

Abstract
Mucopolysaccharidosis type I (MPS I) is a metabolic disorder characterized by a deficiency in α-l-iduronidase (IDUA), leading to impaired glycosaminoglycan degradation. Current approved treatments seek to restore IDUA levels via enzyme replacement therapy (ERT) and/or hematopoietic stem cell transplantation (HSCT). The effectiveness of these treatment strategies in preventing neurodegeneration is limited due to the inability of ERT to penetrate the blood-brain barrier (BBB) and HSCT’s limited CNS reconstitution of IDUA levels. We reprogrammed human cord blood cells into induced pluripotent stem cells (iPSCs), differentiated them into human induced neural stem cells (hiNSCs), and sorted them using fluorescence-activated cell sorting (FACS). Our in vitro studies showed that these hiNSCs can migrate and cross-correct IDUA deficiency. Purified hiNSCs were then transplanted into neonatal immunodeficient MPS I mice (Idua−/−). Analysis of brain tissue obtained 8 months after transplantation showed partially restored IDUA activity, with distribution and differentiation of engrafted hiNSCs throughout the brain into glial cell types. The presence of engrafted hiNSCs was associated with decreased levels of biomarkers commonly elevated in the Idua−/− mouse brain, such as β-hexosaminidase, CD68, and LAMP1, suggesting physiological efficacy. These results highlight the potential of hiNSCs for use as a patient-specific cellular therapy for MPS I.
Keywords: cell- and tissue-based therapy, induced pluripotent stem cell, neural stem cell, NSC, cell differentiation, neurodegenerative diseases, hereditary, CNS, inflammation, CXCR4
Graphical abstract
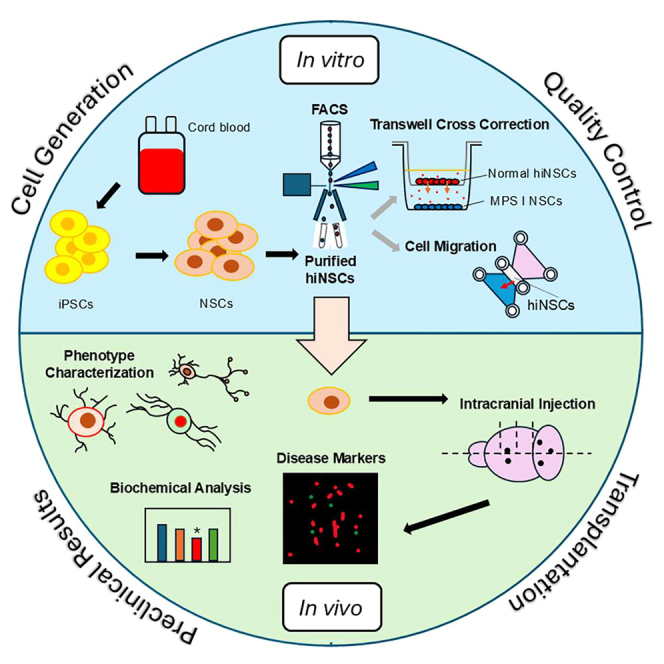
Kan and colleagues conducted an emerging proof-of-concept preclinical study to develop an iPSC-derived neurogenic cellular therapy as a complementary treatment for MPS-IH.
Introduction
Mucopolysaccharidosis type I (MPS I; MIM: 607014-6) is a rare autosomal recessive lysosomal storage disorder caused by a deficiency of the enzyme α-l-iduronidase (IDUA; Enzyme Commission [EC] number: 3.2.1.76). Lack of this enzyme impairs the degradation of complex glycosaminoglycans (GAGs), heparan sulfate (HS), and dermatan sulfate, causing subcellular accumulation, which in turn triggers a cascade leading to tissue damage and organ dysfunction, including neurodegeneration.1,2 The current approved treatments for MPS I include allogeneic hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT) with recombinant human (rh)IDUA.3 Both treatments exogenously supplement the deficient IDUA and enrich enzyme-deficient cells via mannose-6-phosphate receptor-mediated internalization to facilitate GAG clearance.4,5
Thus far, ERT has been the standard of care for MPS I6; however, there is limited or no CNS penetration by the enzyme because of the blood-brain barrier (BBB).3 Additionally, patients may develop immunoglobulin G (IgG) antibodies against rhIDUA from ERT over time.3 While HSCT is considered the standard of care for MPS IH (Hurler syndrome, the severe phenotype of MPS I) and optionally for MPS I-Hurler/Scheie syndrome (the attenuated phenotype of MPS I), HSCT has shown limited neurocognitive benefit, resulting in survivors with continued learning disabilities and below-average cognition scores, even with early transplantation.7,8 The overall efficacy of these treatments in preventing or slowing the progression of neurodegeneration remains constrained by their limited ability to penetrate the BBB.
Consequently, there has been a surge in exploring novel alternative treatments to circumvent the BBB, not only for MPS I but also for other neuropathic MPS diseases such as MPS II (Hunter syndrome), MPS III (Sanfilippo syndrome), and MPS VII (Sly syndrome). The aim for these neurogenic gene and cellular therapies is to repair and regenerate cells in the brain, thereby providing functional improvement in CNS-related diseases.9 These alternative treatments include gene therapy10 and other direct neural stem cell (NSC) transplants that have shown potential to correct disease with varying promising results.11,12,13,14,15,16,17,18 A cord blood-derived, induced pluripotent stem cell (iPSC)-derived neurogenic cellular therapy may provide a more readily available and long-term IDUA supplementation to the CNS for refractory neuropathic MPS IH and would serve as a complement to HSCT therapies to better protect the CNS.19
In our previous studies, human cord blood cells were reprogrammed into iPSCs and differentiated into human induced NSCs (hiNSCs).20 To further explore the potential applications of hiNSCs as a complementary cellular therapy to alleviate lysosomal dysfunction in the CNS, we sought to increase the purity of the hiNSC population. This was accomplished using fluorescence-activated cell sorting (FACS) to enrich a CD184-expressing subset, while excluding cells expressing CD44 and CD271.21 We selected CD184 as a surface marker for hiNSC because CD184 and its ligand, SDF-1α (stromal derived factor 1α, CXCL12), guide hiNSC migration, which is crucial for in vivo applications.22,23 Cells expressing CD44 and CD271 were excluded to mitigate the risk of spontaneous differentiation into other ectodermal lineage cell types, thereby maintaining the sorted cells’ status as NSCs.21
CD184-enriched hiNSCs were then transplanted into neonatal immunodeficient Idua−/− mice24 and allowed to engraft for 8 months. Brain tissue sections of treated mice were stained with a human-specific antibody, and the observed staining pattern showed that transplanted hiNSCs had migrated and engrafted in the mouse brain, differentiated into glial lineage cells, and partially restored IDUA enzymatic activity. This engraftment led to the reduced expression of biomarkers commonly elevated in MPS I mice such as β-hexosaminidase (β-hex), CD68, and LAMP1. Importantly, no evidence of tumors was seen. These outcomes suggest that engrafted hiNSCs may be promising for future clinical applications.
Results
Characterization of purified human iPSC-derived NSCs
hiNSCs generated from CD34+ cord blood cells from a healthy donor were differentiated from iPSC as described previously,20 resulting in a heterogeneous cell population. To refine and enrich a purified homogeneous population of hiNSCs for animal transplantation, the heterogeneous cell populations were analyzed and sorted using FACS for cell surface markers CD184, CD44, and CD271. Among the pre-sorted populations, approximately 75.7% were CD184+/CD44−/CD271−; this subpopulation was further expanded in culture. To test the stability of our CD184+/CD44−/CD271−enriched hiNSCs, the sorted population was re-analyzed after multiple freeze-thaw cycles and culturing after 4 months. The results showed that the hiNSC population was stably maintained, with 89.4% of the population expressing the selection criteria of interest (Figure 1A). Karyotyping of iPSCs prior to hiNSC differentiation and hiNSCs post-FACS enrichment revealed no chromosomal abnormalities.
Figure 1.

Purification and characterization of hiNSCs derived from iPSC
(A) Fluorescence-activated cell sorting (FACS) analysis of hiNSCs. CD184+/CD44−/CD271−sorting captured approximately 75.7% of the hiNSC population (top). After 4 months, approximately 89.44% of these previously sorted hiNSCs retained CD184 expression (bottom), indicating stability in the expression profile over time. (B) Immunocytochemistry staining for NSC markers Nestin, PAX6, SOX1, and SOX2 alongside CD184, a marker for migration capacity. Notably, the absence of OCT3/4, typically found in induced iPSCs, confirms the differentiated nature of the NSCs. Scale bar, 50 μm. (C) Cell migration assay. CD184+ hiNSCs grew and migrated primarily toward chemoattractant SDF-1α (100 ng/mL) (yellow arrow; left chamber) after 72-h incubation. Scale bar, 100 μm. (D) hiNSC transwell co-culture assay. Left graph: intracellular IDUA activity was significantly increased in MPS I-NSCs cross-corrected (light gray) with hiNSCs (dark gray), compared to uncorrected MPS I-NSCs (black). Right graph: GAG levels were significantly reduced in cross-corrected MPS I-NSCs, while GAG levels remained 2-fold higher in uncorrected MPS I-NSCs, with minimal amounts of enzyme activity as seen in the left graph. Data from biochemical assays are presented as mean ± SD. Statistical significance is denoted by asterisks (∗∗p < 0.01; ∗∗∗∗p < 0.0001; ns, not significant).
Post-sorted cells were further evaluated for conventional NSC and iPSC markers. Key NSC protein markers such as Nestin, PAX6, SOX1, and SOX2 were detected, confirming the NSC-differentiated state of these cells. Notably, the absence of OCT3/4, a marker of iPSCs due to its association with pluripotency, in our hiNSC population affirms their differentiated nature.25 Moreover, CD184, the selection marker utilized for hiNSC purification, was also positively expressed (Figure 1B).
Enriched hiNSCs exhibited chemotaxis toward SDF-1α
To confirm the migratory capacity of purified CD184+ hiNSCs toward their chemoattractant, SDF-1α, a gradient of SDF-1α (0–100 ng/mL) was established across the ibidi μ-Slide. Following a 72-h incubation period, a significant portion of proliferating hiNSCs cultured in the center observation chamber demonstrated migration toward the left chamber reservoir containing SDF-1α. This observation underscores the functionality of CD184 receptors on the surface of our hiNSCs, which enabled their migration toward SDF-1α. Conversely, minimal to no growth was noted in the direction of the right chamber reservoir, which lacked SDF-1α (Figure 1C; Video S1).
Reduced GAG levels in NSCs derived from MPS I patient
IDUA and GAG levels were measured in NSCs derived from an MPS I patient fibroblast cell line (GM00415; MPS I-NSCs) and in enriched hiNSCs, were co-cultured in a Transwell culture system. No detectable IDUA was seen in the cells or conditioned medium of cultured MPS I-NSCs. IDUA activity, however, was detectable in both cells and conditioned medium of hiNSCs. The results of Blyscan assays showed a 2-fold increase in intracellular GAG accumulation in MPS I-NSCs, compared to our hiNSCs. After 96-h incubation in the Transwell system, a discernible amount of intracellular IDUA enzymatic activity was detected in co-cultured MPS I-NSCs (approximately 10.2% of the activity observed in hiNSCs; MPS I-NSCs corrected in Figure 1D). Similarly, after cross-correction, GAG levels in MPS I-NSCs significantly decreased, approaching the levels observed in our hiNSCs. These findings suggest that even modest amounts of restored IDUA in MPS I-NSCs are sufficient to mitigate abnormal GAG accumulation levels in vitro (Figure 1D).
Assessment of hiNSC engraftment in immunodeficient Idua−/− (NSGI) mice
To assess the distribution and subsequent impact of hiNSCs on disease-associated neuropathology in vivo, enriched hiNSCs were transplanted into neonatal Idua−/− mice (Figure S1C). No adverse events were observed in the treated animal cohort (n = 8) during the duration of this study. IDUA and β-hex enzymatic activity in brain sections was assessed at 8 months in hiNSC-treated NSGI mice, and in controls from age-matched unaffected Idua+/− and untreated Idua−/− mice. Mean levels of IDUA enzyme activity in each brain section homogenate were undetectable in each untreated Idua−/− mouse, but ranged from 1.09–1.56 U/mg protein, across sections, in unaffected Idua+/− controls (p < 0.001 in brain 4–6, but not significant in brain 1–3 due to the higher variation in a small number of animals in the treated group (Figure 2A). In hiNSC-treated NSGI mice, mean IDUA enzyme activity varied across brain sections Figure 2A). Notably, six of eight samples from the cerebellar area exhibited a significant increase in IDUA activity (mean: 1.09 ± 0.27 U/mg protein) compared to the same region in untreated Idua−/− mice. These levels were comparable to the average range observed in Idua+/− control mice. This observation suggests a potential correlation between the observed increase in IDUA activity and proximity to the three sites used for intraparenchymal transplantation to the cerebellar area (Figure 2A).
Figure 2.

Biochemical evidence of therapeutic effects in immunodeficient Idua−/− mice after hiNSC transplantation
(A) IDUA enzyme activity quantification (U/mg protein) in brain sections (refer to Figure S1D for further details), brain 1 + 2, brain 3, brain 4, and brain 5 + 6 for unaffected carriers (+/−; n = 6, white), untreated Idua−/− (−/−; n = 6, black), and transplanted Idua−/− (−/− Tx; n = 8, gray) cohorts at 8 months post-hiNSC transplantation. Transplanted mice showed higher variability in brain 1 + 2 and brain 3 IDUA activity. Each dot represents the measurement from each animal. (B) β-Hexosaminidase (β-hex) activity quantification (U/mg protein) from the same brain sections in (A). In each brain region examined, β-hex activity is significantly lower in transplanted mice, compared to untreated mice. This decrease suggests functional expression of IDUA by engrafted cells. Data are presented as mean ± SD. Statistical significance is denoted by asterisks (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001).
Measurements of β-hex enzyme activity were also used to assess the effect of hiNSC engraftment on the MPS I disease phenotype. Brain homogenate from untreated Idua−/− mice showed a consistent 1.7- to 2.8-fold increase in β-hex enzyme activity compared to age-matched unaffected Idua+/− controls (p < 0.001). In Idua−/− mice that received a single neonatal hiNSC transplantation, β-hex enzyme activity was significantly reduced in all brain sections, compared to the corresponding brain section in untreated Idua−/− mice (p < 0.01). β-Hex enzyme activity in treated Idua−/− mice approached the level observed in unaffected Idua+/− controls (Figure 2B).
Distribution and differentiation of engrafted hiNSCs
To further investigate the distribution and characteristics of engrafted cells, brain sections harvested from transplanted Idua−/− mice were probed with STEM121, an antibody specific for human cytoplasmic protein. Positive STEM121 staining thus marks hiNSCs that have successfully migrated and differentiated in the Idua−/− mouse brain after the initial transplantation. Strong STEM121 staining was observed throughout the brain in hiNSC-engrafted mice, particularly in areas such as the olfactory bulb, midbrain, cerebellum, pons, and medulla (Figure 3). This pattern of expression was consistent with the observed pattern of restored IDUA enzymatic activity, as shown in Figure 2. Additionally, these human cells in various areas of the brain were observed to display multiple processes, suggesting the potential differentiation of the hiNSCs after engraftment (Figures 3 and 4).
Figure 3.

Distribution of hiNSC in immunodeficient Idua−/− mice 8 months post-transplantation
Immunohistochemical staining of STEM121 is shown in representative animal images. Positive STEM121 signal was observed in olfactory bulb (OB), midbrain (MB), cerebellum (CB), and pons/medulla (P/MD). Full brain scan images (top) and selected regions of interest (bottom) show broad distribution of engrafted hiNSCs across multiple brain regions. For each full-brain image, selected regions below (left) are 10× magnification and further expanded to 40× magnification for detailed examination (right). Scale bars, 50 μm.
Figure 4.

Differentiation of engrafted hiNSCs into glial cells in the brains of immunodeficient Idua−/− mice
Immunohistochemical staining of brain tissue from Idua−/− mice transplanted with hiNSCs shows the differentiation status of engrafted cells. STEM121 staining marks engrafted hiNSCs (red, first column). CNS cell-type markers (green, second column) are designed for use in human or mouse tissue. Co-staining for STEM121 and CNS cell markers reveals the differentiation of hiNSCs into distinct CNS cell types, while single staining for CNS markers identifies resident mouse CNS cells. In merged images (third column), colocalized staining marks hiNSCs that have differentiated into glial cell types, such as astrocytes (GFAP) and oligodendrocytes (Olig2). Selected areas from merged images are expanded to provide additional detail (fourth column). Notably, there is no co-staining observed for neuronal markers (NF, NeuN), indicating the absence of neuronal differentiation. Scale bars, 50 μm.
In addition to STEM121 staining, we explored STEM123, a marker specific to human glial fibrillary acidic protein (GFAP), to assess the differentiation status of hiNSCs in treated mouse brains. Human GFAP+ cells, primarily exhibiting larger cell bodies of star-shaped morphology indicative of typical astrocytes, were observed in brain tissue from Idua−/− mice treated with hiNSCs. Cells with astrocyte-like morphology were most prominent in the cerebellum and medulla areas of the brain (Figure S2).
The differentiation status of engrafted hiNSCs was further characterized by co-staining with STEM121 (red) and various CNS cell markers (green): GFAP and Olig2 for astrocytes and oligodendrocytes; neurofilament (NF) and neuronal nuclear antigen (NeuN) for neural cell phenotypes. Notably, the CNS cell markers utilized in this study can effectively label specific cell types in both human and mouse tissues. Co-staining for STEM121 and a CNS cell marker thus indicated that hiNSCs had differentiated into distinct CNS cell-type lineage, while staining for CNS markers only identified resident mouse CNS cells (Figure 4). In the images captured from representative medulla areas, multiple overlapping cell signals from STEM121 and GFAP staining (yellow) were observed, suggesting differentiation of the hiNSCs into astrocytes by 8 months of engraftment (Figure 4). This co-staining result mirrors the previously observed star-like cell morphology distribution pattern of STEM123 staining in cells that had engrafted in the mouse brain (Figure S2).
We also examined the expression of Olig2, a transcription factor primarily for oligodendrocytes that localizes in the nucleus.26 In representative sections from the cerebellar areas, nuclear staining of Olig2 is surrounded by cytoplasmic human STEM121 signal. While Olig2 may also stain specialized neurons such as motor neurons,26 no colocalization of STEM121 with either NF or NeuN markers was observed, suggesting that transplanted hiNSCs did not differentiate into neuron-lineage cells by 8 months of engraftment (Figure 4).
Engrafted hiNSCs improves MPS I pathology
To evaluate the functional efficacy of our transplanted hiNSCs in host mouse tissues, immunofluorescence (IF) co-staining of hiNSCs was performed using STEM121 with LAMP1 or CD68, respectively. We chose to stain for LAMP1 and CD68 because these markers were significantly elevated in the cortex and pons regions of brains in our Idua−/− mouse model compared to the Idua+/− mouse model. In mice treated with hiNSCs, areas devoid of STEM121 signal (representing no engraftment of hiNSCs) exhibited sustained elevation in LAMP1 and CD68 levels similar to those observed in untreated Idua−/− mice. However, in regions where STEM121 signal was present, expression levels of LAMP1 and CD68 decreased significantly to levels similar to those observed in unaffected Idua+/− mice (Figure 5). This observation suggests that our hiNSCs possess a degree of physiological efficacy in mitigating MPS I neuropathology, as evidenced by the reduced expression of markers such as LAMP1 and CD68.
Figure 5.

Histological evidence of improved MPS I pathology after hiNSC transplantation in Idua−/− mice
Representative immunofluorescence images of cortex and pons brain tissue show elevated expression of common neuropathic markers LAMP1 (A) and CD68 (B) in Idua−/− mice compared to unaffected carriers (Idua+/−). After successful engraftment of transplanted hiNSCs in Idua−/− mice (red), levels of LAMP1 (A) and CD68 (B) (green) are similar to those observed in unaffected carriers. Conversely, in regions devoid of engraftment (absence of red), levels of LAMP1 (A) and CD68 (B) (green) are comparable to those observed in untreated Idua−/− mice. No staining for STEM121 is observed in the first three rows because animals did not receive hiNSCs. Scale bars, 50 μm.
Discussion
Since the pioneering work of Thomson et al., using human blastocyst-derived pluripotent stem cells,27 and Takahashi and Yamanaka, using postnatal derived iPSCs,28 the applications of phenotype-specific cells have been studied to develop as cellular therapies for a host of diseases and injuries. This innovative approach formed the basis of what is now commonly referred to as regenerative medicine.29 Our studies have focused on using a population of iPSC-derived NSCs to treat the neurological manifestations of the severe form of the lysosomal storage disorder, MPS IH. Importantly, the goal is to complement current forms of therapy, such as HSCT and/or ERT, and to extend those therapeutic effects to the CNS.
The results of the present study demonstrate success with the proposed application for brain-engraftable hiNSCs, which included streamlining steps designed to facilitate clinical use. While the prospective transplantation would be preferably autologous, this is not possible for a genetic disease without gene correction. We used cord blood, rather than bone marrow, as a starting material because these cells demonstrate superior engraftment, lower associated risk of graft-versus-host disease,30 and CD34+ cells derived from cord blood are also easily converted into iPSCs, as we have previously shown.20 Since our goal was to generate an add-on therapy to current best practices, we propose that the same cord blood used for allogeneic HSCT also be used to derive the NSCs used for “autologous” CNS transplantation—autologous to the new hematopoietic system derived from the cord blood HSCT.
In this study, we sorted human CD34+ hematopoietic stem cells that were then reprogrammed into iPSCs and further differentiated and sorted into purified hiNSCs. This cell-sorting step was critical not only to enrich a homogeneous NSC population but also to select for the migratory capacity specific to CD184 (CXCR4)+ NSCs. CD184 is known to migrate toward its chemoattractant SDF-1α, or CXCL12, a chemokine actively expressed in the developing brain with a crucial role in engraftment and stem cell progenitor recruitment.31 After birth, SDF-1α exhibits prominent expression in areas such as the brainstem, olfactory bulb, cerebellum, and various areas of the brain vasculature, agreeing with the distribution of engrafted cells in this study.32 SDF-1α is also increased in the presence of neuroinflammation and lesions in brain tissue that require tissue repair.33,34 In the presence of inflammation in the brains of MPS I neonates in this study, there is proposed increased expression of SDF-1α that attracts our hiNSCs to regions of the developing brain.
CD184+ hiNSCs were also negatively sorted based on the presence of CD271 and CD44 phenotypes. CD271 has broad differentiation potential (ectodermal and mesodermal germ layers),21 and CD44 is known to be a marker for glial progenitors and astrocyte cells. Negative selection for these markers aims to minimize the risk of spontaneous differentiation or commitment to other cell-type lineages, thereby maintaining hiNSC status as progenitors capable of SDF-1α-guided migration.21
FACS-sorted hiNSCs showed approximately 90% retention of their characteristic CD184+/CD44−/CD271− phenotype at 4 months after multiple passages and freeze-thaw cycles. They continued to express NSCs markers such as Nestin, PAX6, SOX1, and SOX2, but not the undifferentiated pluripotent stem cell marker OCT3/4 (Figures 1A and 1B). Additionally, hiNSCs showed normal karyotypes, indicating genomic stability during differentiation,35 ensuring the stability and progenitor nature of hiNSCs that are critical for in vivo studies. Our CD184+ hiNSCs also demonstrated the capacity for migration toward SDF-1α on a chemotaxis slide and were able to release functional IDUA, which in turn allowed for the cross-correction of MPS I cells in the transwell system (Figures 1C and 1D). The results of these in vitro assays indicate that these minimally manipulated iPSC-derived hiNSCs are well suited for in vivo testing.
Once cells were observed to exhibit the characteristics and functionality of NSCs in vitro, in vivo studies were conducted in a newly generated immunodeficient Idua−/− mouse strain on the NSG background, which lacks mature T cells, B cells, and functional natural killer cells, and is deficient in cytokine signaling. Our previous study demonstrated that this mouse model can successfully accept human engrafted cells without transplant rejection.24 In addition, these animals have a longer lifespan due to their resistance to lymphoma, which is ideal for the preclinical testing of novel PSC-based therapies.36 At 8 months post-hiNSC transplant, immunostaining with the human-specific antibody STEM121 revealed that the injected cells had migrated away from the injection sites and successfully engrafted in the Idua−/− mouse brain, indicating successful integration of human cells into the host tissues (Figure 3). Engrafted hiNSCs were observed in multiple brain regions, including the olfactory bulb, midbrain, cortex, and cerebellum. However, variability in engraftment among treated mice was noted, which may be attributed to differences in the delivery technique used to apply the hiNSCs at each injection site.
In parallel, the biochemical results from brain sections of the hiNSC-treated Idua−/− mice showed partial restoration of IDUA enzyme activity and near-complete elimination of β-hex levels (Figure 2). β-Hex is among the most abundantly expressed lysosomal enzymes, and secondary elevation of β-hex is observed in the Idua−/− mouse model.37 Normalization of this elevation is commonly employed as a metric to evaluate the efficacy of treatments due to its correlation with rescued lysosomal function.24,38 Our results demonstrate a strong association between the recovery of IDUA expression and the restoration of baseline β-hex levels in hiNSC-treated Idua−/− mice. These findings suggest the existence of a cross-correction mechanism mediated by the engrafted hiNSCs. Therefore, the supplemental IDUA delivered by our hiNSCs is presumed to be functional. However, due to the observed variance between brain regions in levels of IDUA restoration, we believe that the endogenous IDUA from our hiNSCs may exert only local effects.
We conducted further co-staining with human STEM121 antibody in combination with a series of NSC markers to monitor the differentiation of engrafted hiNSCs in Idua−/− mouse brain after 8 months. Our observations revealed elevated levels of engrafted cells expressing GFAP and Olig2 transcription factor. These findings strongly indicate that our hiNSCs differentiated into the glial cell lineage, specifically astrocytes and oligodendrocytes. The glial cell lineage is known for its higher mitotic nature compared to post-mitotic neurons, as seen in Figure 4. In addition, immunohistochemistry (IHC) using STEM123, specific to human GFAP protein, highlighted the presence of human GFAP+ cells in the cerebellum and medulla regions of the brain. The distinct stellate morphology and numerous processes observed in these cells are characteristic of typical astrocytes, providing additional evidence of their differentiation into the glial cell lineage (Figure S2).
No substantial co-staining was observed between STEM121 and neuronal-lineage cell markers NF and NeuN, however. This absence suggests that the transplanted hiNSCs may not have differentiated into neuronal-lineage cells by 8 months of engraftment or, if they did differentiate, they did not survive the long engraftment period. It is important to note that previous studies have indicated the potential for neurons marked by NF and NeuN to form within 4–12 weeks.39,40 Our observation that transplanted cells had differentiated into glial-lineage cells rather than neuronal-lineage cells was anticipated, as glial cells play a more supportive role in the brain,41 and their mitotic nature allows for more stable transplantation as seen with their prevalence after 8 months in our study.
In addition to partially restoring IDUA enzymatic activity and reducing elevated β-hex levels in the brains of Idua−/− neonates treated with hiNSCs (Figure 2), differentiated astrocytes and oligodendrocytes appear to induce observable physiological changes in the expression of markers such as LAMP1 and CD68. LAMP1 is a specific lysosomal marker that is heavily expressed in MPS I due to lysosomal enlargement from GAG accumulation.42 CD68 is a marker associated with inflammation in macrophages and activated microglia.43 Both LAMP1 and CD68 have been shown to be upregulated in MPS I mouse models.24,44 In this study, the observed LAMP1 and CD68 staining patterns demonstrated a reduction or reversal of phenotypes in the presence of hiNSCs similar to those seen in unaffected mice (Idua+/−). However, these co-staining results must be considered with the qualification that our hiNSCs may have exhibited localized efficacy because only thin brain sections in the sagittal plane were analyzed. Therefore, our results may not accurately reflect the distribution of hiNSC in the three-dimensional structure of the brain or the efficacy of hiNSC treatment in tissue surrounding the engrafted cells.
In future preclinical studies, it may be beneficial to assess cell engraftment at several time points post-transplant until end of life. This may help in guiding or estimating future dosing, true efficacy over longer periods of time, and the potential for various cellular fates (e.g., neuronal) in the initial stages of engraftment/differentiation.39 Previous work from other groups have used various transplantation methodologies in animal models, including the introduction of genetically modified cells to enhance expression, uptake, distribution, safety, and CNS delivery.12,13,14,15,16,18 Studies similar to ours that implanted genetically modified mouse iPSC-derived NSCs were conducted in MPS II, MPS IIIB, and MPS VII mouse models. These studies observed partial cross-correction around the grafts in the CNS with varying characterization and maturity of the grafted cells.12,13,14,16 Systemic HSC gene therapy (HSCGT) has been applied in mouse models of MPS I,45 MPS II,46,47 MPS IIIA,48,49 and MPS IIIB,50 with results also showing varying levels of CNS improvement.
In the present study, neonatal MPS I mice were transplanted with hiNSCs shortly after birth to reduce the neuropathic effects of MPS I. Early treatment age is considered critical for NSC transplantation to effectively improve the CNS pathology characteristic of mucopolysaccharidoses. Over time, the accumulation of HS may decrease the SDF-1α-induced migration of NSCs and thus limit the efficacy of stem cell transplantation.47,51 With the inclusion and rise of newborn screening for MPS I in the United States19 and the accessibility of cord blood, earlier intervention is more feasible52; therefore, we believe that early administration of this treatment is essential for MPS I because of the reduced efficacy associated with increased levels of HS.51
Taking our preliminary results and animal studies into account, future studies for MPS I and other related diseases affecting CNS function may benefit from the inclusion of ex vivo gene therapy to upregulate the deficient enzyme in either allogeneic cord blood or autologous patient cells. This approach may allow for a more widespread effect, in contrast to the localized efficacy observed in this study. Currently, several active clinical studies have administered autologous53,54,55 or allogeneic56 CD34+ HSCGT to neuropathic MPS patients with no safety concerns yet reported, with ongoing patient follow-up.3 Numerous other clinical treatment approaches attempt to circumvent the BBB; one such example is AAV gene therapy, which shows promising results in the treatment of MPS with CNS involvement.57,58,59 However, use of the AAV vector is associated with constraints such as smaller packaging capacity and limited opportunity for repeat dosing if a patient does not respond to treatment, due to the risk of immune reactions or a decrease in efficacy from neutralizing antibodies against AAV vectors.60,61 These factors highlight the benefits of the “autologous” NSC transplant that we have described, especially for refractory cases of MPS IH variants.
In this proof-of-concept study, we successfully demonstrated that the engraftment of iPSC-derived hiNSCs from cord blood of a normal donor can partially restore deficient IDUA enzyme and reduce neuropathic phenotypes in the Idua−/− mouse brain. Further modification to expression of the IDUA gene, such as introducing the transgene under a stronger promoter (e.g., EF1 promoter), may significantly increase the expression level of IDUA as well as potentially decrease the dose of genetically modified cells required to reach the same degree of treatment efficacy.
Materials and methods
Human cell culture
Reagents for cell culture were sourced from Thermo Fisher Scientific (Waltham, MA), unless stated otherwise. Human CD34+ cells were purified from umbilical cord blood acquired from the CHOC Cord Blood Bank under institutional review board-approved protocols.20 Mononuclear cells were isolated using Ficoll-Paque PLUS density gradient media (Cytiva, Marlborough, MA) according to the manufacturer’s instructions. Subsequently, CD34+ cells were isolated using the human CD34 MicroBead Kit (Miltenyi Biotec, Auburn, CA). Cells were cryopreserved and later thawed for iPSC reprogramming. Human skin fibroblasts from a Hurler’s patient (GM00415) were obtained from the Coriell Institute for Medical Research (Camden, NJ). Fibroblasts were maintained in DMEM high glucose with 10% fetal bovine serum and passaged with TrypLE Express.
Human iPSC derivation and differentiation
Human iPSCs (hiPSCs) were generated from either CD34+ cord blood cells or skin fibroblasts using the CytoTune-iPS 2.0 Sendai Reprogramming Kit, as previously described.20 hiPSCs were expanded in StemPro hESC SFM (human embryonic stem cell serum-free medium) on Matrigel (Corning Life Sciences, Tewksbury, MA)-coated 6-well plates. On day 0 of induction, hiPSC cultures at 90% confluency were passaged with StemPro Accutase onto a low-attachment plate to encourage the formation of embryoid bodies (EBs). During days 1–12, neural induction in EBs was performed with the STEMdiff SMADi Neural Induction Kit (STEMCELL Technologies, Vancouver, Canada). On day 13, EBs were further plated on Matrigel-coated plates to start neural rosette formation using NSC growth medium (GM) formulated with 10% BIT9500 (STEMCELL Technologies), 20 ng/mL recombinant human fibroblast growth factor (FGF)-basic (Peprotech, Cranbury, NJ), 20 ng/mL recombinant human epidermal growth factor (EGF; R&D Systems; Minneapolis, MN), and 10 μM heparin (Sigma-Aldrich, St. Louis, MO) in DMEM/F12 with 2.5 mM GlutaMAX for 7 days. On day 20, neural rosettes were selected with STEMdiff Neural Rosette Selection Reagent (STEMCELL Technologies), then cultured on Matrigel-coated plates and maintained for ∼3 days before analysis and sorting. Characterization and karyotyping of hiPSCs prior to hiNSC differentiation and post-FACS enrichment was conducted as previously described.20
Flow cytometry and cell sorting
Cells were harvested with non-enzymatic cell dissociation buffer (Sigma-Aldrich) and resuspended cold NSC FACS staining buffer formulated with DMEM/F-12, 15 mM HEPES (R&D Systems), 10% BIT9500, 5 mM EDTA, 2.5 mM GlutaMAX, 20 ng/mL recombinant human FGF-basic (Peprotech), 20 ng/mL recombinant human EGF, and 10 μM heparin. Cells were then counted to a final concentration of 1 × 106 cells/mL and stained for 30 min with conjugated antibodies: CD184 (BD Biosciences, San Diego, CA, catalog no. 555976, RRID: AB_398616), CD44 (BD Biosciences, catalog no. 555479, RRID: AB_395871), and CD271 (BD Biosciences, catalog no. 560927, RRID: AB_10564069) at a dilution of 1 × 106 cells/test, as defined in each manufacturer’s protocol. Cells were washed before resuspension in appropriate NSC FACS buffer. Cells were further strained through a 40-μm cell strainer (CELLTREAT Scientific Products, Pepperell, MA) before being analyzed and sorted with a BD FACSMelody cell sorter (BD Biosciences) using BD FACSChorus Software (BD Biosciences). Sorted CD184+/CD271−/CD44− cells were collected at a concentration of 3.5 × 105 cells/mL in NSC GM and then plated in 96-well plates pre-treated with anti-adherence rinsing solution (STEMCELL Technologies) to encourage neurosphere formation before being plated on Matrigel-coated plates for further expansion.
Immunocytochemistry
Approximately 2.5 × 104 dissociated hiNSCs were cultured on a Matrigel-treated 8-chamber slide for 24 h prior to staining. Cells were stained using Nestin, PAX6, SOX1, and SOX2 antibodies from the Human Neural Stem Cell Immunocytochemistry (ICC) kit (Thermo Fisher) according to the manufacturer’s instructions. CD184 and OCT3/4 antibodies were also included in the study for confirmational purposes, with details summarized in Table 1. After secondary antibodies were applied, cells were mounted with Vectashield Plus Antifade Mounting Medium with DAPI (Vector Laboratories, Newark, CA). Cells were imaged using a Keyence BZ-X810 imaging system (Keyence, Osaka, Japan).
Table 1.
Summary of antibodies used in histology
| Primary antibodies | Source | Isotype | Vendor, catalog no. | RRID | Usage | Dilution |
|---|---|---|---|---|---|---|
| Nestin (Molecular Probes Kit, A24354) | mouse | IgG1 | eBioscience, 14-9843-82 | AB_1548837 | ICC | 1:100 |
| PAX6 (Molecular Probes Kit, A24354) | rabbit | IgG | Thermo Fisher Scientific, 42-6600 | AB_2533534 | ICC | 1:100 |
| SOX1 (Molecular Probes Kit, A24354) | goat | IgG | R&D Systems, AF3369 | AB_2239879 | ICC | 1:100 |
| SOX2 (Molecular Probes Kit, A24354) | rabbit | IgG | Thermo Fisher Scientific, 48-1400 | AB_2533841 | ICC | 1:100 |
| OCT3/4 | mouse | IgG1, κ | BD Biosciences, 560253 | AB_1645304 | ICC | 1:20 |
| STEM 121 | mouse | IgG1 | Takara Bio, Y40410 | AB_2632385 | IHC/IF | 1:50 |
| STEM 123 | mouse | IgG1 | Takara Bio, Y40420 | AB_2833249 | IHC/IF | 1:50 |
| Olig2 | goat | IgG | R&D Systems, AF2418 | AB_2157554 | IF | 1:50 |
| GFAP | rabbit | IgG | Abcam, ab68428 | AB_1209224 | IF | 1:50 |
| NeuN | rabbit | IgG | Cell Signaling Technology, 24307 | AB_2651140 | IF | 1:100 |
| NF | rabbit | IgG | Cell Signaling Technology, 2837 | AB_823575 | IF | 1:200 |
| CD68 | rabbit | IgG | Abcam, ab283654 | AB_2922954 | IF | 1:100 |
| LAMP1 | rabbit | IgG | Abcam, ab24170 | AB_775978 | IF | 1:50 |
| Secondary antibodies | Isotype | Conjugate | Vendor, Catalog no. | RRID | Usage | Dilution |
| Donkey anti-mouse | IgG | AF488 | Molecular Probes, A24354, part no. A24350 | – | ICC | 1:200/1:250 |
| Donkey anti-goat | IgG | AF488 | Molecular Probes, A24354, part no. A24349 | – | ICC | 1:250 |
| Donkey anti-rabbit | IgG | AF594 | Molecular Probes, A24354, part no. A24342 | – | ICC | 1:250 |
| Goat anti-mouse | IgG(H + L) | AF555 | SouthernBiotech, 1031-32 | AB_2794317 | IF | 1:250 |
| Donkey anti-goat | IgG(H + L) | AF488 | Thermo Fisher Scientific, A11055 | AB_2534102 | IF | 1:250 |
| Donkey anti-rabbit | IgG(H + L) | AF488 | Thermo Fisher Scientific, A21206 | AB_2535792 | IF | 1:250–1:1,000 |
| Donkey anti-rabbit | IgG(H + L) | AF647 | Thermo Fisher Scientific, A31573 | AB_2536183 | IF | 1:500 |
Cell migration assay
CD184+ hiNSC migration and functionality assays were conducted using a two-dimensional μ-slide chemotaxis migration assay (ibidi GmbH, Gräfelfing, Germany), according to the manufacturer’s protocol, with minor modifications. The ibidi μ-slide was pre-coated with fibronectin (reservoir, 40 μg/mL; observation chambers, 70 μg/mL; Gibco, Brooklyn, NY) at least 24 h before the application of 10 μL cells (density 6.5 × 106 cells/mL) in the center observation chamber (Figure S1A). NSC GM was equilibrated overnight at 37°C with 5% CO2 in a vented cell culture flask prior to cell seeding. To create the SDF-1α gradient, the left reservoir was filled with medium containing 100 ng/mL SDF-1α (Invitrogen, Waltham, MA), while the right reservoir contained medium without chemoattractant (Figure S1A) inside a BioSpherix ex vivo chamber (Cytocentric , Parish, NY). Images of migration were acquired every 5 min over 72 h with cells at 37°C with 5% CO2 and 80% humidity using an Olympus CKX41 Inverted Phase Contrast Microscope (Olympus Life Science, Waltham, MA). Images were processed with cellSens imaging software (Olympus Life Science).
Transwell and co-culture assays
For transwell cross-correction assays, MPS I-NSCs derived from an MPS I patient (GM00415) were plated at a concentration of 1–1.5 × 106 cells/well in 6-well plates. hiNSCs were plated on 6-well Transwell Permeable Support Inserts (Corning Life Sciences) pre-treated with 10 μg/mL fibronectin, at a concentration of 1 × 106 cells/well (Figure S1B). After 96 h of incubation at 37°C with 5% CO2, MPS I-NSCs were washed three times with PBS before being harvested for biochemical analyses, including an assay of IDUA enzymatic activity and Blyscan quantification.
Enzyme activity and GAG quantification
Unless otherwise specified, all chemicals and reagents used in this study were purchased from Sigma-Aldrich. For biochemical analyses, cell pellets or brain slices were homogenized in CelLytic MT cell lysis reagent. Diluted samples were used to estimate protein concentration using a bicinchoninic acid assay kit (Thermo Fisher Scientific), with BSA as standard. The catalytic activity of IDUA was assessed with 4-methylumbelliferyl substrate α-l-iduronide (Glycosynth, Warrington, UK) at final concentration of 250 μmol/L. The catalytic activity of β-hexosaminidase (combined A and B isoforms) was determined by the hydrolysis of 4-methylumbelliferyl-N-acetyl-β-glucosaminide using 1.25 mM substrate in the incubation mixture.38 Reactions were performed at 37°C for 1 h and quenched with glycine carbonate buffer (pH 10.5). Fluorescence was measured with an Infinite M Plex spectrofluorophotometer (Tecan Systems, Morgan Hill, CA) at excitation and emission wavelengths of 360 and 450 nm, respectively. One activity unit (U) was defined as 1 nmol converted substrate per hour. Specific activity reported in the study was defined as U/mg protein.
GAG quantification was performed using the Blyscan assay (BioVendor, Asheville, NC), a quantitative dye-binding method. Briefly, diluted tissue homogenate, GAG standards, and blank controls were incubated with the dye to form a dye-GAG complex, which precipitated within 30 min. The GAG-bound dye was then recovered using dissociation reagent. Absorbance readings were obtained at 656 nm using an Infinite M Plex spectrofluorophotometer (Tecan Systems). GAG levels were normalized to total protein and expressed as μg GAG/mg protein. Each sample was measured in triplicate for all biochemical assays reported in this study.
Experimental animals
Animal experiments conducted as part of this study were approved by the Institutional Animal Care and Use Committee at Children’s Hospital of Orange County (approval no. 120701). The immunodeficient MPS I knockout mouse model (NOD.Cg-Iduatm1Clk Prkdcscid Il2rgtm1Wjl/SzJ; NSGI) was generated and maintained on an inbred background (NOD.Cg- Prkdcscid Il2rgtm1Wjl/SzJ; NSG) at our institution,24 and is now available at The Jackson Laboratory (RRID: IMSR_JAX:035007). The MPS I genotype was determined using primers: oIMR1451: 5′-GGAACTTTGAGACTTGGAATGAACCAG-3′, oIMR1452: 5′-CATTGTAAATAGGGGTATCCTTGAACTC-3′, and oIMR1453: 5′-GGATTGGGAAGACAATAGCAGGCATGCT-3′. Homozygous males (Idua−/−) and heterozygous females (Idua+/−) were used to generate affected (Idua−/−) and unaffected control (Idua+/−) mice. Animals were housed in sterile cages in high-efficiency particulate air-filtered cage racks and were provided irradiated food and sterile deionized water ad libitum under a 12:12 h light:dark cycle. Experiments were conducted on age-matched mice (usually littermates) of either gender.
Enriched hiNSCs (1 × 105 in 5-μL sterile phosphate-buffered saline [PBS]) were injected bilaterally into the lateral cerebroventricular space, and three sites of cerebellar tegmentum of NSGI neonatal mice (postnatal day 1 or 2) under cryo-anesthesia using 32G needles and Hamilton syringes (Hamilton Company, Reno, NV). The injection sites in lateral ventricles were as described previously.13 Cerebellar injection sites are shown in Figure S1C, and the depth for injections into the cerebellum was 2.5 mm. The lambda and bregma points in mouse brain were used as reference landmarks for injections. A total of eight animals were treated and analyzed for this study.
Tissue harvesting, processing, and histological staining
At 8 months post-transplantation, animals were euthanized using CO2 asphyxiation followed by transcardial perfusion with cold PBS. Brains were dissected sagittally along the midline, and right brain hemispheres were further sectioned into 2-mm-thick coronal slices using an adult mouse brain slicer matrix (Zivic Instruments, Pittsburgh, PA) and then rapidly frozen and stored at −80°C until IDUA and β-hex activity assays were performed (Figure S1D). Left-brain hemispheres were post-fixed overnight at 4°C in 4% paraformaldehyde, then stored in 70% ethanol and sent to the Experimental Tissue Resource Center at University of California, Irvine to be embedded in paraffin blocks and cut in 4-μm-thick sections for further staining.
IHC and IF staining
All slides used for staining were treated with heat-induced antigen retrieval with 10 mM sodium citrate buffer at pH 6.0 and incubated in 0.3% hydrogen peroxide in Tris-buffered saline (TBS) to block endogenous peroxidase activity. Primary antibody was applied after tissue was blocked in 1% BSA in TBS. Details related to antibodies and their dilutions are summarized in Table 1.
IHC was performed with primary antibodies STEM121 and STEM123, followed by horseradish peroxidase-conjugated secondary antibodies. Sections were developed with an ImmPACT DAB Substrate Kit (Vector Laboratories) and mounted using Permount Mounting Medium (Thermo Fisher Scientific). IF co-staining was performed using STEM121 with one of the following primary antibodies: Olig2, GFAP, NeuN, NF, CD68, or LAMP1. Slides were mounted using DAPI Fluoromount-G (SouthernBiotech, Birmingham, AL). Images were acquired using a Keyence BZ-X810 imaging system (Keyence) with 20–40× magnification. Full brain scans were acquired using the advanced observation module (Keyence).
Statistical analyses
All statistical analyses were performed using GraphPad Prism software version 9.0 (GraphPad, La Jolla, CA). One-way ANOVA was used to compare groups greater than two. Results were considered statistically significant when p < 0.05.
Data and code availability
All data and materials are available upon reasonable request.
Acknowledgments
We appreciate the valuable assistance provided by Mr. Anthony Rangel and Ms. Evelyn Torres in managing the animal colony, Dr. Rachel Davis for her professional scientific editing, and the histology preparation performed by the Experimental Tissue Resource Center at University of California, Irvine. We also thank Mr. Michael Neel (Department of Pathology and Laboratory Medicine, University of California, Irvine) for preliminary IHC staining. This work was supported by the California Institute for Regenerative Medicine (TR3-05476), Strategic Grant Program, and the Glass Slipper Guild through the CHOC Foundation, Susan Scott Foundation, CHOC CSO Small Grants Program, Advancing Pediatric Stem Cell Research from the Hoag Foundation (9415), and the Campbell Foundation of Caring.
Author contributions
P.H.S. and S.-H.K. conceptualized and supervised the study. C.C.C., S.-H.K., A.E.S., and P.H.S. designed the experiments. C.C.C., S.-H.K., A.E.S., and J.F.H. performed the experiments. C.C.C., S.-H.K., and A.E.S. performed the data collection. C.C.C., S.-H.K., A.E.S., and E.S.M. performed the data analysis. C.C.C. and S.-H.K. wrote the manuscript and prepared the figures. R.Y.W. and E.S.M. provided clinical guidance. S.-H.K., R.Y.W., and P.H.S. obtained funding for the study. The final draft of the manuscript has been reviewed, edited, and approved by all authors.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101367.
Contributor Information
Shih-Hsin Kan, Email: shih.hsin.kan@choc.org.
Philip H. Schwartz, Email: phil.schwartz60@gmail.com.
Supplemental information
References
- 1.Neufeld E.F., Muenzer J. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C., Beaudet A., Sly W., Valle D., editors. McGraw-Hill; 2001. The mucopolysaccharidoses, 8 Edition; pp. 3421–3453. [Google Scholar]
- 2.Clarke L.A. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev. Mol. Med. 2008;10 doi: 10.1017/S1462399408000550. [DOI] [PubMed] [Google Scholar]
- 3.Gentner B., Tucci F., Galimberti S., Fumagalli F., De Pellegrin M., Silvani P., Camesasca C., Pontesilli S., Darin S., Ciotti F., et al. Hematopoietic Stem- and Progenitor-Cell Gene Therapy for Hurler Syndrome. N. Engl. J. Med. 2021;385:1929–1940. doi: 10.1056/NEJMoa2106596. [DOI] [PubMed] [Google Scholar]
- 4.Kakkis E.D., Muenzer J., Tiller G.E., Waber L., Belmont J., Passage M., Izykowski B., Phillips J., Doroshow R., Walot I., et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N. Engl. J. Med. 2001;344:182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- 5.Whitley C.B., Ramsay N.K., Kersey J.H., Krivit W. Bone marrow transplantation for Hurler syndrome: assessment of metabolic correction. Birth Defects Orig. Artic. Ser. 1986;22:7–24. [PubMed] [Google Scholar]
- 6.Hampe C.S., Wesley J., Lund T.C., Orchard P.J., Polgreen L.E., Eisengart J.B., McLoon L.K., Cureoglu S., Schachern P., McIvor R.S. Mucopolysaccharidosis Type I: Current Treatments, Limitations, and Prospects for Improvement. Biomolecules. 2021;11 doi: 10.3390/biom11020189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor M., Khan S., Stapleton M., Wang J., Chen J., Wynn R., Yabe H., Chinen Y., Boelens J.J., Mason R.W., et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019;25:e226–e246. doi: 10.1016/j.bbmt.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shapiro E.G., Nestrasil I., Rudser K., Delaney K., Kovac V., Ahmed A., Yund B., Orchard P.J., Eisengart J., Niklason G.R., et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: Age, severity, and treatment. Mol. Genet. Metab. 2015;116:61–68. doi: 10.1016/j.ymgme.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermann A., Storch A. Induced neural stem cells (iNSCs) in neurodegenerative diseases. J. Neural. Transm. 2013;120:S19–S25. doi: 10.1007/s00702-013-1042-9. [DOI] [PubMed] [Google Scholar]
- 10.Wood S.R., Bigger B.W. Delivering gene therapy for mucopolysaccharide diseases. Front. Mol. Biosci. 2022;9 doi: 10.3389/fmolb.2022.965089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snyder E.Y., Taylor R.M., Wolfe J.H. Neural progenitor cell engraftment corrects lysosomal storage throughout the MPS VII mouse brain. Nature. 1995;374:367–370. doi: 10.1038/374367a0. [DOI] [PubMed] [Google Scholar]
- 12.Griffin T.A., Anderson H.C., Wolfe J.H. Ex vivo gene therapy using patient iPSC-derived NSCs reverses pathology in the brain of a homologous mouse model. Stem Cell Rep. 2015;4:835–846. doi: 10.1016/j.stemcr.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke D., Pearse Y., Kan S.H., Le S.Q., Sanghez V., Cooper J.D., Dickson P.I., Iacovino M. Genetically Corrected iPSC-Derived Neural Stem Cell Grafts Deliver Enzyme Replacement to Affect CNS Disease in Sanfilippo B Mice. Mol. Ther. Methods Clin. Dev. 2018;10:113–127. doi: 10.1016/j.omtm.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearse Y., Clarke D., Kan S.H., Le S.Q., Sanghez V., Luzzi A., Pham I., Nih L.R., Cooper J.D., Dickson P.I., Iacovino M. Brain transplantation of genetically corrected Sanfilippo type B neural stem cells induces partial cross-correction of the disease. Mol. Ther. Methods Clin. Dev. 2022;27:452–463. doi: 10.1016/j.omtm.2022.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fusar Poli E., Zalfa C., D'Avanzo F., Tomanin R., Carlessi L., Bossi M., Nodari L.R., Binda E., Marmiroli P., Scarpa M., et al. Murine neural stem cells model Hunter disease in vitro: glial cell-mediated neurodegeneration as a possible mechanism involved. Cell Death Dis. 2013;4:e906. doi: 10.1038/cddis.2013.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong J., Cheng Y.S., Yang S., Swaroop M., Xu M., Beers J., Zou J., Huang W., Marugan J.J., Cai X., Zheng W. iPS-derived neural stem cells for disease modeling and evaluation of therapeutics for mucopolysaccharidosis type II. Exp. Cell Res. 2022;412 doi: 10.1016/j.yexcr.2021.113007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rybová J., Ledvinová J., Sikora J., Kuchař L., Dobrovolný R. Neural cells generated from human induced pluripotent stem cells as a model of CNS involvement in mucopolysaccharidosis type II. J. Inherit. Metab. Dis. 2018;41:221–229. doi: 10.1007/s10545-017-0108-5. [DOI] [PubMed] [Google Scholar]
- 18.Robinson A.J., Zhao G., Rathjen J., Rathjen P.D., Hutchinson R.G., Eyre H.J., Hemsley K.M., Hopwood J.J. Embryonic stem cell-derived glial precursors as a vehicle for sulfamidase production in the MPS-IIIA mouse brain. Cell Transplant. 2010;19:985–998. doi: 10.3727/096368910X498944. [DOI] [PubMed] [Google Scholar]
- 19.Arunkumar N., Langan T.J., Stapleton M., Kubaski F., Mason R.W., Singh R., Kobayashi H., Yamaguchi S., Suzuki Y., Orii K., et al. Newborn screening of mucopolysaccharidoses: past, present, and future. J. Hum. Genet. 2020;65:557–567. doi: 10.1038/s10038-020-0744-8. [DOI] [PubMed] [Google Scholar]
- 20.Stover A.E., Brick D.J., Nethercott H.E., Banuelos M.G., Sun L., O'Dowd D.K., Schwartz P.H. Process-based expansion and neural differentiation of human pluripotent stem cells for transplantation and disease modeling. J. Neurosci. Res. 2013;91:1247–1262. doi: 10.1002/jnr.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan S.H., Martin J., Elia J., Flippin J., Paramban R.I., Hefferan M.P., Vidal J.G., Mu Y., Killian R.L., Israel M.A., et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubin J.B., Kung A.L., Klein R.S., Chan J.A., Sun Y., Schmidt K., Kieran M.W., Luster A.D., Segal R.A. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc. Natl. Acad. Sci. USA. 2003;100:13513–13518. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadri F., Rezaei Z., Fereidouni M. The significance of the SDF-1/CXCR4 signaling pathway in the normal development. Mol. Biol. Rep. 2022;49:3307–3320. doi: 10.1007/s11033-021-07069-3. [DOI] [PubMed] [Google Scholar]
- 24.Mendez D.C., Stover A.E., Rangel A.D., Brick D.J., Nethercott H.E., Torres M.A., Khalid O., Wong A.M., Cooper J.D., Jester J.V., et al. A novel, long-lived, and highly engraftable immunodeficient mouse model of mucopolysaccharidosis type I. Mol. Ther. Methods Clin. Dev. 2015;2 doi: 10.1038/mtm.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baek K.H., Choi J., Pei C.Z. Cellular Functions of OCT-3/4 Regulated by Ubiquitination in Proliferating Cells. Cancers. 2020;12 doi: 10.3390/cancers12030663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang K., Chen S., Yang Q., Guo S., Chen Q., Liu Z., Li L., Jiang M., Li H., Hu J., et al. The Oligodendrocyte Transcription Factor 2 OLIG2 regulates transcriptional repression during myelinogenesis in rodents. Nat. Commun. 2022;13:1423. doi: 10.1038/s41467-022-29068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 29.Cerneckis J., Cai H., Shi Y. Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct. Target. Ther. 2024;9:112. doi: 10.1038/s41392-024-01809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dahlberg A., Milano F. Cord blood transplantation: rewind to fast forward. Bone Marrow Transplant. 2017;52:799–802. doi: 10.1038/bmt.2016.336. [DOI] [PubMed] [Google Scholar]
- 31.Mithal D.S., Banisadr G., Miller R.J. CXCL12 signaling in the development of the nervous system. J. Neuroimmune Pharmacol. 2012;7:820–834. doi: 10.1007/s11481-011-9336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng X., Wang H., Zhang X., Zhao S., Zhou Z., Mu X., Zhao C., Teng W. The Role of SDF-1/CXCR4/CXCR7 in Neuronal Regeneration after Cerebral Ischemia. Front. Neurosci. 2017;11:590. doi: 10.3389/fnins.2017.00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guyon A. CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front. Cell. Neurosci. 2014;8:65. doi: 10.3389/fncel.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M., Hale J.S., Rich J.N., Ransohoff R.M., Lathia J.D. Chemokine CXCL12 in neurodegenerative diseases: an SOS signal for stem cell-based repair. Trends Neurosci. 2012;35:619–628. doi: 10.1016/j.tins.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C.X.Q., Abdian N., Maussion G., Thomas R.A., Demirova I., Cai E., Tabatabaei M., Beitel L.K., Karamchandani J., Fon E.A., Durcan T.M. A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types. Methods Protoc. 2021;4 doi: 10.3390/mps4030050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shultz L.D., Lyons B.L., Burzenski L.M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S.D., King M., Mangada J., et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 37.Chung S., Ma X., Liu Y., Lee D., Tittiger M., Ponder K.P. Effect of neonatal administration of a retroviral vector expressing alpha-L-iduronidase upon lysosomal storage in brain and other organs in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2007;90:181–192. doi: 10.1016/j.ymgme.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Kan S.H., Elsharkawi I., Le S.Q., Prill H., Mangini L., Cooper J.D., Lawrence R., Sands M.S., Crawford B.E., Dickson P.I. Biochemical evaluation of intracerebroventricular rhNAGLU-IGF2 enzyme replacement therapy in neonatal mice with Sanfilippo B syndrome. Mol. Genet. Metab. 2021;133:185–192. doi: 10.1016/j.ymgme.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogel S., Schäfer C., Hess S., Folz-Donahue K., Nelles M., Minassian A., Schwarz M.K., Kukat C., Ehrlich M., Zaehres H., et al. The in vivo timeline of differentiation of engrafted human neural progenitor cells. Stem Cell Res. 2019;37 doi: 10.1016/j.scr.2019.101429. [DOI] [PubMed] [Google Scholar]
- 40.Baker E.W., Platt S.R., Lau V.W., Grace H.E., Holmes S.P., Wang L., Duberstein K.J., Howerth E.W., Kinder H.A., Stice S.L., et al. Induced Pluripotent Stem Cell-Derived Neural Stem Cell Therapy Enhances Recovery in an Ischemic Stroke Pig Model. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-10406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen N.J., Lyons D.A. Glia as architects of central nervous system formation and function. Science. 2018;362:181–185. doi: 10.1126/science.aat0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng X.T., Xie Y.X., Zhou B., Huang N., Farfel-Becker T., Sheng Z.H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy. 2018;14:1472–1474. doi: 10.1080/15548627.2018.1482147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chistiakov D.A., Killingsworth M.C., Myasoedova V.A., Orekhov A.N., Bobryshev Y.V. CD68/macrosialin: not just a histochemical marker. Lab. Invest. 2017;97:4–13. doi: 10.1038/labinvest.2016.116. [DOI] [PubMed] [Google Scholar]
- 44.Ohmi K., Greenberg D.S., Rajavel K.S., Ryazantsev S., Li H.H., Neufeld E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J., et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ellison S., Liao A., Gleitz H.F.E., Parker H., Booth L., Robinson J., Wood S., Taylor J., Holley R., Bigger B.W. Sustained long-term disease correction in a murine model of MPSII following stem cell gene therapy. Mol. Ther. Methods Clin. Dev. 2023;31 doi: 10.1016/j.omtm.2023.101127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mandolfo O., Liao A., Singh E., O'leary C., Holley R.J., Bigger B.W. Establishment of the Effectiveness of Early Versus Late Stem Cell Gene Therapy in Mucopolysaccharidosis II for Treating Central Versus Peripheral Disease. Hum. Gene Ther. 2024;35:243–255. doi: 10.1089/hum.2023.002. [DOI] [PubMed] [Google Scholar]
- 48.Ellison S.M., Liao A., Wood S., Taylor J., Youshani A.S., Rowlston S., Parker H., Armant M., Biffi A., Chan L., et al. Pre-clinical Safety and Efficacy of Lentiviral Vector-Mediated. Mol. Ther. Methods Clin. Dev. 2019;13:399–413. doi: 10.1016/j.omtm.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parker H., Ellison S.M., Holley R.J., O'Leary C., Liao A., Asadi J., Glover E., Ghosh A., Jones S., Wilkinson F.L., et al. Haematopoietic stem cell gene therapy with IL-1Ra rescues cognitive loss in mucopolysaccharidosis IIIA. EMBO Mol. Med. 2020;12 doi: 10.15252/emmm.201911185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holley R.J., Ellison S.M., Fil D., O'Leary C., McDermott J., Senthivel N., Langford-Smith A.W.W., Wilkinson F.L., D'Souza Z., Parker H., et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain. 2018;141:99–116. doi: 10.1093/brain/awx311. [DOI] [PubMed] [Google Scholar]
- 51.Watson H.A., Holley R.J., Langford-Smith K.J., Wilkinson F.L., van Kuppevelt T.H., Wynn R.F., Wraith J.E., Merry C.L.R., Bigger B.W. Heparan sulfate inhibits hematopoietic stem and progenitor cell migration and engraftment in mucopolysaccharidosis I. J. Biol. Chem. 2014;289:36194–36203. doi: 10.1074/jbc.M114.599944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayani H., Wagner J.E., Broxmeyer H.E. Cord blood research, banking, and transplantation: achievements, challenges, and perspectives. Bone Marrow Transplant. 2020;55:48–61. doi: 10.1038/s41409-019-0546-9. [DOI] [PubMed] [Google Scholar]
- 53.Jones S., Kinsella J., Holley R.J., Potter J., Booth C., Buckland K., Rust S., Bromley R., Church H.J., Lee H., et al. Clinical outcomes and sustained biochemical engraftment following ex-vivo autologous stem cell gene therapy for mucopolysaccharidosis type IIIA. Mol. Genet. Metab. 2024;141 doi: 10.1016/j.ymgme.2023.107905. [DOI] [Google Scholar]
- 54.Tucci F., Gentner B., Galimberti S., Fumagalli F., Consiglieri G., Filisetti C., Darin S., Sarzana M., Scarparo S., Ciotti F., et al. First-in-human phase I/II clinical trial of hematopoietic stem and progenitor cell gene therapy for Hurler syndrome: Favorable safety profile and extensive metabolic correction. Mol. Genet. Metab. 2022;135:S121–S122. doi: 10.1016/j.ymgme.2021.11.323. [DOI] [Google Scholar]
- 55.Ellison S., Buckland K., Learmonth Y., Day V., Kalra S., Howe L., Roman-Rodriguez F.J., Bonafont J., Booth L., Holley R., et al. Design and validation of a GMP stem cell manufacturing protocol for MPSII hematopoietic stem cell gene therapy. Mol. Ther. Methods Clin. Dev. 2024;32 doi: 10.1016/j.omtm.2024.101271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orchard P.J., Wynn R.F., Kharbanda S., Ahrens-Nicklas R., van Hasselt P.M., Lund T.C., Olson T.S., Tucci F., Martin L., Boeglin N., et al. Design of a multi-center randomized active controlled phase 3 clinical trial (HURCULES) evaluating the safety and efficacy of OTL-203 in patients with MPS IH versus standard of care with allogeneic hematopoietic stem cell transplantation. Mol. Genet. Metab. 2024;141 doi: 10.1016/j.ymgme.2023.107988. [DOI] [Google Scholar]
- 57.Harmatz P., Ficicioglu C., Giugliani R., Rajan D., Hagood J., Fiscella M., Yang L., Gilmor M., Cho Y., Mulatya C., et al. CAMPSIITE™ phase I/II/III: An interim clinical study update of RGX-121, an investigational gene therapy for the treatment of neuronopathic mucopolysaccharidosis type II (MPS II) Mol. Genet. Metab. 2024;141 doi: 10.1016/j.ymgme.2023.107874. [DOI] [Google Scholar]
- 58.Wang R.Y., Movsesyan N., Kan S.-H., Beydoun T., Taylor M., Chang R.C., Phillips D., Burke J., Gilmor M., Cho Y., et al. First in-human, intracisternal dosing of RGX-111, an investigational AAV gene therapy, for a 21-month-old child with mucopolysaccharidosis type I (MPS I): 3.5 year follow-up. Mol. Genet. Metab. 2024;141 doi: 10.1016/j.ymgme.2023.108070. [DOI] [Google Scholar]
- 59.Flanigan K.M., Smith N., Couce M.L., Rajan D., Truxal K., Mcbride K.L., de Castro Lopez M.J., Fuller M., Taylor J., Del Campo A.B., et al. Interim results of Transpher A, a multicenter, single-dose clinical trial of UX111 gene therapy for Sanfilippo syndrome type A (mucopolysaccharidosis IIIA) Mol. Genet. Metab. 2023;138 doi: 10.1016/j.ymgme.2022.107101. [DOI] [Google Scholar]
- 60.Zheng C.X., Wang S.M., Bai Y.H., Luo T.T., Wang J.Q., Dai C.Q., Guo B.L., Luo S.C., Wang D.H., Yang Y.L., Wang Y.Y. Lentiviral Vectors and Adeno-Associated Virus Vectors: Useful Tools for Gene Transfer in Pain Research. Anat. Rec. 2018;301:825–836. doi: 10.1002/ar.23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J.H., Gessler D.J., Zhan W., Gallagher T.L., Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct. Target. Ther. 2024;9:78. doi: 10.1038/s41392-024-01780-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials are available upon reasonable request.