

Abstract
Wieacker–Wolff syndrome (WRWF) is an X-linked genetic disorder characterized by neuromusculoskeletal abnormalities caused by loss-of-function variants of the ZC4H2 gene. Here, we report the case of a male infant with WRWF manifesting as multiple joint contractures and congenital anomalies at birth. He underwent gastrostomy to treat the gastroesophageal reflux disease, which caused mixed apnea and transient bradycardia. The patient subsequently developed hyperinsulinemic hypoglycemia (HH) and was diagnosed with dumping syndrome. Although he underwent multiple treatments, including alpha-glucosidase inhibitors (α-GI) administration, he continued to exhibit HH with seizures and loss of consciousness. Whole-exome sequencing revealed a novel missense variant of ZC4H2 [NM_018684.4: c.557T>G, p.(Met186Arg)] at Xq11.2 in both the patient and his mother. Based on these results and clinical symptoms, the patient was diagnosed with WRWF. Although WRWF is not considered a major cause of HH, we regarded it as a related complication based on previous reports. Diazoxide treatment was initiated, and the hypoglycemic attacks resolved almost entirely without any notable side effects after 18 mo. To the best of our knowledge, this is the first report of WRWF-associated HH treated with low-dose diazoxide and α-GI. Therefore, diazoxide is recommended for the treatment of WRWF-associated HH.
Keywords: Wieacker–Wolff syndrome, ZC4H2, hyperinsulinemic hypoglycemia, diazoxide, alpha-glucosidase inhibitors
Highlights
● Wieacker–Wolff syndrome may complicate hyperinsulinemic hypoglycemia.
● Diazoxide may successfully treat this complication.
● A novel missense variant, c.557T>G, p.(Met186Arg), was identified in ZC4H2.
Introduction
Wieacker–Wolff syndrome (WRWF) is an X-linked inherited arthrogryposis multiplex congenital disorder characterized by intellectual disability, motor developmental delay, poor growth, muscle weakness, and skeletal abnormalities. Patients with WRWF typically have multiple congenital joint contractures at birth due to muscle weakness beginning in utero (1,2,3). In 1985, Wieacker and Wolf reported six male patients with similar manifestations in a family spanning three generations (3). Using next-generation sequencing, Hirata et al. identified that ZC4H2, a zinc-finger gene, affects patients with WRWF (4).
A previous study showed that loss-of-function variants of ZC4H2 can downregulate aberrant oxidative phosphorylation pathways and reduce neural stem cell proliferation (5). Functional analysis in zebrafish has revealed that the ZC4H2 protein is widely expressed in the nervous system, including the forebrain, midbrain, hindbrain, and spinal cord, and that anomalies in the muscle projections of motor neurons are present in individuals lacking functional zc4h2 genes (4). To date, WRWF has been reported in seven families and one male sibling (1, 4, 6,7,8,9); however, its prevalence remains unknown.
In addition to neurological symptoms, patients with WRWF develop complex hypoglycemia caused by hyperinsulinemia or central adrenal insufficiency (8). However, literature discussing the treatment progress for this type of hypoglycemia has not been reported. Here, we report the first case of diazoxide use in a patient with WRWF complicated by hyperinsulinemic hypoglycemia (HH).
Case Presentation
The patient was a male infant born at 40 wk of gestation via emergency cesarean section because of arrested labor, with birth weight and height of 3620 (+1.3 standard deviation [SD]) and 49.5 cm (–0.2 SD), respectively. No abnormalities were observed during pregnancy. At birth, the infant was referred to our hospital because of respiratory disturbances and multiple joint contractures. Laryngomalacia, esophageal hiatal hernia, bilateral ptosis, external rectus muscle defect in the right eye, impaired ocular motility, inguinal hernia, cryptorchidism, multiple joint contractures, knee dislocation, and bilateral hip dislocation were identified as congenital anomalies (Fig. 1). Congenital anomaly syndrome was suspected; however, his karyotype was 46, XY, and no pathogenic variants were detected on the chromosomal microarray.
Fig. 1.

Clinical features of the patient at 10 d of age. (A) Full-length photograph. (B) Bending and overlapping of the fingers. (C) Knee hyperextension and clubfoot.
At 7 mo of age, the patient frequently experienced mixed apnea and transient bradycardia. At 8 mo of age, he underwent gastrostomy and fundoplication to treat gastroesophageal reflux disease, which was presumed to have triggered these attacks. The patient subsequently developed HH following tube feeding and was diagnosed with dumping syndrome. Multiple tube-feeding methods were employed.
At the age of 2 yr, he underwent a TRH loading test and a GH-releasing peptide-2 test due to hypoglycemia and short stature. Based on the results (Table 1), GH deficiency (GHD) was suspected, and adrenal insufficiency was ruled out. The diagnostic criteria for GHD were not clearly met; however, owing to evidence of hypoglycemia, central hypothyroidism, and short stature, treatment was administered based on the guidelines for GHD. Although thyroid-stimulating hormone (TSH) responsiveness to TRH was observed, levothyroxine supplementation was initiated based on persistently low TSH (0.24–0.33 μIU/mL) and free T4 (0.43–0.72 ng/dL) levels. The patient was diagnosed with central hypothyroidism. The patient’s HH associated with dumping syndrome worsened; thus, alpha-glucosidase inhibitors (α-GI), voglibose 0.2 mg/d (three times daily, before each meal), and cornstarch 0.5 g/kg/dose mixed with a small amount of water (twice daily, before breakfast and sleep) were administered. He also underwent nutritional adjustment, but continued to have HH with seizures and loss of consciousness. No convulsions or epileptic seizures were observed in the absence of hypoglycemia. The patient’s growth curve and treatment course are shown in Fig. 2.
Table 1. Results of loading tests.
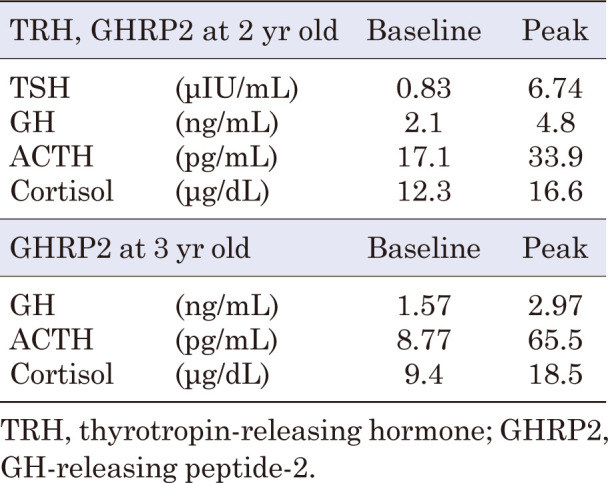
Fig. 2.

Growth chart and medical treatment course of the patient. The black bars indicate the duration of each treatment. Height and weight improved after diazoxide was initiated.
At 4 yr of age, the patient and his mother underwent whole-exome sequencing. When the patient reached 6 yr of age, he and his mother were identified as harboring novel missense variants in the ZC4H2 gene [NM_018684.4: c.557T>G, p.(Met186Arg)] at Xq11.2 (Fig. 3). The patient was hemizygous for this variant, whereas his mother was heterozygous. Based on the American College of Medical Genetics guidelines, the variant was classified as “likely pathogenic” (10). Based on the genetic results and patient’s phenotype, WRWF was diagnosed. His mother had mild symptoms, such as congenital clubfoot, right eyelid ptosis, and right facial nerve palsy. Therefore, she was diagnosed with a milder form of WRWF. However, no abnormalities were detected on blood tests previously.
Fig. 3.

Sanger sequencing of ZC4H2 at Xq11.2 identified a novel missense variant c.557T>G, p.(Met186Arg) in both the patient and his mother. The patient was hemizygous for this variant, whereas his mother was heterozygous.
We considered HH to be associated with WRWF because it continued to be detected in the patient’s blood evaluation (Table 2). Therefore, 50 mg of diazoxide (3 mg/kg/d) was initiated to treat the patient’s HH once he reached 7 yr of age. After diazoxide initiation, continuous glucose monitoring (CGM) showed no hypoglycemia; thus, α-GI and cornstarch were discontinued. However, CGM showed hyperglycemia immediately after tube feeding, followed by subsequent hypoglycemia, which could be considered a recurrence of the dumping syndrome. Therefore, α-GI administration was resumed. The patient’s blood glucose levels stabilized thereafter (Fig. 4). After 18 mo of diazoxide treatment, no side effects, including hypertrichosis, fluid retention, or other cardiovascular signs or symptoms, were observed (11, 12).
Table 2. Laboratory data on admission at 7 yr old (2 hours after meal).

Fig. 4.

Results of continuous glucose monitoring using the FreeStyle Libre Pro. Each rectangle represents one day. The gray areas indicate glucose levels in the range of 70–140 mg/dL. Arrowheads indicate infusion feeding, solid arrows indicate oral feeding, and blue squares indicate continuous infusion feeding. Alpha-glucosidase inhibitors (α-GI) were administered before each meal, and diazoxide was administered before breakfast and dinner. (A) Before diazoxide initiation, both basal and postprandial levels were low. (B) After diazoxide initiation and discontinuation of cornstarch and α-GI treatment, basal values improved but postprandial hyperglycemia and subsequent hypoglycemia persisted. (C) After restarting α-GI, both basal and postprandial levels were within the acceptable range.
Discussion
The patient in the present case was identified as having a novel hemizygous missense variant [c.557T>G, p.(Met186Arg)] in the ZC4H2 gene. On the basis of this variant and the patient’s phenotype, the patient was diagnosed with WRWF. This report describes the case of a mother and son with the same missense variant; it was considered a case of WRWF in the child and mild WRWF in the mother, rather than female-restricted WRWF (WRWFFR). This is because WRWFFR is thought to manifest as a complete loss-of-function variant, and affected males typically do not survive until birth (1, 5, 6). To the best of our knowledge, this is the first report of the use of diazoxide to treat HH associated with WRWF. In the present case, the addition of a small dose of diazoxide stabilized the patient’s refractory glycemic control without causing any side effects.
The prevalence of hypoglycemia with ZC4H2 pathogenic variants ranges from 21.7% to 41.2% in boys, and from 0% to 7.7% in girls (6, 7). In cases with ZC4H2 pathogenic variants, attention should be paid to seizures associated with severe hypoglycemia (8). The six cases in which the detailed course has been reported (including our current case) of pathogenic ZC4H2 variants associated with hypoglycemia are summarized in Table 3 (1, 7, 8). Three of the six cases, including ours, had concomitant HH (7). In one case report, a 1-yr-old child was diagnosed with central adrenal insufficiency and severe recurrent hypoglycemia (8). The causes of the remaining two cases were not specified (1). One documented that a 7-yr-old boy died during sleep, although no association with hypoglycemia was shown (1). Although WRWF is not typically associated with HH, we considered HH as a complication of WRWF in our case based on a review of these previous reports. However, the decision to treat our patient with diazoxide was delayed because he had been treated for dumping syndrome before the age of 1 yr.
Table 3. Our case and previously reported variants, along with detailed hypoglycemia symptoms.

In an experiment using Xenopus, most frogs modified with ZC4H2 substitution variants were unable to stabilize mothers against decapentaplegic homolog (SMAD) in vivo. Loss of ZC4H2 has been suggested to affect bone morphogenetic protein (BMP) signaling by regulating the SMAD protein family (13). SMAD is a signaling factor that works together with transforming growth factor-β, which has demonstrated involvement in β-cell dysfunction by suppressing insulin transcription (14). Similarly, enhanced BMP signaling is also thought to cause β-cell dysfunction by suppressing β-cell proliferation and insulin secretion (15). These findings suggest that ZC4H2 dysfunction results in impaired insulin secretion, particularly hypersecretion. For these reasons, diazoxide, acting directly on β-cells, may be useful for treating HH associated with WRWF.
As previously mentioned, this case of WRWF was complicated by dumping syndrome. In recent years, several cases of improved glucose metabolism in children with dumping syndrome after the introduction of diazoxide have been reported (16,17,18). Nonetheless, diazoxide is associated with side effects such as hypertrichosis, fluid retention, and pulmonary hypertension, which require close monitoring. However, in the present case, the minimum dose of diazoxide was used, enabled by concurrent α-GI administration. The combination of low-dose diazoxide and α-GI may be a better treatment option for patients who have difficulty controlling HH due to dumping syndrome.
Conclusion
We report a case of WRWF with HH that was successfully treated using low-dose diazoxide and α-GI, and identified a novel missense variant in the ZC4H2 gene. Considering that HH can significantly impair the patients’ quality of life, treating it effectively is important. The fact that diazoxide was effective in treating HH in this case suggests that the ZC4H2 variant is associated with β-cell dysfunction. WRWF is a congenital abnormality syndrome that may be associated with HH and responds well to diazoxide treatment.
Conflict of interests
The authors declare no conflicts of interest.
Acknowledgments
We thank the patient and his family for participating in this study. Written informed consent was obtained from the patient’s guardian for genetic testing and publication of this case report.
References
- 1.Wongkittichote P, Choi TI, Kim OH, Riley K, Koeberl D, Narayanan V, et al. Expanding allelic and phenotypic spectrum of ZC4H2-related disorder: A novel hypomorphic variant and high prevalence of tethered cord. Clin Genet 2023;103: 167–78. doi: 10.1111/cge.14248 [DOI] [PubMed] [Google Scholar]
- 2.Godfrey ND, Dowlatshahi S, Martin MM, Rothkopf DM. Wieacker-Wolff syndrome with associated cleft palate in a female case. Am J Med Genet A 2018;176: 167–70. doi: 10.1002/ajmg.a.38527 [DOI] [PubMed] [Google Scholar]
- 3.Wieacker P, Wolff G, Wienker TF, Sauer M. A new X-linked syndrome with muscle atrophy, congenital contractures, and oculomotor apraxia. Am J Med Genet 1985;20: 597–606. doi: 10.1002/ajmg.1320200405 [DOI] [PubMed] [Google Scholar]
- 4.Hirata H, Nanda I, van Riesen A, McMichael G, Hu H, Hambrock M, et al. ZC4H2 mutations are associated with arthrogryposis multiplex congenita and intellectual disability through impairment of central and peripheral synaptic plasticity. Am J Hum Genet 2013;92: 681–95. doi: 10.1016/j.ajhg.2013.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun JJ, Cai Q, Xu M, Liu YN, Li WR, Li J, et al. Loss of protein function causing severe phenotypes of Female-restricted Wieacker Wolff syndrome due to a novel nonsense mutation in the ZC4H2 gene. Genes (Basel) 2022;13: 1558. doi: 10.3390/genes13091558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frints SGM, Hennig F, Colombo R, Jacquemont S, Terhal P, Zimmerman HH, et al. Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita. Hum Mutat 2019;40: 2270–85. doi: 10.1002/humu.23841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kondo D, Noguchi A, Takahashi I, Kubota H, Yano T, Sato Y, et al. A novel ZC4H2 gene mutation, K209N, in Japanese siblings with arthrogryposis multiplex congenita and intellectual disability: characterization of the K209N mutation and clinical findings. Brain Dev 2018;40: 760–7. doi: 10.1016/j.braindev.2018.05.003 [DOI] [PubMed] [Google Scholar]
- 8.Piccolo G, d’Annunzio G, Amadori E, Riva A, Borgia P, Tortora D, et al. Neuromuscular and neuroendocrinological features associated with ZC4H2-related arthrogryposis multiplex congenita in a sicilian family: a case report. Front Neurol 2021;12: 704747. doi: 10.3389/fneur.2021.704747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.May M, Hwang KS, Miles J, Williams C, Niranjan T, Kahler SG, et al. ZC4H2, an XLID gene, is required for the generation of a specific subset of CNS interneurons. Hum Mol Genet 2015;24: 4848–61. doi: 10.1093/hmg/ddv208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17: 405–24. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukutomi M, Shimodera M, Maeda Y, Iwakura M, Hara M. Safety and effectiveness, including intelligence prognosis, of diazoxide in pediatric patients with hyperinsulinemic hypoglycemia: special survey in Japan (long-term, all-case survey). Clin Pediatr Endocrinol 2018;27: 131–43. doi: 10.1297/cpe.27.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brar PC, Heksch R, Cossen K, De Leon DD, Kamboj MK, Marks SD, et al. Management and appropriate use of diazoxide in infants and children with hyperinsulinisms. J Clin Endocrinol Metab 2020;105: 3750–61. doi: 10.1210/clinem/dgaa543 [DOI] [PubMed] [Google Scholar]
- 13.Ma P, Ren B, Yang X, Sun B, Liu X, Kong Q, et al. ZC4H2 stabilizes Smads to enhance BMP signalling, which is involved in neural development in Xenopus. Open Biol 2017;7: 170122. doi: 10.1098/rsob.170122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin HM, Lee JH, Yadav H, Kamaraju AK, Liu E, Zhigang D, et al. Transforming growth factor-β/Smad3 signaling regulates insulin gene transcription and pancreatic islet β-cell function. J Biol Chem 2009;284: 12246–57. doi: 10.1074/jbc.M805379200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urizar AI, Prause M, Ingerslev LR, Wortham M, Sui Y, Sander M, et al. Beta cell dysfunction induced by bone morphogenetic protein (BMP)-2 is associated with histone modifications and decreased NeuroD1 chromatin binding. Cell Death Dis 2023;14: 399. doi: 10.1038/s41419-023-05906-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musthaffa YM, Goyal V, Harris MA, Kapur N, Leger J, Harris M. Dysregulated glucose homeostasis in congenital central hypoventilation syndrome. J Pediatr Endocrinol Metab 2018;31: 1325–33. doi: 10.1515/jpem-2018-0086 [DOI] [PubMed] [Google Scholar]
- 17.Mejia-Otero JD, Grishman EK, Patni N. Diazoxide for the treatment of hypoglycemia resulting from dumping syndrome in a child. J Endocr Soc 2019;3: 1357–60. doi: 10.1210/js.2019-00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldawsari KA, Mattos C, Khan DM, Beckett O, Pagan P. Successful treatment of dumping syndrome with diazoxide in an infant with hypoplastic left heart syndrome. Endocrinol Diabetes Metab Case Rep 2024;2024: 23–0137. [DOI] [PMC free article] [PubMed] [Google Scholar]