

Abstract
Vibrio vulnificus is a significant zoonotic pathogen that causes severe vibriosis in humans and fish. The lack of a national annual surveillance program in China has hindered understanding of its epidemiological characteristics and genetic diversity. This study characterized 150 V. vulnificus isolates collected from diverse sources in China during 2012–2023, including seafood, aquaculture water, migratory birds, marine animals, and clinical patients. Most seafood-derived isolates and all 15 clinical isolates harbored the virulence-related gene vcgC and 16S rRNA type B. The isolates exhibited diverse virulence factors (VFs), including flagella, outer membrane components, RTX toxins, and multiple secretion systems. Genes associated with the Type III secretion system were identified in migratory bird isolates, while a unique Type VI secretion system (T6SS1) were identified exclusively within a specific phylogenetic sub-lineage. T6SS1-positive strains demonstrated an increased number of genomic islands (GIs) and VFs compared to T6SS1-negative strains. Enrichment of genes related to secretion systems and biofilm formation likely facilitated the expansion of the T6SS1-positive population. The novel association between T6SS1 and a specific sub-lineage underscores potential ecological and adaptive advantages. These findings provide new insights into the ecological and evolutionary dynamics of V. vulnificus.
Subject terms: Bacterial genomics, Bacterial infection, Microbial ecology
Isolated Vibrio vulnificus clones from various sources in China showed specific genomic markers linked to human pathogenesis, seafood, and migratory birds, these potentially may confer niche-specific advantages.
Introduction
Vibrio vulnificus is a zoonotic pathogen capable of being transmitted from aquaculture organisms to humans, causing vibriosis outbreaks in both fish and humans1,2. It commonly inhabits in warm coastal environments, including estuarine, and brackish water, particularly in tropical, subtropical, and temperate regions3,4. This pathogen can cause severe bloodstream infection in humans, with mortality rates exceeding 50%4–7. Report indicates a steady rise in annual deaths due to V. vulnificus infections in the United States over the past two decades8. The geographic range of V. vulnificus has expanded significantly northward, driven by climate change and globalization, resulting in increased mortality rates and economic impacts9–11.
Numerous studies have been conducted to classify different V. vulnificus types. V. vulnificus were initially classified into three Biotypes (BT1-BT3) based on biochemical and serological characteristics12–14. Additionally, two distinct subgroups, 16S rRNA_A and 16S rRNA_B, were identified by analyzing 16S rRNA gene polymorphism15. The 16S rRNA_B subgroup was primarily found in clinical isolates, whereas 16S rRNA_A subgroup was primarily observed in environmental samples15. Earlier multilocus sequence analysis (MLSA) of BT1 strains revealed that V. vulnificus strains can be categorized into two genetic lineages (L1 and L2), distinguished by variations in one virulence-associated gene (vcg). Lineage 1 (vcgC) comprises clinical strains, whereas Lineage 2 (vcgE) consists of environmental strains16,17. One report found that over 50% of shrimps derived isolates were of the potential virulent type vcgC-16S rRNA_B (CB)18. Phylogenetic reconstructions for chromosomes I and II, based on core genomes, identified five lineages that did not align with previous biotype classifications19. A recent study analyzed 109 V. vulnificus clinical isolates from USA during 2001-2019 using whole-genome sequencing and MLST-based phylogenetic analysis. The study found that clinical isolates were uniformly distributed across two lineages. There was no correlation found between the severity of the illness and specific phylogenetic lineages20. Despite considerable research, several unresolved questions remain regarding source preferences, ecological interactions, and genetic or virulence potential in V. vulnificus. Notably, no clear association has been established between high levels of pathogenic V. vulnificus and diverse isolated sources. The findings are critical for advancing the understanding of the ecology of pathogenic V. vulnificus.
In China, although individual cases have been documented since 1993, there have been no reports of extensive outbreaks. One possible explanation is the absence of a comprehensive national surveillance strategy for V. vulnificus. Consequently, a thorough understanding of the genetic diversity and epidemiology of this bacterium in China is crucial. This study provides a detailed analysis of 150 newly sequenced isolates from China, significantly contributing to genomic representation from an underreported region. The identification of the lineage-independent distribution of clinical strains and the genomic adaptations of T6SS1-positive sub-lineages present new avenues for understanding the ecological and evolutionary dynamics of V. vulnificus. Most of the 150 V. vulnificus isolates were classified as the highly pathogenic CB type. An extensive analysis of global genome sequences of V. vulnificus isolates was performed. Bioinformatic analysis revealed distinct virulence factor (VF) profiles, with a significantly higher number of VFs exclusively present in a separate phylogenetic cluster. This cluster harbored a distinct T6SS1 with additional VFs, highlighting its crucial role in pathogenicity. In addition, T6SS1-positive strains exhibited a higher quantity of genomic islands in both chromosomes I and II. Further analysis suggests that V. vulnificus strains with T6SS exhibit robust adaptive capacity, potentially contributing to their population expansion. Therefore, closely monitoring highly pathogenic strains in aquatic environments, products, and animals is crucial to mitigate the risk of V. vulnificus infections.
Results
Collection of V. vulnificus in China and worldwide
In this study, we collected and sequenced 150 V. vulnificus isolates in China between 2012 and 2023. The isolates were obtained from various sources, including seafood (n = 106), wounds or blood of clinical patients (n = 15), aquaculture water (n = 13), and other animals, such as marine mammals (spotted seals) (n = 3) and migrating birds (n = 13) Sample sources were categorized into four major groups: seafood, human, environment (aquaculture water) and others. (Fig. 1A and Supplementary Fig. S1A). PCR and WGS analysis confirmed that 94.67% of V. vulnificus isolates, including all 15 clinical isolates, carry the vcgC and 16S rRNA_B (CB type). Our analysis revealed that 92.45% of seafood-derived isolates were of potentially virulent CB type. (Fig. 1B and Supplementary Data 1).
Fig. 1. Global distribution of Vibrio vulnificus strains.

A The number of 150 newly collected V. vulnificus isolates in China over the past decade. B The percentage of isolates from various sources, categorized based on different molecular typing methods used in this study. C The number of V. vulnificus isolates reported by different countries over multiple years. D The geographical distribution of V. vulnificus sources worldwide. In (A) and (B), the term “environment” refers to aquaculture water samples.
As of October 10, 2023, we obtained 1546 globally distributed genome assemblies or raw NGS data of V. vulnificus from the NCBI pathogens database (Supplementary Data 2). Including the 150 isolates in this study, a total of 1696 strains of global V. vulnificus were comprehensively analyzed. A notable portion of these strains were obtained from seafood (49.00%), while human samples accounted for 30.54% and environmental sources contributed 10.79% (Supplementary Fig. S1B). Furthermore, 1595 V. vulnificus isolates were documented across 20 countries on six continents, excluding unspecified collection locations for 101 strains (Fig. 1C, D). A considerable portion of these isolates were reported from the United States (50.16%, 800/1595), followed by China (43.45%, 693/1,595) and South Korea (1.19%, 19/1595). V. vulnificus strains from various sources were found in nine countries, such as the USA, South Korea, Spain, Israel, Germany, and China (Fig. 1D and Supplementary Fig. S1C). Before 1980, only five documented V. vulnificus genomes existed. There were two cases involving seafood from Japan and three cases reported from marine sources and humans in the USA (Fig. 1C). Vibrio vulnificus was reported in France (n = 1) and South Korea (n = 1) between 1980 and 1989. Subsequently, an increasing number of countries reported genomic data on V. vulnificus, which indirectly suggests a growing scietific interest worldwide.
Phylogenomic analysis of global V. vulnificus strains
We utilized 1685 V. vulnificus genomes to construct a maximum-likelihood (ML) phylogenetic tree (Supplementary Fig. S2A) after excluding 11 genomes that had low N50 values. The analysis revealed the existence of two separate Lineages, as depicted in the phylogenetic tree. Lineage 2 (L2) contained 668 strains, with the majority originating From the United States (511 strains, 76.50%). Of the 150 isolates collected in this study, only six belonged to L2. Notably, 67.89% (631/1017) of V. vulnificus strains in Lineage 1 (L1) were derived from China, including 144 newly sequenced strains in this study. Supplementary Fig. S2B illustrates a balanced distribution of specimen hosts between L1 and L2. Our results align with previous studies, indicating that the clinical isolates are distributed across both L1 or L220.
To further explore the VFs and genetic characteristics of 150 recently selected V. vulnificus isolates deeper, we included 30 complete or chromosome-level genomes from the NCBI database as references (Supplementary Data 1). Figure 2 shows the phylogenetic classification of 180 isolates into two distinct Lineages. Figure 2 demonstrates that L1 (163/180, 90.56%) is divided into five sub-Lineages, L1.1 to L1.5. Most of the 163 isolates were found in sub-Lineages L1.4 and L1.5, comprising approximately 79.75% (130 isolates). L2 comprises 17 isolates from various sources and countries, including China, Israel, South Korea, Spain, and the USA (Fig. 2). L2 showed a significant presence of vcgE (88.24%) and 16S rRNA_A (100%), while L1 predominantly contained vcgC (99.39%) and 16S rRNA_B (99.39%).
Fig. 2. Phylogenetic analysis of Vibrio vulnificus strains using the maximum likelihood (ML) method.

The phylogenetic tree was constructed based on core-genome single nucleotide polymorphisms (SNPs). Metadata is displayed in the neighboring tracks (1 to 4) surrounding the tree: Track 1 represents the geographical region, Track 2 indicates the collection year, Track 3 denotes the host organism, and Track 4 includes virulence-associated gene types and 16S rRNA type. The presence of VFs and ARGs is indicated by darker colors. Shared VFs common to all 180 strains are not shown in the figure. Strains with names beginning with CNVV were sequenced explicitly for this study.
A total of 74 VFs were identified, with 37 being common to all strains of V. vulnificus as shown in the Supplementary Table 1. Several VFs linked to pathogenicity were identified, including flagella, outer membrane, RTX toxin, and secretion systems (EPS Type II, Type III, Type IV, and VAS Type VI) (Supplementary Fig. S3C). A majority of V. vulnificus strains (94.44%) displayed at least 50 VFs, as depicted in Supplementary Fig. S3A. Only strains CNVVF081014 and CNVVT081111 were found to contain the vscN2 gene, which is linked to the Type III secretion system (T3SS) (Fig. 2). The two strains were collected from anal swabs of migratory birds in Shanghai. The findings suggest that migratory birds may acquire V. vulnificus through aquaculture water or by eating seafood, such as fish. Interestingly, sub-Lineage L1.5.4, which included 21 aquaculture water and seafood strains, displayed a distinct VF profile with a higher number of VFs than the other sub-Lineage strains. This characteristic is mainly attributed to the substantial prevalence of the VAS Type VI secretion systems (T6SS1). Global analysis revealed that 141 strains harbored the T6SS1, with most belonging to the sub-Lineage of L1 (Supplementary Fig. S2).
The average number of VFs and ARGs in the 180 strains V. vulnificus of were 54.03 and 4.17, respectively (Supplementary Fig. S3A, B). In addition, 21 ARGs conferring resistance to various antibacterial agents were detected (Supplementary Fig. S3D). All 180 strains of V. vulnificus contained CRP, varG, tet(34), and tet(35) genes. Furthermore, varG has demonstrated beta-lactamase activity against penicillin, carbapenems, and cephalosporins in vitro21. The presence of ARGs indicates the potential for resistance to macrolides, carbapenems, tetracyclines, and sulfonamides. Strain CNVV056, obtained from seafood in Guangdong province, exhibited the highest count of 13 ARGs, highlighting the AMR risk associated with seafood-derived V. vulnificus strains.
Comparative genomic analysis of V. vulnificus strains
To enhance our understanding of the genetic structure of V. vulnificus, nine strains of V. vulnificus from different phylogenetic clusters and diverse sample sources (clinical, seafood, environmental, and migratory birds) were selected for long-read sequencing. As showed in Fig. 3A, B, the chromosomal sequences of CNVV120 and CNVV121 exhibited 99.73% identity and 92% coverage. These two strains were classified as belonging to L1 (CNVV121) and L2 (CNVV120), respectively. The T6SS locus (T6SS1), recognized as a VF, was identified in two of the nine isolates (CNVV120 and CNVV121). V. vulnificus strains possessing T6SS1 exhibited a greater abundance of GIs. Two unique GIs (GI1 and GI8) were identified on chromosomes I of strain CNVV120 and CNVV121, but absent in the T6SS1-negative strains (CNVV025, CNVV027, CNVV145, CNVV148, CNVV156, CNVV151 and CNVVF081014). Additionally, these seven T6SS1-negative strains lacked GIs GI3 and GI4, which were consistently found in CNVV120 and CNVV121 on chromosome II. The study found that T6SS1-related proteins are predominantly found at GI3, specifically on chromosome II of V. vulnificus. GI3 has been found to contain key secretory proteins Hcp and VgrG. Furthermore, an additional conserved T6SS loci (T6SS2) can be observed in the chromosome II of all nine complete isolates (Fig. 3B).
Fig. 3. BLAST ring comparison of reference V. vulnificus chromosome and plasmid with homologous sequences from isolates recovered in this study.
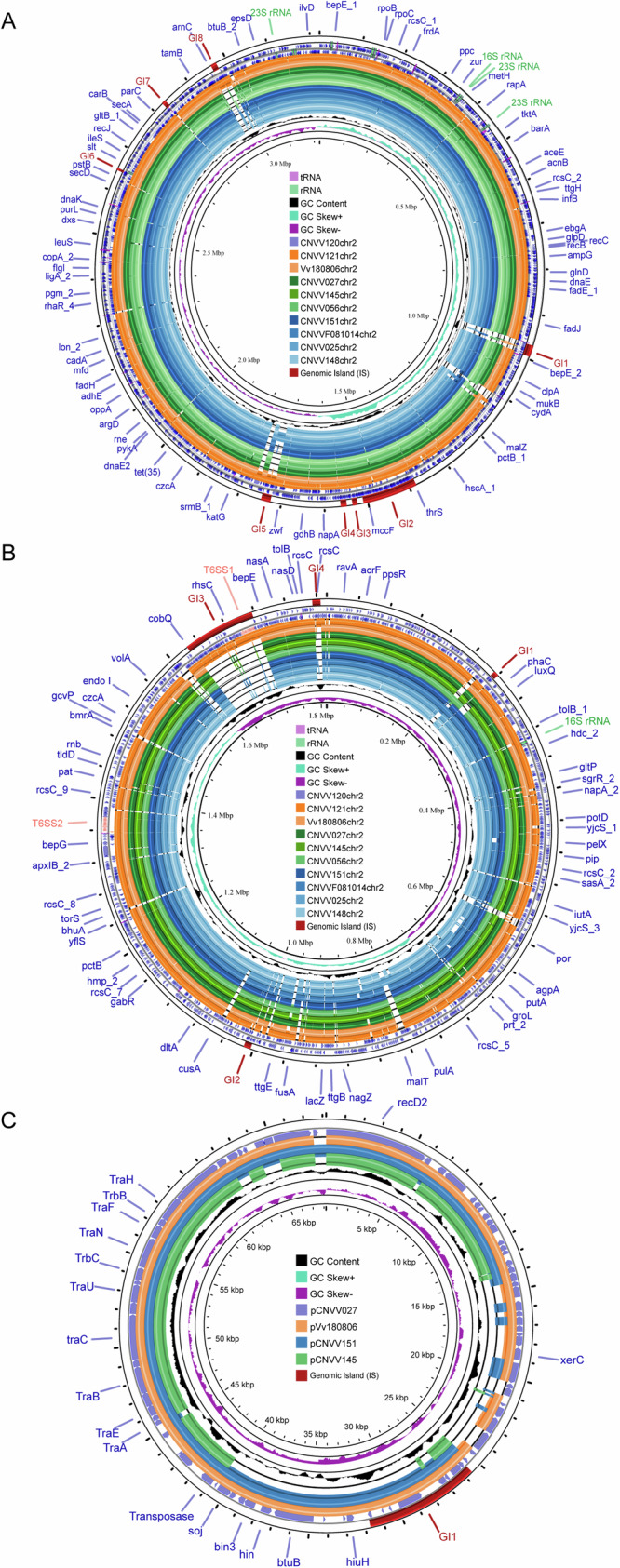
A Chromosome I; B Chromosome II. C Plasmid (reference strain Vv180806: GCA_014107515.1).GIs are highlighted in red in the outermost circle.
The available information on V. vulnificus plasmids is currrently limited. The study revealed that three V. vulnificus strains harbored plasmids, indicating the potentially horizontal gene transfer in this species (Fig. 3C). Plasmid pCNVV027 from seafood samples showed a 99.98% identity and 95% coverage to pVv18086, which was obtained from a clinical mortality case in China (GCA_014107515.1). The findings suggested a strong genetic link between the two plasmids, supporting potential Vibrio transmission between humans and the environment.
Evolutionary history of V. vulnificus strains carrying T6SS1
A comprehensive investigation was conducted to explore the evolutionary context of T6SS1-positive V. vulnificus. We analyzed the global distribution of T6SS1 in V. vulnificus strains. A total of 141 strains were found to possess T6SS1. We collected a dataset of 128 T6SS1-positive strains and 402 T6SS1-negative strains by combining evolutionary branching and excluding strains without recorded sampling times (Supplementary Data 3). The strains were obtained from various sources, including seafood (304 strains), human samples (134 strains), the environment (67 strains), other sources (15 strains), and ten strains with unknown origin. ML phylogenetic tree reconstruction revealed clear clusters formed by T6SS1-positive strains (Fig. 4A). Furthermore, T6SS1-positive V. vulnificus strains exhibit significantly more GIs compared to other strains globally (Fig. 4A, B). Notably, Genomic Island GI8, GI3, and GI4 are located on specific chromosomes (I and II) in T6SS1-positive strains. Lineage L2, represented by the pink background, includes 44 T6SS1-positive strains and 149 T6SS1-negative strains, with their status denoted by specific branch annotations, as detailed in the legend (Fig. 4A). Additionally, metadata of the adjacent tracks on the tree depict a diverse array of collection years, regions, and host sources. Based on sampling time data, the genomic data of T6SS1-positive strains were more likely to be publicly available after 2005.
Fig. 4. Evolutionary history and KEGG enrichment analysis of V.vulnificus strains carrying T6SS1.

A Maximum likelihood (ML) phylogenetic tree of 128 T6SS1-positive strains and 402 T6SS1-negative strains. Branches representing T6SS1-positive strains are highlighted in blue. Adjacent tracks display metadata, including host organism, collection year, and geographical region. B Comparison of GIs carried by V. vulnificus in T6SS1-positive (n = 128) and T6SS1-negative (n = 402) groups. C KEGG enrichment analysis of differentially representative genes conducted on 193 V. vulnificus strains in Lineage L2, with absolute gene counts displayed. A significance threshold of P < 0.05 was applied. D Global epidemic trends of T6SS1-positive strains over time.The heatmap in (A) illustrates the presence of GIs in strain CNVV120 across global V. vulnificus isolates.
We conducted a comprehensive study to identify key genes differentiating between T6SS1-positive and T6SS1-negative strains in Lineage L2 at the pan-genome level. A total of 3184 genes were identified in the core genome, while 393 genes were found in the softcore genome in 95% to 99% of strains. Additionally, 79 genes were absent in T6SS1-negative strains but present in over 50% of T6SS1-positive strains. Besides T6SS-related genes, these differential genes were also associated with T2SS, transferase, and transposase. An in-depth analysis was performed to examine the molecular functions of these genes using KEGG-based annotation and enrichment (Fig. 4C). The most notable distinction between the two groups is the presence of a bacterial secretory system function classified as A09130. Additionally, several genes were linked to membrane transport, biofilm formation, cellular processes, and the production of secondary metabolites (Fig. 4C). The presence of these genes and their associated functions likely contribute to the robust adaptive capabilities of V. vulnificus, potentially driving the emergence of dominant strains over time. Furthermore, the annual percentage of T6SS1-positive strains was comprehensively examined using data from public databases. Certainly, the steady increase in T6SS1-positive strain detection over time (Fig. 4D) likely reflects enhanced surveillance efforts and expanded sampling rather than a true increase in the prevalence of strains.
Genetic contents and organization of T6SS in Vibrio genus
T6SS1 is primarily found in Proteobacteria, with limited occurrences in V. vulnificus. A genomic analysis was conducted to explore the structure of T6SS1. The genetic contexts of T6SS1 were compared across various Vibrio strains. Figure 5A depicts a conserved 20.45 kb core region within the Vibrio genus. This region contains 15 proteins: TssB, TssC, TssE, TssF, TssG, VasC, TssJ, TssK, TssL, TssH, VasH, VasI, TssA, TssM, and ImpA. The genetic structure of T6SS1 shows high similarities within the same Vibrio species, with broad coverage, but relatively lower identity across different species. The major secretion protein Hcp is essential for a functional T6SS in other bacterial species. Hcp genes are often found near the T6SS loci, alongside vgrG gene22,23.
Fig. 5. Linear comparison of the T6SS1 genetic context among different strains in the Vibrio genus.

A Genetic environment of T6SS in various Vibrio strains. B Genetic environment of Hcp protein within T6SS. Arrows indicate open reading frame locations and transcriptional orientation. Blue cross-links highlight regions of significant sequence similarity, with identities exceeding 64%.
Further analysis revealed a clear genetic framework of Hcp, as showed in Fig. 5B. The TssA-TssM-ImpA-Hcp-VgrG-PAAR structure remained consistently intact in both V. vulnificus and V. rotiferianus, consistently linked to the downstream ankyrin repeat domain protein. An interesting exception was observed in V. scophthalmi strain VS-05, linked to a DUF4123 domain-containing protein. In addition, three copies of DUF1851 domain-containing genes were inserted between ImpA and Hcp in the Vibrio sp. SCSIO 43153 (Fig. 5B). Notably, Hcp protein was absent from T6SS1 loci in other Vibrio species and instead located in separate genomic regions.
Discussion
V. vulnificus is a zoonotic pathogen capable of causing disease in both aquatic animals and humans, with approximately 100 cases of V. vulnificus infection reported annually in the United States. Due to its atypical clinical presentation, mild cases of V. vulnificus infection may go undiagnosed, potentially underestimating global infection prevalence24. Unlike other Vibrio species, V. vulnificus can transmit between aquaculture animals and humans. Although V. vulnificus infections are uncommon, they are the leading cause of seafood-related fatalities4,8. Therefore, close monitoring V. vulnificus is essential to better understanding of its prevalence and transmission mode.
Comprehensive understanding regarding the genetic characteristics and virulence pattern of V. vulnificus in China remains limited. In this study, we analyzed 150 V. vulnificus isolates from different sources, including migratory birds—a relative novel finding that suggests potential pathogen transmission within animal food chains. As reported by a meta analysis, there are only eight studies documented V. vulnificus detection in avian species, including wild or domestic birds25–27. To assess global prevalence, we compiled a dataset of 1,696 V. vulnificus strains from 20 countries across six continents. A large proportion of these strains were reported in the United States (50.16%) and China (43.45%). It is acknowledged that the higher isolate counts in countries likely reflect sampling practices, publication output, and access to genomic resources, rather than actual infection rates. V. vulnificus infections are not nationally notifiable in many regions. Some underrepresented areas, such as the European Baltic Sea 28,29, where V. vulnificus infection have been reported several outbreaks despite limited genomic data, and recognized the inherent sampling biases that may lead to data discrepancies across countries.
Consistent with prior findings30, our phylogenomic analysis classified global V. vulnificus strains into two primary lineages. Most isolates from the USA (64.20%, 511/796) were clustered within L1, while the majority of Chinese isolates (91.18%, 631/692) were grouped within L2, indicating a significant evolutionary similarity within each country. Contrary to earlier studies that predominantly associated clinical strains with lineage L1(vcgC)16, our findings reveal a balanced distribution of clinical strains across both L1 and L2 (Supplementary Fig. S2 and Fig. 2), consistent with Stachell’s observations20. Interestingly, the in-depth ML phylogenetic analysis revealed a potential link between L1 and the virulence-related genes vcgC and 16S rRNA_B type, while Lineage 2 was associated with vcgE and 16S rRNA_A, While the statistically percentage values for vcg and 16S rRNA types primarily reflect lineage-associated traits rather than actual virulence markers. In the Supplementary Fig. S5, after incorporating an additional 16 genomes from lineages L3 and L4 as reported by López-Pérez31, none of our 150 isolates clustered within these lineages. This distribution is likely influenced by the geographical isolation of samples.
Further research is essential to deepen our understanding of V. vulnificu’s pathogenesis, as the bacterium harbors a diverse array of VFs and ARGs. Analysis of 180 V. vulnificus strains found that each contained at least 4 ARGs, complicating infection treatment. The mean VFs were 54.03, including flagella, outer membrane, RTX toxin, and different secretion systems. Two strains isolated from migratory birds were found to uniquely harbor genes associated with the T3SS, suggesting that migratory birds may act as potential reservoirs for the transmission of V. vulnificus. Additionally, strains containing the T6SS1 cluster were identified as forming a distinct sub-lineage. While T6SS1-positive strains constitute a small subset of the overall V. vulnificus population, their unique genomic characteristics indicate a potentially significant role in specific ecological niches and clinical scenarios. Extensive genome analysis indicated that these T6SS1-positive strains displayed elevated amounts of GIs across both chromosomes I and II. (Fig. 4). Various studies have confirmed that GIs can transport diverse genetic elements, including antimicrobial resistance cassettes, metabolic operons, and virulence determinants32–34. These genetic elements promote the rapid evolution and ecological success of specific bacterial lineages35. The enrichment of genes associated with biofilm formation and membrane transport highlights the adaptive significance of T6SS1, potentially enabling these strains to thrive and dominate in specific ecological niches. Furthermore, a high similarity was observed between pCNVV027 derived from seafood and pVv18086 obtained from a mortality case, suggesting potential transmission through food or environmental contact. Monitoring V. vulnificus in commercially available seafood is critical, especially in subtropical and tropical regions.
T6SS system is a sophisticated protein nanomachine used by various Gram-negative bacteria, originally identified as a VF. Its primary function is to deliver effector proteins directly into host cells36–38. Recent research has shown that T6SS also plays a key role in inter-bacterial competition and fungal defense39. Bacteria employ T6SS to deliver detrimental effectors into eukaryotic host cells and outcompete other bacteria or fungii, giving them a competitive advantage within bacterial populations22,39–41. Studies have shown that interbacterial competition in K. pneumoniae ST11 relies on the hcp and vgrG genes, with hcp mutants exhibiting changes in cellular transport and localization23. The T6SS substrate Tse2, in P. aeruginosa, functions as a toxin within a toxin-immunity system, capable of inhibiting both prokaryotic and eukaryotic cell growth42. Recent research on the Vibrio genus revealed an active T6SS in V. cholerae strain V52, which exhibited high virulence against Gram-negative bacteria, including Escherichia coli and Salmonella Typhimurium, reducing E. coli survival by up to 100,000-fold43. Lopez-Perez et al. previously reported V. vulnificus strains encoding two T6SS for the first time31. Co-culture experiments demonstrated that V. vulnificus T6SS-positive strains exhibit a clear competitive advantage over T6SS-negative strains, with this advantage specific to T6SS1 rather than T6SS244. These results may shed light on the steady increase in the identification of T6SS1-positive strains over time, emphasizing their global prevalence and potential implications for public health. While no significant differences in host cell viability were observed (Supplementary Fig. S4), this finding does not rule out the involvement of T6SS in other virulence mechanisms. Further studies are needed to investigate the broader impact of T6SS on host cellular components and pathogenicity. However, in conjunction with previous experiments, our analysis including phylogenetic tree reconstruction, KEGG enrichment, and epidemic rate of the global T6SS1-positive strains, shown the T6SS1’s role as a competitive molecular weapon, contributing to its increased prevalence. T6SS1-positive strains exhibit unique functionalities in comparison to T6SS1-negative strains. Distinctions in the secretion system, cellular processes, and metabolism are illustrated in Fig. 4, highlighting their adaptive significance. These functional attributes likely enhance V. vulnificus’s adaptive capacity, potentially contributing to the emergence of dominant strains over time. Therefore, our study contributes additional genomic and structural insights into T6SS-1-positive V. vulnificus using long-read sequencing and KEGG pathway analysis.
In summary, this study advances the understanding of V. vulnificus diversity and adaptation by contributing 150 newly sequenced isolates from diverse sources in China to the global genomic dataset. The novel association between T6SS1 and a specific sub-lineage underscores potential ecological and adaptive advantages, supported by the enrichment of GIs and VFs in these strains. Furthermore, the identification of migratory birds as potential reservoirs highlights a previously unrecognized transmission pathway. Collectively, these findings offer new insights into the evolutionary dynamics and ecological interactions of V. vulnificus, with important implications for public health and seafood safety.
This study has several limitations. The analysis primarily focused on V. vulnificus strains from Guangdong Province, despite examining strains from diverse human, animal, and environmental interfaces. A broader global analysis was also conducted to establish the detailed evolutionary relationships among V. vulnificus strains. Additionally, among the 1696 global genomes, the inclusion of 402 T6SS1-negative strains may introduce a selection bias that could potentially influence observed clustering patterns; however, no such bias was evident in our results (Supplementary Fig. S2). In the future, the release of additional genomes should help mitigate strain selection bias. Lastly, the absence of experimental data from animal models limits our ability to draw definitive conclusions regarding the pathogenic potential of T6SS1-positive V. vulnificus strains. Further research is necessary to elucidate the epidemiological factors and clinical significance of T6SS1 in V. vulnificus infections.
Methods
Strain collections and identifications
Samples were collected from multiple locations in China over ten years (2012–2023), including clinical, seafood, and aquaculture water specimens. Clinical strains were sourced from hospitals in Guangdong and Zhejiang provinces. Isolation of V. vulnificus from seafood and water followed modified protocols from the National Food Safety Risk Monitoring Manual and National Standards of China45. Water samples from aquatic markets were filtered, while seafood samples (e.g., oysters, shrimp, snails, clams) were washed to remove sediment. Each 25 g seafood or water sample was combined with 225 mL alkaline peptone water (3% NaCl) and incubated at 36 °C ± 1 °C for 18 ± 1 h. Cultures were then sub-cultured on Cellobiose-polymyxin (CC) agar and mCPC plates for 18–24 h. Selected colonies were further cultivated on 3% NaCl Tryptic Soy Agar (3% TSA). Colonies were identified as V. vulnificus using MALDI-TOF MS (Autof ms2000, Autobio, China). Additionally, 13 V. vulnificus isolates were collected from bird throat swabs in Shanghai, and three were from marine mammals46. Metadata on collection year, geographic region, host, virulence gene type, and 16S rRNA type is available in Supplementary Data 1. Strains prefixed with “CNVV” represent newly sequenced isolates.
PCR subtyping of V. vulnificus
Each isolated V. vulnificus strain was subtyped by PCR following protocols from previous studies16,18,47. Primer sequences are provided in Supplementary Table 2. PCR conditions included initial denaturation at 95 °C for 5 min, followed by 30 cycles of 94 °C for 1 min and annealing at either 55 °C (vcg) or 58 °C (16S rRNA) for 1 min. For Bt2, 35 cycles were used, with annealing at 64 °C and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 5 min.
Cell viability and MTT assay
The viability of HeLa cells following infection with V. vulnificus strains was assessed using the conventional MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) reduction assay48,49. Five T6SS-positive and five T6SS-negative strains were selected based on their host sources and evolutionary lineages. Bacterial cultures were grown until the optical density at 600 nm (OD600) reached approximately 0.8, after which the supernatant was carefully extracted via centrifugation.
The MTT assay was performed in a 96-well plate, with 3 to 7 × 10³ HeLa cells seeded per well. Cells were treated with bacterial supernatant at a final concentration of 20% (v/v) for varying durations (20–90 min). Subsequently, 10 µL of MTT solution (0.5 mg/mL) was added to each well, and the plate was incubated at 37 °C for 4 h in the dark. After incubation, 100 µL of dimethyl sulfoxide (DMSO) was added to each well to dissolve the resulting formazan crystals. The absorbance of the formazan product was measured at 570 nm using an ELISA plate reader after a 15-min incubation.
Cell viability was calculated as the percentage of surviving cells relative to untreated control cells. Each strain was tested in triplicate, with three independent wells for each experimental condition.
Genomic collection of V. vulnificus isolates
Global V. vulnificus genomic data were obtained from the NCBI Pathogen Database NCBI as of October 10, 2023. From 1796 initial genomes, 250 were excluded due to duplication, laboratory-specific experiments, or species misidentification via fastANI using CMCP6 (GCA_004355205.1) as a reference with a minimum ANI of 95%. This resulted in a dataset of 1546 publicly accessible V. vulnificus strains (Supplementary Data 2), including 30 fully sequenced genomes. Strain types were identified using BLASTn analysis of vcgC, vcgE, 16S rRNA_A, 16S rRNA_B, BT2, and BT3, with a minimum identity threshold of 95%.
Genomic sequencing and bioinformatic analysis
Whole-genome sequencing (WGS) of 150 V. vulnificus isolates was conducted on the NovaSeq 6000 platform (Illumina, USA) with 150 bp paired-end reads. Strains were designated with the prefix “CNVV.” Genomic assembly and annotation followed methods detailed previously50. raw reads were assembled with SPAdes 3.15.3, and annotated using Prokka 1.14.651. ARGs and VFs were were identified with Abricate using NCBI, CARD52, and VFDB databases53, applying default cutoffs (≥75% identity, ≥50% coverage).
Selected strains from each phylogenetic tree branch underwent long-read sequencing on the MinION platform (Oxford Nanopore Technologies, UK). Chromosomal and plasmid maps were created with Proksee54, and genomic islands were identified via IslandViewer455, then annotated on the Proksee map. Genomic comparisons were performed using BLASTn and visualized with Easyfig 2.2.556; gene orientations are marked with arrows, and homologous regions are shaded in gray.
A pan-genome-wide association study was conducted using Panaroo and Scoary to identify core genes in T6SS1-positive and T6SS1-negative strains57–59. Functional pathway analysis was conducted using KEGG-based annotation and enrichment60, with data visualized via a bar graph generated on the Bioinformatics.com.cn platform (accessed February 20, 2024).
Phylogenetic analysis
Phylogenomic analysis was performed on V. vulnificus isolates from diverse regions. Following exclusion of 11 genomes with low N50, the dataset comprised 1535 publicly available genomes and 150 study isolates, resulting in 1685 sequences. Phylogenetic trees were constructed via the maximum likelihood (ML) method, using Roary and FastTree software based on single-copy core genes and core-genome SNPs57,61. An ML tree integrating ARGs and VFs was constructed, including 150 study isolates and 30 NCBI complete genomes. Trees were visualized and annotated with iTOL v662.
Statistics and Reproducibility
Statistical analysis and visualizations were performed using GraphPad Prism 8.0 (GraphPad Prism Software, USA). Error bars represent standard deviation (s.d.). Maps were generated using ArcGIS 10.5 (Esri Inc, USA). Cell viability was tested in triplicate, utilizing three independent wells for each experimental condition.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary File
Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant No. 2024YFE0199000) and Zhuhai Municipal Science Technology Program (No. ZH22036201210189PWC and 2320004000164) and the International Joint Laboratory on Tropical Diseases Control in Greater Mekong Subregion (No. 21410750200).
Author contributions
Both D.L.L. and M.L. contributed equally to this article. D.L.L, M.L, Y.Z.Z. and C.Y. conceived and designed the study. J.W.H, L.C.M, and C.L. conducted the data integration and analysis. Z.L.C. and Y.W.C. conducted the data visualization. C.L., X.K.G., and H.T.H. directed the experiments and made revisions to the manuscript. M.L. conducted the data analysis and wrote the initial version of the manuscript. D.L.L. gathered the isolates and made revisions to the manuscript. The submitted version was approved by all authors.
Peer review
Peer review information
Communications Biology thanks Carmen Amaro and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Christopher LaRock and Tobias Goris.
Data availability
The 150 V. vulnificus genome sequences are deposited in the National Genomics Data Center63, under project number PRJCA025669 at NGDC. Metadata from NCBI are listed in Supplementary Data 2, and data used for Figs. 2 and 4A are available in Supplementary Data 1 and 3.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Dongling Long, Min Li.
These authors jointly supervised this work: Min Li, Chao Yang, Yongzhang Zhu.
Contributor Information
Min Li, Email: minli@shsmu.edu.cn.
Chao Yang, Email: cyang@siii.cas.cn.
Yongzhang Zhu, Email: yzhzhu@sjtu.edu.cn, Email: yzhzhu@hotmail.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-024-07426-5.
References
- 1.Baker-Austin, C. et al. Vibrio spp. infections, Nat. Rev. Dis. Primers, 4, (2018) [DOI] [PubMed]
- 2.Deng, Y. Q. et al. Prevalence, virulence genes, and antimicrobial resistance of Vibrio species isolated from diseased marine fish in South China. Sci. Rep.-Uk10, 14329 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oliver J. D., The Biology of Vibrio vulnificus. Microbiol. Spectr., 3, 10.1128/microbiolspec.VE-0001-2014. (2015). [DOI] [PubMed]
- 4.Heng, S. P. et al. Vibrio vulnificus:An Environmental and Clinical Burden, Front. Microbiol.8, 10.3389/fmicb.2017.00997 (2017). [DOI] [PMC free article] [PubMed]
- 5.Kaspar, C. W. & Tamplin, M. L. Effects of temperature and salinity on the survival of Vibrio vulnificus in seawater and shellfish. Appl Environ. Microbiol.59, 2425–2429 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tao, Z., Larsen, A. M., Bullard, S. A., Wright, A. C. & Arias, C. R. Prevalence and population structure of Vibrio vulnificus on fishes from the northern Gulf of Mexico. Appl. Environ. Microbiol.78, 7611–7618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips, K. E. & Satchell, K. J. F. Vibrio vulnificus: From oyster colonist to human pathogen. Plos Pathog.13, e1006053 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker-Austin, C. & Oliver, J. D. “Vibrio vulnificus: new insights into a deadly opportunistic pathogen”. Environ. Microbiol.20, 423–430 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Sheahan, M. et al. Examining the relationship between climate change and vibriosis in the United States: Projected health and economic impacts for the 21st Century. Environ. Health Perspect.130, 87007 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vezzulli, L. et al. Climate influence on Vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc. Natl Acad. Sci. USA113, E5062–E5071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker-Austin, C., Trinanes, J., Gonzalez-Escalona, N. & Martinez-Urtaza, J. Non-cholera vibrios: the microbial barometer of climate change. Trends Microbiol.25, 76–84 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Tison, D. L., Nishibuchi, M., Greenwood, J. D. & Seidler, R. J.Vibrio-Vulnificus Biogroup-2 - New Biogroup Pathogenic for Eels. Appl Environ. Microbiol.44, 640–646 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amaro, C. & Biosca, E. G. Vibrio vulnificus biotype 2, pathogenic for eels, is also an opportunistic pathogen for humans. Appl Environ. Microbiol.62, 1454–1457 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisharat, N. et al. Clinical, epidemiological, and microbiological features of Vibrio vulnificus biogroup 3 causing outbreaks of wound infection and bacteraemia in Israel. Lancet354, 1421–1424 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Nilsson, W. B., Paranjype, R. N., DePaola, A. & Strom, M. S. Sequence polymorphism of the 16S rRNA gene of vibrio vulnificus is a possible indicator of strain virulence. J. Clin. Microbiol.41, 442–446 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosche, T. M., Yano, Y. & Oliver, J. D. A rapid and simple PCR analysis indicates there are two subgroups of Vibrio vulnificus which correlate with clinical or environmental isolation. Microbiol. Immunol.49, 381–389 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Rosche, T. M., Binder, E. A. & Oliver, J. D. Vibrio vulnificus genome suggests two distinct ecotypes. Environ. Microbiol. Rep.2, 128–132 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Pan, J. H. et al. Molecular characterization and antibiotic susceptibility of Vibrio vulnificus in Retail Shrimps in Hangzhou, People’s Republic of China. J. Food Prot.76, 2063–2068 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Roig, F. J. et al. Phylogeny of Vibrio vulnificus from the Analysis of the Core-Genome: Implications for Intra-Species Taxonomy, Front. Microbiol.8, 10.3389/fmicb.2017.02613 (2018). [DOI] [PMC free article] [PubMed]
- 20.Kling, K. et al. Genetic divergence of Vibrio vulnificus clinical isolates with mild to severe outcomes. Mbio13, e0150022 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin, H. T. V. et al. The regulon encodes a metallo-β-lactamase and an antibiotic efflux pump, which are regulated by VarR, a LysRtype transcription factor. Plos One12, e0184255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell, A. B., Peterson, S. B. & Mougous, J. D. Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Microbiol.12, 137–148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, W. Z. et al. A combination of genomics and transcriptomics provides mRNA expression of type VI secretion system in clinical Msphere, e0082223 (2024). [DOI] [PMC free article] [PubMed]
- 24.Walter, K. What to know about Vibrio vulnificus. JAMA-J. Am. Med. Assoc.329, 772–772 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Ayala, A. J. & Ogbunugafor, C. B. When vibrios take flight: a meta-analysis of pathogenic vibrio species in wild and domestic birds 0065-2598 (Print), (2023). [DOI] [PubMed]
- 26.Zhao, H. R., Sun, R. N., Yu, P. F. & Alvarez, P. J. J. High levels of antibiotic resistance genes and opportunistic pathogenic bacteria indicators in urban wild bird feces. Environ. Pollut.266, 115200 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Páll, E. et al. Assessment and antibiotic resistance profiling in species isolated from wild birds captured in Reserve, Romania. Antibiotics10, 333 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brehm, T. T. et al. Nicht-Cholera-Vibrionen – derzeit noch seltene, aber wachsende Infektionsgefahr in Nord- und Ostsee. Der. Internist62, 876–886 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amato, E. et al. Epidemiological and microbiological investigation of a large increase in vibriosis, northern Europe, 2018. Eurosurveillance27, 2101088 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.López-Pérez, M. et al. Ecological diversification reveals routes of pathogen emergence in endemic populations. Proc. Natl Acad. Sci. USA118, e2103470118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.López-Pérez, M. et al. Evolutionary model of cluster divergence of the emergent marine pathogen: from genotype to ecotype. Mbio10, e02852–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim, J. H. et al. Characteristics of isolates from stool samples of patients with liver abscess caused by hypervirulent. J. Korean Med. Sci.35, e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Regmi, A. & Boyd, E. F. Carbohydrate metabolic systems present on genomic islands are lost and gained in Vibrio parahaemolyticus. BMC Microbiol.19, 112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gal-Mor, O. & Finlay, B. B. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol.8, 1707–1719 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Tan, Y. H. et al. Hypervirulent Klebsiella pneumoniae employs genomic island encoded toxins against bacterial competitors in the gut. ISME J.18, wrae054 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hachani, A., Wood, T. E. & Filloux, A. “Type VI secretion and anti-host effectors. Curr. Opin. Microbiol.29, 81–93 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Mougous, J. D. et al. A virulence locus of encodes a protein secretion apparatus. Science312, 1526–1530 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pukatzki, S. et al. Identification of a conserved bacterial protein secretion system in using the host model system. Proc. Natl Acad. Sci. USA103, 1528–1533 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coulthurst, S. The Type VI secretion system: a versatile bacterial weapon. Microbiol-Sgm165, 503–515 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Diniz, J. A., Liu, Y. C. & Coulthurst, S. J. Molecular weaponry: diverse effectors delivered by the Type VI secretion system. Cell Microbiol.17, 1742–1751 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trunk, K. et al. The type VI secretion system deploys antifungal effectors against microbial competitors. Nat. Microbiol.3, 920–931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hood, R. D. et al. A Type VI secretion system of targets, a toxin to bacteria. Cell Host Microbe7, 25–37 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacIntyre, D. L., Miyata, S. T., Kitaoka, M. & Pukatzki, S. The type VI secretion system displays antimicrobial properties. Proc. Natl Acad. Sci. USA107, 19520–19524 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Church, S. R., Lux, T., Baker-Austin, C., Buddington, S. P. & Michell, S. L. Vibrio vulnificus Type 6 Secretion System 1 contains anti-bacterial properties. PLoS One11, e0165500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.GB4789.44-2020 National food safety standard - Food microbiological examination - Vibrio vulnificus, S. A. f. M. R. National Health Commission of the PRC, Beijing, (2020).
- 46.Li, M. et al. Vibrio vulnificus in aquariums is a novel threat to marine mammals and public health. Transbound. Emerg. Dis.65, 1863–1871 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roig, A. P., Carmona-Salido, H., Sanjuan, E., Fouz, B. & Amaro, C. A multiplex PCR for the detection of Vibrio vulnificus hazardous to human and/or animal health from seafood. Int J. Food Microbiol377, 109778 (2022). [DOI] [PubMed] [Google Scholar]
- 48.Mosmann, T. Rapid colorimetric assay for cellular growth and survival - application to proliferation and cyto-toxicity assays. J. Immunol. Methods65, 55–63 (1983). [DOI] [PubMed] [Google Scholar]
- 49.Oh, Y. J. & Hong, J. Application of the MTT-based colorimetric method for evaluating bacterial growth using different solvent systems. LWT-Food Sci. Technol.153, 112565 (2022). [Google Scholar]
- 50.Li, M. et al. One global disseminated 193 kb high-risk hybrid plasmid harboring tet(X4), mcr or bla(NDM) threatening public health. Sci. Total Environ.876, 162807 (2023). [DOI] [PubMed] [Google Scholar]
- 51.Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics30, 2068–2069 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother.67, 2640–2644 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu, B., Zheng, D. D., Zhou, S. Y., Chen, L. H. & Yang, J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res50, D912–D917 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grant, J. R. et al. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res51, W484–W492 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bertelli, C. et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res.45, W30–W35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: a genome comparison visualizer. Bioinformatics27, 1009–1010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Page, A. J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics31, 3691–3693 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brynildsrud, O., Bohlin, J., Scheffer, L. & Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol.17, 238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tonkin-Hill, G. et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol.21, 180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res.44, D457–D462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: Computing large minimum evolution trees with profiles instead of a distance Matrix. Mol. Biol. Evol.26, 1641–1650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res.49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xue, Y. B. et al. Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res.51, D18–D28 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary File
Data Availability Statement
The 150 V. vulnificus genome sequences are deposited in the National Genomics Data Center63, under project number PRJCA025669 at NGDC. Metadata from NCBI are listed in Supplementary Data 2, and data used for Figs. 2 and 4A are available in Supplementary Data 1 and 3.