human tissues and 41 cancer datasets. It serves as a unique resource for RNA researchers seeking to understand what splicing variations in their transcripts of interest exist across tissues or cancers, and for translational scientists aiming to catalog mRNA isoform usage in tissues/cancers under investigation, all through a user-friendly web-based interface169.
On the experimental side, there is a pressing need to assess which alternatively spliced mRNA isoforms are actually being translated into proteins. Part of the difficulty in addressing this problem lies in the limitations of the methods used to identify proteins or translated mRNAs, particularly for molecules with low expression levels. Theoretically, integrating RNA-sequencing results with large-scale proteomics datasets would be a great way to address this question167,168. However, measurements of protein diversity by mass spectrometry156 are affected by the biases of the digestion enzymes used in the process, which can limit variant detection170. Nonetheless, aberrant peptides produced by cancer-specific splicing alterations can be validated using proteomics14. More recent studies reflecting efforts to increase the diversity of proteases and depth of coverage reveal that 64% of the frame-preserving splicing events of relatively highly expressed genes detected by transcriptomics are translated and present at the protein level157. Additionally, top-down proteomics methods can help identify intact protein isoforms from the same gene171. However, their sensitivity needs to be improved for detecting the full complexity of the proteome, especially in tissues with increased complexities of mRNA isoforms, greater sample-to-sample variability, and limited material availability, such as cancers.
A useful surrogate measurement of translation efficiency is ribosome or polysome profiling. These transcriptome-wide sequencing methods detect RNAs actively engaged by ribosomes and therefore likely to be translated into proteins. However, given that these approaches are typically based on short reads, their outputs often suffer from low coverage and insufficient depth, with only ribosome-protected RNA positions being captured. Thus, profiling translational dynamics at the transcript isoform level is currently challenging. Nevertheless, there is evidence supporting ribosome activity on regions that are unique to spliced isoforms for >50% of the measured events, even after accounting for variation across conditions, such as cell cycle progression, diversity of tissue types, and ‘tumor vs. normal’ comparisons172–174.
While these studies generally support a direct impact of alternative splicing on protein production, additional strategies to address technological challenges are being developed. Their salient features are improve sequencing depths, more precise biochemical selection of ribosome-protected fragments, leveraging multiple datasets to increase consistency across replicates and conditions, combining RNA-seq and Ribo-seq datasets, and generating both comprehensive and cell type-specific transcriptomes172,174. Yet, ribosome or polysome profiling are not routinely used in cancer research, mainly because they are costly and time-consuming, require specialized equipment, and cannot easily be scaled to hundreds of samples.
Functional interrogation of spliced isoforms
Assuming that the majority of polyadenylated mRNA isoforms without retained introns or poison exons are translated into proteins, the next challenge is to assign them functional significance in the context of neoplastic transformation. This task can be approached either at the level of individual transcripts or at scale. One incredibly powerful tool to interrogate the function of specific cancer-associated isoform switches is the use of splice-switching antisense oligonucleotides [G] (ASOs). These short, chemically modified RNA oligonucleotides are complementary to the target sequence in a pre-mRNA, thereby preventing its interaction with the core spliceosome as well as positive or negative regulators of splicing (e.g., SR and HNRNP proteins). Therefore, they can be used to up- or down-regulate any splicing module, including cassette exons, alternative splice sites, mutually exclusive exons, or retained introns. Rational design has made it possible to identify relatively easily ASOs that efficiently modulate splicing at the endogenous level, and demonstrate that a specific splicing event is required for cancer cell growth and/or therapeutic resistance both in vitro and in vivo67,73,163 (Figure 3 and Table 1). For example, in B-ALL cell culture models, it has been demonstrated that ASOs forcing the skipping of the first AUG-containing exon 2 of the CD22 gene makes leukemic cells resistant to the CD22-directed antibody-drug conjugate inotuzumab ozogamycin175. Similarly, the use of ASOs to force inclusion of the poison exon in the FPGS gene renders them resistant to methotrexate61.
Figure 3. Strategies that target mRNA splicing in cancer.
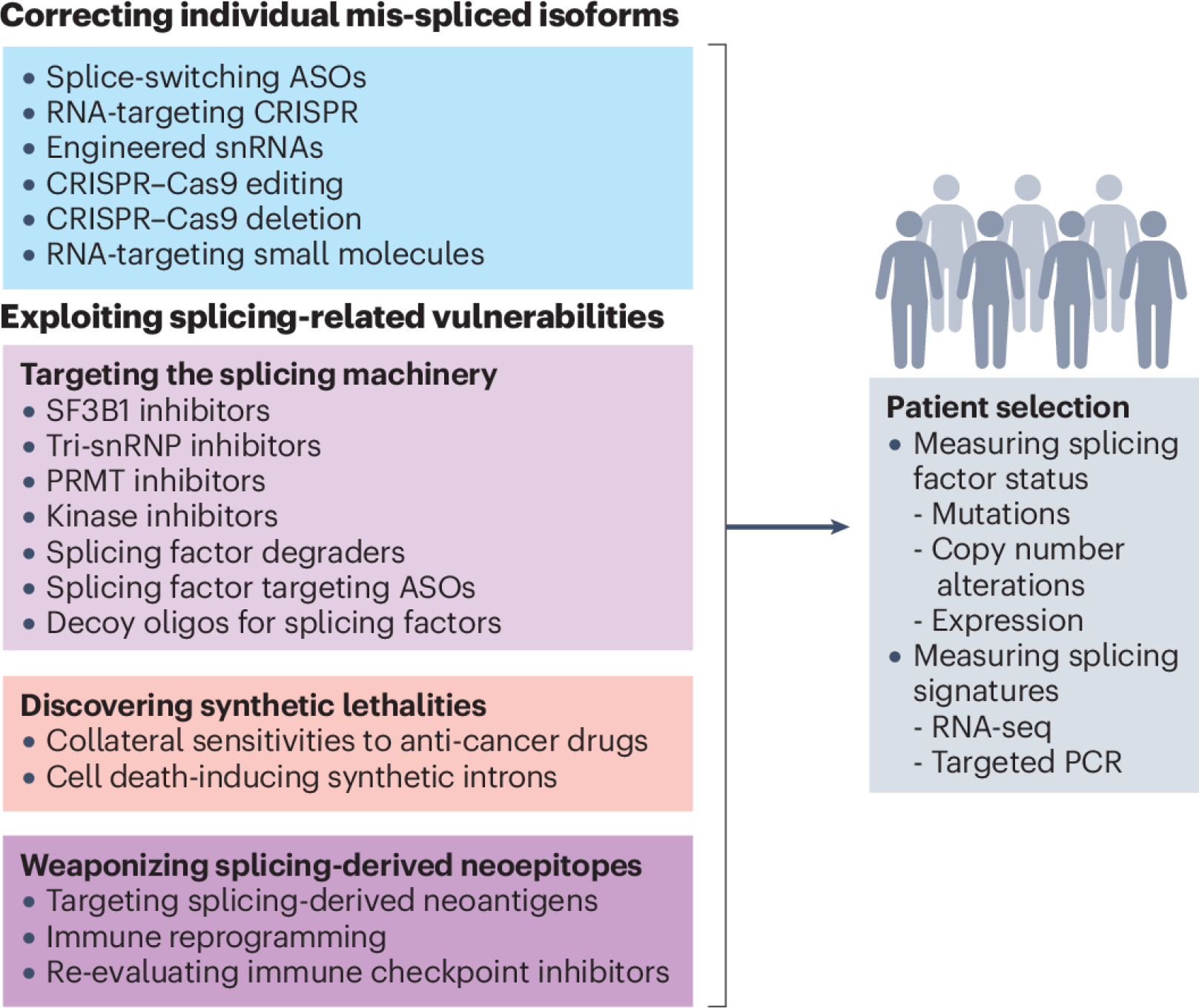
Transcript-centric therapeutic strategies (blue box) target individual mis-spliced mRNAs that are expressed in tumour cells, primarily to alter their splicing patterns. These strategies yield distinct protein isoforms, which either gain tumour-suppressive properties or lose oncogenic ones. Alternatively, they yield non-productive transcripts that are degraded by nonsense-mediated mRNA decay (NMD). Splice-switching can be achieved, for example, by using antisense oligonucleotides (ASOs) that block splicing regulatory elements. Similarly, exogenous small nuclear RNAs (snRNAs) can be engineered to match specific splice sites and in doing so increase inclusion ‘weak’ exons. For genome editing, Cas9 proteins are often paired with single guide RNAs (sgRNAs) to delete a specific exon in a given transcript or to base-edit a specific splice site to restore or to compromise its utilization, leading to exon inclusion or skipping, respectively. Finally, recent studies reported the discovery of small molecules that can recognize a specific RNA sequence or structure and modulate splicing of a specific exon. Spliceosome-centric strategies (light purple box), focus on proteins that controls splicing of multiple downstream transcripts, instead of targeting specific RNA isoforms. These approaches include small-molecule inhibitors that block the activity of the core spliceosomal protein splicing factor 3b subunit 1 (SF3B1), prevent the recruitment of the U4/U5/U6 tri-snRNP, or block the activity of protein kinases or protein arginine methyltransferases (PRMTs) which regulate the activity of splicing factors at the post-translational level. Alternatively, the protein levels of a specific splicing factor can be directly targeted using dedicated protein degraders or splice-switching ASOs that, by modulating poison exon skipping or inclusion, can increase or decrease splicing factor protein levels. Finally, an ASO with sequence complementarity to a splicing factor binding motif can be used as decoy to sequester the splicing factor away from its cognate mRNA targets. In parallel, numerous approaches can exploit synthetic lethalities (pink box), as specific splicing alterations often create new vulnerabilities, including sensitizing cancer cells to conventional anti-cancer drugs. Alternatively, a set of specific introns were found to be spliced exclusively in cancer cells with SF3B1 mutations, opening the door to the use of synthetic intron sequences to enable SF3B1 mutation-dependent expression of exogenous cell death-inducing genes. The last class of splicing-based therapies leverages splicing-derived neo-epitopes produced by cancer cells or even creates new ones (dark purple box). These strategies often involve chimeric antigen receptor (CAR) T cells that target abnormally expressed surface protein isoforms or induce immune reprogramming by selectively promoting spliced isoforms associated with enhanced cytotoxicity, cytokine secretion and the generation of memory T cells. The success of these strategies in the clinic will require appropriate patient selection, which should include more systematic molecular measurements of both the splicing factor status and RNA isoform signatures (grey box). This will help to identify patients who would benefit from therapies targeting specific mRNA isoforms versus those that would respond to more global approaches exploiting splicing-related vulnerabilities. RNA-seq, RNA sequencing; snRNP, small nuclear ribonucleoprotein.
Table 1.
Therapies targeting splicing in preclinical studies and clinical trials
| Target | Approach | Clinical Trial | Tumor Type | Ref. |
|---|---|---|---|---|
| SF3B1 | Small molecule inhibitor (H3B-8800 and E-7107) | Yes | MDS | 228,271 |
| PRMT5 | Small molecule inhibitor (EPZ015666, GSK3326595, JNJ-64619178, PRT543, PRT811 ) | Yes | MDS, blood cancer, solid tumors | 81,272 |
| Pan type-I PRMTs | Small molecule inhibitor (MS023) | No | Blood | 81 |
| RBM39 | Degrader (E7820) | Yes | MDS | 241 |
| U4/U5/U6 tri-snRNP | Small molecule inhibitor (Isoginkgetin) | No | Skin | 229–231 |
| SRPK1 | Small molecule inhibitor (SRPKIN-1, SRPIN340, MVRL09, and SHPINX) | No | Solid tumors | 235 |
| CLK1 | Small molecule inhibitor (TG003) | No | Colon | 234–236 |
| DYRKs & pan-CLK | Small molecule inhibitor (SM08502) | Yes | Solid tumors | 236 |
| U2AF complex | Small molecule stabilizer (NSC-194308) | No | Blood | 273 |
| BRD9 | Splice-switching ASO | No | Skin | 176 |
| ERG | Splice-switching ASO | No | Prostate | 274 |
| FGFR1 | Splice-switching ASO | No | Brain | 275 |
| MCL1 | Splice-switching ASO | No | Skin | 276 |
| MDM4 | Splice-switching ASO | No | Skin | 163 |
| MST1R (RON) | Splice-switching ASO | No | Breast, Stomach | 277 |
| STAT3 | Splice-switching ASO | No | Breast | 278 |
| USP5 | Splice-switching ASO | No | Brain | 279 |
| ERBB4 | Splice-switching ASO | No | Breast | 280 |
| MDM2 | Splice-switching ASO | No | Uterine | 281 |
| ATM | Splice-switching ASO | No | Blood | 282 |
| BRCA2 | Splice-switching ASO | No | Breast | 283 |
| EZH2 | Splice-switching ASO | No | Blood | 81 |
| MKNK2 | Splice-switching ASO | No | Brain | 284 |
| BCL-X | Splice-switching ASO | No | Solid tumors | 285 |
| BIM | Splice-switching ASO | No | Blood | 286 |
| GLDC | Splice-switching ASO | No | Lung | 287 |
| IL5R | Splice-switching ASO | No | Blood | 288 |
| PKM2 | Splice-switching ASO | No | Brain | 289 |
| SRSF3 | Splice-switching and splicing factor inhibition ASO | No | Breast | 290 |
| TRA2β | Splice-switching and splicing factor inhibition ASO | No | Breast | 73 |
| RBFOX | Decoy ASO and splicing factor inhibition | No | Others | 233 |
| SRSF1 | Decoy ASO and splicing factor inhibition | No | Others | 233 |
| PTBP1 | Decoy ASO and splicing factor inhibition | No | Others | 233 |
Abbreviations: ASO, antisense oligonucleotide, ATM, ataxia-telangiectasia mutated; BCL-X, Bcl-2-like protein 1; BIM, Bcl-2-like protein 11; BRCA2, breast cancer type 2 susceptibility protein; BRD9, bromodomain-containing protein 9; CLK1, dual specificity protein kinase CLK1; DYRK, dual-specificity tyrosine phosphorylation-regulated kinase; EZH2, histone-lysine N-methyltransferase; FGFR1, fibroblast growth factor receptor 1; GLDC, Glycine dehydrogenase; IL5R, interleukin 5 receptor; MKNK2, MAP kinase-interacting serine/threonine-protein kinase 2; MDS, myelodysplastic syndrome; MST1R (aka RON), Macrophage Stimulating 1 Receptor; PKM2, Pyruvate kinase PKM; PRMT5, Protein arginine N-methyltransferase 5; PTBP1, polypyrimidine tract-binding protein 1; RBFOX, RNA-binding protein fox-1 homolog; RBM39, RNA-binding motif protein 39; SF3B1, splicing factor 3B subunit 1; SRSF1, serine/arginine-rich splicing factor 1; SRPK1, SRSF protein kinase 1; STAT3, signal transducer and activator of transcription 3; TRA2β, transformer-2 protein homolog beta; USP5, Ubiquitin carboxyl-terminal hydrolase 5.
Several studies also have demonstrated that splice-switching ASOs can delay tumor formation or metastasis in pre-clinical models12 (Table 1). For example, SF3B1-mutant tumors include a poison exon in the bromodomain containing 9 (BRD9) gene leading to BRD9 transcript degradation. An ASO that forces exon skipping increase BRD9 protein levels and decreased tumor volume in uveal melanoma mouse models176. The main benefit of such splice-switching ASOs is that, at least theoretically, they can be used not only for pre-clinical testing in model organisms, but also as viable therapeutic modalities in humans. In fact, several ASOs have been approved by the US Food and Drug Administration (FDA) for non-cancerous genetic diseases177,178 and could one day reach the oncology space. The main drawback of using splice-switching ASOs for functional interrogation is that this approach is not scalable, and candidate ASOs need to be tested individually. Screening hundreds or thousands of them one by one to prioritize candidate splicing events would be time-consuming and expensive, to the point of not being feasible in an academic setting. The same can be said about overexpression or knockdown approaches.
Fortunately, approaches to interrogate splicing isoforms at scale do exist. The impact of hundreds of spliced isoforms can be simultaneously tested in model systems using paired guide RNAs for alternative exon removal74 or single-base editors to program exon skipping in cultured cells or xenograft models of cancer179,180 (Figure 3). However, these approaches target DNA sequences and therefore have the potential to impact both genomic and epigenomic regulatory elements or alter gene transcription. RNA-targeting CRISPR approaches are an attractive alternative strategy that can be used either to sterically block a splice site and prevent its usage, or to guide the recruitment of artificial CRISPR-fused splicing factors to modulate either exon inclusion or skipping. In fact, these approaches have been used successfully for combinatorial targeting of multiple exons73,181–183 (Figure 3).
The main limitations of genome- and transcriptome-editing approaches are three-fold. One is the need for efficient delivery of both the Cas proteins and the guide RNAs. The second is the relatively low efficiency of the current editing approaches. The third and perhaps the most problematic one is the fact that the majority of studies focus on exon-skipping events, which represent less than half of the splicing alterations detected in tumors. Cassette exons studies are also biased in favor of NMD-inducing events, which are easier to model, and given their putative loss-of-function consequences, easier to interpret functionally. Future improvements will be needed to model the impact of other splicing alterations, including intron retention, alternative 5’ or 3’ splice site choices, mutually exclusive exons, as well as combinations of multiple splicing events in the same transcript.
Interpreting splicing signatures
Independently of our ability to pinpoint alternative splicing events of singular significance, there is growing appreciation of the prognostic utility of complex splicing signatures. Several pan-cancer studies demonstrated that alternative splicing-based survival predictors outperform gene expression-based ones. Specifically, splicing signatures can accurately predict survival in breast184, gastric185, pancreatic186, lung187, and ovarian188 cancer patients as well as in adults with high-grade gliomas88. These discoveries underscore the need to integrate clinical and gene expression data with alternative splicing profiles for the most accurate risk stratification and survival prediction in cancer patients189,190. It could also inform treatment decisions, leading to improved patient outcomes.
This being said, the use of splicing signatures in cancer patients has several caveats. One such caveat is the growing evidence suggests that splicing changes can occur in pre-neoplastic tissues. The case in point is clonal hematopoiesis, a common preleukemic condition. Recent data suggest that mutations in splicing factors occur very early during disease initiation and can act as founder mutations27,191,192. Similarly, studies of bladder cancer evolution have reported that mutations in splicing factor RNA binding protein 10 (RBM10) are frequent in most tumor clones, suggesting that splicing alterations could occur early during solid tumor formation and clonal expansion193. It remains unknown whether these events predict progression to frank malignancies more accurately than genomic or epigenetic alterations.
With regard to risk factors, one of the main ones is age, at least in adult cancers. Yet its impact on splicing remains understudied. Interestingly, both spliced isoforms and splicing regulators have been reported to change with age in human and mouse tissues as well as in the context of age-related neurological disorders194, emphasizing splicing dysregulation as a hallmark of aging195,196. While many of the splicing changes seem to be largely tissue specific, a number of splicing patterns, including aberrant intron retention, have been found across aged tissues in multiple studies197–200. Similarly, a loss of splicing fidelity at specific intronic sequences has been seen in age-related neurodegenerative diseases198 and in cancers201. In addition, mutant splicing factors have been causally linked with longevity in model systems199,202–205. Despite the links between aging, cancer and splicing, most studies ignore the aging dimension when modeling and measuring the impact of splicing in cancer. This gap in knowledge has profound implications for our understanding of the mechanistic origins of splicing aberrations in solid tumors and whether they play a role in clonal cell expansion in pre-neoplastic tissues.
Therapeutic targeting of mRNA splicing
Following the identification and functional validation of key splicing aberrations, the development of therapeutic approaches could be approached from two broad angles: mis-splicing normalization and mis-splicing exploitation. The first approach entails identifying individual stand-out “driver” splicing events and reversing them in vivo with ASOs, CRISPR-mediated DNA or RNA editing; or else via modulation of the splicing machinery, as part of systemic splicing normalization (Figure 3). While splicing normalization might sound like an attractive scenario conceptually, many practical hurdles would complicate the development of such therapies, including but not limited to i) the pan-essential function of several components of the spliceosome206; ii) the difficulties in targeting RNA binding proteins, which lack traditional druggable pockets and often contain low-complexity unstructured domains; iii) the loss-of-function nature of many splicing factor mutations (e.g., deep deletions); and iv) the lack of drugs selective for the mutant splicing factor variant in the cases of neomorphic mutations. A radically different second approach is to consider aberrant splicing as a desirable therapeutic vulnerability and to take advantage of it with dedicated targeted or immuno-therapies (Figure 3).
Correcting individual mis-spliced isoforms
The success of this approach is predicated on the assumption that while individual tumors might have thousands of splicing alterations, the vast majority of them are “passenger” events, with only one or two “driver” events being germane to cancerous growth. Such events would likely affect genes already classified as cancer drivers based on accumulation of acquired mutations or copy number variations in tumors of the same origin. Examples of such events may include splicing variants of mutant BRAF with the hotspot V600E amino acid substitution. These splice variants, lacking exons encoding for the Ras binding domain, are often detected in melanoma patients with acquired resistance to B-Raf inhibitor vemurafenib207. Similarly, it has been proposed that the exon-16-skipping HER2 variant has oncogenic activity in breast carcinomas, where HER2 is a known driver208. In these scenarios, one might envision employing therapeutic strategies to target individual spliced isoforms. Such strategies might utilize RNA-based drugs, such as ASOs, to reprogram specific splicing switches and/or to target tumor-associated spliced isoforms that are otherwise undruggable with classical small-molecule compounds209. Because of their potential to restore normal splicing patterns or to selectively target cancer-specific splicing events, these therapies, hold promise for precision oncology.
In preclinical settings, there have been some success stories (Table 1). Splice-switching ASOs have been used to target splicing of the MDM4 transcript. Normally, its translation product suppresses p53 only in highly proliferating cells such as embryonic stem cells, but it is often re-expressed in cancers. ASOs promoting MDM4 exon 6 skipping led to MDM4 protein downregulation, p53 reactivation, and decreased tumor growth in vivo in a TP53 wild-type melanoma PDX model 163. Similarly, splice-switching ASOs can rewire cancer cell metabolism by targeting the M2 pyruvate kinase (PKM2) isoform, which is upregulated in most cancers. In contrast, PKM1 is expressed in terminally differentiated, non-proliferating cells. This is because PKM2, but not PKM1, promotes the Warburg effect, which favors aerobic glycolysis over oxidative phosphorylation for energy metabolism153. Both PKM1 and PKM2 transcripts are derived from the PKM gene through alternative splicing of two mutually exclusive exons: PKM1-specific exon 9 and PKM2-specific exon 10. Splice-switching ASOs promoting a switch from exon 10 to exon 9 usage led to a protein switch from PKM2 to PKM1 and delayed liver cancer cell growth in vitro and in vivo in xenograft models164. Lastly, ASOs modulating myosin phosphatase RHO-interacting protein, a splicing target of the tumor suppressive RNA binding protein fox-1 homolog 2 (RBFOX2), limited pancreatic metastases by altering cytoskeletal organization and focal adhesion162.
The RBFOX2 study also introduced the broader concept of a metastatic signature of alternative splicing, based, at least in part, on the epithelial-mesenchymal transition (EMT)210. Indeed, EMT, broadly recognized as one of the hallmarks of cancer15, is a program activated during cancer cell dissemination to distant organs. Importantly, a combinatorially regulated EMT-associated RNA splicing signature faithfully predicts breast cancer patient survival184. This is because EMT involves multiple transcripts undergoing isoform switching: CD44, FGFR2, Rac1, and ENAH, among others158,211–213. In the case of CD44, the epithelial CD44v isoform sustains Ras-MAPK signaling and in doing so promotes cell proliferation214. Conversely, the alternatively spliced mesenchymal CD44s isoform activates PI3K signaling, leading to enhanced survival, invasion, cancer stem cell properties, and ultimately, cancer metastasis158,160,215,216. Besides RBFOX2217, several regulatory splicing factors have been reported to contribute to EMT, either as activators (hnRNPM and QKI218,219) or as repressors (ESRP1, AKAP8, hnRNPF, and RBM4770,159,161,220,221). ASOs could be very instrumental in determining which of their targets (beyond the usual suspects) might contribute to EMT and metastatic spread.
Beyond ASOs, desirable mRNA-induced changes in splicing might be achieved by targeting distinctive features of RNA sequences at RNA-protein interfaces222,223. This is the mechanism by which Risdiplam, an FDA-approved drug that targets SMN2 splicing to restore the production of viable SMN protein (and the first oral medication approved for the treatment of spinal muscular atrophy), appears to function224. While some small molecules targeting RNA structures have advanced to the clinic225, none of them was designed to modulate splicing for therapeutic use in the oncology space.
In summary, few success stories notwithstanding, both the selection of a single target that would strongly impact the tumor phenotype and the intra-tumoral delivery of splice-switching drugs remains challenging. While splice-switching ASOs have shown promise in pre-clinical models, not a single one has led to complete tumor regression as a monotherapy. It could well be that the underlying issues are merely technical in nature, and that better chemistry and better delivery tools will yield promising results. However, a more sober view is that one target might never be enough, as aberrant splicing contributes to oncogenesis by dysregulating multiple pathways and gene networks, causing the failure of various checkpoint mechanisms. In that case, it might make more sense to consider splicing aberrations a therapeutic vulnerability, where the attainable goal would be not to “fix” but rather to exploit them.
Exploiting splicing-related vulnerabilities
Targeting the splicing machinery
Current strategies to target the core spliceosome in tumors where some of its components are already mutated are based on the idea that there are limits to how much splicing dysregulation a tumor cell can tolerate12. Indeed, there is ample preclinical evidence that cancers with splicing factor mutations and imprecisely functioning splicing machineries can be targeted by direct spliceosome inhibitors and/or inhibition of enzymes that regulate splicing factors via post-translational modifications (Table 1 and references therein). This, in turn, affects both disease pathogenesis and therapeutic responses. By systemically inhibiting the already dysfunctional cancer cell spliceosomes (manifested by alternative 3’SSs, widespread intron retention, etc.), we can find a therapeutic window within which normal tissues with unperturbed splicing will be largely spared.
Clinical-grade small molecules that modulate or inhibit aberrant RNA splicing have been developed and are currently being tested to treat oncological patients in pre-clinical studies and clinical trials, both for blood cancers and solid tumors12 (Table 1). These include molecules that disrupt the splicing reactions, such as inhibitors of the core spliceosome component SF3B1226–228, as well as compounds that prevent the recruitment of the U4/U5/U6 tri-snRNP229–231. H3B-8800 and E-7107 are first-in-class splicing modulators targeting SF3B1, which showed promise in preclinical studies and a phase 1/2 clinical trial, where treatment with H3B-8800 resulted in transfusion independence in a subset of patients with myelodysplastic neoplasms bearing splicing factor mutations228. Other approaches to directly target splicing factors include degraders of the splicing factor RBM39232, as well as ASOs that either act as decoys to sequester splicing factors and attenuate their activity233 or block their production at the level of transcription73 (Table 1).
Moreover, the biogenesis and activity of many splicing factors are regulated by post-translational modifications. Consequently, the activity of select splicing factors could be down-modulated by inhibitors of specific protein kinases (e.g., CLKs, SPRKs, dual specificity tyrosine-phosphorylation-regulated kinases DYRKs)234–236, or arginine methyltransferases PRMTs81,237,238. Ongoing Phase 1 clinical trials are exploring PRMT5 inhibitors and an RBM39 degrader in patients with splicing factor-mutant myelodysplastic neoplasms [see US National Library of Medicine’s https://clinicaltrials.gov/study/NCT03573310 (2018), https://clinicaltrials.gov/study/NCT03886831 (2019), and https://clinicaltrials.gov/study/NCT05024994 (2021)].
Targeting the splicing machinery, although elegant in principle, is not without its challenges. Early clinical trials of SF3B1 inhibitors were terminated due to on-target, off-tumor toxicity in the retina227, and the more recent SF3B1 inhibitor H3B-8800 induced cardiac toxicities228. Follow-up studies of SF3B1 inhibitors in low-risk splicing factor-mutant leukemias have recently been closed after disappointing phase I/II trial results [see US National Library of Medicine’s https://clinicaltrials.gov/study/NCT02841540 (2016)]. While recently developed inhibitors might be more promising, it is still unclear how cytotoxicity can be managed. Moreover, as stem cells share many alternative splicing similarities with cancer cells65, the therapeutic window separating cancer cells from stem cells and other healthy cells remains elusive. Lastly, it remains to be determined whether targeting the regulatory splicing machinery rather than the core spliceosome, would elicit less toxicity.
Discovering splicing-based synthetic lethalities
Given that so far spliceosome inhibitors have not transformed the therapeutic landscape of hematological malignancies, one can ask whether we should turn our attention to discovering unique vulnerabilities arising from splicing, but not directly involving splicing regulators. Indeed, there is emerging evidence that splicing alterations might drive resistance to conventional chemotherapy while creating new vulnerabilities. In B-ALL, inclusion of a microexon into the 5’-nucleotidase, cytosolic II (NT5C2) mRNA was recently shown to confer resistance of leukemic cells to thiopurine while simultaneously rendering them sensitive to the immunosuppressive drug mizoribine (and quite possibly the more widely used mycophenolate mofetil)61,71. Similarly, in refractory AML with U2AF1 mutations there was increased inclusion of a poison exon in transcripts encoding the eukaryotic translation initiation factor 4A2 (EIF4A2). This elicited the integrated stress response (ISR) and rendered AML cells sensitive to the ISR inhibitor ISRIB combined with chemotherapy239. More broadly speaking, there is emerging evidence that splicing factor mutations confer sensitivity to drugs targeting not only the core spliceosome240, but also the RNA binding motif protein 39 (RBM39)241, the Sin3–histone deacetylase (HDAC) pathway38, and protein arginine methyltransferases81. In murine models of AML, mutations in SF3B1 and SRSF2 independently converged on nuclear factor κB (NFκB) signaling242, making this pathway a potential dependency. Conversely, DNA damage repair-deficient cancer cells (e.g., those bearing cohesin mutations) were reported to be exquisitely sensitive to SF3B1 inhibitors, and this treatment can sensitize them to subsequent treatment with poly ADP ribose polymerase (PARP) inhibitors or chemotherapy243.
Other vulnerabilities stemming from aberrant splicing are likely to be discovered through the use of carefully designed CRISPR screens. Alternatively, such vulnerabilities can be artificially created. An elegant approach illustrating this concept was recently developed based on the use of cell-death inducing synthetic introns, which are only processed in cells bearing neomorphic mutations in splicing factors. Inserting these cassettes in the herpes simplex virus-thymidine kinase (HSV-TK) gene allowed TK expression only in SF3B1-mutant cancers, and subsequent treatment with gancyclovir suppressed growth lethal xenografts and improved mouse host survival244. How close such gene therapy-based approaches are to entering clinical trials only the future will show.
Weaponizing splicing-derived neoepitopes
There is considerable evidence that at least in hematological malignancies alternative splicing can contribute to epitope loss and cause immunotherapy failures. Examples include loss of CD19245–248, CD22175, CD20249, and CD33250. One the other hand, alternative splicing has the well-recognized, but still largely untapped propensity to create new epitopes. The approaches closest to clinical translation entail either designing immunotherapies targeting abnormally expressed proteoforms or improving responses to immune checkpoint blockade by increasing the production of splicing-derived neoantigens. Such shared splicing-derived neoantigens have been identified in melanoma251,252, breast and ovarian253 and lung cancers254. However, in most cases identifying shared major histocompatibility complex (MHC)-presented neoantigens for T cell receptor (TCR)-based therapies remains challenging and laborious, in part because human leukocyte antigens (HLAs) are highly polymorphic in the human population and restrict the repertoires of peptides presented to T cells. It is estimated that an average tumor harbors very few, if any, abnormal splicing-derived peptide253. It may therefore be more feasible to devise therapies targeting surface proteins with chimeric antigen receptor (CAR) T cells or antibody drug conjugates, which do not rely on MHC and are HLA-agnostic. A number of computational tools exist to predict splicing-derived neo-epitopes, such as Isoform peptides from RNA splicing for Immunotherapy target Screening (IRIS) and ISOform-guided prediction of epiTOPEs in cancer (ISOTOPE)123,255. There are also examples in the literature of antibodies raised against splice isoforms of surface proteins that have the potential to become good therapeutic targets, such as CD22 in B-cell in human B-ALL175 and collagen 11A in patient-derived xenograft models of pediatric osteosarcoma, Ewing’s sarcoma, and rhabdomyosarcoma256. However, single-cell data is needed to assess intratumoral heterogeneity and to identify neo-epitopes generated by tumor cells themselves and not by other cell types within the tumor microenvironment.
In addition, splicing-based therapies could be aimed at reprogramming the immune system. This approach takes advantage of the fact that alternative splicing plays a crucial role in determining the functional properties of immune cell receptors, cytokines, and signaling molecules257–263. By selectively promoting the generation of splicing isoforms associated with enhanced cytotoxicity, cytokine secretion, and memory formation, therapies altering splicing can augment the ability of immune cells to recognize and eliminate cancer cells. Furthermore, modulating splicing could regulate immune checkpoint pathways to improve immune responses. For example, by targeting alternative splicing events that control the expression of checkpoint receptors (e.g., PD-1, CTLA-4) and their ligands, it might be possible to fine-tune immune responses and to prevent checkpoint blockade264–267. For example, creating decoy PD-1 through forced skipping of exons encoding the transmembrane domain could prevent its cell surface expression264,268. Alternatively, splice-switching ASOs could target key regulatory networks to relieve immunosuppression in cancer. The prototype preclinical study utilized ASOs that mediate knockdown of FOXP3, achieving reduction of suppressive functions of Tregs in mouse models of lymphoma269. A comprehensive understanding of the splicing regulatory landscape in immune cells and its dysregulation in cancer is therefore crucial for rational design and optimization of splicing-based immunotherapies.
Conclusions
The past 15 years witnessed the wide-spread adoption of next-generation sequencing technologies, including short-read RNA-sequencing270. The availability of large transcriptomics datasets along with companion bioinformatics tools led to the cataloging of thousands of non-canonical mRNA isoforms, many of which corresponded to known cancer drivers. This in turn raised hopes that, unlike “hard-wired” somatic mutations and copy number alterations, the “soft-wired” splicing aberration could be reversed with ASOs and other RNA-targeting therapeutics. In parallel, the discovery of splicing factor mutations in human cancers and the concurrent development of SF3B1 inhibitors (E-7107 and its successors) generated a surge in optimism over the clinical use of spliceosome inhibitors, at least against cancers with splicing factor mutations.
So far, both attempts at clinical translation have achieved rather modest successes. Splice-switching ASO drugs proved to be very useful tools for preclinical studies, with greater specificity and less off-target toxicity than traditional small-molecule inhibitors. Moreover, there are documented examples of localized delivery of ASOs to brain, skeletal muscle, and especially liver178. However, whether these nucleic acid-based drugs could bypass the hepatic reticuloendothelial system following intravenous administration or be delivered to disseminated tumors remains to be determined. Small molecule SF3B1 inhibitors have not become FDA-approved cancer medicines, due to their limited efficacy and both on- and off-target toxicities, especially in the retina and the heart.
Despite these setbacks, we are far from being pessimistic and do believe that the time for clinical translation of mRNA splicing research will come. However, before this can happen, both technological and conceptual innovations would need to be implemented, and the two will likely arrive hand in hand. Improvements in long-read single-cell and spatial RNA-sequencing will enrich our understanding of tumor heterogeneity and various neoplastic cell states. Advances in CRISPR-based DNA engineering and especially RNA editing will allow us to interpret with greater confidence the functional significance of individual splicing variations and broad splicing signatures. Creating better in vivo delivery vehicles for ASOs and related compounds will facilitate the credentialing and prioritization of therapeutic targets. It will also enable longitudinal studies of mRNA splicing and the mapping of evolutionary trajectories of clones with splicing aberrations. Large-scale CRISPR/Cas9 and chemical library screens will uncover new vulnerabilities of cancer cells with dysregulated splicing. Finally, new approaches to antibody discovery and protein engineering will allow us to generate and test at scale novel immunotherapeutics directed against splicing-derived proteoforms. On the diagnostic side, better mRNA profiling panels will allow us to identify novel prognostic and predictive biomarkers, beyond somatic mutations in SF3B1 and other core spliceosome components. All this will undoubtedly take time, as the much-needed technologies won’t mature overnight. Nevertheless, we believe that the progress towards clinical translation is best achieved not through experimental and therapeutic shortcuts, but rather through steady investments in cross-pollinating branches of biomedical research.
Supplementary Material
Acknowledgements
The authors thank the Forbeck Foundation for supporting the Forbeck Forum on Therapeutic Targeting of mRNA Splicing in Cancer (co-chairs: A.T.-T. and O.A.). Relevant research in the authors’ laboratories was supported by National Institutes of Health (NIH) grants (R01HL167071 and R01GM140735 to K.M.N.; K08CA245242 to S.X.L.; U01CA232563 to A.T.-T.; R01CA248317, R01GM138541, R21AG080243 and P30CA034196 to O.A.; R35GM136426 to D.B.; R01CA249204 and P30CA196521 to E.G.; R35GM131876 and R01CA182467 to C.C.), CureSearch For Children’s Cancer Acceleration Initiative Grant (to A.T.T.), Emerson Collective Grant (to A.T.T.), Doris Duke Clinical Scientist Development Award (to S.X.L.), Scott R. MacKenzie Foundation Grant (to O.A.), JAX Brooks Scholar award (to B.L.A.), European Innovation Council (to J.V.), European Research Council (to J.V.), Asociación Española Contra el Cáncer (to J.V.) and Australian National Health and Medical Research Council Grant (2018833 to E.E.). C.C. is a CPRIT Scholar in Cancer Research (RR160009).
GLOSSARY
- Antisense oligonucleotide (ASO)
short synthetic chemically modified single-stranded RNA molecules that can bind to specific RNA sequence and alter their function
- Branch point site (BPS)
a nucleotide that performs a nucleophilic attack on the 5’ splice site in the first step of splicing
- Exitrons
non-constitutive introns located within annotated protein-coding exons
- Exon junction complex (EJC
protein complex assembled on the spliced mRNA at the junction of two exons
- Falsitrons
artifactual alternative splicing events characterized by missing exonic fragments, which are only detected by reverse transcription-based protocols
- Heterogeneous nuclear ribonucleoproteins (hnRNPs)
a large family of RNA-binding proteins that regulate multiple RNA processing steps including alternative splicing
- Microexons
a class of exons shorter than 30 nucleotides
- Nonsense-mediated mRNA decay (NMD)
a translation-coupled quality control mechanism that removes mRNAs with premature termination codons
- Polypyrimidine tract
a pyrimidine (C or T)-rich motif upstream of many 3’ splice sites that is bound by U2AF2 to facilitate 3’ splice site recognition
- Poly(A) tail
long chains of adenine nucleotides added to the 3’ of mRNA molecules to increase stability, export and contribute to their translation
- Poison exon
an exon that introduces a premature termination codon when included in the spliced mRNA
- Proteoform
a variant of a protein, including variation due to alternative splicing
- R-loop
a three-stranded nucleic acid structure consisting of an RNA molecule that has invaded duplex DNA
- Serine/arginine-rich (SR) proteins
a large family of RNA-binding proteins that contain one or two serine/arginine-rich domains and regulate multiple RNA processing steps including alternative splicing
- Sm proteins
family of small proteins that bind to RNA and are part of the spliceosome
- Small nuclear ribonucleoprotein (snRNP)
RNA-protein complex that are part of the spliceosome
- Small nuclear RNA (snRNA)
RNA component of the spliceosome
Footnotes
Competing interests
J.V. is a member of the Advisory Boards of Remix Therapeutics, Stoke Therapeutics and IntronX. K.M.W. is an advisor to and holds equity in Ribometrix, ForagR Medicines and A-Form Solutions. O.A. is a member of the Advisory Boards of Caeruleus Genomics.
References
- 1.Berget SM, Moore C & Sharp PA Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci U S A 74, 3171–3175 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chow LT, Gelinas RE, Broker TR & Roberts RJ An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 12, 1–8 (1977). [DOI] [PubMed] [Google Scholar]
- 3.Pan Q, Shai O, Lee LJ, Frey BJ & Blencowe BJ Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40, 1413–1415 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Wang ET et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kjer-Hansen P & Weatheritt RJ The function of alternative splicing in the proteome: rewiring protein interactomes to put old functions into new contexts. Nat Struct Mol Biol 30, 1844–1856 (2023). [DOI] [PubMed] [Google Scholar]
- 6.Marasco LE & Kornblihtt AR The physiology of alternative splicing. Nat Rev Mol Cell Biol 24, 242–254 (2023). [DOI] [PubMed] [Google Scholar]
- 7.Boise LH et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74, 597–608 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Cascino I, Fiucci G, Papoff G & Ruberti G Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol 154, 2706–2713 (1995). [PubMed] [Google Scholar]
- 9.Ueda H et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423, 506–511 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Li X et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat Cell Biol 18, 561–571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han H et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498, 241–245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradley RK & Anczukow O RNA splicing dysregulation and the hallmarks of cancer. Nat Rev Cancer 23, 135–155 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Climente-Gonzalez H, Porta-Pardo E, Godzik A & Eyras E The Functional Impact of Alternative Splicing in Cancer. Cell Rep 20, 2215–2226 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Jayasinghe RG et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep 23, 270–281 e273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discov 12, 31–46 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Berg P, Baltimore D, Brenner S, Roblin RO & Singer MF Summary statement of the Asilomar conference on recombinant DNA molecules. Proc Natl Acad Sci U S A 72, 1981–1984 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papaemmanuil E et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 365, 1384–1395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida K et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Graubert TA et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44, 53–57 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang L et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365, 2497–2506 (2011). References 17–20 revealed that in hematologic malignancies multiple splicing factors accumulate mutations at rates comparable to those of well-established cancer drivers.
- 21.Seiler M et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep 23, 282–296 e284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alsafadi S et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7, 10615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darman RB et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep 13, 1033–1045 (2015). [DOI] [PubMed] [Google Scholar]
- 24. Brooks AN et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 9, e87361 (2014). This study was one of the first to establish direct associations between splicing factor mutations in cancer and alterations in splicing events.
- 25.Ilagan JO et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25, 14–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim E et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630 (2015). This study documented the specificity in the downstream effects of splicing factor mutations.
- 27.Chen S, Benbarche S & Abdel-Wahab O Splicing factor mutations in hematologic malignancies. Blood 138, 599–612 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoue D et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet 53, 707–718 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chathoth KT, Barrass JD, Webb S & Beggs JD A splicing-dependent transcriptional checkpoint associated with prespliceosome formation. Mol Cell 53, 779–790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin S, Coutinho-Mansfield G, Wang D, Pandit S & Fu XD The splicing factor SC35 has an active role in transcriptional elongation. Nat Struct Mol Biol 15, 819–826 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gama-Carvalho M, Barbosa-Morais NL, Brodsky AS, Silver PA & Carmo-Fonseca M Genome-wide identification of functionally distinct subsets of cellular mRNAs associated with two nucleocytoplasmic-shuttling mammalian splicing factors. Genome Biol 7, R113 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ujvari A & Luse DS Newly Initiated RNA encounters a factor involved in splicing immediately upon emerging from within RNA polymerase II. J Biol Chem 279, 49773–49779 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Ma HL et al. SRSF2 plays an unexpected role as reader of m(5)C on mRNA, linking epitranscriptomics to cancer. Mol Cell 83, 4239–4254 e4210 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hegazy YA, Fernando CM & Tran EJ The balancing act of R-loop biology: The good, the bad, and the ugly. J Biol Chem 295, 905–913 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petermann E, Lan L & Zou L Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat Rev Mol Cell Biol 23, 521–540 (2022). [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z & Krainer AR Involvement of SR proteins in mRNA surveillance. Mol Cell 16, 597–607 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Kurosaki T, Popp MW & Maquat LE Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol 20, 406–420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boddu PC et al. Transcription elongation defects link oncogenic SF3B1 mutations to targetable alterations in chromatin landscape. Mol Cell 84, 1475–1495 e1418 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol Cell 68, 745–757 e745 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell 69, 412–425 e416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cusan M et al. SF3B1 mutation and ATM deletion codrive leukemogenesis via centromeric R-loop dysregulation. J Clin Invest 133 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen HD et al. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res 78, 5363–5374 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh S et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 34, 2525–2530 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shkreta L & Chabot B The RNA Splicing Response to DNA Damage. Biomolecules 5, 2935–2977 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahman MA, Lin KT, Bradley RK, Abdel-Wahab O & Krainer AR Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev 34, 413–427 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheruiyot A et al. Nonsense-Mediated RNA Decay Is a Unique Vulnerability of Cancer Cells Harboring SF3B1 or U2AF1 Mutations. Cancer Res 81, 4499–4513 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palangat M et al. The splicing factor U2AF1 contributes to cancer progression through a noncanonical role in translation regulation. Genes Dev 33, 482–497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi PS & Thomas-Tikhonenko A RNA-binding proteins of COSMIC importance in cancer. J Clin Invest 131, e151627 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buccitelli C & Selbach M mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet 21, 630–644 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Shine M et al. Co-transcriptional gene regulation in eukaryotes and prokaryotes. Nat Rev Mol Cell Biol 25, 534–554 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mabin JW, Lewis PW, Brow DA & Dvinge H Human spliceosomal snRNA sequence variants generate variant spliceosomes. RNA 27, 1186–1203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Augspach A et al. Minor intron splicing is critical for survival of lethal prostate cancer. Mol Cell 83, 1983–2002 e1911 (2023). This study argues that the processing of a subset of introns by the minor spliceosome is rate-limiting for cancer cell growth and a potential target for cancer therapies.
- 53.Younis I et al. Minor introns are embedded molecular switches regulated by highly unstable U6atac snRNA. Elife 2, e00780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berg MG et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell 150, 53–64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaida D et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oh JM et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat Commun 11, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suzuki H et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574, 707–711 (2019). This study revealed the accumulation of hotspot mutations of the U1 snRNA in a subset of pediatric medulloblastomas.
- 58. Shuai S et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 574, 712–716 (2019). This study, along with Ref. 58, demonstrated for the first time that mutations in the RNA components of the spliceosome can contribute to cancer progression.
- 59.Bousquets-Munoz P et al. PanCancer analysis of somatic mutations in repetitive regions reveals recurrent mutations in snRNA U2. NPJ Genom Med 7, 19 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Busch A & Hertel KJ Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA 3, 1–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torres-Diz M et al. An Alternatively Spliced Gain-of-Function NT5C2 Isoform Contributes to Chemoresistance in Acute Lymphoblastic Leukemia. Cancer Res (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Karni R et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14, 185–193 (2007). This study was the first demonstration of the oncogenic properties of regulatory splicing factors.
- 63.Park S et al. Differential Functions of Splicing Factors in Mammary Transformation and Breast Cancer Metastasis. Cell Rep 29, 2672–2688 e2677 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen-Eliav M et al. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol 229, 630–639 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Sebestyen E et al. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res 26, 732–744 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu TY et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Koh CM et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100 (2015). References 66–67 uncovered the dependence of MYC-driven cancers on the spliceosome activity and its therapeutic implications.
- 68.Urbanski L et al. MYC regulates a pan-cancer network of co-expressed oncogenic splicing factors. Cell Rep 41, 111704 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ho JS et al. HNRNPM controls circRNA biogenesis and splicing fidelity to sustain cancer cell fitness. Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reinke LM, Xu Y & Cheng C Snail represses the splicing regulator epithelial splicing regulatory protein 1 to promote epithelial-mesenchymal transition. J Biol Chem 287, 36435–36442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Black KL et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res 46, 11357–11369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou Y et al. Posttranslational Regulation of the Exon Skipping Machinery Controls Aberrant Splicing in Leukemia. Cancer Discov 10, 1388–1409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leclair NK et al. Poison Exon Splicing Regulates a Coordinated Network of SR Protein Expression during Differentiation and Tumorigenesis. Mol Cell 80, 648–665 e649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Thomas JD et al. RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat Genet 52, 84–94 (2020). This study used large CRISPR-based screens to assess the function of alternative exons and revealed the contribution of open reading frame-disrupting exons to tumor suppression.
- 75.Dhanasekaran R et al. The MYC oncogene - the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol 19, 23–36 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Levine AJ Spontaneous and inherited TP53 genetic alterations. Oncogene 40, 5975–5983 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Escobar-Hoyos LF et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 38, 198–211 e198 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Das S, Anczukow O, Akerman M & Krainer AR Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 1, 110–117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Anczukow O et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 19, 220–228 (2012). This study demonstrated for the first time direct links between splicing factor dysregulation and tumor phenotypes.
- 80. David CJ, Chen M, Assanah M, Canoll P & Manley JL HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368 (2010). This study demonstrated the existence of an integrated transcriptional/post-transcriptional axis behind the Warburg effect.
- 81. Fong JY et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 36, 194–209 e199 (2019). This study credentialed inhibitors of post-translational modifications of splicing factors as novel anti-cancer therapeutics.
- 82.Muhar M et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sabo A et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511, 488–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin CY et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Blaustein M et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol 12, 1037–1044 (2005). This study demonstrated a direct link between cell signaling pathways and the activity of SR proteins.
- 86.Zhou Z et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47, 422–433 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Claridge SE & Hopkins BD PI3King the Environment for Growth: PI3K Activation Drives Transcriptome Changes That Support Oncogenic Growth. Cancer Res 82, 2216–2218 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song X et al. RNA splicing analysis deciphers developmental hierarchies and reveals therapeutic targets in adult glioma. J Clin Invest 134 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Song X et al. SRSF3-Regulated RNA Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple Cellular Processes. Cancer Res 79, 5288–5301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wan L et al. Splicing Factor SRSF1 Promotes Pancreatitis and KRASG12D-Mediated Pancreatic Cancer. Cancer Discov 13, 1678–1695 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Z et al. Splicing factor BUD31 promotes ovarian cancer progression through sustaining the expression of anti-apoptotic BCL2L12. Nat Commun 13, 6246 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nanjo S et al. Deficiency of the splicing factor RBM10 limits EGFR inhibitor response in EGFR-mutant lung cancer. J Clin Invest 132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Deng L et al. SF3A2 promotes progression and cisplatin resistance in triple-negative breast cancer via alternative splicing of MKRN1. Sci Adv 10, eadj4009 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Closa A et al. A convergent malignant phenotype in B-cell acute lymphoblastic leukemia involving the splicing factor SRRM1. NAR Cancer 4, zcac041 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y et al. Evaluating the impact of sequencing depth on transcriptome profiling in human adipose. PLoS One 8, e66883 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pardo-Palacios FJ et al. Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. Nat Methods 21, 1349–1363 (2024). This study is a comprehensive benchmarking of existing long-read RNA sequencing methods and computational tools.
- 97.Garcia-Ruiz S et al. IntroVerse: a comprehensive database of introns across human tissues. Nucleic Acids Res 51, D167–D178 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marquez Y, Hopfler M, Ayatollahi Z, Barta A & Kalyna M Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res 25, 995–1007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang TY et al. A pan-cancer transcriptome analysis of exitron splicing identifies novel cancer driver genes and neoepitopes. Mol Cell 81, 2246–2260 e2212 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schulz L et al. Direct long-read RNA sequencing identifies a subset of questionable exitrons likely arising from reverse transcription artifacts. Genome Biol 22, 190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Katz Y, Wang ET, Airoldi EM & Burge CB Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7, 1009–1015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li YI et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet 50, 151–158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vaquero-Garcia J et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 5, e11752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trincado JL et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol 19, 40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alamancos GP, Pages A, Trincado JL, Bellora N & Eyras E Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 21, 1521–1531 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shen S et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gohr A et al. Computational Analysis of Alternative Splicing Using VAST-TOOLS and the VastDB Framework. Methods Mol Biol 2537, 97–128 (2022). [DOI] [PubMed] [Google Scholar]
- 108.Vaquero-Garcia J et al. RNA splicing analysis using heterogeneous and large RNA-seq datasets. Nat Commun 14, 1230 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mehmood A et al. Systematic evaluation of differential splicing tools for RNA-seq studies. Brief Bioinform 21, 2052–2065 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Byrne A et al. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun 8, 16027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gazave E, Marques-Bonet T, Fernando O, Charlesworth B & Navarro A Patterns and rates of intron divergence between humans and chimpanzees. Genome Biol 8, R21 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Slaff B et al. MOCCASIN: a method for correcting for known and unknown confounders in RNA splicing analysis. Nat Commun 12, 3353 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun Q et al. Long-read sequencing reveals the landscape of aberrant alternative splicing and novel therapeutic target in colorectal cancer. Genome Med 15, 76 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oka M et al. Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol 22, 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Veiga DFT et al. A comprehensive long-read isoform analysis platform and sequencing resource for breast cancer. Sci Adv 8, eabg6711 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen H et al. Long-Read RNA Sequencing Identifies Alternative Splice Variants in Hepatocellular Carcinoma and Tumor-Specific Isoforms. Hepatology 70, 1011–1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gao Y et al. ESPRESSO: Robust discovery and quantification of transcript isoforms from error-prone long-read RNA-seq data. Sci Adv 9, eabq5072 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Tang AD et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11, 1438 (2020). This study contains the first long-read characterization of transcriptomes in CLL in relation to SF3B1 mutations.
- 119.Han SW, Jewell S, Thomas-Tikhonenko A & Barash Y Contrasting and Combining Transcriptome Complexity Captured by Short and Long RNA Sequencing Reads. bioRxiv (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Irimia M et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523 (2014). This study identified an extensive network of very short alternatively spliced exons regulated in the nervous system by the SRRM4 splcing factor, which also might play a role in CNS tumors.
- 121.Raj B et al. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol Cell 56, 90–103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang S et al. A widespread length-dependent splicing dysregulation in cancer. Sci Adv 8, eabn9232 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pan Y et al. IRIS: Discovery of cancer immunotherapy targets arising from pre-mRNA alternative splicing. Proc Natl Acad Sci U S A 120, e2221116120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mochizuki Y et al. Alternative microexon splicing by RBFOX2 and PTBP1 is associated with metastasis in colorectal cancer. Int J Cancer 149, 1787–1800 (2021). [DOI] [PubMed] [Google Scholar]
- 125.Head SA et al. Silencing of SRRM4 suppresses microexon inclusion and promotes tumor growth across cancers. PLoS Biol 19, e3001138 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Conn VM et al. SRRM4 Expands the Repertoire of Circular RNAs by Regulating Microexon Inclusion. Cells 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Parada GE et al. MicroExonator enables systematic discovery and quantification of microexons across mouse embryonic development. Genome Biol 22, 43 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Z, Zhu C, Steinmetz LM & Wei W Identification and quantification of small exon-containing isoforms in long-read RNA sequencing data. Nucleic Acids Res 51, e104 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang F et al. TEQUILA-seq: a versatile and low-cost method for targeted long-read RNA sequencing. Nat Commun 14, 4760 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Barbieri I & Kouzarides T Role of RNA modifications in cancer. Nat Rev Cancer 20, 303–322 (2020). [DOI] [PubMed] [Google Scholar]
- 131.Gao Y et al. ALKBH5 modulates hematopoietic stem and progenitor cell energy metabolism through m(6)A modification-mediated RNA stability control. Cell Rep 42, 113163 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xiao W et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell 61, 507–519 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Pendleton KE et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 169, 824–835 e814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Barbieri I et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yankova E et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martinez NM et al. Pseudouridine synthases modify human pre-mRNA co-transcriptionally and affect pre-mRNA processing. Mol Cell 82, 645–659 e649 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Acera Mateos P et al. Prediction of m6A and m5C at single-molecule resolution reveals a transcriptome-wide co-occurrence of RNA modifications. Nat Commun 15, 3899 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Arzalluz-Luque A & Conesa A Single-cell RNAseq for the study of isoforms-how is that possible? Genome Biol 19, 110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fu Y et al. Single cell and spatial alternative splicing analysis with long read sequencing. Res Sq (2023). [Google Scholar]
- 140.Buen Abad Najar CF, Yosef N & Lareau LF Coverage-dependent bias creates the appearance of binary splicing in single cells. Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Olivieri JE, Dehghannasiri R & Salzman J The SpliZ generalizes ‘percent spliced in’ to reveal regulated splicing at single-cell resolution. Nat Methods 19, 307–310 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ramskold D et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30, 777–782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Picelli S et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098 (2013). [DOI] [PubMed] [Google Scholar]
- 144.Hagemann-Jensen M et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat Biotechnol 38, 708–714 (2020). [DOI] [PubMed] [Google Scholar]
- 145.Gupta I et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat Biotechnol (2018). [DOI] [PubMed] [Google Scholar]
- 146.Philpott M et al. Nanopore sequencing of single-cell transcriptomes with scCOLOR-seq. Nat Biotechnol 39, 1517–1520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shiau CK et al. High throughput single cell long-read sequencing analyses of same-cell genotypes and phenotypes in human tumors. Nat Commun 14, 4124 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cortes-Lopez M et al. Single-cell multi-omics defines the cell-type-specific impact of splicing aberrations in human hematopoietic clonal outgrowths. Cell Stem Cell 30, 1262–1281 e1268 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dondi A et al. Detection of isoforms and genomic alterations by high-throughput full-length single-cell RNA sequencing in ovarian cancer. Nat Commun 14, 7780 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Joglekar A et al. A spatially resolved brain region- and cell type-specific isoform atlas of the postnatal mouse brain. Nat Commun 12, 463 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hardwick SA et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat Biotechnol 40, 1082–1092 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Al’Khafaji AM et al. High-throughput RNA isoform sequencing using programmed cDNA concatenation. Nat Biotechnol 42, 582–586 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Christofk HR et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233 (2008). [DOI] [PubMed] [Google Scholar]
- 154.Chaffer CL et al. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res 66, 11271–11278 (2006). [DOI] [PubMed] [Google Scholar]
- 155.Ellis JD et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell 46, 884–892 (2012). [DOI] [PubMed] [Google Scholar]
- 156.Tress ML, Abascal F & Valencia A Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem Sci 42, 98–110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sinitcyn P et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat Biotechnol 41, 1776–1786 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Brown RL et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest 121, 1064–1074 (2011). This study revealed the essentiality of splicing isoform switching in the control of cell states and tumor progression.
- 159.Li J et al. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang H et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev 33, 166–179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hu X et al. The RNA-binding protein AKAP8 suppresses tumor metastasis by antagonizing EMT-associated alternative splicing. Nat Commun 11, 486 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jbara A et al. RBFOX2 modulates a metastatic signature of alternative splicing in pancreatic cancer. Nature 617, 147–153 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dewaele M et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest 126, 68–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ma WK et al. ASO-Based PKM Splice-Switching Therapy Inhibits Hepatocellular Carcinoma Growth. Cancer Res 82, 900–915 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.de la Fuente L et al. tappAS: a comprehensive computational framework for the analysis of the functional impact of differential splicing. Genome Biol 21, 119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lee K, Yu D, Hyung D, Cho SY & Park C ASpediaFI: Functional Interaction Analysis of Alternative Splicing Events. Genomics Proteomics Bioinformatics 20, 466–482 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Miller RM et al. Enhanced protein isoform characterization through long-read proteogenomics. Genome Biol 23, 69 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pozo F et al. Assessing the functional relevance of splice isoforms. NAR Genom Bioinform 3, lqab044 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Quesnel-Vallieres M, Jewell S, Lynch KW, Thomas-Tikhonenko A & Barash Y MAJIQlopedia: an encyclopedia of RNA splicing variations in human tissues and cancer. Nucleic Acids Res 52, D213–D221 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang X et al. Detection of Proteome Diversity Resulted from Alternative Splicing is Limited by Trypsin Cleavage Specificity. Mol Cell Proteomics 17, 422–430 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tran JC et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 480, 254–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Calviello L, Hirsekorn A & Ohler U Quantification of translation uncovers the functions of the alternative transcriptome. Nat Struct Mol Biol 27, 717–725 (2020). [DOI] [PubMed] [Google Scholar]
- 173.Weatheritt RJ, Sterne-Weiler T & Blencowe BJ The ribosome-engaged landscape of alternative splicing. Nat Struct Mol Biol 23, 1117–1123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Reixachs-Sole M, Ruiz-Orera J, Alba MM & Eyras E Ribosome profiling at isoform level reveals evolutionary conserved impacts of differential splicing on the proteome. Nat Commun 11, 1768 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zheng S et al. Modulation of CD22 Protein Expression in Childhood Leukemia by Pervasive Splicing Aberrations: Implications for CD22-Directed Immunotherapies. Blood Cancer Discov 3, 103–115 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Inoue D et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 574, 432–436 (2019). This study identified alterations in splicing of BRD9, a component of the BAF chromatin remodeling complex, as key downstream effectors of mutant SF3B1.
- 177.Bennett CF, Krainer AR & Cleveland DW Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu Rev Neurosci 42, 385–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Collotta D, Bertocchi I, Chiapello E & Collino M Antisense oligonucleotides: a novel Frontier in pharmacological strategy. Front Pharmacol 14, 1304342 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gapinske M et al. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol 19, 107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yuan J et al. Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol Cell 72, 380–394 e387 (2018). [DOI] [PubMed] [Google Scholar]
- 181.Du M, Jillette N, Zhu JJ, Li S & Cheng AW CRISPR artificial splicing factors. Nat Commun 11, 2973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li JD, Taipale M & Blencowe BJ Efficient, specific, and combinatorial control of endogenous exon splicing with dCasRx-RBM25. Mol Cell 84, 2573–2589 e2575 (2024). [DOI] [PubMed] [Google Scholar]
- 183.Recinos Y et al. CRISPR-dCas13d-based deep screening of proximal and distal splicing-regulatory elements. Nat Commun 15, 3839 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Qiu Y, Lyu J, Dunlap M, Harvey SE & Cheng C A combinatorially regulated RNA splicing signature predicts breast cancer EMT states and patient survival. RNA 26, 1257–1267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jun Y et al. Comprehensive Analysis of Alternative Splicing in Gastric Cancer Identifies Epithelial-Mesenchymal Transition Subtypes Associated with Survival. Cancer Res 82, 543–555 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ruta V et al. An alternative splicing signature defines the basal-like phenotype and predicts worse clinical outcome in pancreatic cancer. Cell Rep Med 5, 101411 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li Y et al. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett 393, 40–51 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Ouyang Y et al. Alternative splicing acts as an independent prognosticator in ovarian carcinoma. Sci Rep 11, 10413 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shen S, Wang Y, Wang C, Wu YN & Xing Y SURVIV for survival analysis of mRNA isoform variation. Nat Commun 7, 11548 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Trincado JL, Sebestyen E, Pages A & Eyras E The prognostic potential of alternative transcript isoforms across human tumors. Genome Med 8, 85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lawson ARJ et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 370, 75–82 (2020). [DOI] [PubMed] [Google Scholar]
- 194.Angarola BL & Anczukow O Splicing alterations in healthy aging and disease. Wiley Interdiscip Rev RNA 12, e1643 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Schmauck-Medina T et al. New hallmarks of ageing: a 2022 Copenhagen ageing meeting summary. Aging (Albany NY) 14, 6829–6839 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Harries LW Dysregulated RNA processing and metabolism: a new hallmark of ageing and provocation for cellular senescence. FEBS J 290, 1221–1234 (2023). [DOI] [PubMed] [Google Scholar]
- 197.Pabis K et al. A concerted increase in readthrough and intron retention drives transposon expression during aging and senescence. Elife 12 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Adusumalli S, Ngian ZK, Lin WQ, Benoukraf T & Ong CT Increased intron retention is a post-transcriptional signature associated with progressive aging and Alzheimer’s disease. Aging Cell 18, e12928 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Heintz C et al. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature 541, 102–106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wang K et al. Comprehensive map of age-associated splicing changes across human tissues and their contributions to age-associated diseases. Sci Rep 8, 10929 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dvinge H & Bradley RK Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7, 45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Curran SP & Ruvkun G Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 3, e56 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Holly AC et al. Changes in splicing factor expression are associated with advancing age in man. Mech Ageing Dev 134, 356–366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lee BP et al. Changes in the expression of splicing factor transcripts and variations in alternative splicing are associated with lifespan in mice and humans. Aging Cell 15, 903–913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tabrez SS, Sharma RD, Jain V, Siddiqui AA & Mukhopadhyay A Differential alternative splicing coupled to nonsense-mediated decay of mRNA ensures dietary restriction-induced longevity. Nat Commun 8, 306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576 e516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Poulikakos PI et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390 (2011). This study is the first report of a splicing-mediated mechanism of resistance to an approved cancer therapy.
- 208.Castiglioni F et al. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer 13, 221–232 (2006). [DOI] [PubMed] [Google Scholar]
- 209.Crooke ST, Liang XH, Baker BF & Crooke RM Antisense technology: A review. J Biol Chem 296, 100416 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lyu J & Cheng C Regulation of Alternative Splicing during Epithelial-Mesenchymal Transition. Cells Tissues Organs 211, 238–251 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Savagner P, Valles AM, Jouanneau J, Yamada KM & Thiery JP Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol Biol Cell 5, 851–862 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Radisky DC et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436, 123–127 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Melchionna R et al. The pattern of hMENA isoforms is regulated by TGF-beta1 in pancreatic cancer and may predict patient outcome. Oncoimmunology 5, e1221556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cheng C, Yaffe MB & Sharp PA A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev 20, 1715–1720 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhao P et al. The CD44s splice isoform is a central mediator for invadopodia activity. J Cell Sci 129, 1355–1365 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Liu S & Cheng C Akt Signaling Is Sustained by a CD44 Splice Isoform-Mediated Positive Feedback Loop. Cancer Res 77, 3791–3801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Braeutigam C et al. The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 33, 1082–1092 (2014). [DOI] [PubMed] [Google Scholar]
- 218.Xu Y et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev 28, 1191–1203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Pillman KA et al. miR-200/375 control epithelial plasticity-associated alternative splicing by repressing the RNA-binding protein Quaking. EMBO J 37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Warzecha CC, Sato TK, Nabet B, Hogenesch JB & Carstens RP ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33, 591–601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Huang H, Zhang J, Harvey SE, Hu X & Cheng C RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev 31, 2296–2309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Malard F et al. The diversity of splicing modifiers acting on A-1 bulged 5’-splice sites reveals rules for rational drug design. Nucleic Acids Res 52, 4124–4136 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Naryshkin NA et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693 (2014). [DOI] [PubMed] [Google Scholar]
- 224.Ratni H et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J Med Chem 61, 6501–6517 (2018). [DOI] [PubMed] [Google Scholar]
- 225.Childs-Disney JL et al. Targeting RNA structures with small molecules. Nat Rev Drug Discov 21, 736–762 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Seiler M et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24, 497–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hong DS et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs 32, 436–444 (2014). [DOI] [PubMed] [Google Scholar]
- 228.Steensma DP et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 35, 3542–3550 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Vanzyl EJ et al. The spliceosome inhibitors isoginkgetin and pladienolide B induce ATF3-dependent cell death. PLoS One 15, e0224953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Darrigrand R et al. Isoginkgetin derivative IP2 enhances the adaptive immune response against tumor antigens. Commun Biol 4, 269 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yoon SO, Shin S, Lee HJ, Chun HK & Chung AS Isoginkgetin inhibits tumor cell invasion by regulating phosphatidylinositol 3-kinase/Akt-dependent matrix metalloproteinase-9 expression. Mol Cancer Ther 5, 2666–2675 (2006). [DOI] [PubMed] [Google Scholar]
- 232. Han T et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356 (2017). This study reports splicing-mediated anticancer effects of sulfonamides and identifies their targets in the splicing machinery.
- 233.Denichenko P et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat Commun 10, 1590 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Uzor S et al. CDC2-like (CLK) protein kinase inhibition as a novel targeted therapeutic strategy in prostate cancer. Sci Rep 11, 7963 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Wang E et al. Modulation of RNA splicing enhances response to BCL2 inhibition in leukemia. Cancer Cell 41, 164–180 e168 (2023). This study highlights the importance of splicing in modulating the response to cancer therapies.
- 236.Tam BY et al. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett 473, 186–197 (2020). [DOI] [PubMed] [Google Scholar]
- 237.Bezzi M et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 27, 1903–1916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Radzisheuskaya A et al. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol 26, 999–1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jin P et al. Mutant U2AF1-Induced Mis-Splicing of mRNA Translation Genes Confers Resistance to Chemotherapy in Acute Myeloid Leukemia. Cancer Res 84, 1583–1596 (2024). [DOI] [PubMed] [Google Scholar]
- 240.Lee SC et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22, 672–678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bewersdorf JP et al. E7820, an anti-cancer sulfonamide, degrades RBM39 in patients with splicing factor mutant myeloid malignancies: a phase II clinical trial. Leukemia 37, 2512–2516 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Lee SC et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 34, 225–241 e228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wheeler EC et al. Splicing modulators impair DNA damage response and induce killing of cohesin-mutant MDS and AML. Sci Transl Med 16, eade2774 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.North K et al. Synthetic introns enable splicing factor mutation-dependent targeting of cancer cells. Nat Biotechnol 40, 1103–1113 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Asnani M et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia 34, 1202–1207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Sotillo E et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5, 1282–1295 (2015). This study is the first demonstration that aberrant splicing of CD19 contributes to acquired resistance to the CD19-directed CAR T cell immunotherapy in pediatirc patients with acute leukemia.
- 247.Cortes-Lopez M et al. High-throughput mutagenesis identifies mutations and RNA-binding proteins controlling CD19 splicing and CART-19 therapy resistance. Nat Commun 13, 5570 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Fischer J et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J Immunother 40, 187–195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ang Z et al. Alternative splicing of its 5’-UTR limits CD20 mRNA translation and enables resistance to CD20-directed immunotherapies. Blood 142, 1724–1739 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Lamba JK et al. CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children’s Oncology Group Trial AAML0531. J Clin Oncol 35, 2674–2682 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Lu SX et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 184, 4032–4047 e4031 (2021). This study documents the emergence of MHC class I-associated neoantigens in cancer cells followig treatment with splicing inhibitors.
- 252.Li G et al. Splicing neoantigen discovery with SNAF reveals shared targets for cancer immunotherapy. Sci Transl Med 16, eade2886 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Kahles A et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34, 211–224 e216 (2018). This study provides evidence for extensive splicing alterations across cancer types even in the absence of splicing factor mutations.
- 254.Merlotti A et al. Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci Immunol 8, eabm6359 (2023). [DOI] [PubMed] [Google Scholar]
- 255.Trincado JL et al. ISOTOPE: ISOform-guided prediction of epiTOPEs in cancer. PLoS Comput Biol 17, e1009411 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Shaw TI et al. Discovery of immunotherapy targets for pediatric solid and brain tumors by exon-level expression. Nat Commun 15, 3732 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.O’Connor BP et al. Regulation of toll-like receptor signaling by the SF3a mRNA splicing complex. PLoS Genet 11, e1004932 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Shalek AK et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hoss F et al. Alternative splicing regulates stochastic NLRP3 activity. Nat Commun 10, 3238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Martinez NM et al. Alternative splicing networks regulated by signaling in human T cells. RNA 18, 1029–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.McNeill L et al. CD45 isoforms in T cell signalling and development. Immunol Lett 92, 125–134 (2004). [DOI] [PubMed] [Google Scholar]
- 262.Martinez NM & Lynch KW Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev 253, 216–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Ner-Gaon H, Peleg R, Gazit R, Reiner-Benaim A & Shay T Mapping the splicing landscape of the human immune system. Front Immunol 14, 1116392 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Wang X, Yan L, Guo J & Jia R An anti-PD-1 antisense oligonucleotide promotes the expression of soluble PD-1 by blocking the interaction between SRSF3 and an exonic splicing enhancer of PD-1 exon 3. Int Immunopharmacol 126, 111280 (2024). [DOI] [PubMed] [Google Scholar]
- 265.Wahid M et al. Targeting alternative splicing as a new cancer immunotherapy-phosphorylation of serine arginine-rich splicing factor (SRSF1) by SR protein kinase 1 (SRPK1) regulates alternative splicing of PD1 to generate a soluble antagonistic isoform that prevents T cell exhaustion. Cancer Immunol Immunother 72, 4001–4014 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Kennedy PT et al. Soluble CTLA-4 attenuates T cell activation and modulates anti-tumor immunity. Mol Ther 32, 457–468 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hajaj E et al. Alternative Splicing of the Inhibitory Immune Checkpoint Receptor SLAMF6 Generates a Dominant Positive Form, Boosting T-cell Effector Functions. Cancer Immunol Res 9, 637–650 (2021). [DOI] [PubMed] [Google Scholar]
- 268.Ceccarello E et al. Splice-Switching Antisense Oligonucleotides as a Targeted Intrinsic Engineering Tool for Generating Armored Redirected T Cells. Nucleic Acid Ther 31, 145–154 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Revenko A et al. Direct targeting of FOXP3 in Tregs with AZD8701, a novel antisense oligonucleotide to relieve immunosuppression in cancer. J Immunother Cancer 10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Wang Z, Gerstein M & Snyder M RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Eskens FA et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res 19, 6296–6304 (2013). [DOI] [PubMed] [Google Scholar]
- 272.Feustel K & Falchook GS Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors in Oncology Clinical Trials: A review. J Immunother Precis Oncol 5, 58–67 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Chatrikhi R et al. A synthetic small molecule stalls pre-mRNA splicing by promoting an early-stage U2AF2-RNA complex. Cell Chem Biol 28, 1145–1157 e1146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Li L et al. Targeting the ERG oncogene with splice-switching oligonucleotides as a novel therapeutic strategy in prostate cancer. Br J Cancer 123, 1024–1032 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Bruno IG, Jin W & Cote GJ Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum Mol Genet 13, 2409–2420 (2004). [DOI] [PubMed] [Google Scholar]
- 276.Shieh JJ, Liu KT, Huang SW, Chen YJ & Hsieh TY Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J Invest Dermatol 129, 2497–2506 (2009). [DOI] [PubMed] [Google Scholar]
- 277.Ghigna C et al. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol 7, 495–503 (2010). [DOI] [PubMed] [Google Scholar]
- 278.Zammarchi F et al. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci U S A 108, 17779–17784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Izaguirre DI et al. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol Carcinog 51, 895–906 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Nielsen TO, Sorensen S, Dagnaes-Hansen F, Kjems J & Sorensen BS Directing HER4 mRNA expression towards the CYT2 isoform by antisense oligonucleotide decreases growth of breast cancer cells in vitro and in vivo. Br J Cancer 108, 2291–2298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Shiraishi T, Eysturskarth J & Nielsen PE Modulation of mdm2 pre-mRNA splicing by 9-aminoacridine-PNA (peptide nucleic acid) conjugates targeting intron-exon junctions. BMC Cancer 10, 342 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kralovicova J, Moreno PM, Cross NC, Pego AP & Vorechovsky I Antisense Oligonucleotides Modulating Activation of a Nonsense-Mediated RNA Decay Switch Exon in the ATM Gene. Nucleic Acid Ther 26, 392–400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Anczukow O et al. BRCA2 deep intronic mutation causing activation of a cryptic exon: opening toward a new preventive therapeutic strategy. Clin Cancer Res 18, 4903–4909 (2012). [DOI] [PubMed] [Google Scholar]
- 284.Mogilevsky M et al. Modulation of MKNK2 alternative splicing by splice-switching oligonucleotides as a novel approach for glioblastoma treatment. Nucleic Acids Res 46, 11396–11404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Mercatante DR, Bortner CD, Cidlowski JA & Kole R Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells. analysis of apoptosis and cell death. J Biol Chem 276, 16411–16417 (2001). This study provided the initial evidence that modulation of specific alternative splicing events using antisense oligonucleotides can induce cancer cell death.
- 286.Liu J et al. Overcoming imatinib resistance conferred by the BIM deletion polymorphism in chronic myeloid leukemia with splice-switching antisense oligonucleotides. Oncotarget 8, 77567–77585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Lin J et al. Induced-Decay of Glycine Decarboxylase Transcripts as an Anticancer Therapeutic Strategy for Non-Small-Cell Lung Carcinoma. Mol Ther Nucleic Acids 9, 263–273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Karras JG, McKay RA, Lu T, Dean NM & Monia BP Antisense inhibition of membrane-bound human interleukin-5 receptor-alpha chain does not affect soluble receptor expression and induces apoptosis in TF-1 cells. Antisense Nucleic Acid Drug Dev 10, 347–357 (2000). [DOI] [PubMed] [Google Scholar]
- 289.Wang Z et al. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J Mol Cell Biol 4, 79–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sun Y, Yan L, Guo J, Shao J & Jia R Downregulation of SRSF3 by antisense oligonucleotides sensitizes oral squamous cell carcinoma and breast cancer cells to paclitaxel treatment. Cancer Chemother Pharmacol 84, 1133–1143 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.