

Abstract
Preeclampsia (PE) is a significant complication of pregnancy, occurring in approximately 10% of pregnancies. However, the underlying mechanisms of this condition remain unclear. Placentation and tumorigenesis both share many characteristics, but PE is the result of insufficient placentation, in contrast to the overaggression of tumorigenesis. AXL is a biomarker and therapeutic target for multiple metastatic cancers. We hypothesized that its downregulation could play a crucial role in the development of PE. In our study, we demonstrated that pregnant Axl−/− mice exhibited typical PE symptoms, such as hypertension, proteinuria, and inadequate trophoblast invasion and spiral artery remodeling. Cross-mating and embryo transplantation experiments confirmed that these phenotypes were caused by the decidua. RNA sequencing results revealed the abnormal expression of several transcripts in the decidua, including Corin, which encodes a cardiac protease responsible for activating atrial natriuretic peptide (ANP). ANP is a cardiac hormone that regulates sodium homeostasis and blood pressure. Chromatin immunoprecipitation-qPCR analysis indicated that the decreased CORIN in Axl−/− decidua was due to reduced signal transducer and activator of transcription 3 (STAT3) binding. Treatment with ANP successfully alleviated the PE symptoms. Furthermore, we observed that in PE decidua, the level of AXL was significantly lower compared to normal pregnancies. These findings suggest that the dysregulation of decidua-derived AXL-CORIN-ANP signaling disrupts maternal-fetal crosstalk and contributes to the development of PE.
Keywords: MT: RNA/DNA Editing, ANP, AXL, CORIN, Decidua, Placenta, Preeclampsia
Graphical abstract
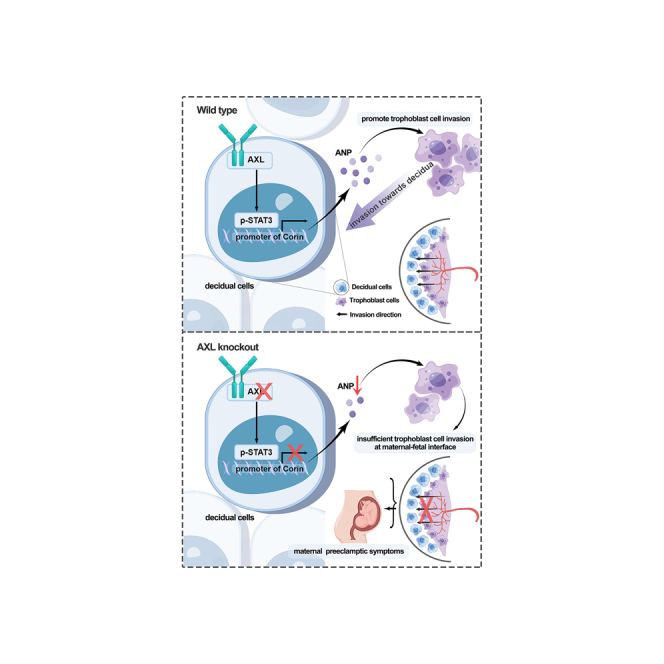
Preeclampsia is a significant complication of pregnancy. AXL serves as both a biomarker and a therapeutic target for cancers. Zhang et al. demonstrate that AXL plays roles in maternal-fetal communication and preeclampsia through the STAT3-CORIN-ANP pathway. ANP, being a cardiac hormone that regulates sodium homeostasis and blood pressure, can alleviate preeclampsia symptoms.
Introduction
Preeclampsia (PE) is a gestational syndrome affecting 5%–8% of all pregnancies and is the leading cause of fetal and maternal morbidity and mortality. PE is characterized by newly occurring hypertension and/or proteinuria after 20 weeks of gestation.1,2 The only treatment currently is the delivery of the placenta.3 The etiology of PE is heterogeneous and complex, with studies in humans identifying endothelial dysfunction, immune dysregulation, and defective decidualization as elements in the pathogenesis of this disease.4,5,6,7,8 Animal models, particularly mice and rats, have been used to study the molecular mechanisms underlying PE.9,10
Shallow trophoblast invasion of the uterine spiral artery (SpA) leads to abnormal placentation, followed by maternal symptoms in clinical manifestation.11 It is widely believed that PE appears in two-stage pathogenesis in that the shallow trophoblast invasion of the uterine decidua leads to abnormal placentation, followed by maternal symptoms.12 The insufficient arterial modification by trophoblasts results in poor perfusion of the placenta with maternal blood, leading to PE and/or intrauterine growth restriction of the fetus.13 Decidua formation precedes conceptus implantation, and decidualized cells are a source of secreted molecules such as growth factors, cytokines, metalloproteases, and protease inhibitors that support/control trophoblast invasion.14,15
AXL is a receptor tyrosine kinase belonging to the tyrosine-protein kinase receptor 3-Axl-c-mer tyrosine kinase family, with growth arrest-specific 6 (GAS6) as the common ligand. The binding of GAS6 to AXL stimulates AXL dimerization and intracellular domain autophosphorylation, thereby transducing extracellular signals to downstream effectors, playing essential roles in cell adhesion, invasion, migration, proliferation, and pro-inflammatory cytokine production.16,17 AXL has been described as a biomarker and therapeutic target for a variety of metastatic cancers.18 It is highly expressed in human ovarian tumors, and the inhibition of AXL signaling results in decreased cell invasion and prevents ovarian tumor progression.19
Placentation and tumorigenesis share many compelling features, including high proliferation, invasion, angiogenesis, and immune tolerance. Numerous studies have revealed that trophoblastic cells and cancer cells share many similar molecular pathways involved in proliferation and differentiation, immunomodulation, apoptosis and survival, migration, angiogenesis, the Warburg effect, the epithelial-mesenchymal transition, extracellular matrix proteases, and vessel remodeling.20,21 In regard to PE, the situation is quite similar to trophoblast cells and cancer cells: only the genes that are upregulated in trophoblast cells and cancer cells are typically downregulated in preeclamptic placentas. Therefore, the pathogenesis of PE is thought to be a result of inadequate placental development, as opposed to the overaggression seen in tumorigenesis.21 Considering the elevated expression of AXL in cancers and the significant decrease in AXL levels in the decidua of PE patients as revealed by our previous RNA sequencing (RNA-seq) analysis,22 we hypothesized that AXL could be an important contributor to the pathogenesis of PE.
In the present study, we investigated the potential physiological role of AXL during pregnancy by examining Axl knockout (Axl−/−) mice. We found that Axl knockout caused PE symptoms in pregnant mice bearing this mutation. Axl knockout affected the interaction between maternal decidual tissue and placental trophoblast, causing compromised trophoblast invasion and SpA remodeling. Furthermore, we discovered that AXL levels were lower in the decidua of women with PE compared to levels in normal pregnancies. Therefore, our study demonstrates that AXL-related decidual dysfunction hinders maternal-fetal communication and is a contributing factor in the pathogenesis of PE.
Results
Axl knockout leads to PE symptoms in pregnant mice
To evaluate the potential impact of AXL during pregnancy, we examined a previously established mouse model that employed homologous recombination to delete exon 9 of the Axl locus.23 Our findings showed that the pregnant Axl−/− mice had significantly higher systolic blood pressure (SBP) compared to pregnant Axl+/+ mice, starting around embryonic day 14.5 (E14.5), with blood pressure increasing further as pregnancy progressed and delivery neared (Figure 1A). The increased pressure is similar to the early onset of gestational hypertension seen in women with PE.2 Elevated urinary albumin/creatinine ratios are a major symptom of PE. Urine collected from pregnant Axl−/− females in metabolic cages between E14.5 and E17.5 for 72 h showed significantly higher urinary albumin/creatinine ratios compared to pregnant Axl+/+ mice (Figure 1B).
Figure 1.

Hypertension, proteinuria, renal pathology, and reduced fetal weight in pregnant Axl knockout mice
(A) Repeated tail-cuff systolic blood pressure (SBP) measurements of Axl+/+(n = 6) and Axl−/− (n = 7) mice before and during pregnancy. (B) Urine albumin/creatinine ratios of Axl+/+ and Axl−/− mice. Urine samples were collected (from E14.5 to E17.5) in metabolic cages (n = 3). (C) H&E and PAS staining of renal tissues from pregnant Axl+/+ and Axl−/− mice. The H&E staining reveals glomerular enlargement in Axl−/− mice. The PAS staining shows increased extracellular matrixes and collapsed glomerular capillaries in the Axl−/− mice. The findings were observed in at least three individual samples. (D) Transmission electron microscopy (TEM) imaging of the narrow glomerular capillary lumen (L), thick glomerular basement membranes (GBM), and the shortened secondary processes of podocytes indicate endothelial hyperplasia and extracellular matrix (EC) expansion in Axl−/− glomeruli (indicated by red lines) (n = 3). X-magnification 15,000× and Y-magnification 6,000×. (E) Uterine artery blood flow velocity waveforms in Axl+/+ and Axl−/− mice, assessed using pulse Doppler ultrasonography starting from E14.5 (n = 3). (F and G) Uterine artery waveforms from Axl+/+ and Axl−/− were analyzed to obtain resistance and pulsatility indexes. Values are means ± SEMs. Data are from 4 to 5 independent litters. (H) Representative photographs of placentas and fetuses of E14.5 Axl+/+ and Axl−/− mice. (I and J) Placental and fetal weights (E14.5) were reduced in Axl−/− females compared to Axl+/+ females. All data are expressed as means ± SDs. Scale bar, 20 μm. np, not pregnant. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Analysis of kidney glomerular sections taken at E14.5 revealed that the Axl−/− mice had ischemic glomeruli, as evidenced by reduced numbers of red blood cells in the enlarged glomeruli (Figure 1C). Furthermore, transmission electron microscopy of kidney glomerular sections revealed signs of glomerular hyperplasia in the pregnant Axl−/− mice, mainly manifested as capillary lumen narrowing, capillary loop occlusion, endothelial cell swelling and hypertrophy, and shortening of podocyte secondary processes (Figure 1D). These histopathology features recapitulate the renal pathology of women suffering from PE.24,25
We also conducted non-invasive Doppler ultrasound of the umbilical artery to assess the intrauterine blood supply, monitoring umbilical blood flow and placental resistance. Pulse wave images were acquired from the SpA in Doppler mode and used to quantify the velocity of umbilical artery blood flow. Axl−/− females had significantly elevated uterine artery resistance index (RI) values and significantly elevated uteroplacental pulsatility index values compared to Axl+/+ females at E14.5 (Figures 1E–1G). Additionally, pregnant Axl−/− mice had significantly reduced placental weight and fetal weight at E14.5 (Figures 1H–1J). These observations indicate that pregnant Axl−/− mice developed PE symptoms.
Impaired trophoblast invasion, SpA remodeling, and glycogen deposition in Axl−/− mice
It has been demonstrated that during pregnancy, trophoblast invasion and uterine SpA remodeling contribute to reducing maternal vascular resistance and increasing uteroplacental blood flow. Impaired trophoblast invasion and uterine SpA remodeling have been linked to PE symptoms.26 Cytokeratin staining of the uteroplacental interface showed that trophoblasts invaded deep into the decidua of Axl+/+ mice at E10.5 (arrows) and that Axl+/+ uteri exhibited enlarged SpAs surrounded by trophoblasts at E14.5 (solid box) (Figures 2Aa and 2Ac). Conversely, trophoblast invasion in Axl−/− uteri was markedly reduced, with only smaller arteries (solid box) present in both superficial and deep decidua (Figures 2Ab and 2Ad). By E18.5, Axl+/+ mice demonstrated a greater number of trophoblasts in the decidua and myometrium compared to Axl−/− mice (Figures 2Ba and 2Bb). Immunofluorescence of smooth muscle actin showed the expected replacement of smooth muscle by invasive trophoblasts in the deep decidua of Axl+/+ mice, a phenomenon not observed in Axl−/− sections (Figures 2Bc and 2Bd). Immunofluorescence of smooth muscle actin indicated the expected replacement of smooth muscle by invasive trophoblasts in the of Axl+/+ mice in Axl−/− sections (Figures 2Bc and 2Bd). Our findings suggest that Axl knockout hinders trophoblast invasion and uterine SpA remodeling in the maternal decidua of mice.
Figure 2.

Utero-placental interfaces of Axl+/+ and Axl−/− mice
(A) Immunohistochemical analysis of the Axl+/+ and Axl−/− interface at E10.5 and E14.5 shows the invasion level of cytokeratin expressing trophoblast cells (brown signal) (a–d). (B) Expression of trophoblast cell marker (cytokeratin, CK), blood vessel endothelial marker (CD31), and smooth muscle marker (smooth muscle actin [SMA]) at E18.5 maternal-fetal interface of Axl+/+ and Axl−/− mice. (C) Comparison of Sirius red, H&E, and PAS staining of Axl+/+ and Axl−/− interfaces shows a compact decidua (blue asterisks) and glycogen deposition in junctional zone (arrows) in Axl−/− mice at E14.5 (a–f). Arrows points to foci of glycogen deposition. (D) Comparison of H&E staining of Axl+/+ and Axl−/− interfaces shows glycogen deposition at E18.5 in Axl−/− mice. Red arrows indicate trophoblast cells of junctional zone. Black arrows point to foci of glycogen deposition. These findings were observed in at least three individual samples. Scale bars, 0.5 mm. DB, decidua basalis; JZ, junctional zone; Lab, labyrinth; Myo, myometrium; SpA, spiral artery.
We also noted that the Axl+/+ decidua basalis was loose (black asterisk), with reduced extracellular matrix content (Figures 2C, 2a, and 2c), while that of Axl−/− was compact (blue asterisk) with a high content of extracellular matrix (marked by arrows) (Figures 2Cb and 2Cd) through Sirius red staining of E14.5 sections. Moreover, H&E staining of Axl−/− mice revealed a noticeable increase in glycogen deposition (marked by arrows) in the junctional zone (Figure 2Cd), resembling the glycogen deposition seen in preeclamptic placentas in human studies and animal models.27,28 Periodic acid-Schiff (PAS) staining confirmed the presence of glycogen deposition in the junctional zone of Axl−/− placentas (marked by green arrows) but not in Axl+/+ samples at E14.5 (Figures 2Ce and 2Cf). The glycogen deposition (black arrows) was still observed in the junctional zone of E18.5 Axl−/− females (Figure 2D). These results demonstrate significant abnormalities in decidua basalis structure and increased glycogen deposition in Axl−/− mice.
To further investigate whether the abnormality of the placenta was confined to the maternal-fetal interface, we conducted dual fluorescence analysis of CD31 and cytokeratin to label blood vessels and trophoblasts in the placental labyrinth (Figures S1A and S1B). No differences were detected between the Axl−/− and Axl+/+ samples. Therefore, we conclude that insufficient trophoblast invasion, SpA remodeling, and aberrant glycogen deposition are confined to the decidua and the junctional zone of the placenta. These factors are responsible for the abnormal maternal-fetal interface observed in the Axl−/− mice.
Maternal Axl knockout causes the PE symptoms that are due to defective decidualization in Axl−/− mice
The debate over whether PE originates paternally or maternally has been ongoing for some time due to mutations that are maternally active and paternally imprinted genes that are normally expressed in placentogenesis.29 To better understand this issue, we conducted various crosses involving paternal or maternal mutations to investigate the maternal-fetal interactions and the individual contributions of each part to the PE symptoms. Females who were either Axl+/− or Axl−/− were crossed with males who were either Axl+/+ or Axl−/−. This led to the creation of interfaces representing four distinct genotypes. H&E staining revealed that regardless of the embryonic genotypes, Axl+/− maternal decidua (black asterisks) displayed a normal incompact phenotype and contained enlarged SpAs (Figures 3Aa and 3Ab). In contrast, Axl−/− decidua (carrying either an Axl+/− or Axl−/− fetus), showed abnormalities such as narrow vessels, compact decidual cells, and a non-degraded extracellular matrix (blue asterisks). The junctional zone of Axl−/− placentas displayed excessive glycogen deposition (arrows) (Figures 3Ac and 3Ad). Additionally, we utilized an embryo transfer strategy to transfer Axl+/+ embryos into Axl−/− uteri, and vice versa (Figure S2A). H&E staining showed glycogen deposition and a thinned junctional zone in Axl−/− mice carrying an Axl+/+ fetus, while no such symptoms were observed in Axl+/+ mice carrying an Axl−/− fetus (Figure S2B). The glomerular structures of the pregnant Axl−/− mice carrying Axl+/+ fetuses exhibited phenotypes similar to those of Axl−/− mice carrying an Axl−/− fetus (Figure S2C). Overall, these findings demonstrate that maternal Axl knockout was the underlying cause of abnormal maternal-fetal crosstalk in the context of PE syndrome.
Figure 3.

Maternal Axl knockout leads to abnormal interface in Axl−/− mice
(A) Utero-placental interface structure of different mating strategies by H&E staining. Black arrows point to foci of glycogen deposition. Black asterisks mark the loosened decidual zone, while blue asterisks point to the compacted decidua. These findings were observed in at least three individual samples. (B) The histology of E7.5 implantation sites and polyploid stromal cells in Axl+/+ and Axl−/− females. (C) The expression of decidual marker PRL8A2 at Axl+/+ and Axl−/− implantation sites on E7.5 (n = 3). (D) DNA content quantification of decidual cells by FACS from Axl+/+ and Axl−/− mice on E7.5. (E) Cellular distribution for 2N, 4N, and >4N populations of stromal cells at Axl+/+ (n = 3) and Axl−/− (n = 3) implantation sites on E7.5. Data represent the means ± SEMs. Scale bars, 0.5 mm; ∗p < 0.05. DB, decidua basalis; Em, embryo; JZ, junctional zone; Lab, labyrinth.
Previous studies have shown that mouse uterine stromal cells do not passively undergo modification by invasive trophoblasts, but rather initiate decidualization and transform into secretory decidual cells when embryos implant. This process is crucial for modulating the maternal-fetal interface.30 To investigate abnormalities in Axl−/− decidual cells, stromal decidualization was examined in early pregnant uteri. On E7.5, histological analysis revealed that implantation sites in Axl−/− mice appeared normal, with multinucleated secretory cells indicating successful transformation of decidual cells (Figure 3B). PRL8A2, a member of the prolactin family, is a well-known marker of mouse decidualization.31The immunohistochemistry (IHC) results showed no obvious difference in the expression of PRL8A2 between Axl+/+ and Axl−/− uterine decidua (Figure 3C). However, the size of the decidual cells was noticeably smaller in the Axl−/− mice compared to their Axl+/+ counterparts (Figures 3B and 3C). Flow cytometric analysis of DNA content indicated a concurrent decrease in >4N cells in Axl−/− decidua on E7.5 (Figures 3D and 3E), indicating inadequate decidualization in the Axl−/−mice and suggesting molecular abnormalities.
Downregulated Corin via the STAT3 pathway contributes to the defective decidualization in Axl−/− mice
To explore the molecular mechanisms underlying the defects in Axl−/− decidua, we employed artificially induced decidualization by oil injection and observed the induction of multinucleated decidual cells in both Axl+/+ and Axl−/− mice (Figures 4A and 4B). Further investigation of the molecules in Axl−/− decidua involved performing RNA-seq of the induced deciduoma samples, taking into account the aforementioned in vivo data of Axl−/− mice. A heatmap illustrating the clustering of differentially expressed genes was established (Figure 4C). Upon comparing the transcriptomes of the decidualized cells in the two groups, 850 dysregulated genes (˃2-fold) were identified among the total 19,486 genes, with 423 downregulated genes and 327 upregulated genes in Axl−/− deciduoma compared to Axl+/+ counterparts as shown in Figure 4D. It was observed that Corin, the gene encoding an atrial natriuretic peptide (ANP)-converting enzyme, was downregulated in Axl−/− deciduoma (Figure 4D). CORIN is a cardiac protease that activates ANP, a cardiac hormone that regulates blood pressure and sodium homeostasis.27 A previous study showed that both CORIN and ANP play essential roles at the maternal-fetal interface, and their abnormalities contribute to PE by impairing trophoblast invasion and SpA remodeling.32 We therefore confirmed differential expression of angiogenesis (Angptl1, Ada) and renin-angiotensin system-related genes (Corin, Ren2) through qPCR analysis (Figure 4E). These findings indicate that the downregulated AXL-CORIN signaling contributes to the defective decidualization observed in Axl−/− decidua.
Figure 4.

Decreased Corin expression in Axl−/− decidua
(A) Gross morphology of deciduoma at E8.5 of pseudopregnancy after artificial decidualization is induced in Axl+/+ and Axl−/− mice. (B) H&E staining of deciduoma sections from the oil-instilled left horn of the uterus showed a similar area in Axl+/+ and Axl−/− pseudopregnant mice 96 h after the oil injection (n = 3). (C) Heatmap of differentially expression genes in E8.5 oil-induced Axl−/− deciduoma compared with Axl+/+ ones. Red to black to blue indicates a gradient of high to low expression. (D) Volcano plot of the expression of altered genes. Genes that are differentially expressed at FDR <0.05 and logFC > 1 are highlighted and labeled. (E) Real-time qPCR validation of five genes selected from the volcano plot. The error bars represent SEM (n = 3). (F) Western blot analysis of CORIN and STAT3 activation level in both Axl+/+ and Axl−/− deciduoma. β-Actin was used as the control. (G) The binding of STAT3 to Corin promoter was decreased in Axl−/− mice. ChIP assays were performed using anti-STAT3 and control IgG antibodies, and fold enrichment of the indicated regions of CORIN promoter was examined using real-time PCR in isolated Axl+/+ and Axl−/− primary decidual cells (n = 3). All ChIP samples were normalized to intergenic input control. Scale bars, 0.5 mm; ∗∗p < 0.01; ∗∗∗p < 0.001.
We subsequently investigated the molecular mechanism by which Axl knockout leads to Corin downregulation. Immunoblotting of decidual tissue from Axl−/− mice showed a significant decrease in CORIN expression compared to control decidua. Activation of the Janus kinase-signal transducer and activator of transcription 3 (JAK-STAT3) pathway, a canonical downstream target of AXL, was impaired in Axl−/− decidua compared to Axl+/+ decidua (Figure 4F). These findings demonstrate that Axl knockout led to the downregulation of CORIN via the JAK-STAT3 pathway in the decidua. Next, we studied the relationship between Corin and JAK-STAT3 through chromatin immunoprecipitation (ChIP)-qPCR using a STAT3 antibody on decidual tissue samples. Our results showed a significant reduction in the binding between STAT3 and the specific region (−782 to −622 bp) of Corin in Axl−/− decidua compared to Axl+/+ decidua, as indicated by the binding peak (Figure 4G). These results further support the conclusion that Axl knockout downregulates CORIN via the JAK-STAT3 pathway in the decidua, leading to impaired signaling that contributes to defective decidualization and poor communication between maternal decidua and fetal trophoblasts.
ANP rescued PE symptoms in Axl knockout mice by promoting trophoblast invasion
As the cardiac protease CORIN activates ANP, a cardiac hormone regulating blood pressure,32,33 the downregulation of CORIN in Axl−/− decidua would be expected to deplete ANP abundance. To test this hypothesis, we injected Axl−/− mice daily with ANP (2 μg/0.1 mL/day per mouse) or vehicle control (saline) by intraperitoneal injection from E8.5 to E18.5 to assess the ability of ANP to rescue PE symptoms. We noticed that compared to the continuously increased SBP before E16.5 observed in vehicle control Axl−/− mice, both vehicle-treated Axl+/+ mice and ANP-treated Axl−/− mice exhibited normotensive blood pressure (Figure 5A). Additionally, proteinuria symptoms in Axl−/− mice were rescued by ANP treatment (Figure 5B). These findings demonstrate that ANP successfully rescues PE symptoms in Axl−/− mice.
Figure 5.

ANP treatment rescues PE symptoms in Axl−/− mice
(A) Mean SBP increased in Axl−/− mice treated with saline but not in those with ANP (2 μg/0.1 mL/day per mouse). (B) Late gestational urine albumin/creatine in Axl−/− mice treated with saline or ANP. n = 3 per group. (C) Immunohistochemical analysis of trophoblast cell marker (cytokeratin, CK) at Axl−/− interfaces with vehicle (saline) or ANP treatment at E10.5. Black triangles mark invasive trophoblast cells. These findings were observed in at least three individual samples. (D) Photomicrographs of trophoblast cell marker (CK) and blood vessel endothelial marker (CD31) at E18.5 maternal-fetal interface of Axl−/− mice treated with vehicle or ANP. (E) H&E staining of Axl−/− mice treated with ANP at E14.5 and E18.5. (F) Quantification of the average area of the junctional zone and labyrinth of placentas from different groups (n = 3). Scale bars, 0.5 mm. Data are means ± SEMs. ∗p < 0.05; ∗∗p < 0.01. DB, decidua basalis.
To determine whether ANP can alleviate the placental phenotype in Axl−/− mice, in addition to reducing blood pressure and proteinuria symptoms, we explored the uteroplacental interface of Axl−/− mice using IHC, immunofluorescence, and H&E staining. The IHC results demonstrated that ANP promoted trophoblast invasion into the decidua at E10.5 in Axl−/− mice, similar to the invasion observed in the Axl+/+ line (Figure 5C). Furthermore, immunofluorescence staining for CD31 and cytokeratin revealed that the SpA lumen was enlarged, and the number of invaded trophoblasts increased in Axl−/− decidua treated with ANP at E18.5 (Figure 5D). These results indicate that ANP enhanced trophoblast invasion and SpA remodeling, thus improving maternal and fetal interaction. Additionally, H&E staining of the placenta showed glycogen deposition (red arrows) in the junctional zone of Axl−/− mice at E14.5 after ANP treatment, with significant improvement observed by E18.5 (Figure 5E). This suggests that the condition of Axl−/− mice continuously improved with ANP treatment.
AXL expression is decreased in the decidual tissues of preeclamptic women
Decidual tissue sections of patients with severe PE (sPE) and normal pregnancies (NPs) (Table S1) were subjected to IHC against AXL (Figure 6A). Using vimentin as a marker for decidual cells quantified with Image-Pro Plus 6.0, we observed decreased AXL expression in the sPE group compared to the NP group (Figures 6A and 6B). In addition, we examined AXL mRNA levels using qPCR and found a significant downregulation in the sPE group compared to the NP group (Figure 6C). Western blotting of the decidual tissue further confirmed a significant decrease in AXL protein levels in the sPE group compared to the NP group (Figures 6D and 6E). These results suggest that decidual AXL expression is reduced in patients with sPE.
Figure 6.

The expression of AXL in decidua of normal pregnancy and sPE patients
(A and B) Representative images and quantification of AXL intensity in the decidual tissue of normal pregnancies (NP) and severe preeclampsia (sPE). (a, b) AXL; (c, d) vimentin. Dec, decidual cell-enriched area; T, trophoblast cell-enriched area. (C) Relative expression of AXL mRNA to GAPDH in utero-fetal interfaces of NP and sPE. (D) Western blotting showed that the protein level of AXL was decreased in sPE utero-fetal interfaces. GAPDH was used as the loading control. (E) Western blotting results were quantified and are shown in bar graphs (NP vs. sPE, 17 vs. 16). (F) Pathway model in which Axl knockout leads to insufficient placental trophoblast invasion through STAT3-CORIN-ANP signaling, thus causing maternal preeclamptic symptoms. Our study highlights the role of aberrant interactions between the maternal decidua and fetal placenta in the pathogenesis of PE. Data are presented as means ± SDs. Scale bars, 0.2 mm; ∗p < 0.05; ∗∗p < 0.01.
Discussion
AXL is highly expressed in cancers. Recent studies have shown that reducing AXL expression can weaken the malignancy and drug resistance of cancer cells,34 while AXL levels are significantly decreased in the decidua of patients with PE discovered in our previous study.22 This expression difference aligns with the opposing characteristics and similar regulatory mechanisms observed in various studies comparing PE and cancers.20,21 Our present study further demonstrated that the downregulation of AXL can impact placental development, angiogenesis, invasion, and other processes, potentially leading to the development of PE. We believe that the function of ALX in PE shares many principles with its role in cancer metastasis. The main difference lies in the varying levels of expression between PE and cancers, which result in different consequences.
Glomerular endotheliosis is characterized by enlarged glomeruli that lack blood cells due to the swollen endothelial cells and occluded capillary lumina.35 In our study, analysis of kidney sections revealed that Axl−/− pregnant females displayed abnormal glomerular structures, including a narrow capillary lumen, thickened basement membrane, and shorted foot process. These abnormalities decrease glomerular filtration and result in proteinuria due to endothelial destruction. Our findings are similar to a previous report that renal tissue in PE exhibits hallmarks of glomerular endotheliosis.36
At the maternal-fetal interface, we observed that the junctional zone of Axl−/− mice exhibited increased glycogen deposition. This localized glycogen deposition is also commonly seen in other murine models of PE, including a model induced by injecting interleukin-10 knockout mice with PE patient sera.27,28 Placental glycogen deposition is believed to stem from either impaired utilization or as a means of compensating for placental dysfunction in PE.27 Numerous studies have delved into the potential role of glycogen cells in the placenta and their connection to pregnancy complications. In human pregnancies, two medical conditions have been linked to altered glycogen deposition and/or metabolism: PE and diabetes.28 The presence of abnormal carbohydrate deposition in our research offers further compelling evidence for a causal relationship between glycogen deposition and PE.
By comparing the interfaces of Axl+/+ and Axl−/−, we have observed that the decidua of Axl−/− mice was more compact and contained a non-degraded extracellular matrix. The extracellular matrix serves various functions, such as regulating the formation of new vessel sprouts and providing vital contacts between endothelial cells and surrounding tissue, which helps prevent vessel collapse.37 When vascular cells migrate to form new sprouts, the extracellular matrix is proteolytically degraded, leading to changes in its composition. The reduction of the extracellular matrix during vessel sprouting is crucial for SpA remodeling and angiogenesis. We can infer that the persistence of non-degraded extracellular matrix in the decidua of Axl−/− mice plays a role in the observed PE symptoms in this model.
The maternal-fetal interfaces resulting from crossmating and embryo transfer strategies employing different genotypes demonstrated that Axl−/− maternal decidua is the cause of the abnormality. Our trials comparing gestational decidua and artificial oil-induced decidua show that decidualization occurs morphologically normally in the Axl−/− mouse relative to the control counterpart. The principal difference is that the decidua is more compact. Our subsequent RNA-seq, real-time qPCR, and western blot analysis of oil-induced deciduoma revealed the downregulated expression of CORIN in decidual tissue in the Axl−/− mouse, consistent with women with complications of PE.32 The downstream consequence of AXL signaling is decreased activation of the STAT3 pathway. There is a pronounced reduction in the activation of this pathway in Axl−/− decidua. Our further studies using ChIP-qPCR found that STAT3 can bind directly to the Corin promoter, and the downregulation of the STAT3 pathway in Axl−/− decidua inactivated the Corin promoter. The deficiency of Corin or its product, ANP, impairs SpA remodeling, causing gestational hypertension and proteinuria, the major PE symptoms.32,38 Moreover, ANP has been reported in promoting trophoblast invasion in a tumor necrosis factor-related apoptosis-inducing ligand-dependent way and decidualization,39 which helps us to prove that ANP rescues the symptoms of Axl−/− pregnant mice. Our discovery of the disturbance of the AXL-STAT3-CORIN pathway sheds light on how ANP could rescue the Axl−/− symptoms. The reduced expression of Corin in Axl−/− females due to JAK-STAT3 dysregulation in the uterine decidua contributes to PE, even though their decidualization is morphologically not affected.
ANP has been shown to facilitate trophoblast cell invasion in vitro.32,40 In HTR8/SVneo cells, a cell line of human invasive cytotrophoblasts, knockdown of the ANP receptor, NPR-A with small interfering RNA impaired its invasive ability, but had no effect on its proliferation or frequency of apoptosis.41 These findings are concordant with our observations in the mouse decidua. Previous reports have also shown that the invasion of trophoblast cells into the uterus is regulated in situ by a number of locally produced molecules, such as transforming growth factor-β and decorin, which are essential for maintaining healthy maternal-fetal homeostasis.13,40 Our study provides convincing evidence that the decidua-derived cascade of AXL-CORIN-ANP signaling is essential for proper trophoblast invasion, ensuring well-modified vascular connections between the mother and fetus. Collectively, our study provides convincing evidence that the decidua-derived cascade of AXL-CORIN-ANP signaling is essential to proper trophoblast invasion, ensuring well-modified vascular connections between the mother and the fetus. However, decidual defects induced by AXL deficiency impair decidualization, contributing to this life-threatening disease, thus emphasizing the essential role of decidual signaling in regulating pregnancy.
Materials and methods
Animal care and genotyping
Axl−/− mice on C57BL/6J background were purchased from The Jackson Laboratory (Bar Harbor, ME), and genotyping was performed as previously reported.23 Axl−/− mice were crossed to obtain the Axl+/− mice, and the Axl+/+ littermates were used as controls. All mice were housed under controlled temperatures (22°C ± 3°C) and light conditions (14 h light, 10 h darkness; lights on at 7 a.m.) and allowed free access to chow and water at the Experimental Animal Center of Shandong Normal University. All animal experiments were approved by the Animal Ethics Committee of Shandong Normal University.
Analysis of pregnancy and the artificial decidualization model
Two-month-old Axl+/+ and Axl−/− female mice were mated with fertile or vasectomized males of the same strain to induce pregnancy or pseudopregnancy, respectively. The morning of finding a vaginal plug was designated E0.5 of pregnancy. To induce artificial decidualization, one uterine horn of pseudopregnant mice was infused with sesame oil (25 μL) on E3.5; the non-infused contralateral horn was taken as a control. The tissue of infused and non-infused uterine horns was collected 4 days after the oil infusion. A section of each uterus was snap frozen in liquid nitrogen for subsequent RNA-seq analysis. A minimum of three mice for each genotype were used for every individual experiment.
Blastocyst transfer
Axl+/+ and Axl−/− female mice (donor mice) were injected with 7.5 U pregnant mare serum gonadotropin (Tianjin Laboratory Animal Center, Tianjin, China) and 48 h later with 7.5 U human chorionic gonadotropin (Tianjin Laboratory Animal Center) to induce ovulation; injected female mice were then mated. Meanwhile, Axl+/+ and Axl−/− female mice (receiver mice) were mated with Axl+/+ vasectomized males to induce pseudopregnancy. At E3.5, donor females were killed by cervical dislocation, and blastocysts were collected and transferred into the uterine horn of receiver mice. At least three mice of each mouse model were used for individual experiments.
Flow cytometry
E7.5 decidual cells were digested, centrifuged, and suspended in PBS; 1 mL cold 80% ethanol was added dropwise with continued gentle vertexing and incubated for 30 min on ice before staining. Cell pellets were suspended in staining solution (5 mg/mL propidium iodide and 2 mg/mL DNase-free RNase A in PBS) for 30 min at 37°C in the dark before flow cytometry was performed using Beckman Cytoflex. Flow cytometry data were analyzed using FlowJo software (version 10, Tree Star, Ashland, OR). Fluorescence-activated cell sorting (FACS) intensity values for side scatter-A and forward scatter-A (FSC-A) were used for loose gates. The intensity values of FSC-A and FSC-H by FACS were used to gate individual cells. Polyploid cells were differentiated by FSC-A intensity values proportional to cell size, and propidium iodide staining intensity values were used to measure DNA content. The experiments were repeated three times.
Blood pressure monitoring
Blood pressure was measured by a noninvasive blood pressure system for mice and rats (Softron Biotechnology, Beijing, China), as described previously.42 The investigators were blinded during blood pressure measuring. SBPs from six Axl+/+ mice and seven Axl−/− mice were calculated from three consecutive recordings. The data presented were from continuous recordings of at least 6 h/day (10 a.m.–4 p.m.).
Urinary albumin/creatinine measurements
Urine samples from three mice were collected for 72 h in metabolic cages from E14.5 to E17.5 in the presence of a mix of proteinase inhibitors (Roche, Hvidovre, Denmark). Urine creatinine concentrations were determined using the Creatinine Companion kit (Cayman Chemical, Ann Arbor, MI), and albumin concentrations were determined using a mouse albumin ELISA quantification kit (Bethyl Labs, Montgomery, TX).
Uterine artery Doppler ultrasound
More than three pregnant mice of both Axl+/+ and Axl−/− genotypes on E14.5 were imaged transcutaneously using Doppler ultrasound and a 30- or 40-MHz transducer operating at 30 frames per second (model Vevo 660, VisualSonics, Toronto, Canada). Studies were performed between 1 and 5 p.m. The high-pass filter was set at 6 Hz, and the pulsed repetition frequency was set between 4 and 48 kHz, to detect low to high blood flow velocities, respectively. A 0.2- to 0.5-mm pulsed Doppler gate was used, and the angle between the Doppler beam and the vessel was adjusted from 0° to 60° to provide the best alignment. Waveforms were saved for later offline analysis. The duration of anesthesia was limited to ∼1 h. The RI and the pulsatility index of the uterine arteries were calculated.
Transmission electron microscopy
For transmission electron microscopy (TEM) analysis, E14.5 Axl+/+ and Axl−/− kidneys (n = 3 for each genotype) were fixed with 2.5% glutaraldehyde at room temperature for 2–3 h, placed at 4°C. Then, they were fixed in 1% osmic acid for 1.5 h in the dark. The samples were then graded dehydrated in ethanol and infiltrated with acetone:resin = 1:1 for 1 h, acetone:resin = 1:2 overnight, and finally infiltrated with resin twice for 4 h each time. After embedding in resin, the samples were trimmed manually, and sections were cut (65 nm) using a Leica UC7 Ultramicrotome (Hitachi, Tokyo, Japan). The sections were stained with 2% uranyl acetate solution for 25 min, washed with water, and then stained with lead citrate for 7 min. The sections were subsequently baked under infrared light for 10 min and then observed under an 80-kV transmission electron microscope (Hitachi HT-7800), and photographs were taken at different magnifications.
Immunofluorescence
Pregnant uteri were snap frozen in liquid nitrogen and then cryosectioned at 10 μm at −20°C. The slides were fixed with ice acetone for 30 min, cleaned with PBS, and then permeabilized for 10 min in 0.1% Triton X-100. After blocking with 10% normal goat serum in PBS for 1 h at room temperature, the sections were incubated with primary antibodies in blocking solution overnight at 4°C. Specific secondary antibodies were used to detect the antigen, and DAPI was applied to identify the cell nucleus. All samples were imaged using a Leica confocal microscope (TCS SP8 MP, Leica Microsystems, Wetzlar, Germany).
H&E and IHC
Isolated tissues were fixed overnight in 4% paraformaldehyde in PBS, dehydrated using an ethanol series, embedded in paraffin, and sectioned (5 μm). For H&E staining, the tissues were deparaffinized in xylene, rehydrated through a series of ethanol solutions, and stained with H&E. For IHC, deparaffinized sections were subjected to antigen retrieval by boiling for 1 h in Tris/EDTA (pH 9.0) and cooling to room temperature, followed by peroxidase blocking for 10 min at room temperature. The blocking and antibodies were used for the immunofluorescence process described above. Sections were then incubated overnight at 4°C with the primary antibodies, incubated for 20 min at room temperature with horseradish peroxidase-labeled goat anti-rabbit immunoglobulin G (IgG) (Zhongshan Company, Beijing, China), and rinsed with PBS. The antibody complexes were detected using 3,3′-diaminobenzidine reagent according to the manufacturer’s instructions (Zhongshan Company). H&E staining was then performed per standard protocols. The antibodies that we used are listed in Table S2. All samples were imaged on the Pannoramic Scan (3DHISTECH, Budapest, Hungary). The staining quantification was performed using Image-Pro Plus 6.0 image analysis software (Media Cybernetics, Silver Spring, MD). For each group, a total of 30 fields from 10 different women (three fields of view for each woman) were photographed and examined.
PAS staining
To detect the corpora amylacea, paraffin sections were incubated in 0.5% PAS (Solarbio, Beijing, China) for 5 min at room temperature, followed by washing in tap water for 1 min. Then, the sections were incubated in Schiff reagent (Solarbio) for 10 min, followed by washing in tap water for 10 min. After the sections were rinsed in PBS for 10 min, the nuclei were stained with H&E.
SDS-PAGE and western blotting
The decidual tissues were extracted with Pierce radioimmunoprecipitation assay buffer (Beyotime, Shanghai, China). The lysates containing 30 μg protein were loaded on a 10% SDS-polyacrylamide gel, separated with 1× Tris-glycine running buffer, and then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Burlington, MA) in a wet electroblotting system. PVDF membranes were blocked for 1 h in 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 detergent (TBS-T), then incubated overnight at 4°C with antibodies. After washing in TBS-T, the membranes were incubated with secondary antibody (ZSGB-BIO, Beijing, China) in TBS-T and then detected with the SuperSignal chemiluminescent detection system (Thermo Scientific, Carlsbad, CA). The antibodies we used are listed in Table S2. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal loading control.
RNA isolation and qPCR
Total RNA was isolated with the TRIzol reagent (Invitrogen, Waltham, MA) and quantified with a NanoDrop 2000 (Thermo Scientific). An aliquot of 1 μg RNA was used to synthesize cDNA. Expression levels of the genes were validated by real-time qPCR analysis with SYBR Green (Takara Bio, San Jose, CA). Cycling parameters were 95°C for 3 min, followed by 40 cycles of 95°C for 30 s, 60°C for 20 s, 72°C for 30 s, and a final extension for 4 min at 72°C. The efficiency of all SYBR Green primer sets was determined and validated using serial dilutions to generate a five-point curve. The primers used for test genes are listed in Table S3.
Construction of RNA-seq libraries and sequencing
Total RNA was extracted from the samples (n = 3 for each genotype) using TRIzol reagent (Invitrogen). The RNA quality was detected with Agilent 2200 and stored at −80°C. The RNA integrity number >7.0 was used for the construction of cDNA libraries. A cDNA library was constructed for each RNA sample using the TruSeq Strand mRNA Library Prep Kit (Illumina, San Diego, CA), according to the manufacturer’s instructions. In brief, mRNA was purified from 1 μg total RNA using oligomeric (dT) magnetic beads and cleaved into 200- to 600-bp RNA fragments using divalent cations at 85°C for 6 min. The cleaved RNA fragments were used for the synthesis of the first and second strands. The cDNA fragment was end repaired and attached to the index adapter. The ligated cDNA product was purified and treated with uracil DNA glycosylase to remove the second strand cDNA. The purified first-strand cDNA was enriched by PCR to establish a cDNA library. The libraries were quality controlled with Agilent 2200 and sequenced with NovaSeq 6000 on a 150-bp paired-end run.
RNA-seq data analysis
Clean reads are obtained from raw reads by removing adapter sequences and low-quality reads prior to read mapping. The clean reads were then aligned to the mouse genome (mm10, NCBI) using Hisat2.43 HTseq44 was used to obtain gene counts and the reads per kilobase million method was used to determine gene expression. The DESeq2 algorithm45 was used to filter the differentially expressed genes. After significant analysis, p value and fase discovery rate (FDR) analysis46 were subjected to the following criteria: (1) fold change (FC) >2 or <0.5 and (2), p < 0.05, FDR <0.05. Gene Ontology (GO) analysis was performed to elucidate the biological implications of the differentially expressed genes.47 Fisher’s exact test was applied to identify the significant GO categories (p < 0.05).
ChIP‒qPCR
STAT3 binding to the Corin promotor was assessed by ChIP assay using Axl+/+ and Axl−/− decidua (n = 3 for each genotype). ChIP analysis was performed as previously described.48 In brief, the isolated tissues were fixed with formaldehyde for 10 min at room temperature. The chromatin was sonicated using the Bioruptor Pico sonication device (Diagenode Diagnostics, Liege, Belgium) to fragment the DNA-protein complexes. This supernatant was then incubated overnight at 4°C with STAT3 antibody, followed by protein A/G beads to pull down the complex. After several washes, the resulting protein/DNA complexes were subjected to crosslink reversal and DNA extraction. Specific primers were used to detect immunoprecipitated chromatin fragments, as well as input chromatin, using qPCR assay (the primers we used are listed in Table S3).
In vivo treatment with ANP
Pregnant Axl−/− mice were treated with ANP (2 μg/0.1 mL/day per mouse) or vehicle control (saline) (n = 3 for each treatment) intraperitoneal injection daily from E8.5 to E18.5. The blood pressure was measured every other day after E6.5. Mice were sacrificed at E18.5 to collect utero-placental tissues.
Human subjects
Sixteen women with sPE and 17 women with NPs were recruited in the Department of Obstetrics and Gynecology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University between May 2019 and July 2021. sPE is defined as SBP ≥160 mmHg on at least two occasions 6 h apart or with proteinuria >3+ protein on dipstick after 20 weeks of gestation. Only cesarean pregnancies were included, and none of the included mothers were in labor prior to cesarean section. Multiparous pregnancies, renal disease, maternal diabetes, chromosomal aberrations, and fetal and placental structural abnormalities were excluded from our study. Decidual tissues were obtained by scrubbing the uterine wall at the site of the placental bed with gauze after the placenta was delivered. The decidua was washed with sterilized water to remove blood, snap frozen in liquid nitrogen, and stored at −80°C until use. The study was approved by the Renji Hospital Research and Ethics Committees. Informed consent was obtained from all participants before the collection of decidual samples.
Statistical analysis
GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA) was used for statistical analysis. Student’s unpaired two-tailed t tests were used for statistical analyses. p < 0.05 was considered statistically significant. Sample sizes were selected based on current and previous experiments, and no statistical method was applied to predetermine sample size. All experiments were repeated at least three times.
Data and code availability
All data included in this study are available upon request by contacting the corresponding author.
Acknowledgments
This study was supported by the National Key R&D Program of China (2019YFA0802600) and the National Natural Science Foundation of China (32170863 and 31871512) to C. Zhang. Support was also received from grants from the Shanghai Commission of Science and Technology (20DZ2270900).
Author contributions
C. Zhou, Y.Z., and C. Zhang planned the mouse experiments, which were carried out by C. Zhou, Y.Z., L.Z., and M.Z. C. Zhou and C. Zhang wrote the manuscript. C. Zhou and C. Zhang planned and executed the human decidua experiments and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102408.
Supplemental information
References
- 1.Souza J.P., Gülmezoglu A.M., Vogel J., Carroli G., Lumbiganon P., Qureshi Z., Costa M.J., Fawole B., Mugerwa Y., Nafiou I., et al. Moving beyond essential interventions for reduction of maternal mortality (the WHO Multicountry Survey on Maternal and Newborn Health): a cross-sectional study. Lancet. 2013;381:1747–1755. doi: 10.1016/S0140-6736(13)60686-8. [DOI] [PubMed] [Google Scholar]
- 2.Rana S., Lemoine E., Granger J.P., Karumanchi S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019;124:1094–1112. doi: 10.1161/CIRCRESAHA.118.313276. [DOI] [PubMed] [Google Scholar]
- 3.Burton G.J., Redman C.W., Roberts J.M., Moffett A. Pre-eclampsia: pathophysiology and clinical implications. BMJ. 2019;366 doi: 10.1136/bmj.l2381. [DOI] [PubMed] [Google Scholar]
- 4.Ho L., van Dijk M., Chye S.T.J., Messerschmidt D.M., Chng S.C., Ong S., Yi L.K., Boussata S., Goh G.H.Y., Afink G.B., et al. ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science. 2017;357:707–713. doi: 10.1126/science.aam6607. [DOI] [PubMed] [Google Scholar]
- 5.Garrido-Gomez T., Quiñonero A., Dominguez F., Rubert L., Perales A., Hajjar K.A., Simon C. Preeclampsia: a defect in decidualization is associated with deficiency of Annexin A2. Am. J. Obstet. Gynecol. 2020;222:376. doi: 10.1016/j.ajog.2019.11.1250. [DOI] [PubMed] [Google Scholar]
- 6.Zhou C.C., Zhang Y., Irani R.A., Zhang H., Mi T., Popek E.J., Hicks M.J., Ramin S.M., Kellems R.E., Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat. Med. 2008;14:855–862. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rong M., Yan X., Zhang H., Zhou C., Zhang C. Dysfunction of Decidual Macrophages Is a Potential Risk Factor in the Occurrence of Preeclampsia. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.655655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lv H., Tong J., Yang J., Lv S., Li W.P., Zhang C., Chen Z.J. Dysregulated Pseudogene HK2P1 May Contribute to Preeclampsia as a Competing Endogenous RNA for Hexokinase 2 by Impairing Decidualization. Hypertension. 2018;71:648–658. doi: 10.1161/HYPERTENSIONAHA.117.10084. [DOI] [PubMed] [Google Scholar]
- 9.Blois S.M., Dechend R., Barrientos G., Staff A.C. A potential pathophysiological role for galectins and the renin-angiotensin system in preeclampsia. Cell. Mol. Life Sci. 2015;72:39–50. doi: 10.1007/s00018-014-1713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winship A.L., Koga K., Menkhorst E., Van Sinderen M., Rainczuk K., Nagai M., Cuman C., Yap J., Zhang J.G., Simmons D., et al. Interleukin-11 alters placentation and causes preeclampsia features in mice. Proc. Natl. Acad. Sci. USA. 2015;112:15928–15933. doi: 10.1073/pnas.1515076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jim B., Karumanchi S.A. Preeclampsia: Pathogenesis, Prevention, and Long-Term Complications. Semin. Nephrol. 2017;37:386–397. doi: 10.1016/j.semnephrol.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 12.Fisher S.J. Why is placentation abnormal in preeclampsia? Am. J. Obstet. Gynecol. 2015;213:S115–S122. doi: 10.1016/j.ajog.2015.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siddiqui M.F., Nandi P., Girish G.V., Nygard K., Eastabrook G., de Vrijer B., Han V.K.M., Lala P.K. Decorin over-expression by decidual cells in preeclampsia: a potential blood biomarker. Am. J. Obstet. Gynecol. 2016;215:361.e1–361.e15. doi: 10.1016/j.ajog.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 14.Garrido-Gomez T., Dominguez F., Lopez J.A., Camafeita E., Quiñonero A., Martinez-Conejero J.A., Pellicer A., Conesa A., Simón C. Modeling human endometrial decidualization from the interaction between proteome and secretome. J. Clin. Endocrinol. Metab. 2011;96:706–716. doi: 10.1210/jc.2010-1825. [DOI] [PubMed] [Google Scholar]
- 15.Jovanović M., Vićovac L. Interleukin-6 Stimulates Cell Migration, Invasion and Integrin Expression in HTR-8/SVneo Cell Line. Placenta. 2009;30:320–328. doi: 10.1016/j.placenta.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Graham D.K., DeRyckere D., Davies K.D., Earp H.S. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer. 2014;14:769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 17.Linger R.M.A., Keating A.K., Earp H.S., Graham D.K. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holland S.J., Powell M.J., Franci C., Chan E.W., Friera A.M., Atchison R.E., McLaughlin J., Swift S.E., Pali E.S., Yam G., et al. Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res. 2005;65:9294–9303. doi: 10.1158/0008-5472.CAN-05-0993. [DOI] [PubMed] [Google Scholar]
- 19.Rankin E.B., Fuh K.C., Taylor T.E., Krieg A.J., Musser M., Yuan J., Wei K., Kuo C.J., Longacre T.A., Giaccia A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010;70:7570–7579. doi: 10.1158/0008-5472.CAN-10-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louwen F., Muschol-Steinmetz C., Reinhard J., Reitter A., Yuan J. A lesson for cancer research: placental microarray gene analysis in preeclampsia. Oncotarget. 2012;3:759–773. doi: 10.18632/oncotarget.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y.-Q., Yan X.-Y., Li H., Zhang C. Insights into the Pathogenesis of Preeclampsia Based on the Features of Placentation and Tumorigenesis. Reproductive and Developmental Medicine. 2021;5:97–106. [Google Scholar]
- 22.Tong J., Zhao W., Lv H., Li W.P., Chen Z.J., Zhang C. Transcriptomic Profiling in Human Decidua of Severe Preeclampsia Detected by RNA Sequencing. J. Cell. Biochem. 2018;119:607–615. doi: 10.1002/jcb.26221. [DOI] [PubMed] [Google Scholar]
- 23.Lu Q., Gore M., Zhang Q., Camenisch T., Boast S., Casagranda F., Lai C., Skinner M.K., Klein R., Matsushima G.K., et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398:723–728. doi: 10.1038/19554. [DOI] [PubMed] [Google Scholar]
- 24.Mol B.W.J., Roberts C.T., Thangaratinam S., Magee L.A., de Groot C.J.M., Hofmeyr G.J. Pre-eclampsia. Lancet. 2016;387:999–1011. doi: 10.1016/S0140-6736(15)00070-7. [DOI] [PubMed] [Google Scholar]
- 25.Strevens H., Wide-Swensson D., Hansen A., Horn T., Ingemarsson I., Larsen S., Willner J., Olsen S. Glomerular endotheliosis in normal pregnancy and pre-eclampsia. BJOG. 2003;110:831–836. [PubMed] [Google Scholar]
- 26.Ridder A., Giorgione V., Khalil A., Thilaganathan B. Preeclampsia: The Relationship between Uterine Artery Blood Flow and Trophoblast Function. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20133263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cubro H., Nath K.A., Suvakov S., Garcia-Valencia O., Parashuram S., White W.M., Weissgerber T.L., Nath M.C., Milic N.M., Sontag F., et al. Mechanisms of vascular dysfunction in the interleukin-10-deficient murine model of preeclampsia indicate nitric oxide dysregulation. Kidney Int. 2021;99:646–656. doi: 10.1016/j.kint.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akison L.K., Nitert M.D., Clifton V.L., Moritz K.M., Simmons D.G. Review: Alterations in placental glycogen deposition in complicated pregnancies: Current preclinical and clinical evidence. Placenta. 2017;54:52–58. doi: 10.1016/j.placenta.2017.01.114. [DOI] [PubMed] [Google Scholar]
- 29.Graves J.A. Genomic imprinting, development and disease--is pre-eclampsia caused by a maternally imprinted gene? Reprod. Fertil. Dev. 1998;10:23–29. doi: 10.1071/r98014. [DOI] [PubMed] [Google Scholar]
- 30.Menkhorst E.M., Van Sinderen M., Correia J., Dimitriadis E. Trophoblast function is altered by decidual factors in gestational-dependant manner. Placenta. 2019;80:8–11. doi: 10.1016/j.placenta.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 31.Bao H., Sun Y., Yang N., Deng N., Ni Z., Tang Y., Li G., Du L., Wang Y.L., Chen D., et al. Uterine Notch2 facilitates pregnancy recognition and corpus luteum maintenance via upregulating decidual Prl8a2. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui Y., Wang W., Dong N., Lou J., Srinivasan D.K., Cheng W., Huang X., Liu M., Fang C., Peng J., et al. Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature. 2012;484:246–250. doi: 10.1038/nature10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S., Cao P., Dong N., Peng J., Zhang C., Wang H., Zhou T., Yang J., Zhang Y., Martelli E.E., et al. PCSK6-mediated corin activation is essential for normal blood pressure. Nat. Med. 2015;21:1048–1053. doi: 10.1038/nm.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Y., Zang H., Wen Q., Fan S. AXL in cancer: a modulator of drug resistance and therapeutic target. J. Exp. Clin. Cancer Res. 2023;42:148. doi: 10.1186/s13046-023-02726-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stillman I.E., Karumanchi S.A. The glomerular injury of preeclampsia. J. Am. Soc. Nephrol. 2007;18:2281–2284. doi: 10.1681/ASN.2007020255. [DOI] [PubMed] [Google Scholar]
- 36.Phipps E.A., Thadhani R., Benzing T., Karumanchi S.A. Pre-eclampsia: pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019;15:275–289. doi: 10.1038/s41581-019-0119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmeliet P. Angiogenesis in health and disease. Nat. Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 38.Armstrong D.W.J., Tse M.Y., Wong P.G., Ventura N.M., Meens J.A., Johri A.M., Matangi M.F., Pang S.C. Gestational hypertension and the developmental origins of cardiac hypertrophy and diastolic dysfunction. Mol. Cell. Biochem. 2014;391:201–209. doi: 10.1007/s11010-014-2003-9. [DOI] [PubMed] [Google Scholar]
- 39.Zhang W., Li S., Lou J., Li H., Liu M., Dong N., Wu Q. RBMS1 regulates lung cancer ferroptosis through translational control of SLC7A11. J. Clin. Invest. 2021;131:e152067. doi: 10.1172/JCI152067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C., Wang Z., He M., Zhou T., Niu Y., Sun S., Li H., Zhang C., Zhang S., Liu M., et al. Kruppel-like factor 17 upregulates uterine corin expression and promotes spiral artery remodeling in pregnancy. Proc. Natl. Acad. Sci. USA. 2020;117:19425–19434. doi: 10.1073/pnas.2003913117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan H., Lin L., Huang L., Yu Y. Is Atrial Natriuretic Peptide (ANP) and Natriuretic Peptide Receptor-A (NPR-A) Expression in Human Placenta and Decidua Normal? Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2019;25:2868–2878. doi: 10.12659/MSM.915449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei Y.L., Li X.H., Zhou J.Z. Prenatal exposure to lipopolysaccharide results in increases in blood pressure and body weight in rats. Acta Pharmacol. Sin. 2007;28:651–656. doi: 10.1111/j.1745-7254.2007.00593.x. [DOI] [PubMed] [Google Scholar]
- 43.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benjamini Y., Drai D., Elmer G., Kafkafi N., Golani I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 47.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Meng N., Bao H., Jiang Y., Yang N., Wu K., Wu J., Wang H., Kong S., Zhang Y. Circadian gene PER1 senses progesterone signal during human endometrial decidualization. J. Endocrinol. 2019;243:229–242. doi: 10.1530/JOE-19-0284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data included in this study are available upon request by contacting the corresponding author.