

Abstract
This study developed a prognostic model for patients with colon adenocarcinoma (COAD) based on glycosylation-associated genes. By analyzing TCGA-COAD data, 110 key genes were identified, and a prognostic model incorporating five glycosylation-related genes was constructed. The model exhibits good predictive performance and is significantly associated with clinical features such as age, N stage, M stage, and lymph node count. The prognostic genes are involved in various biological processes and pathways, influence T cell differentiation, and may contribute to CRC development. High-risk patients show a higher degree of immune cell infiltration. This model aids in the early diagnosis, prognosis assessment, and treatment planning for CRC, and offers a direction for further research.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-84300-8.
Keywords: Colon cancer, Bio-informatics, Prognostic, Single cell
Subject terms: Computational biology and bioinformatics, Gastroenterology, Oncology
Introduction
Colon cancer is the most common type of gastrointestinal tumor with a high morbidity and mortality rate1. Colonic adenocarcinoma (COAD) is the main pathological type of colon cancer2, and many evidences support the correlation between the prognosis of COAD and glycosylation3,4. In recent years, more and more research evidence shows that the prognosis of cancer is closely related to the glycosylation process5–7.
Glycosylation is a key post-translational modification process of proteins and plays a decisive role in the regulation of protein function8. This modification makes glycosylated proteins play a crucial role in many biological processes such as cell recognition, signal transduction, and immune response5,8,9. Glycosylation is also essential for the regulation of immune cell function. The alteration of glycosylation pattern affects the recognition ability of receptors on the surface of immune cells, and thus affects the activation state of immune cells7,10. For example, the glycosylation status of glycoprotein on the surface of T cells is closely related to the activity of T cells11,12. Abnormal glycosylation may enhance the immunosuppressive ability of tumor cells, reduce the phagocytosis, and promote immune escape3,13,14. This leads to immune invasion, tumor recurrence, metastasis and drug resistance15.
During the occurrence and development of COAD, the abnormal glycosylation of cancer cells not only intensifies the invasion and metastasis ability of the tumor, but also has a significant impact on the therapeutic effect16. Abnormal glycosylation has a profound impact on tumor progression and clinical targeted therapy8,16,17.
Therefore, we develop a prognostic model for COAD using glycosylation-related genes based on the data of TCGA-COAD patients, and verified the clinical application of this model by GEO datasets. Through the development and application of this model, it is expected to provide a strong scientific basis for the early diagnosis, prognosis assessment and treatment strategy formulation of COAD, so as to bring more accurate and effective treatment plans for patients.
Materials and methods
Database download
The data analyzed in this study were all obtained from public databases. Transcriptomic data and clinical data were downloaded from The Cancer Genome Atlas (TCGA, https://www.cancer.gov/ccg/research/genome-sequencing/tcga) database as our training cohort datasets. GSE12945 data were obtained from Gene Expression omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo) as our validation datasets. The training cohort datasets included clinical 512 samples, including 41 normal and 471 tumor samples. Chi-square test and Fischer test showed no significant difference between tumor and normal samples (Supplementary Table 1). Verification datasets include 62 samples that all are tumor. Single cell datasets GSE178318 also download from GEO database.
Glycosylation-related genes were obtained from the database GGDB (https://acgg.asia/ggdb2/), and a total of 242 glycosylation-related genes were obtained.
Identification of DE genes
The Wilcox test was used to conduct differential analysis. R-package “DESeq2” was used to analyze Count data of TCGA tumors and normal patients with differentially expressed genes (fol change |FC|>0.5, p < 0.05). ggplot2 was used to map the volcano of differential gene expression.
Development and validation of a prognostic risk model
Intersection genes of glycosylated-related genes and differentially expressed genes (DEGs) in TCGA dataset as our Candidate gene (Candidate Gene1, CG1). R-package “rms” was used for Univariate Cox analysis for CG1, and p < 0.05 gene was selected as our CG2. CG2 were analyzed by LASSO and CG3 was obtained. In order to select more critical genes from CG3 for developing prognostic models, we construct four machine learning (RF, SVM, GLM, PLS) models through R packages “caret”, “DALEX”, “e1071”, “glmnet”, “plyr”, and evaluate the model performance. The top 60% genes with the smallest residual error of each machine learning model are intersected, and the resulting genes are used as our prognostic genes for the construction of prognostic models. In order to evaluate the prognostic risk model, The following calculation formula was used for the optimum cutoff:
Risk Score = 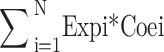
N, Expi, and Coei represented gene number, level of gene expression, and coefficient value, respectively. The optimum cutoff value (1.240599 in train cohort, 11.81231 in test corhort) was set as the cutoff value to divided all colon cancer patients into high-risk and low-risk groups. A high-risk score shows poor survival for colon cancer patients. R packages “survivalROC” and “timeROC” were used to conduct a ROC analysis. Time-dependent receiver operating characteristic (ROC) analysis for overall survival (OS) was used to evaluate the accuracy of the prognostic model. An area under the ROC (AUC) > 0.6 was treated as an acceptable prediction value. To further test the prognostic model, GEO datasets GSE12945 was used to validate the prognostic model by Survival analysis and time-dependent ROC analysis.
Correlation analysis
Spearman method was used for correlation analysis, and Wilcox test was used to test significance. The correlation heat map is drawn by R package “ggplot2” and “corrplot” for display.
Clinical utility of the model
To evaluate the prediction ability of the model in colon cancer patients, we assessed the relationships between our model (level of risk genes and the risk score) and the clinical features in the entire cohort. The patients were divided into high-low risk group by the best truncation value of the risk score, and the overall survival rate of patients in the high-low risk group was compared, which was displayed by the KM curve. Using the age, sex, race, the number of lymph nodes (0, 1–9, >=10) of patients and pathological stage, T, N and M in the TCGA dataset, clinical factors with p < 0.05 were selected as candidate clinical factors by univariate Cox analysis. Using PH tests, clinical factors with p > 0.05 were selected to construct the nomogram, and the accuracy and reliability of the nomogram were assessed.
Visualization of chromatin signals
We obtained H3K27me3 (ID: 82733), H3K27ac (ID: 69792), ATAC-seq (ID: 92855), H3K36me3 (ID: 69778), TP53 (ID: 82544) from CistromDB (http://cistrome.org/db/). IGV software is used for data visualization. Chromatin interaction information from 3D Genome Browser (http://3dgenome.fsm.northwestern.edu/), select colorectal cancer cells of HCT-116 cell line of Hi-C data for display.
GO/KEGG analysis
The R package “org.Hs.eg.db” and “clusterProfiler package was used to conduct gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis18–20. GO terms and KEGG terms were identified as significantly enriched when p.adjust < 0.05.
Gene set enrichment analysis (GSEA)
Use the R package “psych” to calculate the correlation coefficients for each core gene and all genes in the training set separately. Then, all genes are sorted according to the correlation coefficient of genes, and the list of related genes corresponding to each core gene is obtained. The sequencing results are used (reference gene set is from MSigDB database; The threshold was set to p < 0.05), and the “clusterProfiler” R package was used for GSEA path enrichment analysis.
PPI network
In order to demonstrate the interactions between CG1, String database (https://cn.string-db.org/) was used to identify gene interaction network, interaction genes with high confidence (0.9) were selected, and PPI network was mapped using Cytoscape software.
Somatic mutation analysis
In order to understand the mutations of prognostic genes, somatic mutations R-package “TCGAmutations” were used to identify somatic mutations in TCGA COAD samples.
Medicine predict and analysis
TCGA patients were divided into high and low risk groups using the optimal cut-off value, and R packages “pRRophetic” was used to predict the IC50 values of 138 drugs in the high and low risk group, and the significance test was performed by wilcox test. The box diagram is drawn by “ggplot2” for display. Spearman method was used to calculate the correlation between drugs and prognostic genes.
Drugs and prognostic genes with cor > 0.4 and has significant were as potentially effective drug. Spatial structure information of gene encoded proteins was obtained from protein database (PDB) (http://www.rcsb.org). PubChem database (https://pubchem.ncbi.nlm.nih.gov) to obtain the 3D molecular structure of potentially effective drug. Used for the structure of the information, use the CB-Dock2 (https://cadd.labshare.cn/cb-dock2/php/index.php) for molecular docking simulation and the analysis of the interaction.
Immunoinfiltration analysis
The R packages “ssgsea” was used to assess immunoinfiltration in TCGA samples, and the difference in the abundance of immunoinfiltration of 28 types of immune cells was examined by wilcox test in patients with high or low risk.
Single cell data processing
Given that our investigation centered on colorectal cancer (CRC), we meticulously selected a subset of six primary CRC samples—namely COL07_CRC, COL12_CRC, COL15_CRC, COL16_CRC, COL17_CRC, and COL18_CRC—from the comprehensive single-cell dataset identified as GSE178318. To screen out low-quality cells, we used R packages “Seurat” to exclude cells with nFeature_RNA < 200, nFeature_RNA > 6000, per.mt > 0.05, nCount_RNA < 200, nCount_RNA > 30,000 in the sample.
In the subsequent cell clustering process, we again enlisted “Seurat,” opting for dimensionality reduction and clustering algorithms with parameters tailored to our specific needs. Specifically, we set the number of dimensions Dim to 40 and the resolution to 0.8, employing both Uniform Manifold Approximation and Projection UMAP and t-distributed Stochastic Neighbor Embedding tSNE techniques for clustering analysis. For annotating distinct cell types, we referenced established marker genes gleaned from literature21 (Supplementary Table 2).
A particular emphasis was placed on analyzing T cells as critical cellular elements within this context. For this purpose, we adjusted the dimensionality selection using standard deviation “Dim” set at 12, and used a “resolution” value of 0.6 by R package “harmony”. The T cell markers utilized for identification are detailed in Supplementary Table 2, derived from pertinent academic resources.
Cell communication and pseudotime series analysis
Cell communication and pseudotime series analysis cell chat between 10 cell types annotated in the single cell dataset GSE178318 was analyzed using R packages “CellChat”. Employing the sophisticated capabilities of the “monocle” R package, we reconstructed a pseudo-temporal series, simulating the dynamic landscape of T cell development.
R packages and version
All R packages details were used are described in Supplementary Table 3. The R version is 4.4.1. And Python (V = 3.7.16) were used to help tidy up the file and results.
Results
Obtain candidate gene of glycosylation related
We download from TCGA (https://portal.gdc.cancer.gov/), colorectal adenocarcinoma as our training sample data set. The TCGA samples included 41 normal samples and 471 tumor samples. The data set included 266 men and 244 women. The tumor group included 246 men and 223 women, and the normal group included 20 men and 20 women. Supplementary Table 1 summarizes the demographic profiles of patients.
We obtained a total of 9635 differentially expressed genes (DEGs), of which 5065 were up-regulated and 4570 down-regulated (Fig. 1A-B). A total of 242 glycosylation-related genes (GRGs) were obtained from the database (GGDB, https://acgg.asia/ggdb2/). By taking the intersection of these 242 genes and DEG, a total of 110 genes (CG1) were obtained as candidate genes for subsequent analysis (Supplementary Fig. 1A). GO/KEGG analysis was used to understand the biological processes and pathways involved in these 110 genes (Fig. 1C-D). The results showed that the biological pathways involved in these genes were mainly related to glycosylation such as glycosphingolipid biosynthesis. The PPI network shows how they interact (Supplementary Fig. 1B). The results showd that most of the protein code by these genes were have function or physical interaction with each other. Among them, B3GALT5, B3GNT6 and B4GALT1 had the most interactions. Finally, tissue enrichment analysis showed the expression of these 110 candidate genes in tissues (Supplementary Fig. 1C). There was no significant difference in the enrichment results of these genes in tissues.
Fig. 1.

Refined annotations for comprehensive gene expression analysis in COAD. (A) Identification of Differentially Expressed Genes (DEGs) in TCGA-COAD Dataset. Red dots signify genes that have undergone significant up-regulation, while green dots correspond to those experiencing down-regulation. (B) Heat Maps Depicting Top 10 DEGs. Blue represents samples from the Normal cohort, whereas red stands for Tumor specimens. Below, the heat map’s lower section uses cyan to represent genes showing decreased expression, juxtaposed with red highlighting those exhibiting up-regulation. (C) KEGG Pathway Enrichment Analysis. On the right, diverse colors code for various KEGG enrichment pathways, revealing the complex interplay of biochemical processes affected. To the left, green denotes genes whose expressions have been down-regulated, with red indicating their up-regulated counterparts. (D) Gene Ontology (GO) Enrichment Analysis. The inner circle portrays the number of genes clustered under specific GO terms, acting as a visual metric for functional grouping. Outside the circle, individual dots embody single genes, where green signifies up-regulated status, and blue points to down-regulated states.
Construction of the prognostic GRGs model
The TCGA transcriptome data were used as a training cohort. Univariate Cox regression analysis of these CG1, 14 potential prognostic GRGs remained (p < 0.05, CG2, Fig. 2A). The results showed that there are 5 protect factor genes (SLC35D1, GALNT7, HS2ST1, B4GALT6, B3GNT6), and 9 genes (MFNG, UST, CHST1, CHPF, DPM2, ALG3, CHST8, HS6ST3, EXTL1) are risk factor. LASSO Cox proportional hazards regression were used to define the model on the CG2 and 10 GRGs were retained (CG3, Fig. 2B-C, Supplementary Table 4). To make our prognostic GRGs model more effective, we developed four machine learning models (RF, SVM, GLM, PLS) to further screen CG3. Model performance evaluated by receiver operating characteristic and residual (Fig. 2D, Supplementary Fig. 2A-C). In order to exclude the risk of overfitting of a single model, we integrated the four machine learning models by taking the top 60% gene intersection of the four models with the smallest residuals. As the result, 5 gene were obtained as our prognostic genes (PGs) for construct prognostic model (Fig. 2E).
Fig. 2.

Advanced prognostic gene analysis and machine learning model evaluation in oncological research. (A) Univariate Cox Regression Forest Plot. The forest plot illustrates the outcome of univariate Cox regression analysis applied to 110 candidate genes, identifying 14 potential prognostic genes (GRGs). Among them, 5 genes exhibit hazard ratios (HR) less than 1, suggesting protective roles, while 9 others are identified as risk factors, reflecting their detrimental impact on patient outcomes. (B,C): Least Absolute Shrinkage and Selection Operator (LASSO) Regression. (B) Each colored line represents a unique gene, showcasing its coefficient profile during LASSO regularization path. The optimal lambda (λ), determined by minimizing prediction error, is visually indicated, highlighting the regularization strength that achieves the best trade-off between bias and variance. (C) The LASSO model optimized via 10-fold cross-validation. (D) Receiver Operating Characteristic (ROC) Curve Comparison. Graphical representation of ROC curves for GLM (Generalized Linear Model), PLS (Partial Least Squares), SVM (Support Vector Machines), and RF (Random Forest). (E) Venn Diagram Illustrating Overlapping Genes. Highlights the common subset of top 60% genes with the least residual errors selected by all four machine learning algorithms. (F) Kaplan-Meier Survival Analysis. The KM plot compares survival probabilities between high-risk and low-risk patient cohorts. The blue line indicates low-risk individuals’ survival rates over time, contrasted sharply by the red line representing high-risk counterparts. (G) Time-Dependent ROC Curves. Time-dependent ROC analysis for predicting OS at 1, 3, and 5 years.
Based on the formula of optimal cutoff, the optimal cutoff value for the risk score was set to 1.240599. This value successfully stratified the patients in the training corhort into high- and low-risk groups (Supplementary Fig. 2D-E). Specifically, the overall survival (OS) of the low-risk group was significantly higher than that of the high-risk group (Fig. 2F). The Area Under the Curve (AUC) values of time-dependent receiver operating characteristic (ROC) curve analysis for the prognostic GRGs model at 1-, 3-, 5-year of OS was 0.671, 0.673 and 0.699, respectively (Fig. 2G). This results demonstrate that prognostic GRGs model based on 5 PGs are effective.
Validation of the prognostic GRGs model
To corroborate the precision and reliability of our prognostic GRGs model, we subjected it to rigorous validation within an independent testing cohort derived from the GSE12945 database. Utilizing a refined cut-off threshold of 11.81231 for the risk score, we meticulously segregated patients into high- and low-risk categories (Supplementary Fig. 3A-3B).
Kaplan-Meier survival analyses revealed stark disparities in survival trajectories between these two risk strata within the testing cohort (Fig. 3A), thereby echoing the model’s efficacy even when applied externally. To further fortify confidence in its predictive capabilities, we conducted Time-dependent ROC curve analyses. Impressively, the Area Under the Curve (AUC) metrics stood at robust levels of 0.897, 0.696, and 0.773 for 1-, 3-, and 5-year survival predictions, respectively, within the external cohort (Fig. 3B). This results suggest that our model have a good performance in outer datasets.
Fig. 3.

Detailed analysis of risk stratification and function exploration of prognostic genes in GSE12945 dataset. (A) Kaplan-Meier (KM) Survival Analysis. The KM plot depicts survival trajectories distinguishing high-risk from low-risk patients in the GSE12945 dataset. The blue trajectory signifies prolonged survival times in the low-risk population, markedly diverging from the red line, indicative of shorter survival periods in high-risk individuals. (B) Time-Dependent Receiver Operating Characteristic (ROC) Curves-Training Data (GSE12945). Shows the temporal accuracy of the model in forecasting 1-, 3-, 5-year Overall Survival (OS) outcomes using training set data. (C) Correlation Heatmap of Five Prognostic Genes (GRGs). Demonstrates correlations among five selected GRGs. Red dot represent positive correlations, signified by asterisks for statistically significant relationships, with blue hues denoting negative correlations. (D) Functional Similarity of Five GRGs. Elucidates the extent of shared functionalities among the GRGs, highlighting their collaborative involvement in similar biological pathways or processes, contributing to disease etiology or patient prognosis. (E,F): Chromatin Landscape of Two Genes (ALG3 and DPM2) Around Transcription Start Sites (TSS). Reveals the epigenetic modifications surrounding the TSS of ALG3 and DPM2, potentially influencing gene expression regulation. (G) Expression Profiles of Five GRGs in Tumor vs. Normal Samples. Compares the expression levels of the five GRGs in tumor tissues versus healthy controls, indicating their altered states in carcinogenesis. (H) Immunohistochemical Staining of Gene Expression. Provides direct evidence of protein expression in tissue sections, correlating mRNA abundance with immunohistochemical signals, thereby bridging genomic findings with phenotypic manifestations. (I,J): Mutation Profiling in High and Low-Risk Groups. Bar charts summarize mutation occurrences per gene in respective risk categories, emphasizing mutational landscapes distinctive to each group. Color codes identify types of mutations, with the accompanying bar chart depicting relative frequencies, illuminating genetic mutation contributing to risk stratification.
These compelling results collectively affirm the robustness and generalizability of our GRGs model, illustrating its proficiency in accurately forecasting clinical outcomes for Colorectal Adenocarcinoma (COAD) patients across diverse datasets. This achievement underscores the model’s potential utility as a reliable prognostic tool in real-world applications.
PGs interactions and functions analysis
To elucidate the interconnectivity and homogeneity of functionality amongst the quintet of Prognostic Genes (PGs), an exhaustive Spearman correlation analysis was conducted using samples from TCGA-COAD (Fig. 3C-D). The resultant data existence of parallel performance profile across these five PGs, indicative of potential cooperative action within underlying biological mechanisms.
To supplement this discovery, GeneMANIA, an online platform designed for gene network exploration (https://genemania.org/), was employed to identify and visualize interacting partners and associations between the aforementioned PGs (Supplementary Fig. 3C). Intriguingly, the outcomes revealed an intricate web of physical connectivity and shared protein domains among the majority of the genes under scrutiny.
Eager to delve deeper into the functional repertoire of our PGs, we performed a comprehensive examination through Gene Set Enrichment Analysis (GSEA) to pinpoint the pivotal pathways influenced by each PG individually. Our findings unveiled the top-five significantly impacted signaling pathways for every PG: namely, the spliceosome pathway, ribosome assembly, proteasome regulation, and protein export machinery (Supplementary Fig. 3D-3 H). This compelling evidence points towards the central role played by our PGs in the orchestration of protein synthesis and post-translational modifications, thereby cementing their reliance on protein dynamics as a cornerstone for predicting patient prognosis.
Collectively, these observations not only highlight the synergistic impact of our PGs in facilitating protein transcription and modification but also imply a coordinated effort during their functional execution. Such insights underscore the collective contribution of our PG ensemble to the broader framework of cellular proteomics, thereby enriching our understanding of their significance in prognosis prediction.
TF netwrok, histone signal and mutation analysis of PGs
Transcription Factors (TFs), crucial for orchestrating gene regulation, were explored for their interplay with PGs. Employing FunRich software, we uncovered 42 TFs that govern four out of the five PGs, excluding ALG3 (Supplementary Table 5). A regulatory network connecting TFs and core genes was visually rendered using Cytoscape (Supplementary Fig. 4A), offering a panoramic view of transcriptional control.
To gain deeper insights into the dynamic chromatin landscape governing the activity of our PGs, specifically focusing on ALG3 and DPM2, we leveraged Chromatin Immunoprecipitation (ChIP) sequencing data for H3K27ac, H3K27me3, H3K36me3, ATAC-seq, and TP53. Visual inspection through the Integrative Genomics Viewer (IGV v2.18.0) revealed a dense accumulation of histone marks around the ALG3 and DPM2 loci, indicative of heightened epigenetic accessibility and gene activity (Fig. 3E-F). Mirroring this finding, gene expression profiles from TCGA samples showcased a parallel upregulation pattern in tumors versus normal tissues (Fig. 3G-H), reinforcing the notion of elevated gene activity.
Furthermore, utilizing Hi-C data, we delineated the spatial proximity of ALG3 and DPM2 to neighboring genes, revealing substantial interaction strength with adjacent genomic elements (Supplementary Fig. 4B-C). This highlights the potential role of these PGs in complex regulatory networks.
To scrutinize mutational landscapes within high- and low-risk COAD patients, we mined TCGA mutation databases. Missense mutations emerged as the dominant variant type (Fig. 3I-J, Supplementary Fig. 4D-G), with the median somatic mutation burden reaching 2.27 mutations per megabase pair (Supplementary Fig. 4H). Additionally, we charted the temporal expression dynamics of prognostic genes across various COAD stages alongside correlating methylation patterns (Supplementary Fig. 4I-L). Copy number alterations were characterized by amplification of HS6ST3 and deletion of B4GALT6 (Supplementary Fig. 4M).
In conclusion, our integrative analyses reveal multifaceted layers of gene regulation and interaction among PGs in COAD, encompassing epigenetic marks, mutations, expression patterns, and TF binding. These findings underscore the complexity and interconnectedness of PGs in disease pathogenesis and offer novel avenues for targeted therapeutic intervention.
Clinical utility of the model
To explore the differential distribution of risk scores in relation to clinical attributes and discern variations among them in both high- and low-risk group, we applied the Wilcoxon rank-sum test to dissect the following patient characteristics: gender, age, T-stage, N-stage, M-stage, race, and lymph node count (Supplementary Fig. 5A-B). A univariate Cox proportional hazards regression analysis, anchored in the risk score and clinical descriptors from the TCGA cohort, served to elucidate the associations between various factors and patient prognosis (Fig. 4A). Amongst the clinical features investigated, riskScore, age, N-stage, M-stage, and Lymphnode number surfaced as statistically significant predictors. The Proportional Hazards Assumption Test confirmed the validity of including these four clinical attributes (riskScore, age, N-stage, Lymphnodes number) in the model (Fig. 4B), ensuring alignment with the foundational hypothesis and validating their selection for model construction.
Fig. 4.

Clinical risk analysis and prognostic modeling in oncological studies. (A) Univariate Cox Proportional Hazards Analysis. Through rigorous statistical assessment, RiskScore, Age, N-stage, M-stage, and Lymphnodes number emerge as potent predictors, uniformly classified as risk-enhancing elements impacting patient outcomes. (B) Proportional Hazards Assumption Verification. Confirms that RiskScore, Age, N-stage, and Lymphnode number adhere to the proportional hazards assumption, a fundamental requirement for Cox regression validity (p > 0.05). (C) Construction of Nomogram Incorporating Selected Clinical Risks. (D) Calibration Curve of Nomogram. Demonstrates alignment between predicted and observed survival rates, validating the nomogram’s precision in forecasting long-term outcomes. (E) Kaplan-Meier Survival Analysis Based on Optimal Cut-off Value. Divides subjects into high and low-risk groups using a calculated cut-off of 1.240599, delineated by red (high risk) and blue (low risk) lines. (F) ROC Curves for Predictive Performance. Evaluates the five-year prognostic capability of identified variables, depicting sensitivity against specificity at varied thresholds, ensuring comprehensive evaluation of prediction quality. (G) Time-Dependent ROC Curves. Illustrates the effectiveness of combining risk scores with clinical features (Age, N-stage, and Lymphnode number) in predicting survival at 1, 3, and 5 years, providing longitudinal insights into forecasting accuracy. (H) Immunoscore Distribution in High vs. Low Risk Groups. (I) Correlation Matrix of Differentially Expressed Immune Cells. (J) Relationship Between Immune Cells and Prognostic Genes. The interplay between immune components and prognostic genes. (K) Molecular Docking Simulation. Simulates the binding affinity between drug NVP-TAE684 and HS2ST1-encoded protein.
Capitalizing on the identified predictors—riskScore, age, N-stage, and lymph node number—we developed a nomogram tailored for forecasting 1-, 3-, and 5-year survival probabilities in COAD patients (Fig. 4C-D). This nomogram exhibited commendable performance in estimating overall survival, as reflected by its calibration and discrimination abilities. Patients were categorized into high and low-risk cohorts based on an optimal cutoff of 1.240599, illustrated vividly through Kaplan-Meier survival curves (Fig. 4E), which indicated inferior survival rates in the high-risk segment compared to the low-risk counterpart.
Through ROC curves, we gauged the predictive accuracy of riskScore, age, N-stage, and lymph node number in forecasting patient outcomes (Fig. 4F). Each variable displayed promising predictive power. Subsequently, integrating these factors with the risk score, we established a composite predictive model and appraised its robustness via ROC curves. Strikingly, the AUC values for 1-, 3-, and 5-year predictions surpassed 0.75 (Fig. 4G), outperforming the singular risk score model, thereby underscoring the enhanced precision of our integrated approach.
In summation, the amalgamation of clinical parameters with genetic risk assessment yields a more sophisticated predictive algorithm for colorectal adenocarcinoma prognosis, providing clinicians with a dependable decision-support tool that surpasses traditional models in terms of reliability and accuracy. This approach may indicate personalized treatment strategies and improved patient care management.
Immune cell infiltration analysis
To comprehensively understand the immunological microenvironment characterizing COAD, we evaluated essential metrics such as the ESTIMATE score, immune score, stromal score, and tumor purity score across high- and low-risk patient groups (Fig. 4H). Comparative analysis of 28 distinct immune cell populations between these risk strata unveiled statistical disparities (p < 0.05) in 21 of those, along with their respective correlations with one another and the Prognostic Genes (PGs) (Fig. 4I-J, Supplementary Fig. 5C). Notably, the high-risk group consistently displayed elevated immune scores relative to the low-risk cohort—a finding resonant with prior reports linking increased immunogenicity to poorer prognosis22.
Consistent with previous literature, our study also catalogued the expression profiles of 79 immune checkpoint molecules within the high-low risk spectrum, identifying 42 markedly divergent entities (Supplementary Fig. 5D). Spearman correlation analyses further probed the interplay between these distinctive immune checkpoints and the risk score, as well as their associations with PGs (Supplementary Fig. 5E-F).
These findings collectively illuminate the intricate interplay between immune infiltration, checkpoint modulation, and COAD progression, underscoring the significance of the immune milieu in disease trajectory and clinical outcome. The pronounced immune response in high-risk individuals suggests a complex interplay between immunobiology and adverse prognosis, warranting future research aimed at elucidating potential immunotherapeutic targets or interventions. Our work reinforces the critical role of immune surveillance in shaping cancer evolution and patient survival, while highlighting the need for integrated approaches to fully harness the immune system’s potential against COAD.
Drug sensitivity analysis and predict potential drugs
Aim to evaluate the drug sensitivity of differences between high and low risk group from cancer genomics database (GDSC) drug sensitivity (https://www.cancerrxgene.org/) for 138 drugs and half maximum inhibitory concentration (IC50), The sensitivity of patients in the high-low risk group of TCGA-COAD to 138 drugs was calculated. We found significant differences in IC50 values for 92 drugs in the high-low risk group and calculated correlation between the 10 drugs with the lowest p-values and prognostic genes (Supplementary Fig. 5G-H, Supplementary Table 6). Molecular docking is used to investigate the effectiveness of drugs in drug sensitivity analysis. We combined the results of drug sensitivity analysis and correlation analysis, and selected three pairs of drug and protein interactions to demonstrate. The results showed that all three pairs of drugs and proteins had good interactions, suggesting that these drugs may be effective drugs for the treatment of COAD (Fig. 4K, supplementary Fig. 5I-J).
Decoding the colorectal cancer tumor microen viroment through single-cell transcriptiomics
To deepen our understanding of the intricate dynamics within colorectal cancer’s tumor microenvironment, we harnessed the extensive single-cell dataset GSE178318, meticulously selecting six primary colon cancer samples encompassing a vast array of cellular constituents. Initially, the cohort comprised 55,291 cells; following stringent quality control measures, 41,622 cells met the criteria for inclusion in downstream analyses (Supplementary Fig. 6A-B), ensuring robustness and reliability in subsequent investigations.
The selection process continued with the identification of the top 2,000 highly variable genes through variance stabilization transformation (Supplementary Fig. 6C), laying the groundwork for in-depth exploratory analyses. Principal Component Analysis (PCA) served as a critical step for dimensionality reduction on this curated list of high-variation genes (Supplementary Fig. 6D-G). Employing cutting-edge clustering methodologies—Uniform Manifold Approximation and Projection (UMAP) and t-Distributed Stochastic Neighbor Embedding (tSNE)—we achieved precise segregation, yielding a total of 28 distinct cell clusters (Supplementary Fig. 6H).
Drawing upon established literature (Supplementary Table 2), we leveraged marker genes to assign identity to each cluster (Fig. 5A). Leveraging these markers, ten cell types were annotated, and T-cells are the most abundance cell type in all samples, with a total of 23,130 T cells in all samples (Fig. 5B-C). This finding resonates harmoniously with our prior immunoinfiltration assessments, demonstrating a marked elevation in T-cell prevalence amongst the high-risk cohort vis-à-vis the low-risk group.
Fig. 5.

Single cell annotation and cell chat. (A) Dotplot of marker genes in different cell clusters in all samples. The dot size present percent expression and red means that the gene is high express in the cluster. (B) UMAP and cell annotation results for cell type. In left, every color present a cluter and right is the annotated results. (C) Proportion of cell numb in samples. Every color present a cell type. (D) Cell chat in the 10 type cells. The node size present the number of cell and line thickness degree present number chat (left) and strength (right), separately. (E) Receptor and ligand interaction plot, red dot present have a high interaction. (F) Marker expression in T cells subtypes. The dot size present percent expression and red means that the gene is high express in the cluster. (G) UMAP of T cell subtype annotated results, each color present a cell type. (H) T cell pseudo-time analysis results. In top left plot, dark color mean original cell. Cell differentiate from dark area to light blue area. Top right and bottom are present subtype T cells differentiation and bottom plot color present cell cluster. (I) Heat map of pseudo-time results.
Further, employing Reactome Gene Set Analysis (reactomeGSA), we embarked on enriching the biological landscape of these ten cellular populations (Supplementary Fig. 6I). The results illuminated that T-cells gravitated towards the FGFR1c and Klotho ligand binding and activation pathways. These pathways are known to spur tumor cell proliferation, bolster survival capabilities, amplify malignancy, and facilitate metastatic spread, while also impacting responsiveness to chemotherapeutic regimens and immunotherapies23,24. Intriguingly, our study proposes that aberrant engagement of the FGFR signaling axis could disrupt T-cell functionality, potentially derailing tumor immune surveillance and dictating therapeutic outcomes.
T cell analysis and cell chat
To understand cell chat among these cell types, we demonstrate the number and strength of communication between these 10 types of cells (Fig. 5D). The results showed that T cells had strong interactions with all other cell types, and the communication intensity between T cells and CAFs was the strongest in terms of communication intensity. To understand the ligand-receptor interactions of cell communication, we mapped the ligand-receptor interactions bubble map of TCs interacting with other cells (Fig. 5E). This results showed that T cells interact with other cells most by MIF and CD99.
We selected T cells as our key cells for further analysis. We demonstrated the expression of marker gene in T cells and UMAP were used to cluster T cells, and annotated cell ypes by markers from literatures (Fig. 5F-G). As a result, we annotated 9 T cell subtypes - Stem-like memory T cells (Tscm), Naive T cells (naive), Central memory T cells (Tcm), Regulatory T cells (Tregs), Type 1 cytotoxic T cells (Tc1), Effector memory T cells (Tem), Type 17 cytotoxic T cells (Tc17), Recent thymic emigrants (RTE), Type 1 helper T cells (Th1) (Fig. 5G). We note that there are some annotated T cells like Tregs, Tcm, Tem, Th1 are have a significantly higher immune score in high risk group than low in our immune results (Supplementary Fig. 5C).
We used pseudo-time analysis to further understand the pseudo-time differentiation of T cells (Fig. 5H). We first used R package “monocle” to select 473 highly variable genes, and then used these highly variable genes to perform a pseudo-time analysis of T cells. The results showed that the number of Th1, Tem and Tc1 cells increased in the late stage of T cell differentiation.
Finally, we understood the expression changes of prognostic genes in T cell differentiation. The results showed that the expression of prognostic genes B4GALT6 was less in the early stage, while DPM2, ALG3 were more expressed in the early stage and DPM2 expression gradually increased in the later stage (Fig. 5I). This result is consistent with the correlation between immune cells and prognostic genes.
Discussion
Colon cancer is one of the most common cancers worldwide, with about 1.1 million new cases and 550,000 deaths in 20182. In this study, we use bioinformatics techniques to explore the relationship between GRGs and diseases, and to construct prognostic models. We established and validated a prognostic model based on five GRGs, which can be used as an independent prognostic variable.
In this study, TCGA COAD samples were used as a training set, and 9635 DEGs were obtained between tumor and normal samples. A total of 242 GRGs were obtained from GGDB database, and 110 genes were obtained from the intersection of DEGs and GRGs. We understood the major enrichment pathways of these 110 genes through GO/KEGG enrichment analysis results demonstrate that these genes function are major in N- and O-glycan biosynthesis. Combined with other results, like PPI networks and PGs correlation, we believed that these genes may cooperate for each other to participate in N- and O-glycan biosynthesis related biologic process.
We selected five genes (DPM2, ALG3, B4GALT6, HS2ST1, HS6ST3) to construct a prognostic model by combined univariate Cox regression analysis, LASSO, and machine learning. Then, KM curve and ROC curve were used to test the model performance. The results showed that the prognosis model had accurate reliable predictive performance. And we also considered the clinical characteristic factors as independent prognostic factor in our model and select three clinical characteristic factors (Age, N-stage, Lymphnodes_numb). As the results, these clinical characteristic factors performance in predict prognostic are poor than our models. Finally, we added these clinical character factors in our models, as a complex model that combined genes and clinical features, have a better performance than any single feature model. But sadly for that we have enough data to test the performance in other datasets.
For understood these 5 PGs biological processes, we utilized GSEA enrichment analysis. We found that these 5 PGs biologic function are major in spliceosome, proteasome and other related to protein. This results suggest that our PGs may focus on the protein synthesis, modification and trans. Based on this, we select some drugs for COAD by IC50 and use molecular dock to predict the effectiveness of these drugs. As the results, we found some drugs could target the PGs code protein and would have some effectiveness in cure COAD.
The tumor immune microenvironment (TIMV) of the samples was observed and the TIMV score was calculated. There were significant differences in the enrichment scores of 21 kinds of immune cells between high- and low-risk groups. Correlation analysis was used to explore the correlation between differential immune cells in high- and low-risk group samples, as well as the correlation between prognostic genes and differential immune cells.
In order to understand the TIMV in single-cell insight, we used a scRNA-seq dataset GSE178318 and annotated the types of cells types. A total of 10 cell types were annotated. As the results, we found that T cells are the most abundance cell type in all samples and accord with our TIMV results. Then we focused on T cells and get more details in the component of T cells by annotate T cells subtypes. Because the T cells is too similar to each other to difficult to annotated. So, we used many markers from literature (Supplementary Table 2) and test many times to select a better and effective markers to annotate. Finally, we got 9 T cell subtypes (Tscm, naive, Tcm, Tregs, Tc1, Tem, Tc17, RTE, Th1). In pseudo-time analysis results, we found that Th1, Tem and Tc1 cells increased in the late stage of T cell differentiation. On the contrary, naive and Tregs are major in early stage of T cell differentiation.
We used the first two thousand high-side genes to simulate the reverse temporal differentiation of T cells. Since HS6ST3 was not recognized by the high-variable genes, the correlation analysis of HS6ST3 was not performed. Our results showed that the expression of genes B4GALT6 and DPM2 increased during late differentiation of single-cell T cells. Previously research reported that genes B4GALT6 and DPM2 are mainly involved in the synthesis of N-linked glycans25–27. N-glycans play a multi-sided role in tumor, which can affect the growth, invasion, metastasis and interaction with the immune system of tumor cells17. N-glycans on the surface of tumor cells may exhibit increased branching, altered sugar chain structure, and abnormal expression of glycosyltransferase and glycosidase5. These changes may promote tumor cells’ ability to evade immune surveillance, enhance intercellular adhesion and migration, and affect tumor cells’ sensitivity to drugs28. This suggests that genes B4GALT6 and DPM2 may influence patient outcomes by influencing the synthesis of N-glycans. ALG3 gene code an enzyme involved in N-linked glycan biosynthesis29. It catalyzes the transfer of mannose in the endoplasmic reticulum of cells and is one of the key steps in the formation of correctly folded N-glycan structures10,29,30. During glycosylation, the role of ALG3 is essential for the proper folding and function of proteins. Our results show that ALG3 gene expression is reduced in late T cell differentiation, which may lead to the production of misfolded N-glycan structures, thereby affecting patient prognosis. At the same time, this study also has some shortcomings. The first is that our sample does not cover all relevant groups, which may affect the generality and reliability of the findings. Since this study relies on bioinformatics methods and instruments, a large number of external validations are still needed to verify the accuracy and reliability of the model.
In total, this study combined the scRNA-seq data and TCGA database, and used bioinformatics research methods to study the prognostic significance of glycosylation-related genes in COAD. Five prognostic genes were used to establish a prognostic model, and the molecular regulatory mechanism and immune characteristics of this genetic prognostic model were analyzed to promote the development of COAD targeted therapy and improve the survival rate of patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Not applicable.
Author contributions
R.L., S.H., T.Q. and Y.M. made contribution to the conception and design; K.X. and S.L. analyzed and interpreted data; R.L. and S.H. drafted the article; W.Z. revisied it critically for important intellectual content; All authors approved the final version to be published.
Funding
The research was supported by National Natural Science Foundation of China (No. 82260959). Fund of Doctor from Beijing Jishuitan Hospital Guizhou Hospital (No. JGYBS[2024]01). 2023 Excellent Reserve Talents of Affiliated Hospital of Guizhou Medical University (No. gyfyxkrc-2023-082021). Cultivate Project 2021 for National Natural Science Foundation of China from Affiliated Hospital of Guizhou Medical University (No. gyfynsfc-2021-9). National Natural Science Foundation of China (No: 82060523).
Data availability
The datasets analysed during the current study are available in the Gene Expression Omnibus (GEO database, https://www.ncbi.nlm.nih.gov/geo/, GSE12945) and The Cancer Genome Atlas (TCGA, https://www.cancer.gov/ccg/research/genome-sequencing/tcga). We obtained H3K27me3 (ID: 82733), H3K27ac (ID: 69792), ATAC-seq (ID: 92855), H3K36me3 (ID: 69778), TP53 (ID: 82544) from CistromDB (http://cistrome.org/db/).Cell markers were put in Supplementary Table S2.All of data in this paper are open access.The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rui Li and Sha He contributed equally to this manuscript.
References
- 1.Fabregas, J. C., Ramnaraign, B. & George, T. J. Clinical updates for Colon Cancer Care in 2022. Clin. Colorectal Cancer. 21, 198–203 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Center, M. M., Jemal, A., Smith, R. A. & Ward, E. Worldwide variations in colorectal cancer. CA Cancer J. Clin.59, 366–378 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Alymova, I. V. et al. Aberrant cellular glycosylation may increase the ability of Influenza viruses to escape host Immune responses through modification of the viral glycome. mBio13, e0298321 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold, J. N. et al. Novel glycan biomarkers for the detection of lung cancer. J. Proteome Res.10, 1755–1764 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Pinho, S. S. & Reis, C. A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer. 15, 540–555 (2015). [DOI] [PubMed] [Google Scholar]
- 6.González-Vallinas, M. et al. Clinical relevance of the differential expression of the glycosyltransferase gene GCNT3 in colon cancer. Eur. J. Cancer. 51, 1–8 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Büll, C., den Brok, M. H. & Adema, G. J. Sweet escape: Sialic acids in tumor immune evasion. Biochim. Biophys. Acta. 1846, 238–246 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Lin, Y. & Lubman, D. M. The role of N-glycosylation in cancer. Acta Pharm. Sinica B. 14, 1098–1110 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Minh, G., Esquea, E. M., Young, R. G., Huang, J. & Reginato, M. J. On a sugar high: role of O-GlcNAcylation in cancer. J. Biol. Chem. 299. (2023). [DOI] [PMC free article] [PubMed]
- 10.Bangarh, R. et al. Aberrant protein glycosylation: implications on diagnosis and immunotherapy. Biotechnol. Adv. 66. (2023). [DOI] [PubMed]
- 11.Earl, L. A. & Baum, L. G. CD45 glycosylation controls T-cell life and death. Immunol. Cell. Biol.86, 608–615 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Daniels, M. A., Hogquist, K. A. & Jameson, S. C. Sweet ‘n’ sour: The impact of differential glycosylation on T cell responses. Nat. Immunol.3, 903–910 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Xu, Y. et al. PD-L2 glycosylation promotes immune evasion and predicts anti-EGFR efficacy. J. Immunother Cancer 9. (2021). [DOI] [PMC free article] [PubMed]
- 14.Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol.15, 346–366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li, J., Li, X. & Guo, Q. Drug resistance in cancers: A free pass for bullying. Cells 11. (2022). [DOI] [PMC free article] [PubMed]
- 16.Alexander, K. L. et al. Modulation of glycosyltransferase ST6Gal-I in gastric cancer-derived organoids disrupts homeostatic epithelial cell turnover. J. Biol. Chem.295, 14153–14163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krug, J. et al. N-glycosylation regulates intrinsic IFN-γ resistance in Colorectal Cancer: Implications for Immunotherapy. Gastroenterology164, 392–406e5 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci.28, 1947–1951 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res.51, D587–d92 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Che, L. H. et al. A single-cell atlas of liver metastases of colorectal cancer reveals reprogramming of the tumor microenvironment in response to preoperative chemotherapy. Cell. Discov. 7, 80 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruni, D., Angell, H. K. & Galon, J. The immune contexture and immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer. 20, 662–680 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Helsten, T. et al. The FGFR landscape in cancer: Analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res.22, 259–267 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Liu, Q. et al. FGFR families: biological functions and therapeutic interventions in tumors. MedComm 2023; 4, e367. (2020). [DOI] [PMC free article] [PubMed]
- 25.Rostami, A. & Ciric, B. Astrocyte-derived lactosylceramide implicated in multiple sclerosis. Nat. Med.20, 1092–1093 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Mayo, L. et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med.20, 1147–1156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radenkovic, S. et al. Expanding the clinical and metabolic phenotype of DPM2 deficient congenital disorders of glycosylation. Mol. Genet. Metab.132, 27–37 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung, P. et al. Metabolic homeostasis and tissue renewal are dependent on beta1,6GlcNAc-branched N-glycans. Glycobiology17, 828–837 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Sun, X. et al. ALG3 contributes to stemness and radioresistance through regulating glycosylation of TGF-β receptor II in breast cancer. J. Exp. Clin. Cancer Res.40, 149 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bloch, J. S. et al. Structure and mechanism of the ER-based glucosyltransferase ALG6. Nature579, 443–447 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analysed during the current study are available in the Gene Expression Omnibus (GEO database, https://www.ncbi.nlm.nih.gov/geo/, GSE12945) and The Cancer Genome Atlas (TCGA, https://www.cancer.gov/ccg/research/genome-sequencing/tcga). We obtained H3K27me3 (ID: 82733), H3K27ac (ID: 69792), ATAC-seq (ID: 92855), H3K36me3 (ID: 69778), TP53 (ID: 82544) from CistromDB (http://cistrome.org/db/).Cell markers were put in Supplementary Table S2.All of data in this paper are open access.The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.