

SUMMARY
Inactivating mutations in chromatin modifiers, like the α-thalassemia/mental retardation, X-linked (ATRX)-death domain-associated protein (DAXX) chromatin remodeling/histone H3.3 deposition complex, drive the cancer-specific alternative lengthening of telomeres (ALT) pathway. Prior studies revealed that HIRA, another histone H3.3 chaperone, compensates for ATRX-DAXX loss at telomeres to sustain ALT cancer cell survival. How HIRA rescues telomeres from the consequences of ATRX-DAXX deficiency remains unclear. Here, using an assay for transposase-accessible chromatin using sequencing (ATAC-seq) and cleavage under targets and release using nuclease (CUT&RUN), we establish that HIRA-mediated deposition of new H3.3 maintains telomeric chromatin accessibility to prevent the detrimental accumulation of nucleo-some-free single-stranded DNA (ssDNA) in ATRX-DAXX-deficient ALT cells. We show that the HIRA-UBN1/UBN2 complex deposits new H3.3 to prevent TERRA R-loop buildup and transcription-replication conflicts (TRCs) at telomeres. Furthermore, HIRA-mediated H3.3 incorporation into telomeric chromatin links productive ALT to the phosphorylation of serine 31, an H3.3-specific amino acid, by Chk1. Therefore, we identify a critical role for HIRA-mediated H3.3 deposition that ensures the survival of ATRX-DAXX-deficient ALT cancer cells.
Graphical Abstract
In brief
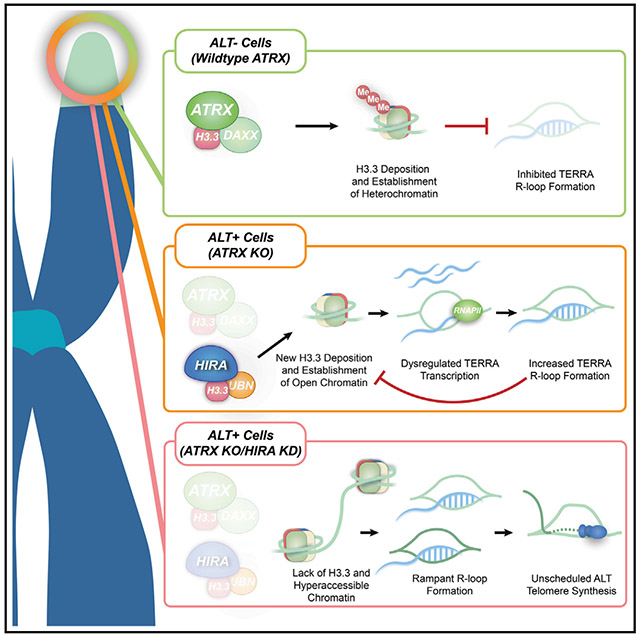
Lynskey et al. report links between HIRA-mediated histone H3.3 deposition and R-loop homeostasis at ALT telomeres. In most ALT cancer cells, after ATRX-DAXX is inactivated, HIRA regulates telomeric chromatin assembly. HIRA limits ssDNA accumulation and mitigates out-of-control R-loop formation and transcription-replication conflicts at telomeres, threatening cancer cell survival.
INTRODUCTION
Telomere length maintenance confers the proliferative immortality that is a hallmark of cancer cells. In most cases, telomerase, a specialized ribonucleoprotein (RNP) complex, directly synthesizes and adds new telomere repeat (TTAGGG) sequences at chromosome ends. However, in cancer cells that do not express telomerase, a homology-directed repair (HDR) mechanism named alternative lengthening of telomeres (ALT) is responsible for telomere extension and cancer cell survival.1,2 ALT usually arises in the advanced stages of cancer progression, correlating with the inactivation of chromatin modifiers, including theATRX (α-thalassemia/mental retardation, X-linked) and DAXX (death domain-associated protein) chromatin remodeling/histone deposition complex.3 ATRX-DAXX has been implicated in preserving telomere integrity through histone H3.3 deposition and resolving G4-quadruplexes and RNA:DNA hybrids (R-loops) at telomeres.4-6 The latter is attributed to the increased abundance of the telomere repeat-containing large intergenic non-coding RNA (lincRNA) TERRA at telomeres after ATRX-DAXX loss.7 Unresolved TERRA R-loops cause instability, replication stress, and a strong telomere DNA damage response.8 However, TERRA can also invade and hybridize with telomeric DNA, forming R-loop and D-loop structures9-11 that prime the assembly of the ALT replisome comprised of PCNA (Proliferating Nuclear Antigen), RFC1-5 (Replication Factor C), and DNA Polδ. Importantly, ALT-HDR is active primarily12,13 in the G2 cell cycle phase and restricted to neoplastic cells, seldom detected in healthy somatic cells. This engagement of HDR pathways and factors is a crucial cellular adaptation that preserves telomere length and integrity in ATRX-DAXX-mutated ALT cancer cells.
ATRX-DAXX loss also disrupts DAXX-mediated deposition of H3.3 at telomeres.14 Proteomic studies have uncovered other chromatin modifiers that fill this void and re-establish functional telomeric chromatin.15,16 These alternative chromatin modifiers include HIRA, a specialized histone H3.3 chromatin assembly factor.17,18 Though linked with repressing the expression of specific developmentally regulated genes in progenitor and stem cells, HIRA typically regulates H3.3 deposition within transcriptionally active chromatin at genes, promoters, and enhancer elements.19 HIRA was also implicated in re-priming transcription following the repair of ultraviolet-C (UVC)-induced DNA lesions and in the genome-wide deposition of H3.3 at nucleosome-free DNA to preserve global chromatin integrity.20,21 Our prior studies uncovered that HIRA mobilizes to telomeres following ATRX -DAXX inactivation in ALT cancer cells, facilitating productive telomere extension by ALT.22 Notably, depletion of HIRA provoked the cytotoxicity of ATRX-DAXX-mutated ALT cancer cells,22,23 which was reversed by restoring wild-type (WT) ATRX protein expression.22 Therefore, the mobilization of HIRA is imperative for the survival of ATRX-DAXX-mutated ALT cancer cells. However, the precise molecular basis for the dependency of these ALT cells on HIRA remained to be determined.
This study shows that HIRA depletion leads to the hyper-accessibility of telomeric chromatin in cells that have acquired ALT through the targeted disruption of ATRX. Our data attribute this to the defective deposition of newly synthesized histone H3.3 by the HIRA/UBN (Ubinuclein)subcomplex, and it is accompanied by greater telomeric single-stranded DNA (ssDNA), consistent with nucleosome-free telomeres. This coincides with striking increases in TERRA R-loop formation, transcription-replication conflicts (TRCs) at telomeres, and a switch to non-productive ALT activity. Notably, pre-existing TERRA R-loops block new H3.3 deposition at telomeres by HIRA.
Strikingly, we also find that HIRA-mediated H3.3 deposition is integral in the phosphorylation of H3.3-specific serine 31 by CHK1. Our data indicate that this unique histone mark may be crucial for suppressing aberrant TERRA R-loop formation and maintaining optimal ALT activity. Thus, our study links HIRA-mediated histone H3.3 deposition with antagonizing TERRA R-loop homeostasis, revealing an unexpected but crucial protective function for HIRA at telomeres in ALT cancer cells following ATRX-DAXX loss.
RESULTS
HIRA maintains a functional telomeric chromatin configuration following ATRX loss
Prior studies found that HIRA and its cognate interacting factors, like ASF1a (anti-silencing factor 1a), represent synthetic lethal interactions with ATRX-DAXX inactivation.22,23 As HIRA is the major remaining histone H3.3 chaperone in ATRX-DAXX-mutated ALT cancer cells, we envisioned that it could adopt essential roles in managing telomeric chromatin that underly the dependency of those cells on HIRA. To further evaluate HIRA’s role in organizing telomeric chromatin, we depleted HIRA by small interfering RNA (siRNA) knockdown in control (IMR90 CTRL) and ATRX knockout (KO) SV40 large T antigen (IMR90 ATRX-KO ALT) immortalized IMR90 cells (Figure S1A), the latter having activated ALT through ATRX disruption.24 ATAC-seq (assay for transposase-accessible chromatin using sequencing) confirmed prior studies by finding that ATRX disruption and ALT activation are accompanied by significant increases in Tn5 transposase accessibility at telomeres (Figure 1A). Interestingly, we found that Tn5 accessibility at telomeres in the IMR90 ATRX-KO ALT cells and not their IMR90 CTRL counterparts increased further after HIRA depletion (Figures 1A and S1B). Changes, and increases in particular, in telomere length are unlikely the cause of this. Furthermore, our prior examination of HIRA indicated that its loss acutely reduces telomere DNA synthesis.22 Rather, such acute changes in telomere accessibility would likely be linked to the loss of HIRA-directed assembly of H3.3-containing nucleosomes.
Figure 1. HIRA maintains chromatin accessibility in ATRX-deficient ALT cells.

(A) Schematic of the ATAC-seq methodology. Tn5 transposase is active at accessible chromatin regions, enabling the isolation and detection of DNA sequences at open chromatin through next-generation sequencing (NGS) and downstream analysis. The graph shows the normalized count of total telomeric motifs (TTAGGG) that contain ≥3 tandem telomeric repeats in CTRL and IMR90 ATRX-KO ALT cells expressed as means ± standard error of the mean (SEM) from three biological replicates (n = 3). p values were calculated using a paired t test (one tailed).
(B) Schematic of the CUT&RUN methodology. Protein A/G-MNase is recruited to the H3.3-bound primary antibody and cleaves around the binding site upon activation with Ca2+, followed by downstream solubilization and sequencing of isolated DNA fragments underlying H3.3. The graph shows normalized telomere repeat counts from H3.3-associated reads in CTRL and IMR90 ATRX-KO ALT cells expressed as means ± SEM from three biological replicates (n = 3). p values were calculated using a paired t test (one tailed).
(C) Left: representative images of ssTelo detected by non-denaturing (native) FISH after siRNA knockdown of HIRA or H3.3 in IMR90 ATRX-KO ALT cells. Right: the graph displays the number of ssTelo signals detected per cell in each condition. Individual data points of ≥600 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown.
(D) Left: telomeric western dot blot of ssDNA, double-stranded DNA (dsDNA), and Southern blot detection of telomeric and Alu repeat DNA in CTRL and IMR90 ATRX-KO ALT cells following knockdown of HIRA. Right: the fold change and SEM of detected telomeric ssDNA calculated relative to telomeric dsDNA derived from three biological replicates (n = 3).
(E) Representative images of TERRA RNA-FISH and TRF2 IF in IMR90 ATRX-KO ALT cells following HIRA and H3.3 knockdown. The graph displays the percentage of TERRA-positive telomeres as the percentage of co-localization of TERRA and TRF2. Individual data points of ≥200 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown.
(F) Top: relative levels of R-loops at telomeres in CTRL and IMR90 ATRX-KO ALT cells after knockdown of HIRA or H3.3. Bottom: relative levels of R-loops at telomeres of samples pre-treated with RNaseH1 before pull-down.
All data in (C)–(E) represent the mean ± SEM from three biological replicates (n = 3) except the data in (F), which represent the mean ± SEM from four biological replicates (n = 4). p values in (C) and (E) were calculated using a one-way ANOVA, in (D) by unpaired t test, and in (F) by a two-way ANOVA. Scale bar: 5 μM.
See also Figure S1.
To profile H3.3 on native chromatin in IMR90 CTRL and IMR90 ATRX-KO ALT cells with or without HIRA depletion, we performed CUT&RUN (cleavage under targets and release using nuclease).25,26 First, compared with the IMR90 CTRL, H3.3 occupancy at telomeres was significantly greater in IMR90 ATRX-KO ALT cells (Figure 1B). This may reflect the requirement to replenish telomeric chromatin with H3.3 following telomere hyper-extension by the ALT mechanism during G2 phase, which only occurs on the IMR90 ATRX-KO ALT cells after ATRX inactivation. Second, whereas histone H3.3 levels were unaltered by HIRA depletion in IMR90 CTRL cells, H3.3 occupancy at telomeres was reduced by ~30%–40% in HIRA-deficient IMR90 ATRX-KO ALT cells (Figure 1B). Furthermore, using the SNAP-tag in vivo histone labeling assay, we verified that newly synthesized histone H3.3 deposition is reduced following HIRA depletion in IMR90 ATRX-KO ALT cells (Figures S1C and S1D). Intriguingly, we found that chromatin accessibility at transcription start sites (TSSs) was unaltered even though H3.3 occupancy was notably reduced following HIRA depletion in both IMR90 CTRL and IMR90 ATRX-KO ALT cells (Figures S1E and S1F). Though counterintuitive given HIRA’s established role in coordinating nucleosome dynamics during transcription, recent and emerging studies have described how H3.3 and HIRA deficiency can reduce promoter accessibility in mouse embryonic stem cells (ESCs)27 and has little effect on chromatin accessibility of promoter-proximal regulatory elements in HeLa cells,28 respectively. Perturbations in transcription factor/chromatin re-modeler complex recruitment, reduced nucleosome turnover, and assembly of nucleosomes with histone H3.1 were suggested to enable cells to tolerate the loss of H3.3 deposition and preserve chromatin at these crucial regulatory regions. However, our observations highlight the dependency on HIRA for chromatin assembly at telomeres that these cells acquire following inactivation of the ATRX-DAXX histone H3.3 deposition pathway.
HIRA loss provokes ssDNA, replicative stress, and elevated TERRA R-loops
Perturbations in chromatin assembly complex expression or histone dynamics in S-phase can cause post-replicative nucleo-some-free gaps that may later be replenished with H3.3-containing nucleosomes by HIRA.18,29 Template-directed DNA synthesis during ALT generates high levels of new telomeric DNA repeats that, in principle, must be chromatinized. While HIRA typically does not mediate H3.3 deposition and chromatin assembly during DNA synthesis, prior studies revealed that HIRA’s association with telomeres occurs during the G2 cell cycle phase, coinciding with when ALT-directed telomere extension is active.22 Thus, we postulated that failure to deposit histone H3.3 on telomere DNA could cause ssDNA to accumulate and persist in telomeres. We first employed the ssTelo assay, which enables the visualization of C-rich single-stranded telomeric DNA using fluorophore-conjugated (TTAGGG)3 peptide nucleic acid (PNA) probes by non-denaturing native fluorescence in situ hybridization (FISH).30 These experiments revealed a robust increase in telomeric ssDNA abundance following HIRA or histone H3.3 depletion (Figure 1C).
Considering the abundance of extrachromosomal telomere DNA species in ALT cancer cells, we asked whether the increased ssDNA detected after HIRA knockdown emanated from chromosomal telomeric DNA. Duplex telomeric DNA was isolated from restriction-enzyme-digested genomic DNA by streptavidin pull-down using biotinylated PNA probes annealed to complementary telomere DNA sequences.31 Probing of the telomere DNA isolates with anti-single- or double-stranded DNA antibodies in dot-western blot revealed more ssDNA specifically in telomere DNA derived from HIRA-depleted IMR90 ATRX-KO ALT cells but not IMR90 CTRL cells (Figure 1D). Immunofluorescence (IF) and western analyses showed an enhanced telomeric presence and serine phosphorylation of the ssDNA sensor protein RPA2, respectively, indicating elevated localized replicative stress following HIRA and histone H3.3 disruption in these IMR90 ATRX-KO ALT cells (Figures S1G and S1H). Like ssDNA and RPA2, increased TERRA and TERRA R-loops are a source of replicative stress at telomeres.8 By RNA-FISH using fluorescently labeled (CCCTAA)7 oligonucleotide probes, we detected a more significant association of TERRA RNA with telomeres in HIRA- and histone H3.3-depleted cells (Figure 1E). Lastly, we conducted DNA-RNA immunoprecipitation (DRIP)-qPCR assays with the S9.6 antibody.32 The sensitivity of the immunoprecipitated RNA:DNA hybrids to the RNaseH endonuclease confirmed the R-loop-specific recovery by the DRIP assay using the S9.6 antibody (Figure 1F). Interestingly, DRIP-qPCR indicated that HIRA or histone H3.3 depletion causes a substantial increase in TERRA R-loop abundance only in the IMR90 ATRX-KO ALT cells (Figures 1F and S1I). These results provided evidence of an unexpected link between HIRA-mediated H3.3 deposition and the homeostasis of TERRA R-loops at telomeres in ATRX-deficient ALT cells.
ATRX loss links HIRA-mediated histone H3.3 deposition to R-loop homeostasis
Since HIRA mobilizes to telomeres to maintain telomeric chromatin following ATRX-DAXX inactivation, restoring functional ATRX protein should rescue the observed detrimental impacts of HIRA depletion. For this, we turned to the previously characterized inducible ATRX U2OS cell line system (referred to as iATRX), where full-length and functional ATRX protein can be re-expressed by adding doxycycline (dox) to the culture media4 (Figure 2A). Following 10 days of dox treatment, bands corresponding to ATRX were detected by western blot. We noted that when either HIRA or H3.3 was depleted by siRNA, the abundance of ATRX was notably increased (Figure 2A). This might reflect either a gain of function of ATRX when a functionally related histone H3.3 chaperone is absent33 or that histone H3.3 availability limits ATRX protein stability by an unknown mechanism. Our prior studies using this iATRX U2OS cell line demonstrated that, upon its re-expression, ATRX usurps HIRA for histone H3.3 deposition at telomeres and telomere DNA breaks generated by the TRF1-FokI endonucleolytic fusion protein.22,34 By conducting DRIP-qPCR assays following the expression of WT or inactive (D450A) DA-TRF1-FokI, we again observed that HIRA and histone H3.3 depletion increased TERRA R-loops at telomere breaks in uninduced iATRX U2OS cells. However, this induction of TERRA R-loops following HIRA or histone H3.3 depletion was abolished in dox-treated iATRX U2OS cells that re-express ATRX (Figures 2B and S2A).
Figure 2. HIRA-mediated H3.3 deposition suppresses TERRA R-loops and transcription-replication conflicts.

(A) Western blot analysis of ATRX protein expression with doxycycline for 10 days in iATRX U2OS cells expressing wild-type (WT) or the catalytically inactive D450A (DA) TRF1-FokI after siRNA knockdown of HIRA or histone H3.3. γTUB is the loading control.
(B) Relative levels of telomeric R-loops after ATRX re-expression in iATRX U2OS cells expressing WT or DA TRF1-FokI after siRNA knockdown of HIRA or H3.3. Data represent mean ± SEM from four biological replicates (n = 4). p values were calculated using a two-way ANOVA.
(C) Top: western blot analysis of the re-expression of ATRX protein with doxycycline for 10 days in iATRX U2OS cells after siRNA knockdown of FANCM. γTUB is the loading control. Bottom: relative levels of R-loop signals at telomeres after FANCM depletion with or without ATRX re-expression in iATRX U2OS cells. All data represent mean ± SEM from three biological replicates (n = 3). p values were calculated by a two-way ANOVA.
(D) Left: representative images of SNAP-H3.3 at telomere DNA breaks in control and HIRA or FANCM-depleted U2OS cells expressing WT or inactive (DA) TRF1-FokI.
(E) The graph displays the percentage of SNAP-histone H3.3-positive TRF1-FokI telomeres. Individual data points of ≥200 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown. p values were calculated using a one-way ANOVA. Scale bar: 5 μM.
(F) Representative images of PCNA and RNAPII proximity ligation assay (PLA) foci at TRF1-FokI telomere DNA breaks after HIRA or histone H3.3 knockdown in iATRX U2OS cells with (+dox) and without (—dox) ATRX expression. Arrows indicate co-localized PLA and TRF1-FokI foci.
(G) The graph displays the percentage of PCNA-RNAPII PLA-positive TRF1-FokI telomeres in iATRX cells after siRNA knockdown of HIRA or H3.3. Individual data points of ≥200 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown. p values were calculated using a two-way ANOVA. Scale bar: 5 μM.
See also Figure S2.
The increased accessibility of telomeric chromatin in the IMR90 ATRX-KO ALT cell line indicated that HIRA cannot fully restore the closed repressive chromatin configuration typically found at telomeres, such as in the IMR90 CTRL cells. We hypothesized that unresolved or stochastic R-loops at telomeres might represent obstacles that prevent HIRA-mediated chromatin assembly. Indeed, genome-wide mapping studies showed that R-loops preferentially accumulate within nucleosome-free regions.35 Furthermore, though R-loops are linked with targeting chromatin modifier complexes during transcription regulation,35-37 their non-B form structure makes R-loops poor substrates for in vitro nucleosome assembly.38,39 With this precedent, we tested this hypothesis by examining histone H3.3 deposition in IATRX U2OS following FANCM depletion, which leads to a robust induction of TERRA R-loops40 in iATRX U2OS cells. We first confirmed R-loop accumulation by DRIP-qPCR after FANCM knockdown in iATRX U2OS cells (Figures 2C and S2B). Interestingly, dox-induced ATRX expression suppressed this induction of TERRA R-loops (Figure 2C). We also observed by DRIP-qPCR that depleting DAXX in iATRX U2OS cells did not increase TERRA R-loop levels, regardless of ATRX expression (Figures S2C-S2E). Then, using the SNAP-tag histone H3.3 tracking assay, we observed strongly impaired new histone H3.3 deposition at TRF1-FokI-generated DNA breaks in FANCM-depleted cells, mimicking the effect of HIRA loss (Figure 2D). This observation suggested that TERRA R-loops, which persist at telomeres in ATRX-deficient ALT cancer cells, may impede H3.3 deposition by HIRA, potentially compromising the complete restoration of chromatin at telomeres. One prediction, therefore, was that removing TERRA R-loops by ectopic RNaseH1 expression could restore H3.3 deposition in FANCM-depleted cells. Due to technical constraints, this experiment could not be conducted in U2OS cells expressing WT-TRF1-FokI and SNAP-H3.3. Thus, we switched to IMR90 ATRX-KO ALT cells, where the robust incorporation of SNAP-tagged histone H3.3 can be visualized (Figures S1G and 2F). As in the U2OS cell line, SNAP-H3.3 detection at telomeres was impaired following FANCM knockdown (Figures S2F and S2G). Strikingly, SNAP-H3.3 foci that co-localized with telomeric foci (marked by TRF2) were visible following transfection of FANCM-depleted IMR90 ATRX-KO ALT cells with GFP-WT-RNaseH1, indicating that H3.3 deposition is restored after the R-loops are removed (Figures S2F and S2G). Notably, GFP-RNaseH1 foci that co-localized with telomeres were also observed at these foci. These data provide further support for the scenario that TERRA R-loops can impede H3.3 deposition at telomeres by HIRA.
Unresolved R-loops and replicative stress provoke genome-destabilizing TRCs between RNA polymerase II (RNAPII) and the DNA replication machinery.41 Telomere TRCs can be visualized by proximity ligation assay (PLA), which detects the vicinal contacts between RNAPII and PCNA at telomere DNA breaks generated by TRF1-FokI.11 We treated U2OS cells with flavopiridol, a CDK9 inhibitor that blocks transcription elongation by preventing phosphorylation of RNAPII-C-terminal domain (CTD) serine 2, and triptolide, an XPB/TFIIH transcription initiation inhibitor that stimulates the proteasomal degradation of RNAPII, as controls. We validated the reported effects of these inhibitors on RNAPII levels and post-translational modifications (PTMs)s by western blot (Figure S2F). We also validated their efficacy as transcriptional inhibitors using the 5’-ethynyluridine (EU)-click-IT assay, which detects nascent RNA in individual cells (Figure S2G). TRCs were robustly induced at telomeres following RNAPII inhibition with flavopiridol. TRCs were largely abolished after triptolide treatment (Figures S2J and S2K). After comprehensively validating the parameters of the PLA assay, we observed that HIRA elicited a greater frequency of PLA signals at TRF1-FokI breaks in the uninduced iATRX cells. This was comparable to the robust increase in TRC frequency observed following flavopiridol treatment (Figures S2C-S2E). However, these increases were dwarfed by a relatively enormous increase in TRCs at TRF1-FokI breaks following histone H3.3 depletion (Figures 2E and 2F). Notably, many additional non-telomeric PLA signals were also visible after histone H3.3 depletion and, to a lesser degree, following HIRA depletion, implicating HIRA and H3.3 in preventing TRCs elsewhere in the genome (Figures 2E and 2F). Remarkably, the PLA signals at TRF1-FokI breaks and those at non-telomeric sites were significantly diminished in the induced iATRX U2OS cells (Figures 2E and 2F).
These results highlight the multifunctionality of ATRX in coordinating H3.3 deposition and alleviating replicative stress at telomeres and non-telomeric regions of the genome. When these ATRX activities are lost in ALT cancer cells, HIRA can partly compensate by fulfilling histone H3.3 deposition at telomeres. However, HIRA lacks ATRX’s capacity to resolve R-loops, so telomeres remain prone to replicative stress and TRCs.
R-loop suppression depends on the HIRA-UBN new histone H3.3 deposition pathway
Cognate interactions between HIRA and ASF1a or UBN1-2 determine whether previously incorporated histone H3.3 will be recycled and deposited into chromatin or if newly synthesized histone H3.3 will be used to replenish nucleosome-free chromatin gaps, respectively42,43 (Figure 3A). We found that YFP-tagged WT HIRA and the ASF1a-binding (I461D) mutant43,44 localize to TRF1-FokI breaks. However, a HIRA-R227K mutant that cannot interact with either UBN1 or its lesser-studied paralog UBN242,45,46 ultimately failed to localize to TRF1-FokI breaks (Figures S3A-S3C). We next performed knockdown experiments to disrupt the HIRA-UBN and HIRA-ASF1a histone H3.3 deposition pathway (Figure 3B). HIRA interactions have a stabilizing effect on UBN1/2 (as well as on CABIN1, which is not examined here). Thus, as previously described,18 HIRA knockdown also diminished the levels of UBN1 protein as detected in western blot analysis (Figure 3B). Since the R227K mutation disrupts HIRA interactions with UBN1 and UBN2,42,45,46 UBN1 and UBN2 were co-depleted in U2OS cells (indicated as UBN; Figures 3B and S3D). By examining new H3.3 deposition with SNAP-tagged histone H3.3, we found that H3.3 deposition at TRF1-FokI-generated telomere break sites strongly depended on UBN (Figure 3C). In contrast, ASF1a was dispensable for new H3.3 deposition (Figure 3B). Likewise, the depletion of DAXX did not alter new H3.3 accumulation at telomerase break sites (Figures S3E-S3G). Thus, we conclude that the HIRA-UBN pathway is required for new H3.3 deposition at telomeres in ALT cancer cells.
Figure 3. Deposition of new histone H3.3 by HIRA-UBN is required for productive ALT-HDR.

(A) Cartoon of HIRA-mediated recycling of old H3.3 via HIRA-ASF1a and the deposition of newly synthesized histone H3.3 via HIRA-UBN.
(B) Western blot analysis of HIRA, UBN1, H3.3, and ASF1a in U2OS cells after siRNA knockdown of the indicated corresponding proteins. γTUB is the loading control.
(C) Representative images of SNAP-H3.3 at telomere DNA breaks in control and HIRA-, UBN-, and ASF1a-depleted U2OS cells expressing WT or inactive (DA) TRF1-FokI.
(D) The graph displays the percentage of SNAP-H3.3-positive TRF1-FokI telomeres in each condition. Individual data points of ≥200 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown. p values were calculated using a one-way ANOVA. Scale bar: 5 μM.
(E) Left: representative images of ssTelo detected by non-denaturing (native) FISH after siRNA knockdown of HIRA, histone H3.3, UBN, and ASF1a in U2OS cells. Right: the graph displays the number of ssTelo signals detected per cell in each condition. Individual data points of ≥1,200 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown. p values were calculated using a one-way ANOVA. Scale bar: 5 μM.
(F) Western dot blot of ssDNA and Southern blot detection of telomeric DNA in U2OS cells following knockdown of HIRA, histone H3.3, UBN, and ASF1a. The graph shows the fold change and the SEM of detected ssDNA calculated relative to telomeric dsDNA derived from three biological replicates (n = 3). p values were calculated using a one-way ANOVA.
(G) Relative levels of R-loop signal at telomeres after HIRA and UBN depletion in U2OS cells expressing either WT or inactive (DA) TRF1-FokI. All data represent mean ± SEM from four biological replicates (n = 4). p values were calculated using a one-way ANOVA.
(H) Representative images of telomeric FISH on metaphase chromosomes from HIRA-, H3.3-, or UBN-depleted U2OS cells. Purple and orange sections display examples of telomere loss (also termed signal-free ends) and telomere fragility, respectively.
(I) Graphs on the left and right display the percentages of telomere loss (signal-free ends) and fragility (multiple FISH signals per chromatid) observed per metaphase, respectively. Individual data points of ≥55 metaphases counted and the mean ± SEM derived from three biological replicates (n = 3) are shown. p values were calculated using a one-way ANOVA. Scale bar: 5 μM.
(J) Representative images of focal EdU accumulation at telomere DNA breaks in control and HIRA-, histone H3.3-, and UBN-depleted U2OS cells expressing WT or inactive (DA) TRF1-FokI. White arrows indicate co-localizing signals.
(K) The graph displays the percentage of EdU-positive TRF1-FokI telomeres in the indicated conditions. Individual data points of ≥150 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown. p values were calculated using a one-way ANOVA. Scale bar: 5 μM.
See also Figure S3.
Using the ssTelo assay, we found that, like HIRA and histone H3.3 depletion, UBN loss leads to significantly more single-stranded C-rich telomeric DNA (Figure 3D). The greater abundance of telomeric ssDNA after UBN loss was corroborated by the isolation of telomeric DNA by biotin capture and streptavidin pull-down (Figure 3E). Even though elevated ssTelo signals were also seen following ASF1a depletion, greater ssDNA was not evident within telomeric DNA isolated by the pull-down assay (Figures 3D and 3E). ASF1a loss might substantially impact telomere replication during the S-phase when ASF1a and its paralog ASF1b coordinate histone transfer in conjunction with the DNA replisome.29,47 HIRA-UBN, on the other hand, may act specifically to deposit new H3.3 during G2, when ALT-associated telomere extension occurs.
Focusing on the impact of the HIRA-UBN complex on telomeres and ALT, by DRIP-qPCR, we found that, like HIRA and histone H3.3 depletion, UBN loss in U2OS cells led to increased TERRA R-loops (Figures 3F and S3H). Increases in the frequency of TRCs were observed by PLA following UBN depletion, like HIRA depletion (Figures S3I and S3J). Next, we evaluated the effect of disrupting the HIRA-UBN pathway on telomere integrity and ALT activity. Defective histone H3.1 and H3.3 transfer through interfering with ASF1a and ASF1b was shown to cause telomere fragility.48 This phenotype has been linked with incomplete telomere replication and the formation of post-replicative ssDNA gaps within telomeres. The inadequate re-chromatinization of telomeres could contribute to chromatin gap formation that manifests in telomere fragility. In a metaphase spread analysis, we found that disrupting new histone H3.3 deposition by depleting histone H3.3 itself or UBN elevated the frequency of telomere fragility (Figures 3G and 3H). This contrasted with HIRA loss, which had a minimal effect on fragility but did elicit moderately more telomere loss (i.e., no detected telomere FISH signal) (Figures 3G and 3H).
We evaluated the impact of disrupting new H3.3 deposition on telomere DNA synthesis. UBN and histone H3.3 loss increased the frequency of ALT-associated promyelocytic leukemia (PML) bodies (APBs) in U2OS cells and VA13, another ATRX-deficient ALT cell line (Figures S3K and S3L). Like the metaphase analysis, HIRA loss acted differently from histone H3.3 or UBN depletion and reduced the frequency of APBs (Figure S3L). We also found that, as in U2OS cells and IMR90 ATRX-KO ALT cells, loss of HIRA, histone H3.3, and UBN increased the frequency of ssTelo signals in individual VA13 cells (Figure S3M). Finally, by monitoring the incorporation of the nucleotide analog 5-ethyl-2’deoxyuridine (EdU) at telomere DNA breaks, we observed that in contrast to HIRA depletion, which diminished EdU incorporation, disrupting the UBN histone H3.3 deposition pathway robustly increased EdU incorporation at telomere DNA break sites (Figure 3I). Notably, the same effects were observed at telomeric sites marked by the inactive TRF1-FokI, demonstrating that disrupting new histone H3.3 deposition can stimulate telomere DNA synthesis without DNA breaks. This evaluation of several indicators of telomere recombination and ALT-associated HDR suggested that the HIRA-UBN-mediated histone H3.3 pathway suppresses unscheduled ALT activity. This was unexpected, as prior studies showed that HIRA loss impairs telomere DNA synthesis in response to TRF1-FokI breaks. The distinction may be due to HIRA’s dual function in depositing recycled and newly synthesized H3.3. While interfering in both pathways imposes a robust block in telomere DNA synthesis and cytotoxicity,22 these data indicate that disrupting new H3.3 deposition hyperactivates ALT, which can be highly detrimental to ALT cancer cells.
HIRA enables CHK1-driven serine 31 phosphorylation to mitigate R-loop formation
The phosphorylation of a serine at position 31 is unique to the histone H3.3 variant.49 Since HIRA is essential for H3.3 deposition in ATRX-DAXX-deficient ALT cancer cells, we reasoned that HIRA loss could adversely affect the levels of this specialized modification. In western blot analysis of chromatin-associated histones isolated from asynchronously proliferating U2OS cells, there was no significant reduction in H3.3S31 phosphorylation (H3.3S31ph) after the depletion of HIRA (Figure 4A). We also assessed trans (on endogenous H3) modifications like H3K27 acetylation and H3S10ph. Like H3.3S31ph, these were largely unaltered (Figure 4A). However, H3.3S31ph is cell cycle regulated and generally restricted to pericentromeric regions in mitosis.49,50 Prior cytogenetic studies showed that H3.3S31ph is deregulated in ALT cancer cells and was shown to spread and decorate mitotic chromosome arms from centromeres to telomeres.49,50 The ATR effector kinase CHK1 was implicated in catalyzing mitotic H3.3S31ph in ALT cancer cells.50 Our metaphase chromosome analysis confirmed the widespread distribution of this mark along chromosomes (Figures 4B and 4C). The robust loss of H3.3S31ph on metaphase chromosomes after 6 h treatment with CHK1i (20 mM, SB-218078) confirmed its CHK1 dependency in U2OS cells (Figures 4B and 4C). Strikingly, we found that depleting HIRA strongly diminished H3.3S31ph across U2OS metaphase chromosomes (Figures 4B and 4C). This was not due to any appreciable changes in cell cycle progression following HIRA depletion as inferred by flow cytometry analysis (Figure S4A). Thus, HIRA-mediated delivery of H3.3 to telomeres might ensure S31 phosphorylation by CHK1.
Figure 4. Histone H3.3 serine 31 is essential to suppress TERRA-associated telomere dysfunction.

(A) Western blot analysis of chromatin-associated HIRA, H3.3, phospho-H3.3 S31 (H3.3S31ph), acetyl-H3K27 (H3K27ac), H3S10ph, and cyclin B1 after siRNA knockdown of HIRA in U2OS cells. Ponceau-stained histones are shown as loading.
(B) Representative images of metaphase chromosomes stained with DAPI and anti-H3.3S31ph antibodies.
(C) The graph displays the frequency of metaphase spreads displaying H3.3S31ph on chromosome arms observed in the indicated conditions. ≥500 metaphases were analyzed in each condition, and the mean ± SEM derived from three biological replicates (n = 3) is shown. p values were calculated using a Student’s t test. Scale bar: 5 μm.
(D) Left: representative images of ssTelo detected by non-denaturing (native) FISH after siRNA knockdown of endogenous histone H3.3 and complementation with exogenous WT and mutant histone H3.3. Scale bar: 5 μM.
(E) The graph displays the number of ssTelo signals detected per cell in each condition. Individual data points of ≥300 cells and the mean ± SEM derived from three biological replicates (n = 3) are shown. p values were calculated using a two-way ANOVA. Scale bar: 5 μm.
(F) Top: representative TERRA RNA-FISH and TRF2 immunofluorescence images in DMSO- and CHK1i-treated U2OS cells. Bottom: the graph displays the percentage of TERRA-positive TRF2 foci detected per cell in each condition. Individual data points of ≥500 cells and the mean ± SEM derived from two biological replicates (n = 2) are shown. p values were calculated using an unpaired t test. Scale bar: 5 μM.
(G) Top: relative levels of R-loops at telomeres in DMSO- and Chk1i-treated U2OS cells. Bottom: relative levels of R-loops at telomeres of samples pre-treated with RNaseH1 before pull-down. p values were calculated using an (top) unpaired t test and (bottom) a two-way ANOVA.
(H) Summary model of the contribution of HIRA to telomeric chromatin in ATRX-DAXX-deficient ALT cells.
See discussion and Figure S4.
How H3.3S31ph contributes to ALT remains unclear. The dependence of this unique H3.3 modification on HIRA prompted us to examine whether CHK1-dependent H3.3S31ph participates in regulating R-loop homeostasis at telomeres in ALT cancer cells. Using the ssTelo assay, we screened the ability of WT, S31-to-alanine (S31A), and phospho-mimetic glutamine (S31E) GFP-tagged H3.3 to complement the effect of histone H3.3 loss (Figures S4B and S4C). Endogenous H3.3 was depleted in U2OS cells using a siRNA pool targeting the 3’untranslated regions of histone H3.3 genes H3F3a and H3F3b (Figure S4B). GFP-positive cells were then scored in the ssTelo assay. As previously mentioned, depleting H3.3 increased the ssTelo foci phenotype. This was suppressed by the expression of WT H3.3 (Figures 4D, 4E, and S4C). The non-phosphorylatable S31A mutant, where alanine is the corresponding residue in canonical H3.1/2, did not rescue H3.3 loss, with even greater ssTelo signals detected in GFP-positive cells (Figures 4D, 4E, and S4C). However, the increased number of ssTelo signals was suppressed in cells expressing the phospho-mimetic S31E mutant (Figures 4D, 4E, and S4C). Therefore, H3.3S31ph may be required to sustain optimal ALT activity. H3.3S31ph can influence the trans acetylation of lysine 27 (H3K27)51 and the cis methylation of lysine 36 (H3.3K36) (Figure S4B). We expressed GFP-H3.3-harboring substitutions K27A and G34R (Figure SB). We chose to express G34R due to its inhibitory effect on H3K36 methylation but primarily because this oncohistone mutation has been strongly associated with ALT and ATRX loss in pediatric glioma.52-55 Western blot analysis validated the expression of these histone H3.3 mutants (Figure S4B). While the K27A mutant suppressed the increase in ssTelo signals due to endogenous H3.3 loss, the G34R mutant had an intermediary effect that, while not statistically significant, was more consistent with a modest suppression of ssTelo signals (Figures S4C and S4D). Therefore, unlike substitutions that interfere with H3.3S31ph, these substitutions and their associated modifications are dispensable for optimal ALT activity, as inferred by the ssTelo assay.
Even though several kinases, including IKKα and Aurora B, have been implicated in H3.3S31ph in specialized contexts,56-58 prior studies and our metaphase analysis show that CHK1 is chiefly responsible for H3.3S31ph in ALT cancer cells. Accordingly, inhibiting CHK1 and ATR kinase activity, but not IKK or Aurora B, induced the ssTelo phenotype in U2OS cells. (Figures S4E and S4F). Combining CHK1 inhibition with H3.3 depletion did not yield a further increase in ssTelo signals, indicative of epistasis (Figures 4D and 4E). Moreover, expressing WT H3.3 rescued the loss of endogenous H3.3 and restored the induction of ssTelo signals following CHK1i. This induction of ssTelo signals by CHK1i was lost in cells expressing the non-phosphorylatable S31A or phospho-mimetic S31E mutants (Figures 4D and 4E). These observations reinforce the possibility that CHK1-driven H3.3S31ph could be vital for maintaining productive ALT. The premise was bolstered by experiments showing that CHK1i treatment caused a striking association of TERRA at telomeres and substantial increases in TERRA R-loop levels (Figures 4F and 4G). These results are consistent with the conclusion that HIRA-mediated histone H3.3 deposition is required to limit the aberrant formation of TERRA R-loops at telomeres. We envision that HIRA achieves this function through its chromatin assembly role, thereby preventing nucleosome gaps. This activity may also facilitate the establishment of unique histone marks, namely H3.3S31ph, that are also integral in enacting this role during ALT.
DISCUSSION
Chromatin-directed control of “good” and “bad” R-loops during ALT
Prior studies have connected R-loops to chromatin, though usually in the context of transcriptional regulation at promoters, enhancers, and transcription termination sites.35,37 DNA hybrids were shown to biochemically prevent in vitro nucleosome assembly,38 suggesting that R-loops could profoundly affect chromatin structure in vivo. Accordingly, the dynamic formation and resolution of R-loops are implicated in targeting chromatin modifier complexes that regulate transcription. Rapid R-loop turnover was also correlated with increased histone exchange rates. Furthermore, the genetic disruption of the FACT histone H2A-H2B chaperone elicits R-loop-linked genome instability in yeast and human cells.59
Our study expands on the links between R-loop and chromatin assembly in the context of pathological ALT telomere extension in cancer cells lacking ATRX-DAXX. When present, ATRX protects telomeres by limiting aberrant DNA secondary structure formation and managing telomeric chromatin by recruiting DAXX to deposit H3.3. Upon inactivation of the ATRX-DAXX histone H3.3 deposition complex, HIRA becomes essential for ensuring histone H3.3 deposition at telomeric chromatin and ALT cancer cell survival.22 Here, we determined that disrupting HIRA renders telomeric chromatin hyper-accessible and susceptible to adverse ssDNA and TERRA R-loop accumulation. Left unabated, this ultimately leads to TRCs (Figure 4H). We uncovered that replenishing telomeric chromatin with newly synthesized histone H3.3 by the HIRA-UBN subcomplex is required to suppress excess TERRA R-loop formation. Intriguingly, when TERRA R-loop formation is stimulated through FANCM depletion, histone H3.3 deposition by HIRA is impeded. Remarkably, RNaseH1 expression, which removes R-loops, was sufficient to restore H3.3 deposition at telomeres in FANCM-depleted cells. Notably, disrupting new histone H3.3 deposition, through depleting HIRA, UBN, or FANCM, leads to non-productive ALT and challenges ALT cell viability. Therefore, borrowing the analogy put forth by Aguilera and colleagues,60 we envision that from the perspective of ALT cancer cells, HIRA (and UBN)-mediated deposition of new H3.3 suppresses the unscheduled formation of bad TERRA R-loops that threaten cancer cell viability. In contrast, good R-loops that form before H3.3 can be deposited or formed, as homologous recombination (HR) intermediates remain in place to stimulate the required level of telomere instability needed for productive ALT-HDR (Figure 4H). Indeed, the buildup of bad TERRA R-loops in nucleosome-deprived chromatin in HIRA-depleted cells coincided with increased ssDNA, R-loops, and TRCs within the nucleosome-deprived telomeric chromatin of HIRA-deficient cells (Figure 4H). When concurrent, these are associated with alterations in chromatin compaction and common fragile site (CFS) instability,61 like the telomere fragility observed after HIRA-UBN and H3.3 depletion.
While TERRA R-loop formation could be favored in the more accessible chromatin, as in HIRA-depleted cells, histone mark patterns are also influential. R-loops have been implicated in anchoring chromatin modifiers to establish pro-transcriptional H3K36 and H3K4 methylation patterns at enhancer and promoter-proximal regions.35,37 In contrast, histone H3 phosphorylation exacerbated the destabilizing effects of R-loops on genome integrity.62 Our investigation of H3.3-specific modifications revealed an unexpected role for HIRA in facilitating the downstream CHK1-dependent phosphorylation of serine 31 (Figure 4H). Our data indicated that CHK1-dependent H3.3S31ph is critical for suppressing aberrant ssDNA and TERRA and that CHK1, like HIRA, suppresses aberrant TERRA R-loops. This specialized and unique histone mark accumulates at telomeres in late G2 and mitosis49,50 when ALT-HDR is active, so it may contribute to ensuring efficient telomere-extension-coupled chromatin dynamics.
In conclusion, when efficiently deposited by HIRA, histone H3.3 might represent a barrier that limits TERRA R-loop formation and associated instability at telomeres. This protective act could be due to a block imposed by assembled nucleosomes that prevent invading or nascent TERRA RNA from hybridizing with telomeric DNA. HIRA might dislodge factors that promote TERRA R-loops or recruit factors that resolve them. The physical proximity of HIRA to the RNAPII complex19 might favor its action to suppress aberrant co-transcriptional R-loop formation. Furthermore, by delivering H3.3 bearing histone modifications not regulated by ATRX-DAXX, HIRA might profoundly change the chromatin signaling at telomeres. These observations highlight the antagonistic dynamics between HIRA, histone deposition, and TERRA R-loops at telomeres that support productive ALT-HDR and ALT cancer cell survival. Acquiring deeper insights into HIRA’s role(s) could substantially improve our knowledge of the compensatory response to ATRX-DAXX loss in ALT cancer cells and reveal new therapeutic opportunities.
Limitations of the study
This study does not directly evaluate how individual H3.3S31 histone mutants impact TERRA R-loop homeostasis. While the ssTelo assay was used as a proxy for ALT activity in H3.3-depleted and H3.3-mutant-complemented cells, superior strategies to disrupt histone H3.3 and express individual histone mutant transgenes or introduce mutations through genome editing are required. Furthermore, our study does not investigate those cis and trans effects of H3.3S31ph or the crosstalk with other histone marks may cooperate with CHK1-directed H3.3S31ph in regulating TERRA R-loop homeostasis. Conceivably, while newly deposited H3.3 physically blocks aberrant R-loop formation, H3.3S31ph could modulate the establishment and reading of proximal modifications like H3K27 and H3K36, as shown in different contexts.51,57 With mounting evidence linking R-loops to the cytotoxic impact of FANCM disruption or DNA-damage-associated factor inhibition (ATR, CHK1, WEE1, PARP1),23,40,63-66 understanding the impact of histone mutants and histone-modifying enzymes on TERRA R-loop homeostasis could offer substantial insights into whether targeting these factors and pathways could be harnessed to kill ALT cancer cells, particularly those lacking ATRX-DAXX.
STAR ★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
U2OS (ATCC, HTB-96, Female) cell lines were obtained, authenticated by STR profiling, and confirmed mycoplasma-free by ATCC cell line authentication services. WT/DA TRF1-FokI U2OS and VA13 (female) cell lines were generously provided by Roger Greenberg (University of Pennsylvania). Doxycycline inducible ATRX expressing U2OS (iATRX) cells were previously described4 and generously provided by David Clynes (University of Oxford). SV40 Large T-antigen immortalized control and IMR90 ATRX-KO ALT (female) were previously described24 and generously provided by Hongwu Zheng and Jihye Paik (Weill Cornell Medicine). Each cell line was cultured in Glutamax-DMEM supplemented with 10% fetal growth serum. Cells were cultured at normal oxygen conditions of 20% O2 and 7.5% CO2 at 37°C.
METHOD DETAILS
ATAC-seq
ATAC-seq was performed according to published protocols.74-79 Briefly, 50,000 nuclei per replicate were extracted from fresh harvested cells by incubating in lysis buffer (Resuspension buffer [10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2], 0.1% v/v NP-40, 0.1% v/v Tween 20, and 0.01% v/v digitonin [Promega]) on ice for 15 min. Wash buffer (Resuspension buffer, 0.1% v/v Tween 20) was added to nuclei extraction suspension at the end of this incubation period, samples centrifuged at 600 x g for 10 min at 4°C, supernatant discarded, and nuclei flash frozen for storage at −80°C. Nuclei were thawed on ice, resuspended in 50 μL transposition mix (2X Tagmentation Buffer [Diagenode C01019043], 1X PBS, 0.1% v/v Tween 20, 0.01% v/v Digitonin, 2.5 μL loaded Tn5 tagmentase (Diagenode C01070012), and incubated at 37°C for 30 min with 1000 rpm shaking on a thermomixer. Following the transposition reaction, DNA was purified by loading over a Zymo DNA Clean and Concentrator kit column and following the manufacturer’s instructions (Zymo D4004). Libraries were prepared using Nextera i5 (50*) and i7 (70*) primers (each at 25 μM) with NEBNext High-Fidelity 2X PCR Master Mix (NEB M0541S). Fragments were first amplified for 5 cycles with high-fidelity PCR. After 5 initial amplification cycles, 1 μL of partially amplified libraries was used as input to qPCR with NXT PCR Primers 1 and 2 (2 μM) and KAPA SYBR Green for 20 cycles to determine the number of additional PCR cycles needed, as previously described.74-79 Libraries were run on 1.5% agarose gel, DNA extracted using the QG Buffer Method (Qiagen 1014876), purified over an EconoSpin DNA column purification (Epoch Life Science 1920-250), and quantified using the Qubit dsDNA HS Assay Kit (ThermoFisher Q32851) and fluorometer. The final libraries were paired-end sequenced on a NextSeq2000 obtaining ~30 million uniquely mapped reads per sample.
CUT&RUN
CUT&RUN experiments were performed as previously described using 100,000 cells.74-79 Nuclei were extracted from each cell line using nuclear extraction (NE) buffer (20 mM HEPES-KOH pH 7.9, 10 mM KCl, 0.5 mM spermidine, 0.1% Triton X-100, 20% glycerol, freshly added protease inhibitors), flash frozen and stored at −80°C. Nuclei were thawed on ice for ~5 min and resuspended in 600 μL of NE buffer. Nuclei were bound to pre-washed lectin-coated ConA beads (Polysciences). Samples were pre-blocked by adding blocking buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine, 0.1% BSA, 2 mM EDTA, fresh protease inhibitors) followed by resuspension in wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine, 0.1% BSA, fresh protease inhibitors). Bead-bound nuclei were incubated in wash buffer containing either a primary antibody targeting H3.3 (Mouse, Active Motif Cat No. 91191, Lot No. 25820004) or a no primary antibody (no ab) negative control for 1 h at room temperature with rotation. Following the primary antibody incubation period, bead-bound nuclei were washed and resuspended in wash buffer containing purified recombinant pA/G-MNase for 30 min at room temperature incubation with rotation. Samples were temperature equilibrated in an ice-water bath before initiation of digestion by pA/G-MNase via the addition of 3 mM CaCl2 for 30 min. The digestion reaction was quenched by adding STOP Buffer (20 mM EDTA, 4 mM EGTA, 1.5 pg MNase-digested S. cerevisiae spike-in DNA). CUT&RUN enriched fragments were solubilized and used for input for library preparation. Libraries were prepared as described80 and paired-end sequenced on a NextSeq2000, obtaining ~10 million uniquely mapped reads per sample.
Direct immunofluorescence (IF)
Cells on glass coverslips were washed twice in PBS and fixed with 2% paraformaldehyde (PFA) for 10mins. Cells were permeabilized with 0.1% (w/v) sodium citrate and 0.1% (v/v) Triton X-100 for 5mins and incubated with fresh blocking solution (1 mg/mL BSA, 10% normal goat serum, 0.1% Tween) for 30mins. Primary antibodies were diluted in blocking solution and added to cells for 1 h at RT or overnight refrigerated. Next, cells were washed three times with PBS for 5mins and incubated with Alexa-coupled secondary antibodies (488nm, 555nm, 647nm) (Life Technologies) for 1 h at RT. Then, cells were washed three times with PBS and mounted on slides with Prolong Gold Anti-fade reagent with DAPI (Life Technologies). Once the Prolong Anti-fade polymerized and cured, cells were visualized by conventional fluorescence with 60X Plan λ objective (1.4 oil) using a Nikon Eclipse Ti2-E.
IF-FISH
After secondary antibody incubation, cells were washed, and then the IF staining was fixed with 2% paraformaldehyde (PFA) for 10mins. PFA was washed off with PBS and coverslips dehydrated with successive washes in 70%, 95%, and 100% EtOH for 3mins, allowed to air dry completely. Next, the coverslips were mounted on glass slides with 15 ml per coverslip of hybridization mix (70% deionized Formamide, 1 mg/ml of Blocking Reagent [Roche], 10mM Tris-HCl pH 7.4) containing Alexa 488-(CCCTAA)4 PNA probe. DNA was denatured by setting the slides on a heating block set to 72°C for 10 min and then incubating overnight at RT in the dark. The coverslips were then washed twice for 15mins with Wash Solution A (70% deionized formamide and 10mM Tris-HCl pH7.2) and three-time with Solution B (0.1M Tris-HCl pH7.2, 0.15M NaCl, and 0.08% Tween) for 5 mins at RT. EtOH dehydration was repeated as above, and finally, the samples were mounted and analyzed as mentioned above.
Proximity ligation assay (PLA)
Cells on glass coverslips were washed twice in PBS and fixed with 2% paraformaldehyde (PFA) for 10mins. Cells were permeabilized with 0.1% (w/v) sodium citrate and 0.1% (v/v) Triton X-100 for 5mins and blocked for 1h at 37°C with blocking solution (Sigma). The coverslips were incubated in primary antibody overnight at 4°C (1:1000 mouse RNAPII, 1:8000 rabbit PCNA). Cells were incubated in a pre-mixed solution of PLA probe anti-mouse minus and PLA probe anti-rabbit plus (Sigma) for 1 h at 37°C. According to the manufacturer’s instructions, the Duolink in situ Detection Reagents (Green) were used to perform the PLA reaction. Then, cells were washed three times with PBS, and the samples were mounted and analyzed as mentioned above.
TERRA RNA-FISH
Following IF, the cells were washed, and then the IF staining was fixed with 2% paraformaldehyde (PFA) for 10mins. Coverslips were dehydrated in 70%, 95%, and 100% EtOH for 3mins, and allowed to air dry completely. Next, the coverslips were mounted on glass slides with 40mL per coverslip of hybridization mix (50% deionized Formamide, 2X SSC, 2 mg/mL BSA, 10% Dextran Sulfate) containing Alexa 488-(CCCTAA)7 PNA probe. Glass slides were incubated in a hybridization chamber at 40°C overnight. The coverslips were washed three times for 5mins with Wash Buffer 1 (2X SSC, 50% deionized formamide) and three times for 5mins with Wash Buffer 2 (2X SSC) at 40°C. EtOH dehydration was repeated as above, and finally, the samples were mounted and analyzed as mentioned above.
Metaphase spreads (FISH)
Colcemid (Gibco) was added for ~2hrs, cells were harvested by trypsinization, swelled in 75mM KCl and fixed in 70% Methanol: 30% Acetic Acid. Samples are stored at −20°C overnight. Metaphase chromosomes were spread onto washed slides, then RNase A (0.5 mg/mL) and pepsin treated. The slides were then washed in PBS, dehydrated by EtOH washes and allowed to air dry completely. The telomeres were hybridized with fluorescently labeled by a specific PNA probe for the positive telomere strand (TTAGGG)4 (Alexa 488, green color). Before hybridization of the PNA, DNA is denatured by heating at 72°C for 10mins, as in IF-FISH, and then incubated for 2 hrs at RT. Slides were washed twice for 10mins with Wash Solution A (see IF-FISH), dried and then incubated with the second PNA for 2 hrs at RT. The slides were then re-washed twice for 15 mins with Wash Solution A and 3 times with Wash Solution B (see IF-FISH) for 5 mins at RT. Finally, cells were dehydrated in EtOH and mounted on slides with Prolong Gold Anti-fade reagent with DAPI (Life Technologies). Metaphase chromosomes were visualized with a 60X Plan λ objective (1.4 oil) on a Nikon Eclipse Ti2-E microscope.
SNAP histone H3.3 labeling assay
U2OS cells were transfected with indicated siRNAs. 48hrs later, these cells were transfected with SNAP-H3.3-HA, Flag-WT, or DA-TRF1-FokI. The next day (24hrs later), the quenching of old parental histone H3.3 and labeling of newly synthesized histone H3.3 was conducted as follows. Pre-existing histones were first quenched by incubating cells with 10μM SNAP-cell Block (New England Biolabs) for 30min followed by a 30min wash and a 2hr chase in fresh growth medium. Newly synthesized SNAP-tagged histone H3.3 that were synthesized during the chase were labeled for detection by immunofluorescence by incubating cells with 2μM SNAP-cell TMR star (New England Biolabs) for 15min (pulse) followed by a 20min incubation in fresh medium. PBS-washed cells were incubated in CSK buffer for 10mins before fixation and subsequent staining.
Telomere DNA synthesis detection by EdU
Following siRNA knockdown, TRF1-FokI U2OS cells were induced with doxycycline (40 ng mL−1) and synchronized in G2 with RO-3306 (Sigma) (10μM) 24hrs before harvest, followed by tamoxifen (4-OHT, Sigma) (1μM) and Shield1 Ligand (Takara Clontech) (1μM) 3hrs before harvest. Cells were pulsed with EdU (10μM) 1 h before harvest. Cells on glass coverslips were washed twice in PBS and fixed with 2% paraformaldehyde (PFA) for 10mins. Cells were permeabilized with 0.1% (w/v) sodium citrate and 0.1% (v/v) Triton X-100 for 5mins. The Click-IT Plus EdU Cell Proliferation Kit with Alexa Flour 488 (Invitrogen) was used to detect EdU.
Telomeric DNA isolation
Approximately 2 × 106 cells per condition were harvested by scraping in PBS on ice. Cells were washed with 1X PBS and centrifuged (500G, 5mins, 4°C). Cell pellets were resuspended in 350mL TNE (10mM Tris pH 7.4, 100mM NaCl, 10mM EDTA), and 35μL TNES/proteinase K (10mM Tris pH 7.4, 100mM NaCl, 10mM EDTA, 1% SDS, 100 mg/mL proteinase K) overnight at 37°C. Genomic DNA was then extracted using Phenol-Chloroform. 250mg of DNA was incubated with HinFI, HphI, MnlI and RE buffer (330 mM Tris-HCl, pH 8.0, 100 mM magnesium acetate, 1 M LiCl, 5 mM DTT) overnight at 37°C. The digested DNA was then combined with RSE-H buffer (polymerase and dNTPs) and the telomere capture oligo (Biotin-5’-ATCC(CCCTAA)3−3’). The samples were then placed at 64°C for 20 min for denaturation and primer extension. The RSE reaction was then combined with 90μL of RSE-B beads and incubated for 1hr rotating at RT. The beads were then subjected to 2 washes of 500mL RSE Wash Buffer for 5 min rotating at RT. Beads were resuspended in 45mL of RSE Resuspension Buffer R, eluted in an 82°C water bath for 5 min, and let to sit overnight to come to RT. DNA was then quantified and prepped for dot blotting.
DNA-RNA immunoprecipitation (DRIP)
DRIP was conducted as described.11 siRNAs were transfected twice (0 and 3 days) in (WT)- or (D450A)- TRF1-FokI cells. Cells were induced with doxycycline (40 ng μL−1) 24hrs before harvest at day 6, followed by tamoxifen (1μM) and SHIELD (1μM) 3hrs before harvest. Approximately 107 cells per condition were harvested by trypsinization and placed on ice. Cells were washed with 1X PBS and centrifuged (500G, 5mins, 4°C). Cells were dissolved in 200μL cold RLN buffer (50 mM Tris-HCl pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 1 mM dithiothreitol (DTT), and 100 U ml–1 RNasIN PLUS) on ice for 5mins. Cells were centrifuged (500G, 5mins, 4°C), and the liquid was carefully discarded. Cell pellets were lysed with 500μL RLT lysis buffer (RNAeasy Plus, Qiagen) and homogenized through a 20G × 11/2 syringe (0.9 mm × 40 mm). The homogenized extracts were mixed with 250μL H2O and 750μL ultra-pure phenol:chloroform: isoamyl alcohol (25:24:1). The samples were transferred to a heavy phase-lock tube and centrifuged (13,000G, 5mins). The top aqueous phase was poured into a tube containing 750μL ice-cold propanol and 50mM NaCl. The samples were inverted until nucleic acids precipitated. The samples were centrifuged (13,000G, 30mins, 4°C) to form nucleic acid pellets. The pellets were washed twice with 70% ice-cold EtOH and air-dried. The pellets were dissolved in 130μL of H2O and sonicated on a Covaris system (Peak power- 50, Duty Factor- 20, Cycles/Burst- 200, 150sec) to obtain fragments of approximately 200bp. 30μg of nucleic acids was incubated with RNaseH or H2O in RNaseH buffer (New England Biosystems) at 37°C overnight. Protein G beads (Dynabeads) were washed three times in DIP1 Buffer (10 mM HEPES-KOH pH 7.5, 275 mM NaCl, 0.1% Na-deoxycholate, 0.1% SDS, 1% Triton X-100). Samples were diluted by ten in DIP1 Buffer, and 40μL of Protein G beads were added for 1 h of pre-clearing rotating at 4°C. One percent of the sample was saved as input. Samples were transferred to a new tube, and 40μL of Protein G beads with 3μg of S9.6 antibody were added to each sample and incubated overnight, rotating at 4°C. Next, beads were washed with DIP2 Buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.1% Na-deoxycholate), DIP3 Buffer (50 mM HEPES-KOH pH 7.5, 500 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.1% Na-deoxycholate), and DIP4 Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 250 mM LiCl, 1% NP-40, 1% Na-deoxycholate). The sample was eluted in 100μL of Elution Buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA pH 8.0, 0.5% SDS, Proteinase K) shaking at 55°C for 30mins twice. DNA was purified (PCR cleanup, NucleoSpin Macherey-Nagel) and eluted in 100μL H2O. For DRIP coupled with qPCR, input and IP samples were analyzed by qPCR using telomeric primers (see Table S1).
Western blotting
Cells were harvested with trypsin, quickly washed in PBS, counted with Cellometer Auto T4 (Nexcelom Bioscience), and directly lysed in 4X NuPage LDS sample buffer at 10000 cells per mL. Proteins were gently homogenized using a Nuclease (ThermoFisher), denatured for 10 mins at 70°C, and resolved by SDS-Page electrophoresis, transferred to nitrocellulose membranes, blocked in 5% milk in TBST for 30mins and probed. HRP-linked anti-rabbit or mouse (Amersham) was used for secondary antibodies, and the HRP signal was visualized with SuperSignal ECL substrate (Pierce).
Isolation of chromatin-associated histones
Approximately 15 min before collection, 100nM Calyculin A (Cell Signaling Technology) was added to cell cultures. Cells were harvested using ice-cold PBS, scraped from the dish, and transferred to pre-cooled tubes. Cell pellets were resuspended in 200mL ice-cold RIPA buffer (10mM Tris-HCl (pH 7.5), 500mM NaCl, 0.5mM EDTA, 0.1% SDS, 1% Sodium deoxycholate, 1% Triton) with 1mM PMSF, 2.5mM MgCl2,1 mg/ml Benzonase (Pierce), protease inhibitor cocktail (Thermo). Calyculin A (Cell Signaling Technology) was added to PBS wash, RIPA, and elution buffer to preserve histone phosphorylation. Tubes were placed on ice for 30mins with extensive pipetting every 10mins before clarification by centrifugation at 20,000x g for 10 min at 4°C. Cleared lysates were transferred to pre-cooled tubes. 4X LDS buffer was added. Samples were then boiled at 68°C for 5mins.
siRNA transfections
siRNA Smartpools from Dharmacon (GE) were used (see Table S1). Briefly, 200,000 and 700,000 cells were seeded per well of a 6-well plate and 10cm dish containing growth medium without antibiotics. ~6hrs later, cells were transfected. siRNAs and Dharmafect were diluted in OptiMEM (Life Technologies). A working siRNA concentration of 50nM was used. We used 2.5 μL and 10 μL Dharmafect transfection reagent per well and 10cm plate. Transfection medium was replaced with complete culture media 24hrs later, or cells were split for the desired application and harvested at 72hrs post-transfection.
TERRA ASO knockdown
TERRA knockdown was conducted as described.11 Approximately 150,000 cells were seeded per well of a 6-well plate containing 2mL of growth medium without antibiotics. ~Six hours later, cells were transfected with 6pmol of either Scramble or TERRA ASO and 4 μL of RNAiMax (ThermoFisher) diluted in OptiMEM (Life Technologies). The transfection media was replaced with complete culture media 24hrs later, and cells were harvested 72hrs post-transfection.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data in this study were analyzed in GraphPad Prism, ImageJ, and Microsoft Excel. Detection, colocalization, and quantification of cells were performed using the ComDet v.0.5.3 plugin for ImageJ. Statistical tests used are indicated in the figure legend accompanying each figure. In most cases, one-way analysis of variance (ANOVA) was used. Typically, unless otherwise stated, n refers to the# of independent experiments and SEM refers to the standard error of means. The sample size was not pre-determined. For telomere motif quantification, paired t-tests were used to calculate statistical significance between samples from the same cell line background (i.e., ALT NT vs. ALT siHIRA). Unpaired t-tests were used to calculate statistical significance across conditions between cell line backgrounds (i.e., Control siHIRA vs. ALT siHIRA).
ATAC-seq analysis
Raw sequencing reads were first trimmed to 25bp and then aligned to the human T2T genome assembly (chm13v2.0) using bowtie2 aligner with a mismatch parameter allowance of 1bp to account for telomeric repeat variants (–dovetail -N 1 -I 10 -X 1000 -no-unal-S), generating a sam file output. Picard was used to coordinate sort aligned reads and filter out PCR duplicates from sam file. Next, the command grep -vwE was used to remove mitochondrial reads and generate a bam output file. Bam files were sorted into 1-100bp size class using SAMtools.69 The merge command from SAMtools was used to create a single merged bam file from the three 1-100bp size class sorted bam file replicates for each experimental condition. RPGC normalized bigWig files were generated from the merged 1-100bp size class sorted and indexed bam files using the bamCoverage command from deepTools73 with options – normalizeUsing RPGC –effectiveGenomeSize 2725240337 –ignoreDuplicates -bs 1. Heatmaps were generated from RPGC normalized bigWig files over RefSeq annotated chm13v2.0 genome assembly transcription start sites (TSSs) genome wide using the computeMatrix (–missingDataAsZero -b 2000 -a 2000 -bs 20) and plotHeatmap commands from deepTools.16 To enable comparison across experimental conditions and replicates, a sorted matrix bed file was generated for the IMR90 control siRNA sample using the plotHeatmap -m command over TSSs with option -descend –outFileSortedRegions bed. Next, this control sample matrix coordinate bed file was used as the reference file -R for sorting all other experimental samples using computeMatrixOperations sort -keep. All matrices were then visualized over T2T annotated RefSeq TSSs using plotHeatmap.
CUT&RUN analysis
Raw sequencing read files were first trimmed to 25bp and then mapped to the human T2T genome assembly (chm13v2.0) using bowtie2 aligner with a mismatch parameter allowance of 1bp (–dovetail -N 1 -I 10 -X 1000) to generate a sam file. Aligned reads were coordinated and sorted, and PCR duplicates quality mapping score reads (MAPQ ≥10) were removed from the sam file using Picard. Reads were sorted into a 150-500bp size class file representing nucleosome-sized fragments using SAMtools and output as a bam file. Bam files were sorted and indexed before merging across replicates of the same cell line and experimental condition using the merge command from SAMtools. RPGC normalized bigWig files were generated from the 150-500bp size class sorted and indexed merged bam files using the bamCoverage command from deepTools16 with options –normalizeUsing RPGC –effectiveGenomeSize 2725240337 –ignoreDuplicates -bs 1 -bl CUT&RUN specific T2T assembly blacklist regions.81 Heatmaps were generated from RPGC normalized bigWig files over RefSeq annotated chm13v2.0 genome assembly transcription start sites (TSSs) genome-wide using the computeMatrix and plotHeatmap commands from deepTools. To enable comparison across experimental conditions, a sorted matrix bed file was generated for the IMR90 control siRNA sample using the plotHeatmap -m command over TSSs with option -descend –outFileSortedRegions bed. Next, this control matrix coordinate bed file was used as the reference file -R for sorting all other experimental samples using computeMatrixOperations sort -keep. All matrices were then visualized over T2T annotated RefSeq TSSs using plotHeatmap.
Telomere-specific ATAC-seq and CUT&RUN analyses
The filtered bam file output from Picard (https://broadinstitute.github.io/picard/) was used as input to qMotif73 to quantify telomere motif counts. The BuildBamIndex command from Picard was used on the filtered bam file to generate an index (bai) for use with qMotif. qMotif was run in fast mode (includes_only = true) so that only the reads aligned to known telomeric regions were read from the input bam file. The qMotif configuration.ini file used is included as part of the supplementary materials and was written with a stage 1 string pass command threshold of 3 consecutive canonical telomeric repeats (stage1_motif_string = TTAGGGTTA GGGTTAGGG) and the second regular expressions pass command to capture reads with telomere motif variants containing variation in the second bp position (stage2_motif_regex=(… GGG){2,}∣(CCC …){2,}). Raw qMotif telomeric motif counts were normalized to account for differences in sequencing depth across samples as follows: qMotif input bam files were first converted to bed files using the bamtobed command from BEDTools.73 Then, the coverageBed command from BEDTools was applied to calculate the average genome coverage for each replicate across all biological conditions (excluding mitochondrial reads, X and Y chromosome reads). The average fraction genome coverage was normalized between samples by generating a normalization scaling factor calculated from setting the sample with the lowest fraction of genome coverage to 1, and then multiplying the raw telomere motif count for each sample by the respective normalization scaling factor to calculate a normalized telomere motif count.
TelSeq was used to quantify average estimated telomere length and cumulative telomeric reads (%) within each sample from the same bam file used as input for qMotif analysis.70,71 Telomeric repeats were defined as copies of TTAGGG. The parameter -k 2 was used, meaning that reads that contained at least 2 telomeric repeats were considered as telomeric reads for average estimated telomere length calculations and cumulative telomeric reads quantification. Telomere length was calculated from the TelSeq output report. Cumulative telomeric reads for each read length (i.e., the number of telomere repeats) was calculated by dividing the number of telomeric reads per telomeric read length by the total number of reads in the TelSeq input bam file.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TRF2 | Novus | Cat# NB110-57130; RRID:AB_844199 |
| FLAG (D6W5B) | Cell Signaling Technology | Cat# 14793S; RRID:AB_2572291 |
| γ-Tubulin | Sigma-Aldrich | Cat# T-6557; RRID:AB_477584 |
| RNAPII | Millipore | Cat# 05-962; RRID:AB_11213782 |
| RNAPII phospho serine-5 | Cell Signaling Technology | Cat# 13523; RRID:AB_2798246 |
| RNAPII phospho serine-2S2 | Cell Signaling Technology | Cat# 13499; RRID:AB_2798238 |
| PCNA | Abcam | Cat# ; RRID:AB_2798238 |
| S9.6 | Kerafast | Cat# ENH001; RRID:AB_2798238) |
| RPA2 | Abcam | Cat# ab2175; RRID:AB_302873 |
| phospho-S4/S8 RPA2 | Bethyl | Cat# A300-245A; RRID:AB_210547 |
| phospho-S33 RPA2 | Bethyl | Cat# A300-246A; RRID:AB_210547 |
| γ-H2AX | Millipore | Cat# 05-636; RRID:AB_309864 |
| H2AX | Cell Signaling Technology | Cat# 2595; RRID:AB_10694556 |
| UBN1 | Abcam | Cat# ab101282; RRID:AB_10671644 |
| FANCM | Novus | Cat# NBP2-50418; RRID:AB_2716711 |
| HIRA | Active Motif | Cat# 39557; RRID:AB_2793256 |
| ATRX | Cell Signaling Technology | Cat# 14820; RRID:AB_2798630 |
| DAXX | Cell Signaling Technology | Cat# 4533; RRID:AB_2088778 |
| Histone H3.3 (WB) | Abcam | Cat# ab176840; RRID:AB_2715502 |
| Histone H3.3 (C&R) | Active Motif | Cat# 91191; RRID:AB_2793796 |
| Phospho-Histone H3.3 S31 | Active Motif | Cat# 39637; RRID:AB_2793286 |
| GFP-HRP | Miltenyi | Cat# 130-091-833; RRID:AB_247003 |
| H3K27ac | Thermo Fisher | Cat# MA5-23516; RRID:AB_2608307 |
| H3.3 G34R | Discovery Antibodies | Cat# cCRB2005185 |
| Cyclin B | Cell Signaling Technology | Cat# 4138; RRID:AB_2072132 |
| Phospho-Histone H3 S10 | Cell Signaling Technology | Cat# 9701; RRID:AB_331535 |
| Auto anti-dsDNA | University of Iowa, Developmental Studies Hybridoma Bank | Cat# autoanti-dsDNA; RRID:AB_10805293 |
| Auto anti-ssDNA | University of Iowa, Developmental Studies Hybridoma Bank | Cat# autoanti-ssDNA; RRID:AB_10805144 |
| PML | Santa Cruz Biotechnology | Cat# sc-966; RRID:AB_628162 |
| Chemicals, peptides, and recombinant proteins | ||
| Benzonase | Merck Millipore | Cat# 70746 |
| 4-hydroxytamoxifen | Sigma | Cat# H7904 |
| Doxycycline | Clontech | Cat# 631311 |
| Shield Ligand | Clontech | Cat# 632189 |
| Lipofectamine 3000 | Thermo Fisher | Cat# L3000 |
| Dharmafect Reagent 1 | Dharmacon | Cat# T-2001 |
| RNAiMAX | Thermo Fisher | Cat# 13778075 |
| Tel C PNA probe | PNA Bio | Cat# F1004 |
| Tel G PNA probe | PNA Bio | Cat# F1008 |
| TERRA PNA Probe | IdT | (TAACCC)5-Alexa488 |
| Flavopiradol | Selleck Chem | Cat# S1230 |
| Triptolide | Selleck Chem | Cat# S3604 |
| Critical commercial assays | ||
| Duolink® In Situ PLA® Probe Anti-Rabbit PLUS | Millipore | Cat# DUO92002; RRID:AB_2810940 |
| Duolink® In Situ PLA® Probe Anti-Mouse MINUS | Millipore | Cat# DUO92004; RRID:AB_2713942 |
| SNAP-Cell® TMR-Star | NEB | Cat# S9105 |
| SNAP-Cell® Block | NEB | Cat# S9106 |
| Click-iT™ RNA Alexa Fluor™ 488 Imaging Kit | Invitrogen | Cat# 10329 |
| Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit | Invitrogen | Cat# C10337 |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat# 740609.50 |
| Deposited data | ||
| CUT&RUN data | This study | GSE269261 |
| ATAC-seq data | This study | GSE269268 |
| Experimental models: Cell lines | ||
| U2OS | ATCC | Cat# HTB-96; RRID:CVCL_0042 |
| VA13 | ATCC | Cat# CCL-75.1; RRID:CVCL_2759 |
| WT/DA TRF1-FokI U2OS | Dilley et al.12 | N/A |
| Doxycycline-inducible ATRX expressing U2OS (iATRX) | Clynes et al.4 | N/A |
| SV40 Large T-antigen immortalized control and ATRX KO ALT IMR90 | Li et al.24 | N/A |
| Oligonucleotides | ||
| Primer sequences | N/A | Table S1 |
| siRNA sequences | N/A | Table S1 |
| Recombinant DNA | ||
| SNAP-H3.3-3xHA | Ray-Gallet et al.18 | N/A |
| GFP-H3.3 | Prokhorova et al.67 | N/A |
| GFP-H3.3-K27A | This study | N/A |
| GFP-H3.3-G34R | This study | N/A |
| GFP-H3.3-S31A | This study | N/A |
| GFP-H3.3-S31E | This study | N/A |
| YFP-HIRA | Torné et al.42 | N/A |
| YFP-HIRA-I461D | Torné et al.42 | N/A |
| YFP-HIRA-R227K | Torné et al.42 | N/A |
| EGFP-RNaseH1 | Suzuki et al.68 | N/A |
| Software and algorithms | ||
| FastQC, V0.11.7 | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Bowtie 2, v2.4.5 | N/A | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Picard, v2.18.12 | Broad Institute | https://broadinstitute.github.io/picard/ |
| SAMtools, v1.10 | Li et al.69 | http://www.htslib.org/download/ |
| qMotif, v1.2 | Holmes et al.70 | https://adamajava.readthedocs.io/en/latest/qmotif/qmotif_1_2/ |
| TelSeq, v0.0.2 | Ding et al.71 | https://github.com/zd1/telseq |
| bedtools, v2.29.0 | Quinlan et al.72 | https://bedtools.readthedocs.io/en/latest/ |
| deepTools, v3.3.0 | Ramírez et al.73 | https://deeptools.readthedocs.io/en/develop/index.html |
| Scripts and analysis pipeline | This study | https://doi.org/10.5281/zenodo.13942196 |
| ComDet plugin for ImageJ | N/A | https://github.com/UU-cellbiology/ComDet |
Highlights.
HIRA establishes greater telomeric chromatin accessibility after ATRX-DAXX loss
Deposition of new H3.3 by HIRA-UBN restricts telomeric ssDNA and TERRA R-loops
Unresolved TERRA R-loops block new H3.3 deposition by HIRA-UBN
CHK1 phosphorylation of H3.3 is critical to prevent ssDNA and TERRA R-loop buildup
ACKNOWLEDGMENTS
We thank the Genome Stability Program at UPMC Hillman for helpful discussions and are grateful to Genevieve Almouzni (Curie Institute), Lars Jansen (Oxford University), Hongwu Zheng, and Jihye Paik (Weill Cornell Medicine) for providing SNAP-H3.3 plasmids and cell lines, respectively. We are grateful to Will MacDonald at the University of Pittsburgh HSCRF Genomics Research Core for assistance with sequencing and the University of Pittsburgh Center for Research Computing for providing access to the HTC cluster. We thank Jeff Ward for technical advice on image processing. We acknowledge Eros Denchi (NCI), Travis Stracker (NCI), and Kyle Miller (Emory) for discussing complementary data before submission. This work was funded by grants #R01CA207209 (NCI), #R01262316 (NCI), and #R37CA263622 (NCI) to R.J.O.; grant #R35GM133732 (NIGMS) to S.J.H.; Ruth L. Kirschstein NRSA grant #F30CA278287 (NCI) to A.R.W.; and T32 training grant #EB001026 to E.E.B. R.J.O.’s laboratory at UPMC Hillman Cancer Center is supported by Cancer Center Support grant #P30CA047904 (NCI). This work used the HTC cluster at the University of Pittsburgh Center for Research Computing, supported by NIH award #S10OD028483.
Footnotes
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Roderick J. O’Sullivan (osullivanr@upmc.edu).
Materials availability
All reagents generated in this study are available upon request to the lead contact and upon signature of the corresponding material transfer agreement, if necessary.
- Original code has been deposited and is publicly available as of the publication date. The DOI is listed in the key resources table.
- Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114964.
REFERENCES
- 1.Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, and Reddel RR (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med 3, 1271–1274. 10.1038/nm1197-1271. [DOI] [PubMed] [Google Scholar]
- 2.Dunham MA, Neumann AA, Fasching CL, and Reddel RR (2000). Telomere maintenance by recombination in human cells. Nat. Genet 26, 447–450. 10.1038/82586. [DOI] [PubMed] [Google Scholar]
- 3.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. (2011). Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 333, 425. 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, and Gibbons RJ (2015). Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun 6, 7538. 10.1038/ncomms8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen DT, Voon HPJ, Xella B, Scott C, Clynes D, Babbs C, Ayyub H, Kerry J, Sharpe JA, Sloane-Stanley JA, et al. (2017). The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 18, 914–928. 10.15252/embr.201643078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. (2010). Distinct Factors Control Histone Variant H3.3 Localization at Specific Genomic Regions. Cell 140, 678–691. 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suvà ML, Benes CH, et al. (2015). Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273–277. 10.1126/science.1257216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva B, Arora R, Bione S, and Azzalin CM (2021). TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun 12, 3760. 10.1038/s41467-021-24097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feretzaki M, Pospisilova M, Fernandes RV, Lunardi T, Krejci L, and Lingner J (2019). RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 587, 303–308. 10.1038/s41586-020-2815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yadav T, Zhang J-M, Ouyang J, Leung W, Simoneau A, and Zou L (2022). TERRA and RAD51AP1 promote alternative lengthening of telomeres through an R- to D-loop switch. Mol. Cell 82, 3985–4000.e4. 10.1016/j.molcel.2022.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaminski N, Wondisford AR, Kwon Y, Lynskey ML, Bhargava R, Barroso-González J, García-Expósito L, He B, Xu M, Mellacheruvu D, et al. (2022). RAD51AP1 regulates ALT-HDR through chromatin-directed homeostasis of TERRA. Mol. cell 82, 4001–4017.e7. 10.1016/j.molcel.2022.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, and Greenberg RA (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58. 10.1038/nature20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J-M, Yadav T, Ouyang J, Lan L, and Zou L (2019).Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 26, 955–968.e3. 10.1016/j.celrep.2018.12.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis PW, Elsaesser SJ, Noh K-M, Stadler SC, and Allis CD (2010). Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 107, 14075–14080. 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Déjardin J, and Kingston RE (2008). Purification of proteins associated with specific genomic Loci. Cell 136, 175–186. 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Exposito L, Bournique E, Bergoglio V, Bose A, Barroso-Gonzalez J, Zhang S, Roncaioli JL, Lee M, Wallace CT, Watkins SC, et al. (2016). Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase η in the Alternative Lengthening of Telomeres. Cell Rep. 17, 1858–1871. 10.1016/j.celrep.2016.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tagami H, Ray-Gallet D, Almouzni G, and Nakatani Y (2004). Histone H3.1 and H3.3 Complexes Mediate Nucleosome Assembly Pathways Dependent or Independent of DNA Synthesis. Cell 116, 51–61. 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- 18.Ray-Gallet D, Woolfe A, Vassias I, Pellentz C, Lacoste N, Puri A, Schultz DC, Pchelintsev NA, Adams PD, Jansen LET, and Almouzni G (2011). Dynamics of Histone H3 Deposition In Vivo Reveal a Nucleosome Gap-Filling Mechanism for H3.3 to Maintain Chromatin Integrity. Mol. Cell 44, 928–941. 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Pchelintsev NA, McBryan T, Rai TS, van Tuyn J, Ray-Gallet D, Almouzni G, and Adams PD (2013). Placing the HIRA Histone Chaperone Complex in the Chromatin Landscape. Cell Rep. 3, 1012–1019. 10.1016/j.celrep.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adam S, Polo SE, and Almouzni G (2013). Transcription Recovery after DNA Damage Requires Chromatin Priming by the H3.3 Histone Chaperone HIRA. Cell 155, 94–106. 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 21.Bouvier D, Ferrand J, Chevallier O, Paulsen MT, Ljungman M, and Polo SE (2021). Dissecting regulatory pathways for transcription recovery following DNA damage reveals a non-canonical function of the histone chaperone HIRA. Nat. Commun 12, 3835. 10.1038/s41467-021-24153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoang SM, Kaminski N, Bhargava R, Barroso-González J, Lynskey ML, García-Expósito L, Roncaioli JL, Wondisford AR, Wallace CT, Watkins SC, et al. (2020). Regulation of ALT-associated homology-directed repair by polyADP-ribosylation. Nat. Struct. Mol. Biol 27, 1152–1164. 10.1038/s41594-020-0512-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang J, Zhao H, Diplas BH, Liu S, Liu J, Wang D, Lu Y, Zhu Q, Wu J, Wang W, et al. (2020). Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 80, 510–523. 10.1158/0008-5472.can-18-3374. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Deng Z, Zhang L, Wu C, Jin Y, Hwang I, Vladimirova O, Xu L, Yang L, Lu B, et al. (2019). ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 38, e96659. 10.15252/embj.201796659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856. 10.7554/elife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patty BJ, and Hainer SJ (2021). Transcription factor chromatin profiling genome-wide using uliCUT&RUN in single cells and individual blastocysts. Nat. Protoc 16, 2633–2666. 10.1038/s41596-021-00516-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tafessu A, O’Hara R, Martire S, Dube AL, Saha P, Gant VU, and Banaszynski LA (2023). H3.3 contributes to chromatin accessibility and transcription factor binding at promoter-proximal regulatory elements in embryonic stem cells. Genome Biol. 24, 25. 10.1186/s13059-023-02867-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karagyozova T, Gatto A, Forest A, Quivy J-P, Marti-Renom MA, Mirny L, and Almouzni G (2024). HIRA-dependent provision of histone H3.3 in active chromatin ensures genome compartmentalisation. bioRxiv. 10.1101/2024.08.27.609896. [DOI] [Google Scholar]
- 29.Jasencakova Z, Scharf AND, Ask K, Corpet A, Imhof A, Almouzni G, and Groth A (2010). Replication Stress Interferes with Histone Recycling and Predeposition Marking of New Histones. Mol. Cell 37, 736–743. 10.1016/j.molcel.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 30.Loe TK, Li JSZ, Zhang Y, Azeroglu B, Boddy MN, and Denchi EL (2020). Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 34, 650–662. 10.1101/gad.333963.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parikh D, Fouquerel E, Murphy CT, Wang H, and Opresko PL (2015). Telomeres are partly shielded from ultraviolet-induced damage and proficient for nucleotide excision repair of photoproducts. Nat. Commun 6, 8214. 10.1038/ncomms9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bou-Nader C, Bothra A, Garboczi DN, Leppla SH, and Zhang J (2022). Structural basis of R-loop recognition by the S9.6 monoclonal antibody. Nat. Commun 13, 1641. 10.1038/s41467-022-29187-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes AP, Ilter D, Low V, Rosenzweig A, Shen Z-J, Schild T, Rivas MA, Er EE, McNally DR, Mutvei AP, et al. (2019). Dynamic Incorporation of Histone H3 Variants into Chromatin Is Essential for Acquisition of Aggressive Traits and Metastatic Colonization. Cancer Cell 36, 402–417.e13. 10.1016/j.ccell.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho NW, Dilley RL, Lampson MA, and Greenberg RA (2014). Inter-chromosomal Homology Searches Drive Directional ALT Telomere Movement and Synapsis. Cell 159, 108–121. 10.1016/j.cell.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, and Chédin F (2016). Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 63, 167–178. 10.1016/j.molcel.2016.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen PB, Chen HV, Acharya D, Rando OJ, and Fazzio TG (2015). R-loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol 22, 999–1007. 10.1038/nsmb.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skourti-Stathaki K, Kamieniarz-Gdula K, and Proudfoot NJ (2014). R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 516, 436–439. 10.1038/nature13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunn K, and Griffith JD (1980). The presence of RNA in a double helix inhibits its interaction with histone protein. Nucleic Acids Res. 8, 555–566. 10.1093/nar/8.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chedin F, and Benham CJ (2020). Emerging roles for R-loop structures in the management of topological stress. J. Biol. Chem 295, 4684–4695. 10.1074/jbc.rev119.006364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva B, Pentz R, Figueira AM, Arora R, Lee YW, Hodson C, Wischnewski H, Deans AJ, and Azzalin CM (2019). FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun 10, 2253. 10.1038/s41467-019-10179-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786.e19. 10.1016/j.cell.2017.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torné J, Ray-Gallet D, Boyarchuk E, Garnier M, Le Baccon P, Coulon A, Orsi GA, and Almouzni G (2020). Two HIRA-dependent pathways mediate H3.3 de novo deposition and recycling during transcription. Nat. Struct. Mol. Biol 27, 1057–1068. 10.1038/s41594-020-0492-7. [DOI] [PubMed] [Google Scholar]
- 43.Ray-Gallet D, Ricketts MD, Sato Y, Gupta K, Boyarchuk E, Senda T, Marmorstein R, and Almouzni G (2018). Functional activity of the H3.3 histone chaperone complex HIRA requires trimerization of the HIRA subunit. Nat. Commun 9, 3103. 10.1038/s41467-018-05581-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang Y, Poustovoitov MV, Zhao K, Garfinkel M, Canutescu A, Dun-brack R, Adams PD, and Marmorstein R (2006). Structure of a human ASF1a–HIRA complex and insights into specificity of histone chaperone complex assembly. Nat. Struct. Mol. Biol 13, 921–929. 10.1038/nsmb1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricketts MD, Frederick B, Hoff H, Tang Y, Schultz DC, Singh Rai T, Grazia Vizioli M, Adams PD, and Marmorstein R (2015). Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat. Commun 6, 7711. 10.1038/ncomms8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ricketts MD, Dasgupta N, Fan J, Han J, Gerace M, Tang Y, Black BE, Adams PD, and Marmorstein R (2019). The HIRA histone chaperone complex subunit UBN1 harbors H3/H4- and DNA-binding activity. J. Biol. Chem 294, 9239–9259. 10.1074/jbc.ra119.007480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groth A, Corpet A, Cook AJL, Roche D, Bartek J, Lukas J, and Almouzni G (2007). Regulation of Replication Fork Progression Through Histone Supply and Demand. Science 318, 1928–1931. 10.1126/science.1148992. [DOI] [PubMed] [Google Scholar]
- 48.O’Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, and Karlseder J (2014). Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol 21, 167–174. 10.1038/nsmb.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hake SB, Garcia BA, Kauer M, Baker SP, Shabanowitz J, Hunt DF, and Allis CD (2005). Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc. Natl. Acad. Sci 102, 6344–6349. 10.1073/pnas.0502413102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang FTM, Chan FL, R McGhie JD, Udugama M, Mayne L, Collas P, Mann JR, and Wong LH (2015). CHK1-driven histone H3.3 serine 31 phosphorylation is important for chromatin maintenance and cell survival in human ALT cancer cells. Nucleic Acids Res. 43, 2603–2614. 10.1093/nar/gkv104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng Y-C, Tastemel M, and Banaszynski LA (2019). Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat. Genet 51, 941–946. 10.1038/s41588-019-0428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tönjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 53.Voon HPJ, Hughes JR, Rode C, De La Rosa-Velázquez IA, Jenuwein T, Feil R, Higgs DR, and Gibbons RJ (2015). ATRX Plays a Key Role in Maintaining Silencing at Interstitial Heterochromatic Loci and Imprinted Genes. Cell Rep. 11, 405–18. 10.1016/j.celrep.2015.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voon HPJ, Udugama M, Lin W, Hii L, Law RHP, Steer DL, Das PP, Mann JR, and Wong LH (2018). Inhibition of a K9/K36 demethy-lase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun 9, 3142. 10.1038/s41467-018-05607-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Udugama M, Hii L, Garvie A, Cervini M, Vinod B, Chan F-L, Das PP, Mann JR, Collas P, Voon HPJ, and Wong LH (2021). Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat. Commun 12, 2584. 10.1038/s41467-021-22543-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li M, Dong Q, and Zhu B (2017). Aurora Kinase B Phosphorylates Histone H3.3 at Serine 31 during Mitosis in Mammalian Cells. J. Mol. Biol 429, 2042–2045. 10.1016/j.jmb.2017.01.016. [DOI] [PubMed] [Google Scholar]
- 57.Armache A, Yang S, Martínez de Paz A, Robbins LE, Durmaz C, Cheong JQ, Ravishankar A, Daman AW, Ahimovic DJ, Klevorn T, et al. (2020). Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 583, 852–857. 10.1038/s41586-020-2533-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sitbon D, Boyarchuk E, Dingli F, Loew D, and Almouzni G (2020). Histone variant H3.3 residue S31 is essential for Xenopus gastrulation regardless of the deposition pathway. Nat. Commun 11, 1256. 10.1038/s41467-020-15084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herrera-Moyano E, Mergui X, García-Rubio ML, Barroso S, and Aguilera A (2014). The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription–replication conflicts. Genes Dev. 28, 735–748. 10.1101/gad.234070.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.García-Pichardo D, Cañas JC, García-Rubio ML, Gómez-González B, Rondón AG, and Aguilera A (2017). Histone Mutants Separate R Loop Formation from Genome Instability Induction. Mol. Cell 66, 597–609.e5. 10.1016/j.molcel.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 61.Helmrich A, Ballarino M, Nudler E, and Tora L (2013). Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol 20, 412–418. 10.1038/nsmb.2543. [DOI] [PubMed] [Google Scholar]
- 62.Castellano-Pozo M, Santos-Pereira JM, Rondón AG, Barroso S, Andújar E, Pérez-Alegre M, García-Muse T, and Aguilera A (2013). R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. cell 52, 583–590. 10.1016/j.molcel.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 63.Lu R, O’Rourke JJ, Sobinoff AP, Allen JAM, Nelson CB, Tomlinson CG, Lee M, Reddel RR, Deans AJ, and Pickett HA (2019). The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun 10, 2252. 10.1038/s41467-019-10180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan X, Chen Y, Biju B, Ahmed N, Kong J, Goldenberg M, Huang J, Mohan N, Klosek S, Parsa K, et al. (2019). FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep 9, 19110. 10.1038/s41598-019-55537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zimmermann M, Bernier C, Kaiser B, Fournier S, Li L, Desjardins J, Skeldon A, Rimkunas V, Veloso A, Young JTF, et al. (2022). Guiding ATR and PARP inhibitor combinationswith chemogenomic screens. Cell Rep. 40, 111081. 10.1016/j.celrep.2022.111081. [DOI] [PubMed] [Google Scholar]
- 66.Petropoulos M, Karamichali A, Rossetti GG, Freudenmann A, Iaco-vino LG, Dionellis VS, Sotiriou SK, and Halazonetis TD (2024). Transcription-replication conflicts underlie sensitivity to PARP inhibitors. Nature 628, 433–441. 10.1038/s41586-024-07217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prokhorova E, Agnew T, Wondisford AR, Tellier M, Kaminski N, Beijer D, Holder J, Groslambert J, Suskiewicz MJ, Zhu K, et al. (2021). Unrestrained poly-ADP-ribosylation provides insights into chromatin regulation and human disease. Mol. Cell 81, 2640–2655.e8. 10.1016/j.molcel.2021.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suzuki Y, Holmes JB, Cerritelli SM, Sakhuja K, Minczuk M, Holt IJ, and Crouch RJ (2010). An Upstream Open Reading Frame and the Context of the Two AUG Codons Affect the Abundance of Mitochondrial and Nuclear RNase H1. Mol. Cell Biol 80, 5123–5134. 10.1128/mcb.00619-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holmes O, Nones K, Tang YH, Loffler KA, Lee M, Patch A-M, Dagg RA, Lau LMS, Leonard C, Wood S, et al. (2022). qmotif: determination of telomere content from whole-genome sequence data. Bioinform. Adv 2, vbac005. 10.1093/bioadv/vbac005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ding Z, Mangino M, Aviv A, Spector T, and Durbin R; UK10K Consortium (2014). Estimating telomere length from whole genome sequence data. Nucleic Acids Res. 42, e75. 10.1093/nar/gku181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013).Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Arm-strong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klein DC, Lardo SM, McCannell KN, and Hainer SJ (2023). FACT regulates pluripotency through proximal and distal regulation of gene expression in murine embryonic stem cells. BMC Biol. 21, 167. 10.1186/s12915-023-01669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zou H, Poore B, Brown EE, Qian J, Xie B, Asimakidou E, Razska-zovskiy V, Ayrapetian D, Sharma V, Xia S, et al. (2023). A neurodeve-lopmental epigenetic programme mediated by SMARCD3-DAB1-Reelin signalling is hijacked to promote medulloblastoma metastasis. Nat. Cell Biol 25, 493–507. 10.1038/s41556-023-01093-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klein DC, Lardo SM, and Hainer SJ (2024). The ncBAF complex regulates transcription in AML through H3K27ac sensing by BRD9. Cancer Res. Commun 4, 237–252. 10.1158/2767-9764.crc-23-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klein DC, and Hainer SJ (2020). Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 28, 69–85. 10.1007/s10577-019-09619-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nordin A, Zambanini G, Pagella P, and Cantù C (2023). The CUT&RUN suspect list of problematic regions of the genome. Genome Biol. 24, 185. 10.1186/s13059-023-03027-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.