

Summary
The de novo generation of molecules with targeted properties is crucial in biology, chemistry, and drug discovery. Current generative models are limited to using single property values as conditions, struggling with complex customizations described in detailed human language. To address this, we propose the text guidance instead, and introduce TextSMOG, a new text-guided small molecule generation approach via diffusion model, which integrates language and diffusion models for text-guided small molecule generation. This method uses textual conditions to guide molecule generation, enhancing both stability and diversity. Experimental results show TextSMOG’s proficiency in capturing and utilizing information from textual descriptions, making it a powerful tool for generating 3D molecular structures in response to complex textual customizations.
Subject areas: Chemistry, Molecular modelling, Artificial intelligence
Graphical abstract
Highlights
-
•
Combines language models and diffusion models into text-guided 3D molecule generation
-
•
Multi-modal conversion module translates text conditions into reference geometries
-
•
Outperforms existing methods, yielding more stable and diverse subject molecules
-
•
Initializes understanding general text descriptions to explore molecular space
Chemistry; Molecular modelling; Artificial intelligence
Introduction
De novo molecule design, the process of generating molecules with specific, chemically viable structures for target properties, is a cornerstone in the fields of biology, chemistry, and drug discovery.1,2,3,4 It not only allows for the creation of subject molecules but also provides insights into the relationship between molecular structure and function, enabling the prediction and manipulation of biological activity. Constrained by the immense diversity of chemical space, manually generating property-specific molecules remains a daunting challenge.5 However, the generation of molecules that precisely meet specific requirements, including the creation of tailor-made molecules, is a complex task due to the vastness of the chemical space and the intricate relationship between molecular structure and function. Overcoming this challenge is crucial for advancing our understanding of biological systems and for the development of new therapeutic agents. In recent years, machine and deep learning methods have initiated a paradigm shift in the molecule generation,6,7,8,9,10,11 which enable the direct design of 3D molecular geometric structures with the desired properties.8,12,13 Notably, diffusion models,14,15 specifically equivariant diffusion models,16,17 have gradually entered the center of the stage with its outstanding performance. The core of this method is to introduce diffusion noise on molecular data, and then learn a reverse process in either unconditional or conditional manners to denoise this corruption, thereby crafting desired 3D molecular geometries. Meanwhile, some conditional inputs (e.g., polarizability ) could be applied for constraining the model to generate more specific molecules types.
However, despite the promise of these methods, a significant proportion of molecules generated by diffusion models do not meet the practical needs of researchers. For instance, they may lack the desired biological activity, exhibit poor pharmacokinetic properties, or be synthetically infeasible. This would be due to the fact that, on one hand, searching for suitable molecules in drug design typically requires consideration of multiple properties of interest (e.g., simultaneously characterized by specific polarizability, orbital energy, properties like aromaticity, and distinct functional groups).18,19,20 On the other hand, humans seem to struggle with conveying their needs precisely to the model. While a text segment such as “this molecule is an aromatic compound, with small HOMO-LUMO gaps and possessing at least one carboxyl group” can accurately describe human requirements and facilitate communication among humans, it is still challenging to directly convey this “thoughts” to the model. Therefore, we aspire to develop a method that allows for the interactive inverse design of 3D molecular structures through natural language. In other words, we aim to create a system where researchers can describe the properties they want in a molecule using natural language, and the system will generate a molecule that meets these requirements. This aspiration prompts us to explore text guidance in diffusion models, emphasizing the necessity for models adept at precise language understanding and molecule generation.
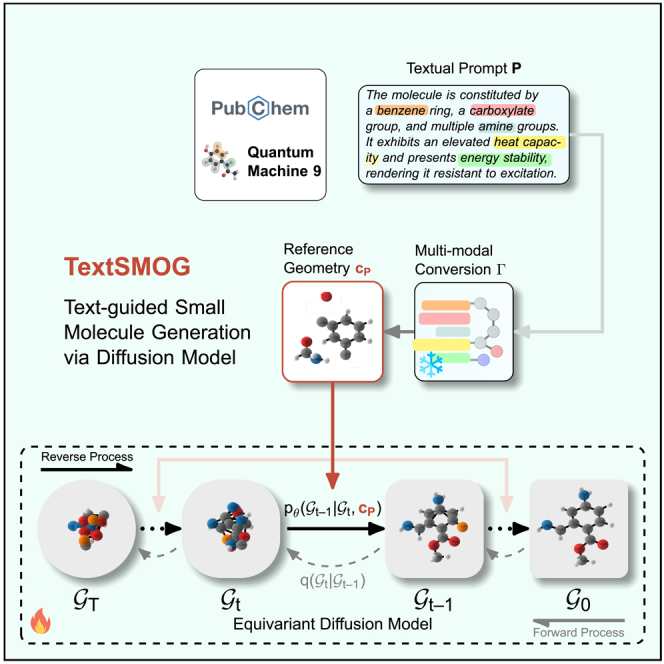
Toward this end, we propose TextSMOG, a new text-guided small molecule generation approach. The basic idea is to combine the capabilities of the advanced language models21,22,23,24,25,26 with high-fidelity diffusion models, enabling a sophisticated understanding of textual prompts and accurate translation into 3D molecular structures. TextSMOG accomplishes this through integrating textual information with a conversion module that conditions a pre-trained equivariant diffusion model (EDM)16 following the multi-modal fusion fashion.9,27,28,29 Specifically, at each denoising step, TextSMOG first generates reference geometry, an intermediate conformation that encapsulates the textual condition signal, through a multi-modal conversion module. Equipped with language and molecular encoder-decoder, corresponding to the textual condition. Then the reference geometry guides the denoising of each atom within the pre-trained unconditional EDM, gradually modifying the molecular geometry to match the condition while maintaining chemical validity. By incorporating valuable language knowledge into the pre-trained diffusion model, TextSMOG enhances the generation of valid and stable 3D molecular conformations that align with a spectrum of diverse directives. This is achieved without the need for exhaustive training on each specific condition, demonstrating the model’s ability to generalize from the language input. This integration allows for the incorporation of valuable language knowledge in the high-fidelity pre-trained diffusion model, thereby enabling the conditional generation contingent upon a spectrum of diverse directives, while enhancing the generation of valid and stable 3D molecular conformations, without specific exhaustive training of the condition.
We applied TextSMOG to the standard quantum chemistry dataset QM930 and a real-world text-molecule dataset from PubChem.31 The experimental results show that TextSMOG accurately captures single or multiple desired properties from textual descriptions, thereby aligning the generated molecules with the desired structures. Notably, TextSMOG outperforms leading diffusion-based molecule generation baselines (e.g., EDM,16 EEGSDE17) in terms of both the stability and diversity of the generated molecules. This is evidenced by higher scores on metrics such as the Tanimoto similarity to the target structure, the synthetic accessibility of the generated molecules, and the diversity of the generated molecule set. Furthermore, when applied to real-world textual excerpts, TextSMOG demonstrates its generative capability under general textual conditions. These findings suggest that TextSMOG constitutes a versatile and efficient text-guided molecular diffusion framework. As an advanced intelligent agent, it can effectively comprehend the meaning of textual commands and accomplish generation tasks, thereby paving the way for a more in-depth exploration of the molecular space.
Results
In this section, we present the architecture and the experimental results of our proposed TextSMOG model, showcasing its ability to generate molecules with desired properties.
Architecture
To evaluate our model, we employ the QM9 dataset,30 which is a standard benchmark containing quantum properties and atom coordinates of over 130K molecules, each with up to 9 heavy atoms (C, N, O, F). For the purpose of training our model under the condition of textual descriptions, we have curated a subset of molecules from QM9 and associated them with real-world descriptions. These descriptions are sourced from PubChem,31 one of the most comprehensive databases for molecular descriptions, and are linked to the molecules in QM9 based on their unique SMILES.
PubChem aggregates extensive annotations from a diverse array of sources, such as ChEBI,32 LOTUS,33 and T3DB.34 Each of these sources offers an emphasis on the physical, chemical, or structural attributes of molecules. Additionally, we have employed a set of textual templates to generate corresponding descriptions based on the quantum properties of the molecules, thereby enriching the content of the dataset and supplementing textual context for those molecules lacking real-world descriptions. This process has enriched QM9 into a dataset of chemical molecule-textual description pairs. Our proposed TextSMOG model, illustrated in Figure 1, is built upon the pre-trained unconditional diffusion model EDM.16 It integrates the textual information into the conditional signal of diffusion models by employing a reference geometry that is updated at each step based on the textual prompt. The final molecular geometry is generated by gradually denoising an initial geometry, while noise is added at each step during the forward process until the molecular geometry is fully noise-corrupted.
Figure 1.

Architecture of our text-guided small molecule generation via diffusion model (TextSMOG)
The model starts with an initial geometry and gradually denoises it to generate the final molecular geometry. The reference geometry , updated at each step based on the textual prompt , is employed to integrate the textual information into the conditional signal of diffusion models. Flame  denotes tunable modules, while snowflake
denotes tunable modules, while snowflake  indicates frozen modules.
indicates frozen modules.
Experiment on single quantum properties conditioning
Following EDM,16 we first evaluate our TextSMOG on the task of generating molecule conditioning on a single desired quantum property in QM9. Then we compare our TextSMOG with several baselines to demonstrate the effectiveness of our model on single quantum properties conditioning molecule generation.
Setup
We follow the same data preprocessing and partitions as in EDM,16 which results in 100K/18K/13K molecule samples for training/validation/test, respectively. In order to assess the quality of the conditional generated molecules w.r.t. to the desired properties, we use the property classifier network introduced by.35 Then for the impartiality, the training partition is further split into two non-overlapping halves and of 50K molecule samples each. The property classifier network is trained on the first half , while our TextSMOG is trained on the second half . This ensures that there is no information leak and the property classifier network is not biased toward the generated molecules from TextSMOG. Then is evaluated on the generated molecule samples from TextSMOG as we introduce in the following.
Metrics
Following,16 we use the mean absolute error (MAE) between the properties of generated molecules and the ground truth as a metric to evaluate how the generated molecules align with the condition (see the supplemental information for details). We generate 10K molecule samples for the evaluation of , following the same protocol as in EDM. Additionally, we then measure novelty,36 atom stability,16 and molecule stability16 to demonstrate the fundamental molecule generation capacity of the model (also see the supplemental information for details).
Baseline
We compare our TextSMOG with a direct baseline conditional EDM16 and a recent work EEGSDE which takes energy as guidance.17 We also compare two additional baselines “U-bound” and “#Atoms” introduced by.16 In the “U-bound” baseline, any relation between molecule and property is ignored, and the property classifier network is evaluated on with shuffled property labels. In the “#Atoms” baseline, the properties are predicted solely based on the number of atoms in the molecule. Furthermore, we report the error of on as a lower bound baseline “L-Bound”.
Results
We generate molecules with textual descriptions targeted to each one of the six properties in QM9, which are detailed in the supplemental information. As presented in Figure 2, our TextSMOG has a lower MAE than other baselines on five out of the six properties, suggesting that the molecules generated by TextSMOG align more closely with the desired properties than other baselines. The result underscores the proficiency of TextSMOG in exploiting textual data to guide the conditional de novo generation of molecules. Moreover, it highlights the superior congruence of the text-guided molecule generation via the diffusion model with the desired property, thus showing significant potential. Furthermore, as indicated in Figure 3, our proposed TextSMOG exhibits commendable performance in terms of novelty and stability. The text guidance we introduced has transformed the exploration of the model in the molecule generation space, generally enhancing the novelty of the generated molecules while maintaining their stability.
Figure 2.

Comparison of MAE for the generated molecules targeted to desired property
Statistics of baselines are from their original papers. The performance of EEGSDE varies depending on the scaling factor, and we report its best results. The numerical values are provided in the supplemental information.
Figure 3.

Comparison of novelty (Novel, %), atom stability (A. Stable,%), and molecule stability (M. Stable,%) on generated molecules targeted to the desired property
Statistics of baselines are from EEGSDE. The performance of EEGSDE varies depending on the scaling factor, and we report its best results.
Experiment on multiple quantum properties conditioning
The capacity to generate molecules, guided by multiple conditions, is a crucial aspect of the molecule generation model. When guided by textual descriptions, characterizing the condition with multiple desired properties is highly intuitive and flexible. Following the same setup and metrics in the previous section, we evaluate our TextSMOG on the task of generating molecules with multiple desired quantum properties in QM9. Then we compare TextSMOG with two baselines to showcase the effectiveness of our model in generating molecules conditioned on multiple quantum properties.
As shown in Table 1, our TextSMOG has a remarkably lower MAE than the other two baselines, thereby demonstrating the superiority of our model in generating molecules with multiple desired properties. This also further substantiates that, without necessitating additional targeted interventions, textual conditions can be utilized in our model to guide molecule generation that conforms to multiple desired properties.
Table 1.
Comparison of MAE on the generated molecules targeted to the multiple desired properties
| Method | MAE1 | MAE2 |
|---|---|---|
| Condition | and | |
| EDM | 1.079 | 1.156 |
| EEGSDE | 0.981 | 0.912 |
| TextSMOG | 0.645 | 0.836 |
| Condition | and | |
| EDM | 2.76 | 1.158 |
| EEGSDE | 2.61 | 0.855 |
| TextSMOG | 2.27 | 0.809 |
| Condition | and | |
| EDM | 683 | 1.130 |
| EEGSDE | 563 | 0.866 |
| TextSMOG | 489 | 0.843 |
Statistics of baselines are from EEGSDE. Boldface indicates the best performance.
Additionally, as highlighted in Table 2, our proposed TextSMOG maintains superior performance in terms of novelty and stability, when generating molecules targeted at multiple desired properties. The results indicate that the flexible integration of multiple conditions through textual description does not compromise the stability of the generated molecules. Furthermore, this approach enhances novelty when compared to the baseline.
Table 2.
Comparison of novelty (Novel, %), atom stability (A. Stable, %), and molecule stability (M. Stable,%) on the generated molecules targeted to the multiple desired properties
| Method | Novel | A. Stable | M. Stable |
|---|---|---|---|
| Condition | and | ||
| EDM | 85.31 | 98.00 | 77.42 |
| EEGSDE | 85.62 | 97.67 | 74.56 |
| TextSMOG | 85.79 | 97.89 | 77.33 |
| Condition | and | ||
| EDM | 85.06 | 97.96 | 75.95 |
| EEGSDE | 85.56 | 97.61 | 72.72 |
| TextSMOG | 85.64 | 98.01 | 75.97 |
| Condition | and | ||
| EDM | 85.18 | 98.00 | 77.96 |
| EEGSDE | 85.36 | 97.99 | 77.77 |
| TextSMOG | 85.44 | 98.06 | 78.03 |
Statistics of baselines are from EEGSDE. Boldface indicates the best performance.
Generation on general textual descriptions
To further assess our model, we undertake additional training on a vast dataset of over 330K text-molecule pairs we gleaned from PubChem.31 Then, we generate molecules based on general textual descriptions to observe the capacity of our model to generate from generalized textual conditions.
Visual observations, as depicted in Figure 4, illuminate the impressive aptitude of our TextSMOG in aligning molecule structures with the desired property within the textual descriptions. For instance, when the textual description includes affirmatively mentioned terms such as “simple chain structure”, “at least one carboxyl group”, and “soluble in water”, the generated molecules consistently exhibit chain structures with at least one carboxyl group, and characteristics indicative of water solubility.
Figure 4.

Generated molecules targeted to text description excerpts
Moreover, when the textual description includes “polycyclic heteroarene” and specifies the solubility and heat capacity of the molecule, TextSMOG generates a variety of polycyclic aromatic hydrocarbon molecules. The ubiquitously present amino and nitro groups attest to a certain degree of solubility of the molecules. Referring to structurally similar molecules, their expected specific heat capacity is also relatively low.
Lastly, when the text description explicitly demands multiple nitrogen atoms and a low energy gap, the molecules generated by TextSMOG not only possess the required polycyclic structure and multiple nitrogen atoms, but also the rings on the same plane denote the low-energy structures of these molecules that are difficult to excite.
The remarkable alignment between the conditions and the generated molecule stands as a testament to the exceptional generative capabilities of TextSMOG. The result demonstrates that TextSMOG is equipped to deeply explore the chemical molecular space in a text-guided manner, thereby generating prospective molecules for subsequent applications. This capability could potentially expedite drug design and the discovery of materials.
The results highlight TextSMOG’s versatility in generating a wide variety of molecular structures, from simple chain structures to complex polycyclic compounds, under the guidance of general text descriptions. This underscores the model’s potential to perform well even when the conditions deviate significantly from the distribution of the training set.
Discussion
The translational impacts of TextSMOG are particularly significant for the field of drug discovery and materials science. By enabling the generation of molecular structures directly from textual descriptions, TextSMOG can streamline the early stages of drug design where rapid prototyping and iterative testing are crucial. This approach can facilitate the discovery of subject drug candidates by allowing researchers to quickly generate and evaluate provided molecules based on specific desired properties mentioned in literature or derived from expert knowledge.
Furthermore, TextSMOG can aid in the development of materials with tailored properties by generating molecules that meet specific criteria. This capability is valuable in industries such as polymers, nanomaterials, and catalysts, where precise molecular structures can significantly influence material performance.
In drug discovery, TextSMOG’s ability to generate molecules that align with complex textual prompts can accelerate the identification of compounds with potential therapeutic effects. This is especially relevant for targeting diseases with well-characterized biochemical pathways, where detailed descriptions of molecular interactions and desired properties are available. By generating candidate molecules that meet these criteria, TextSMOG can help narrow down the pool of potential drugs, reducing the time and cost associated with experimental validation.
Additionally, TextSMOG’s flexibility in handling diverse textual inputs can facilitate interdisciplinary research, where insights from different fields can be integrated into the molecule generation process. For instance, combining insights from biology, chemistry, and pharmacology can lead to more informed and effective drug design strategies.
Despite the complexity of translating textual prompts into accurate molecular structures, we have successfully integrated advanced language models with high-fidelity diffusion models in TextSMOG, a text-guided diffusion approach for 3D molecule generation. Our experiments on the QM9 and PubChem datasets demonstrate the superior performance of TextSMOG over leading baselines, affirming its efficacy in capturing desired properties from textual descriptions and generating corresponding valid molecules.
Limitations of the study
The integration of textual information with the denoising process of a pre-trained EDM allows TextSMOG to generate valid and stable molecular conformations that closely align with diverse textual directives. This initial success paves the way for significant advancements in the exploration of chemical space and the development of compounds. Nevertheless, our findings are not without limitations.
Our work was constrained by the scarcity of high-quality data linking real-world 3D molecules to their corresponding textual descriptions. This limitation impacted our ability to fully train the model on a diverse set of text-3D molecule pairs, potentially affecting the accuracy of the generated molecules in generating molecules that accurately align with complex textual descriptions. Moreover, the relative slowness of the sampling process due to the iterative nature of the total diffusion steps can pose a challenge in scenarios requiring rapid molecule generation, such as high-throughput drug discovery or material design.
In addition to these limitations, the current design of TextSMOG necessitates that the properties to condition on must be known upfront during the training phase. This might not always be feasible in practical settings, where specific properties linked to a particular drug discovery target may only become available later on, and often with very limited sample data. The generalization of TextSMOG to more complex and real-world scenarios also needs further exploration.
Looking ahead, we are optimistic about the potential of text-guided 3D molecule generation to revolutionize drug discovery and related fields. Future work will focus on overcoming these challenges by expanding and enhancing the quality of datasets linking textual descriptions to molecular structures, improving the efficiency of the sampling process, and making TextSMOG more adaptable to real-world applications. Addressing these limitations will not only enhance the performance of TextSMOG but also contribute significantly to the advancement of text-guided molecule generation technology.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wenjie Du (duwenjie@mail.ustc.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The datasets generated during this study are available at HuggingFace and are publicly available as of the date of publication. The url is listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. The url is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
This research is supported by the National Natural Science Foundation of China (92270114). This research was also supported by the advanced computing resources provided by the Supercomputing Center of the USTC.
Author contributions
Conceptualization, Y.L. and J.F.; methodology, Y.L. and S.L.; investigation, Z.L. and S.L.; writing—original draft, Y.L. and J.F.; writing—review and editing, J.W., A.Z., and W.D.; funding acquisition, X.W.; resources, J.W., A.Z., and W.D.; supervision, X.W.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Processed PubChem Dataset | This paper, PubChem | https://pubchem.ncbi.nlm.nih.gov/ |
| QM9 Dataset | Quantum Machine | http://quantum-machine.org/datasets/ |
| Software and algorithms | ||
| TextSMOG Model | This paper | https://github.com/lyc0930/TEDMol |
Method details
In this section, we elaborate on the proposed text-guided small molecule generation approach via diffusion model (TextSMOG), as illustrated in Figure 1. It integrates the textual information (i.e., text guidance) into the conditional signal of diffusion models by employing the reference geometry that is described in the first subsection following. Subsequently, we introduce an efficient learning approach that incorporates both the encoded conditional signal and pre-trained unconditional signal in the reverse process, to generate molecules that are not only structurally stable and chemically valid but also align well with the specified conditions, as presented in the second subsection.
Notation and background
We begin with a background of diffusion-based 3D molecule generation, introducing the fundamental concepts of the diffusion model and delving into equivariant diffusion models. See the comprehensive literature review on these topics in the Section Related works in supplemental information. In accordance with prior studies,12,16,17 we use the variable to represent the 3D molecular geometry. Here signifies the atom coordinates, while denotes the atom features. These features encompass atom types and atom charges, characterizing the atomic properties within the molecular structure.
Diffusion model
The diffusion model14,15 emerges as a leading generative model, having achieved great success in various domains.59,60,61,62,63,64 Typically, it is formulated as two Markov chains: a forward process (aka. noising process) that gradually injects noise into the data, and a reverse process (aka. denoising process) that learns to recover the original data. Such a reverse process endows the diffusion model with enhanced capabilities for effective data generation and recovery.
Forward Process
Given the real 3D molecular geometry , the forward process yields a sequence of intermediate variables using the transition kernel in alignment with a variance schedule . Formally, it is expressed as:
| (Equation 1) |
where is a Gaussian distribution and is the identity matrix. This defines the joint distribution of conditioned on using the chain rule of the Markov process:
| (Equation 2) |
Let and . The sampling of at time step is in a closed form:
| (Equation 3) |
Accordingly, the forward process posteriors, when conditioned on , are tractable as:
| (Equation 4) |
where
| (Equation 5) |
Reverse Process
To recover the original molecular geometry , the diffusion model starts by generating a standard Gaussian noise , then progressively eliminates noise through a reverse Markov chain. This is characterized by a learnable transition kernel at each reverse step , defined as:
| (Equation 6) |
where the variance and the mean is parameterized by deep neural networks with parameters :
| (Equation 7) |
where is a noise prediction function to approximate the noise from .
With the reverse Markov chain, we can iteratively sample from the learnable transition kernel until to estimate the molecular geometry .
Equivariant diffusion models
The molecular geometry is inherently symmetric in 3D space — that is, translating or rotating a molecule does not change its underlying structure or features. Previous studies65,66,67 underscore the significance of leveraging these invariances in molecular representation learning for enhanced generalization. However, the transformation of these higher-order representations usually requires computationally expensive approximations or coefficients.16,35 In contrast, equivariant diffusion models16,68,69 provide a more efficient approach to ensure both rotational and translational invariance. The approach rests on the assumption that, with the model distribution remaining invariant to the Euclidean group E(3), identical molecules, despite being in different orientations, will correspond to the same distribution. Based on this assumption, translational invariance is achieved by predicting only the deviations in coordinate with a zero center of mass, i.e., . On the other hand, rotational invariance is accomplished by making the noise prediction network equivariant to orthogonal transformations.16,35 Specifically, given an orthogonal matrix representing a coordinate rotation or reflection, the conformation output from the network is equivariant to , if the following condition holds for all orthogonal matrices :
| (Equation 8) |
A model exhibiting rotational and translational equivariance means a neural network can avoid learning orientations and translations of molecules from scratch.16,35 In this paper, we parameterize the noise prediction network using an E(n) equivariant graph neural network as introduced by,35 which is a type of Graph Neural Network70 that satisfies the above equivariance constraint to E(3).
Equivariant diffusion model for molecule generation
Diffusion models, formulated as two Markov chains—a forward process that gradually injects noise into the data and a reverse process that learns to recover the original data—have been successfully applied to various domains, including molecule generation. This process is particularly effective in the context of molecule generation, where the forward process adds noise to the molecular geometry at each step until it is fully noise-corrupted. The reverse process then gradually denoises the initial geometry to generate the final molecular geometry .
However, molecular geometries are inherently symmetric in 3D space—translations or rotations do not change their underlying structure or features. To take advantage of these invariances for improved generalization, we employ an equivariant diffusion model (EDM). The EDM ensures both rotational and translational invariance by predicting only the deviations in coordinate with a zero center of mass and making the noise prediction network equivariant to orthogonal transformations. This allows the model distribution to remain invariant to the Euclidean group E(3), meaning identical molecules in different orientations correspond to the same distribution.
In this work, the integration of textual information into the conditional signal of the equivariant diffusion model is achieved by employing a reference geometry that is updated at each step based on the textual prompt .
Integrating textual prompts into 3D molecular reference geometry
To ensure high-fidelity 3D molecule generation, the reverse process of the diffusion model is typically guided by tailored conditional information representing desired properties like unique polarizability. We represent this conditional information as , which allows us to formulate the conditional reverse process as:
| (Equation 9) |
Unlike previous approaches relying on limited value guidance (i.e., property values), in this work, we aim to steer the reverse process with text guidance (i.e., informative textual descriptions), which can convey a broader range of conditional requirements. Intuitively, utilizing textual descriptions to specify conditional generation criteria not only provides greater expressivity but also better aligns the resulting 3D molecules with diverse and complex expectations.
Practically, we first introduce a textual prompt P describing desired 3D molecule properties. A multi-modal conversion module , pre-trained on 300K text-molecule pairs from PubChem, is then employed. This module is comprised of a GIN molecular graph encoder71,72 and a language encoder-decoder extended from BERT.21,73 It converts P into a reference geometry , extracting specific information from the target conditions and refining the textual condition signal:
| (Equation 10) |
Nevertheless, we should emphasize that valid and stable 3D molecules can hardly be obtained directly from . The chemical fidelity in 3D molecular space may not be guaranteed. In what follows, we describe how to utilize for conditioning a pre-trained diffusion model to generate molecules that align with the desired properties, meanwhile alleviating the exhaustive training from scratch.
Conditioning with the reference of text guidance
To leverage for text-guided conditional generation while preserving the validity and stability of the synthesized molecule, TextSMOG employs the iterative latent variable refinement (ILVR)74 to condition a pre-trained unconditional diffusion model meanwhile maintaining inherent domain knowledge in the unconditional model.
With the pre-trained unconditional diffusion model EDM,16 we could perform a step-by-step reverse process. Formally, at step , we can sample an unconditional proposal molecular geometry:
| (Equation 11) |
where is the fixed parameters of the pre-trained unconditional diffusion model.16 Then, to incorporate the condition signal in the reverse process, we introduce a linear operation . Therefore the conditional denoising for one step at step can be formulated as:
| (Equation 12) |
where is the identity operation and is the residual operation w.r.t. 75. Accordingly, the condition signal is projected into the reverse denoising process by , thus is obtained as the generated 3D molecular geometry conditioned on . Conceptually, the proposal geometry from unconditional generation tries to push the atoms into a chemically valid position, while the reference geometry pulls the atoms toward the structure targeted to the condition.
By matching latent variables following Equation 12, we enable text-guided conditional generation with the unconditional diffusion model. Accordingly, the one-step denoising distribution conditioned on textual guidance at each step can be reformulated as:
| (Equation 13) |
Training objective
To guarantee the quality of the generated molecules, the key lies in optimizing the variational lower bound (ELBO) of negative log likelihood, which equals minimizing the Kullback-Leibler divergence between the joint distribution of the reverse Markov chain and the forward process :
| (Equation 14) |
where is a constant independent of .
Note that we set as a discrete decoder following.15 Further adopting the reparameterization from,15 can be simplified to:
| (Equation 15) |
Evaluation metrics
Mean absolute error (MAE).76 is a measure of errors between paired observations. Given the property classifier network , and the set of generated molecules , the MAE is defined as:
| (Equation 16) |
where is the generated molecule, and of which is the desired property.
Novelty.36 is the proportion of generated molecules that do not appear in the training set. Specifically, let be the set of generated molecules, the novelty in our experiment is calculated as:
| (Equation 17) |
Atom stability.16 is the proportion of the atoms in the generated molecules that have the right valency. Specifically, the atom stability in our experiment is calculated as:
| (Equation 18) |
where is the set of atoms in the generated molecule , and is the set of atoms in that have the right valency.
Molecule stability.16 is the proportion of the generated molecules where all atoms are stable. Specifically, the molecule stability in our experiment is calculated as:
| (Equation 19) |
where is the set of generated molecules where all atoms have the right valency.
Quantification and statistical analysis
The quantum properties in QM9 dataset
We consider 6 main quantum properties in QM9:
-
•
: Heat capacity at 298.15K.
-
•
: Dipole moment.
-
•
: Polarizability, which represents the tendency of a molecule to acquire an electric dipole moment when subjected to an external electric field.
-
•
: Highest occupied molecular orbital energy.
-
•
: Lowest unoccupied molecular orbital energy.
-
•
: The energy gap between HOMO and LUMO.
Published: September 19, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110992.
Contributor Information
Wenjie Du, Email: duwenjie@mail.ustc.edu.cn.
Xiang Wang, Email: xiangwang1223@gmail.com.
Supporting citations
The following references appear in the Supplemental Information:37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58.
Supplemental information
References
- 1.Hajduk P.J., Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 2.Mandal S., Moudgil M., Mandal S.K. Rational drug design. Eur. J. Pharmacol. 2009;625:90–100. doi: 10.1016/j.ejphar.2009.06.065. [DOI] [PubMed] [Google Scholar]
- 3.Pyzer-Knapp E.O., Suh C., Gómez-Bombarelli R., Aguilera-Iparraguirre J., Aspuru-Guzik A. What Is High-Throughput Virtual Screening? A Perspective from Organic Materials Discovery. Annu. Rev. Mater. Res. 2015;45:195–216. doi: 10.1146/annurev-matsci-070214-020823. [DOI] [Google Scholar]
- 4.Barakat K.H., Houghton M., Tyrrel D.L., Tuszynski J.A. Rational Drug Design: One Target, Many Paths to It. Int. J. Comput. Model Algorithm. Med. 2014;4:59–85. doi: 10.4018/ijcmam.2014010104. [DOI] [Google Scholar]
- 5.Gaudelet T., Day B., Jamasb A.R., Soman J., Regep C., Liu G., Hayter J.B.R., Vickers R., Roberts C., Tang J., et al. Utilizing graph machine learning within drug discovery and development. Briefings Bioinf. 2021;22 doi: 10.1093/bib/bbab159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alcalde M., Ferrer M., Plou F.J., Ballesteros A. Environmental biocatalysis: from remediation with enzymes to novel green processes. Trends Biotechnol. 2006;24:281–287. doi: 10.1016/j.tibtech.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Anand N., Eguchi R., Mathews I.I., Perez C.P., Derry A., Altman R.B., Huang P.-S. Protein sequence design with a learned potential. Nat. Commun. 2022;13:746. doi: 10.1038/s41467-022-28313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mansimov E., Mahmood O., Kang S., Cho K. Molecular geometry prediction using a deep generative graph neural network. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-56773-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zang C., Wang F. KDD. ACM; 2020. MoFlow: An Invertible Flow Model for Generating Molecular Graphs; pp. 617–626. [DOI] [Google Scholar]
- 10.Satorras V.G., Hoogeboom E., Fuchs F., Posner I., Welling M. NeurIPS. 2021. E(n) Equivariant Normalizing Flows; pp. 4181–4192. [Google Scholar]
- 11.Gebauer N.W.A., Gastegger M., Schütt K. NeurIPS. 2019. Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules; pp. 7564–7576. [DOI] [Google Scholar]
- 12.Huang L., Zhang H., Xu T., Wong K. AAAI. AAAI Press; 2023. MDM: Molecular Diffusion Model for 3D Molecule Generation; pp. 5105–5112. [DOI] [Google Scholar]
- 13.Luo S., Shi C., Xu M., Tang J. NeurIPS. 2021. Predicting Molecular Conformation via Dynamic Graph Score Matching; pp. 19784–19795. [DOI] [Google Scholar]
- 14.Sohl-Dickstein J., Weiss E.A., Maheswaranathan N., Ganguli S. Vol. 37. JMLR; 2015. Deep Unsupervised Learning using Nonequilibrium Thermodynamics; pp. 2256–2265. (JMLR Workshop and Conference Proceedings). ICML. [DOI] [Google Scholar]
- 15.Ho J., Jain A., Abbeel P. NeurIPS. 2020. Denoising Diffusion Probabilistic Models. [DOI] [Google Scholar]
- 16.Hoogeboom E., Satorras V.G., Vignac C., Welling M. Vol. 162. PMLR; 2022. Equivariant Diffusion for Molecule Generation in 3D; pp. 8867–8887. (Proceedings of Machine Learning Research). ICML. [DOI] [Google Scholar]
- 17.Bao F., Zhao M., Hao Z., Li P., Li C., Zhu J. ICLR. OpenReview; 2023. Equivariant Energy-Guided SDE for Inverse Molecular Design. [DOI] [Google Scholar]
- 18.Honório K., Moda T., Andricopulo A. Pharmacokinetic properties and in silico ADME modeling in drug discovery. Med. Chem. 2013;9:163–176. doi: 10.2174/1573406411309020002. [DOI] [PubMed] [Google Scholar]
- 19.Gebauer N.W.A., Gastegger M., Hessmann S.S.P., Müller K.R., Schütt K.T. Inverse design of 3d molecular structures with conditional generative neural networks. Nat. Commun. 2022;13:973. doi: 10.1038/s41467-022-28526-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee M., Min K. MGCVAE: Multi-Objective Inverse Design via Molecular Graph Conditional Variational Autoencoder. J. Chem. Inf. Model. 2022;62:2943–2950. doi: 10.1021/acs.jcim.2c00487. [DOI] [PubMed] [Google Scholar]
- 21.Devlin J., Chang M., Lee K., Toutanova K. NAACL-HLT (1) Association for Computational Linguistics; 2019. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding; pp. 4171–4186. [DOI] [Google Scholar]
- 22.Liu Y., Ott M., Goyal N., Du J., Joshi M., Chen D., Levy O., Lewis M., Zettlemoyer L., Stoyanov V. RoBERTa: A Robustly Optimized BERT Pretraining Approach. CoRR. 2019 doi: 10.48550/arXiv.1907.11692. [DOI] [Google Scholar]
- 23.Beltagy I., Lo K., Cohan A. EMNLP/IJCNLP (1) Association for Computational Linguistics; 2019. SciBERT: A Pretrained Language Model for Scientific Text; pp. 3613–3618. [DOI] [Google Scholar]
- 24.Raffel C., Shazeer N., Roberts A., Lee K., Narang S., Matena M., Zhou Y., Li W., Liu P.J. Exploring the Limits of Transfer Learning with a Unified Text-to-Text Transformer. J. Mach. Learn. Res. 2020;21:1–67. doi: 10.5555/3455716.3455856. [DOI] [Google Scholar]
- 25.Brown T.B., Mann B., Ryder N., Subbiah M., Kaplan J., Dhariwal P., Neelakantan A., Shyam P., Sastry G., Askell A., et al. NeurIPS. 2020. Language Models are Few-Shot Learners. [DOI] [Google Scholar]
- 26.OpenAI GPT-4 Technical Report. CoRR. 2023 doi: 10.48550/arXiv.2303.08774. [DOI] [Google Scholar]
- 27.Su B., Du D., Yang Z., Zhou Y., Li J., Rao A., Sun H., Lu Z., Wen J. A Molecular Multimodal Foundation Model Associating Molecule Graphs with Natural Language. CoRR. 2022 doi: 10.48550/ARXIV.2209.05481. [DOI] [Google Scholar]
- 28.Edwards C., Zhai C., Ji H. EMNLP (1) Association for Computational Linguistics; 2021. Text2Mol: Cross-Modal Molecule Retrieval with Natural Language Queries; pp. 595–607. [DOI] [Google Scholar]
- 29.Edwards C., Lai T.M., Ros K., Honke G., Cho K., Ji H. EMNLP. Association for Computational Linguistics; 2022. Translation between Molecules and Natural Language; pp. 375–413. [DOI] [Google Scholar]
- 30.Ramakrishnan R., Dral P.O., Rupp M., von Lilienfeld O.A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data. 2014;1 doi: 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B., et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–D1395. doi: 10.1093/NAR/GKAA971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degtyarenko K., de Matos P., Ennis M., Hastings J., Zbinden M., McNaught A., Alcántara R., Darsow M., Guedj M., Ashburner M. ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008;36:344–350. doi: 10.1093/NAR/GKM791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rutz A., Sorokina M., Galgonek J., Mietchen D., Willighagen E., Gaudry A., Graham J.G., Stephan R., Page R., Vondrášek J., et al. The LOTUS initiative for open knowledge management in natural products research. Elife. 2022;11 doi: 10.7554/eLife.70780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wishart D., Arndt D., Pon A., Sajed T., Guo A.C., Djoumbou Y., Knox C., Wilson M., Liang Y., Grant J., et al. T3DB: The toxic exposome database. Nucleic Acids Res. 2015;43:D928–D934. doi: 10.1093/nar/gku1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satorras V.G., Hoogeboom E., Welling M. Vol. 139. PMLR; 2021. E(n) Equivariant Graph Neural Networks. (Proceedings of Machine Learning Research). ICML. [DOI] [Google Scholar]
- 36.Simonovsky M., Komodakis N. Vol. 11139. Springer; 2018. GraphVAE: Towards Generation of Small Graphs Using Variational Autoencoders; pp. 412–422. (ICANN (1)). Lecture Notes in Computer Science. [DOI] [Google Scholar]
- 37.Chen N., Zhang Y., Zen H., Weiss R.J., Norouzi M., Chan W. ICLR. OpenReview; 2021. WaveGrad: Estimating Gradients for Waveform Generation. [DOI] [Google Scholar]
- 38.Kong Z., Ping W., Huang J., Zhao K., Catanzaro B. ICLR. OpenReview; 2021. DiffWave: A Versatile Diffusion Model for Audio Synthesis. [DOI] [Google Scholar]
- 39.Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988;28:31–36. doi: 10.1021/CI00057A005. [DOI] [Google Scholar]
- 40.Kotsias P.C., Arús-Pous J., Chen H., Engkvist O., Tyrchan C., Bjerrum E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020;2:254–265. doi: 10.1038/s42256-020-0174-5. [DOI] [Google Scholar]
- 41.Jin W., Barzilay R., Jaakkola T.S. Vol. 80. PMLR; 2018. Junction Tree Variational Autoencoder for Molecular Graph Generation; pp. 2328–2337. (Proceedings of Machine Learning Research). ICML. [DOI] [Google Scholar]
- 42.Jing B., Corso G., Chang J., Barzilay R., Jaakkola T.S. NeurIPS. 2022. Torsional Diffusion for Molecular Conformer Generation. [DOI] [Google Scholar]
- 43.Nesterov V., Wieser M., Roth V. 3DMolNet: A Generative Network for Molecular Structures. CoRR. 2020 doi: 10.48550/arXiv.2010.06477. [DOI] [Google Scholar]
- 44.Hoffmann M., Noé F. Generating valid Euclidean distance matrices. CoRR. 2019 doi: 10.48550/arXiv.1910.03131.eprint:1910.03131. [DOI] [Google Scholar]
- 45.Kusner M.J., Paige B., Hernández-Lobato J.M. Vol. 70. PMLR; 2017. Grammar Variational Autoencoder; pp. 1945–1954. (Proceedings of Machine Learning Research). ICML. [DOI] [Google Scholar]
- 46.Dai H., Tian Y., Dai B., Skiena S., Song L. ICLR (Poster) OpenReview; 2018. Syntax-Directed Variational Autoencoder for Structured Data. [DOI] [Google Scholar]
- 47.Liu Q., Allamanis M., Brockschmidt M., Gaunt A.L. NeurIPS. 2018. Constrained Graph Variational Autoencoders for Molecule Design; pp. 7806–7815. [DOI] [Google Scholar]
- 48.Madhawa K., Ishiguro K., Nakago K., Abe M. GraphNVP: An Invertible Flow Model for Generating Molecular Graphs. CoRR. 2019 doi: 10.48550/arXiv.1905.11600. [DOI] [Google Scholar]
- 49.Luo Y., Yan K., Ji S. Vol. 139. PMLR; 2021. GraphDF: A Discrete Flow Model for Molecular Graph Generation; pp. 7192–7203. (Proceedings of Machine Learning Research). ICML. [DOI] [Google Scholar]
- 50.Bian Y., Wang J., Jun J.J., Xie X.-Q. Deep Convolutional Generative Adversarial Network (dcGAN) Models for Screening and Design of Small Molecules Targeting Cannabinoid Receptors. Mol. Pharm. 2019;16:4451–4460. doi: 10.1021/acs.molpharmaceut.9b00500. [DOI] [PubMed] [Google Scholar]
- 51.Assouel R., Ahmed M., Segler M.H.S., Saffari A., Bengio Y. DEFactor: Differentiable Edge Factorization-based Probabilistic Graph Generation. CoRR. 2018 doi: 10.48550/arXiv.1811.09766. [DOI] [Google Scholar]
- 52.Shi C., Xu M., Zhu Z., Zhang W., Zhang M., Tang J. ICLR. OpenReview; 2020. GraphAF: a Flow-based Autoregressive Model for Molecular Graph Generation. [DOI] [Google Scholar]
- 53.Popova M., Shvets M., Oliva J., Isayev O. MolecularRNN: Generating realistic molecular graphs with optimized properties. CoRR. 2019 doi: 10.48550/arXiv.1905.13372. [DOI] [Google Scholar]
- 54.Flam-Shepherd D., Zhu K., Aspuru-Guzik A. Language Models can learn Complex Molecular Distributions. Nat. Commun. 2022;13:3293. doi: 10.1038/s41467-022-30839-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu L., Gong C., Liu X., Ye M., Liu Q. NeurIPS. 2022. Diffusion-based Molecule Generation with Informative Prior Bridges. [DOI] [Google Scholar]
- 56.Kang S., Cho K. Conditional Molecular Design with Deep Generative Models. J. Chem. Inf. Model. 2019;59:43–52. doi: 10.1021/acs.jcim.8b00263. [DOI] [PubMed] [Google Scholar]
- 57.Yang M., Sun H., Liu X., Xue X., Deng Y., Wang X. CMGN: a conditional molecular generation net to design target-specific molecules with desired properties. Briefings Bioinf. 2023;24 doi: 10.1093/bib/bbad185. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez-Lengeling B., Aspuru-Guzik A. Inverse molecular design using machine learning: Generative models for matter engineering. Science. 2018;361:360–365. doi: 10.1126/science.aat2663. [DOI] [PubMed] [Google Scholar]
- 59.Dhariwal P., Nichol A.Q. NeurIPS. 2021. Diffusion Models Beat GANs on Image Synthesis; pp. 8780–8794. [DOI] [Google Scholar]
- 60.Rombach R., Blattmann A., Lorenz D., Esser P., Ommer B. CVPR. IEEE; 2022. High-Resolution Image Synthesis with Latent Diffusion Models; pp. 10674–10685. [DOI] [Google Scholar]
- 61.Ruiz N., Li Y., Jampani V., Pritch Y., Rubinstein M., Aberman K. CVPR. IEEE; 2023. DreamBooth: Fine Tuning Text-to-Image Diffusion Models for Subject-Driven Generation; pp. 22500–22510. [DOI] [Google Scholar]
- 62.Song Y., Sohl-Dickstein J., Kingma D.P., Kumar A., Ermon S., Poole B. ICLR. OpenReview; 2021. Score-Based Generative Modeling through Stochastic Differential Equations. [DOI] [Google Scholar]
- 63.Saharia C., Ho J., Chan W., Salimans T., Fleet D.J., Norouzi M. Image Super-Resolution via Iterative Refinement. IEEE Trans. Pattern Anal. Mach. Intell. 2023;45:4713–4726. doi: 10.1109/TPAMI.2022.3204461. [DOI] [PubMed] [Google Scholar]
- 64.Schneider F. ArchiSound: Audio Generation with Diffusion. CoRR. 2023 doi: 10.48550/ARXIV.2301.13267. [DOI] [Google Scholar]
- 65.Thomas N., Smidt T.E., Kearnes S., Yang L., Li L., Kohlhoff K., Riley P. Tensor Field Networks: Rotation- and Translation-Equivariant Neural Networks for 3D Point Clouds. CoRR. 2018 doi: 10.48550/arXiv.1802.08219. [DOI] [Google Scholar]
- 66.Fuchs F., Worrall D.E., Fischer V., Welling M. NeurIPS. 2020. SE3-Transformers: 3D Roto-Translation Equivariant Attention Networks. [DOI] [Google Scholar]
- 67.Finzi M., Stanton S., Izmailov P., Wilson A.G. Vol. 119. PMLR; 2020. Generalizing Convolutional Neural Networks for Equivariance to Lie Groups on Arbitrary Continuous Data; pp. 3165–3176. (Proceedings of Machine Learning Research). ICML. [DOI] [Google Scholar]
- 68.Köhler J., Klein L., Noé F. Vol. 119. PMLR; 2020. Equivariant Flows: Exact Likelihood Generative Learning for Symmetric Densities; pp. 5361–5370. (Proceedings of Machine Learning Research). ICML. [DOI] [Google Scholar]
- 69.Xu M., Yu L., Song Y., Shi C., Ermon S., Tang J. ICLR. OpenReview; 2022. GeoDiff: A Geometric Diffusion Model for Molecular Conformation Generation. [DOI] [Google Scholar]
- 70.Hamilton W.L., Ying Z., Leskovec J. NIPS. 2017. Inductive Representation Learning on Large Graphs; pp. 1024–1034. [DOI] [Google Scholar]
- 71.Xu K., Hu W., Leskovec J., Jegelka S. ICLR. 2019. How Powerful are Graph Neural Networks? [DOI] [Google Scholar]
- 72.Liu S., Wang H., Liu W., Lasenby J., Guo H., Tang J. ICLR. OpenReview; 2022. Pre-training Molecular Graph Representation with 3D Geometry. [DOI] [Google Scholar]
- 73.Zeng Z., Yao Y., Liu Z., Sun M. A Deep-learning System Bridging Molecule Structure and Biomedical Text with Comprehension Comparable to Human Professionals. Nat. Commun. 2022;13 doi: 10.1038/s41467-022-28494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi J., Kim S., Jeong Y., Gwon Y., Yoon S. ICCV. IEEE; 2021. ILVR: Conditioning Method for Denoising Diffusion Probabilistic Models; pp. 14347–14356. [DOI] [Google Scholar]
- 75.James A.T., Wilkinson G.N. Factorization of the residual operator and canonical decomposition of nonorthogonal factors in the analysis of variance. Biometrika. 1971;58:279–294. doi: 10.2307/2334516. [DOI] [Google Scholar]
- 76.Willmott C.J., Matsuura K. Advantages of the mean absolute error (MAE) over the root mean square error (RMSE) in assessing average model performance. Clim. Res. 2005;30:79–82. doi: 10.3354/cr030079. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The datasets generated during this study are available at HuggingFace and are publicly available as of the date of publication. The url is listed in the key resources table.
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication. The url is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.