

Abstract
Objectives
To investigate the effects of suberoylanilide hydroxamic acid (SAHA) on lung fibroblast activation and to examine the role of p66Shc in this process.
Methods
An in vitro pulmonary fibrosis model was established using transforming growth factor‐β (TGF‐β)‐induced MRC‐5 lung fibroblasts. The proliferation and migration capacities of MRC‐5 cells, along with the expression of fibrosis‐related genes, were assessed following treatment with SAHA and/or silence of p66Shc.
Results
In TGF‐β‐induced MRC‐5 lung fibroblasts, SAHA treatment significantly inhibited cell proliferation and migration, as well as the expression of fibrosis‐related genes, including collagen I and α‐smooth muscle actin (SMA). Western blot and immunofluorescence assays revealed that SAHA increased p66Shc expression in both whole cells and mitochondria. Additionally, mito‐SOX assay confirmed that SAHA treatment led to a marked accumulation of mitochondrial reactive oxygen species (ROS). However, silencing of p66Shc significantly reversed the aforementioned effects of SAHA on MRC‐5 cells. Furthermore, chromatin immunoprecipitation (ChIP) assays demonstrated that SAHA enhanced active histone markers, H3K9Ac and H3K4Me3, in the p66Shc gene region.
Conclusions
SAHA alleviates lung fibroblast activation and migration by increasing p66Shc expression and mitochondrial ROS generation through epigenetic modifications of histone 3.
Keywords: histone modification, lung fibroblasts, p66Shc, pulmonary fibrosis, SAHA
Suberoylanilide hydroxamic acid (SAHA) inhibited proliferation, activation, and migration of lung fibroblasts by increasing mitochondrial reactive oxygen species (ROS) and promoting p66Shc expression via epigenetic modifications of histone 3, including H3K4Me3 and H3K9Ac.
1. INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and fatal lung disease characterized by chronic, repetitive alveolar epithelium injuries, excessive activation of lung fibroblasts, and uncontrolled deposition of extracellular matrix (ECM), for which there are currently no effective therapeutic options. 1 IPF is closely associated with aging, 2 and aging is a major non‐modifiable risk factor for IPF. 3 It has been demonstrated that mutations in the telomerase gene and telomere shortening are major pathogenic causes of IPF, 4 with telomeres being closely linked to human aging and lifespan. An analysis of the British Thoracic Society electronic registry, covering the years 2013 to 2019, showed that the proportion of patients with IPF aged 70 and older increased to 80%. 5 Transforming growth factor‐β (TGF‐β) plays a central role in mediating lung fibroblast activation and pulmonary fibrosis and is therefore considered a potential therapeutic target. 6 However, directly targeting TGF‐β presents many challenges and may lead to unexpected side effects. 7 Unraveling alternative mechanisms of lung fibroblast activation is crucial for the development of new anti‐fibrotic agents.
Numerous studies have demonstrated that epigenetics plays a critical role in pulmonary fibrosis. 8 , 9 The well‐researched mechanisms of histone acetylation and DNA methylation are classical modifications of histone tails that regulate chromatin accessibility. 10 Histone deacetylases (HDACs) remove acetyl groups from histone tails, leading to chromatin compaction. 9 Multiple studies have confirmed that the expression of various HDACs changes significantly with aging. 11 , 12 , 13 Preclinical studies have also demonstrated that HDAC inhibitors can significantly delay aging, extend lifespan, and reduce the occurrence of aging‐related phenotypes in various organs and tissues in model animals. 11 , 12 , 13 Additionally, in animal models and clinical specimens, increased expression of HDACs has been positively correlated with the severity of fibrosis in several organs, including the kidneys, 14 , 15 heart, 16 , 17 and lungs. 18 , 19 , 20 , 21 The broad‐spectrum HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) has been approved by the U.S. Food and Drug Administration (FDA) as an anti‐cancer drug. Previous studies have suggested the therapeutic potential of SAHA in treating organ fibrosis. 22 , 23 , 24 Our preliminary work also revealed that SAHA can downregulate collagen 3A1 in primary fibroblasts isolated from patients with IPF, as well as in a bleomycin‐induced pulmonary fibrosis mouse model. 25 However, the specific mechanisms remain unclear.
P66Shc is a member of the Src homology and collagen (Shc) family, involved in various pathophysiological processes, including reactive oxygen species (ROS) production, cell proliferation, apoptosis, 26 as well as organ fibrosis, such as liver and renal fibrosis. 27 , 28 Furthermore, there is substantial evidence indicating a complex relationship between p66Shc and aging. 26 Trichostatin A, an HDAC inhibitor, has been shown to enhance p66Shc promoter activity and induce p66Shc expression. 29 Numerous studies have highlighted the potential of HDAC inhibitors in preventing pulmonary fibrosis. However, no studies to date have investigated the effects of SAHA on p66Shc expression in lung fibrosis.
In this study, we aimed to investigate the potential of SAHA in preventing fibroblast activation. To achieve this, we used MRC‐5 lung fibroblasts treated with TGF‐β to establish an in vitro pulmonary fibrosis model. We then focus on the specific mechanisms by which SAHA exerts its anti‐fibrotic effects, particularly its regulation of p66Shc expression and the associated histone modification changes.
2. MATERIALS AND METHODS
2.1. Cell culture and treatment
Human embryonic lung fibroblasts (MRC‐5) were purchased from the Chinese Academy of Sciences (Cat. no. GNHu41, Shanghai, China). The cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco, USA) and antibiotics (penicillin and streptomycin, Life Technologies/Gibco, Gaithersburg, MD). The cells were maintained at 37°C in a 5% CO2 atmosphere. MRC‐5 cells were treated with 2 ng/mL TGF‐β1 and SAHA for 24 h.
2.2. Western blot analysis
RIPA buffer was used to extract total cellular protein. A BCA Protein Assay Kit (Thermo Scientific, United States) was employed to determine the protein concentration of the cell lysates. Equal amounts of protein were loaded onto 10% SDS‐PAGE gels and separated via electrophoresis. The separated proteins were then transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% fat‐free milk for 1 h at room temperature, followed by overnight incubation with primary antibodies at 4 °C. Subsequently, the membranes were incubated with horseradish peroxidase‐conjugated secondary antibodies for 1 h at room temperature. Immunoblots were visualized using an Amersham Biosciences 600 imager and quantified through ImageJ software. The antibodies used in this study included anti‐α‐smooth muscle actin (SMA) (Cell Signaling Technology, #19245), anti‐collagen I (Cell Signaling Technology, #84336), anti‐GAPDH (Cell Signaling Technology, #2118), anti‐p66Shc (Cell Signaling Technology, #610878), anti‐H3K27Me3 (Cell Signaling Technology, #3108), anti‐H3K9Ac (Cell Signaling Technology, #61251), anti‐H3K18ac (Cell Signaling Technology, #13998), anti‐H3K27ac (Cell Signaling Technology, #8173), anti‐H3K4Me3 (Cell Signaling Technology, #9751), anti‐H3K9Me3 (Cell Signaling Technology, #13969), anti‐H3K79Me3 (Cell Signaling Technology, #74073), anti‐H3 (Cell Signaling Technology, #4499), goat anti‐rabbit IgG H&L (Beyotime, A0208), and rabbit anti‐mouse IgG H&L (Beyotime, A0216).
2.3. RNA extraction and qRT‐PCR
TRIzol reagent (Sangon Biotech, Shanghai, China) was used to extract messenger RNA, which was then reverse transcribed into cDNA using a Revert Aid First Stand cDNA synthesis Kit (Thermo Scientific, United States). qRT‐PCR was performed using a SYBR Green/qPCR Master Mix kit (Thermo Scientific, United States). All results were normalized to GAPDH using the ΔΔCt method. Primers used for qRT‐PCR analysis are listed in Table 1.
TABLE 1.
Primers used for qRT‐PCR analysis.
| Gene name | Sequence |
|---|---|
| P66Shc | F: 5′‐TGAGGGTGTGGTTCGGACTAAGG‐3′ |
| R: 5′‐CCGCAGAGATGATGGGCAAGTG‐3′ | |
| α‐SMA | F: 5′‐CTATGAGGGCTATGCCTTGCC‐3′ |
| R: 5′‐GCTCAGCAGTAGTAACGAAGGA‐3′ | |
| Collagen I | F: 5′‐GAGGGCCAAGACGAAGACATC‐3′ |
| R: 5′‐CAGATCACGTCATCGCACAAC‐3′ | |
| GAPDH | F: 5′‐CCCATGTTCGTCATGGGTGT‐3′ |
| R: 5′‐TGGTCATGAGTCCTTCCACGATA‐3′ |
2.4. Immunofluorescence Staining
MRC‐5 cells were cultured on coverslips and treated with TGF‐β1 in the absence or presence of SAHA for 24 h. After treatment, the cells were fixed with 4% paraformaldehyde (PFA) at room temperature to preserve structure, permeabilized with 0.1% Triton X‐100, blocked with 10% normal goat serum, and incubated with anti‐collagen I or anti‐α‐SMA antibodies overnight at 4°C. After washing with TBS containing 0.1% Tween, the cells were incubated with goat anti‐rabbit secondary antibodies (Thermo Scientific, United States, Alexa Fluor 594 or 488 conjugate). Fluorescence images were captured using a fluorescence microscope.
2.5. 5‐Ethynyl‐2′‐deoxyuridine (EdU) Incorporation Assay
MRC‐5 cells were treated with different concentrations of SAHA for 24 h and then incubated with EdU for 2 h. Subsequently, 4% PFA was used to fix the cells, followed by incubation with a Click Additive Solution for 30 min. Immunofluorescence signals were captured using a laser scanning confocal microscope (Leica, TCS SP8, Wetzlar, Germany).
2.6. Wound‐healing assay
Cells were seeded in six‐well plates and cultured until they reached 70–80% confluence. A linear gap was created by scratching the cell monolayer with a sterile 200‐μL pipette tip under microscope. After treatment, images of the wound were captured using a microscope and quantitatively evaluated with Image J software.
2.7. Chromatin immunoprecipitation (ChIP) assay
MRC‐5 cells were treated with 1% formaldehyde for 10 min to crosslink proteins and then lysed. Sonication was performed to shear the chromatin in the lysate. Extracts were immunoprecipitated with H3K27Me3 and H3K9Ac antibodies or normal rabbit IgG (Santa Cruz Biotechnology), and protein–DNA complexes were pulled down. After reversing crosslinks, DNA was purified using a PCR purification kit and dissolved in 100 μL of elution buffer. A 2‐μL DNA sample was used for each qPCR reaction with the real‐time PCR primers.
2.8. Statistical analysis
The results of at least three independent replicate experiments for each assay are expressed as means ± standard deviation. Statistical analysis was performed using Student's unpaired t test for two‐group comparisons and one‐way ANOVA test for multi‐group comparisons, both conducted with GraphPad Prism.
3. RESULTS
3.1. SAHA inhibits activation and migration of lung fibroblasts
MRC‐5 cells were treated with different concentrations of SAHA (0, 100, 200, 500, 1000, and 2000 nmol/L) for 24 h to determine the appropriate dose. Cell viability showed no significant differences at SAHA concentration below 500 nmol/L compared with the control. Therefore, 100, 200, and 500 nmol/L of SAHA were used in subsequent experiments (Figure 1A).
FIGURE 1.
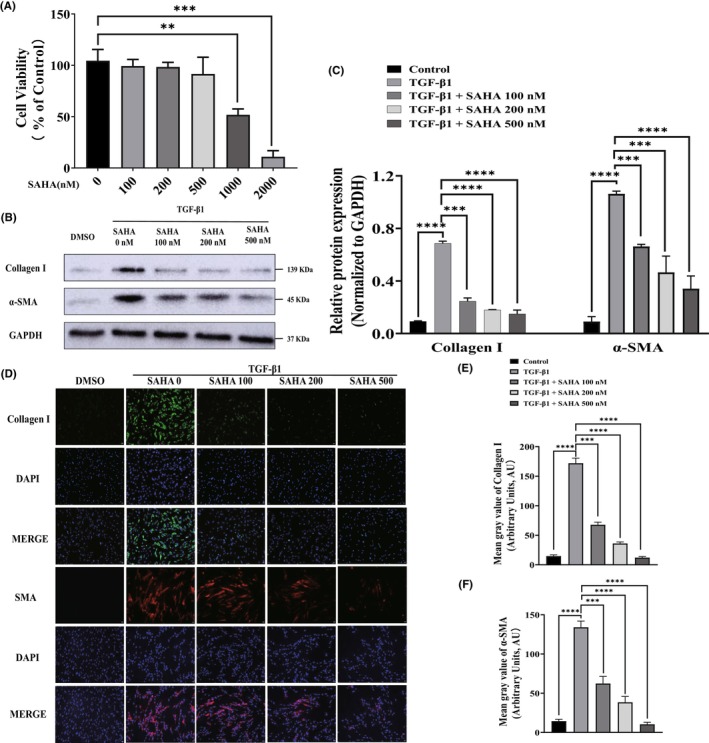
SAHA inhibited TGF‐β1‐induced activation of lung fibroblasts. (A) The viability of MRC‐5 cells incubated with different concentrations of SAHA for 24 h (n = 3). (B, C) Western blot analysis and quantification of collagen I and α‐SMA protein levels in MRC‐5 cells treated with different concentrations of SAHA for 24 h in the presence of TGF‐β1 (n = 3). (D, E, and F) Immunofluorescence images and quantification of collagen I (green) and α‐SMA (red) in TGF‐β1‐induced MRC‐5 cells treated with different concentrations of SAHA (n = 3). The bars represent the mean ± SD of three separate experiments. * indicates p < 0.050, ** indicates p < 0.010, *** indicates p < 0.001, **** indicates p < 0.0001.
To investigate whether SAHA could inhibit the activation of lung fibroblasts after TGF‐β stimulation, the expression of fibrosis‐related genes, collagen I, and α‐SMA were analyzed via Western blot in MRC‐5 cells treated with 100, 200, and 500 nmol/L SAHA for 24 h in the presence of 2 ng/mL TGF‐β1. The results showed that SAHA treatment significantly downregulated the increased expression of collagen I and α‐SMA compared to TGF‐β group (Figure 1B,C), which was further confirmed by immunofluorescence assays (Figure 1D).
Wound healing and tissue fibrosis are driven by cell proliferation, migration, and ECM deposition. 30 The effect of SAHA on the proliferation of MRC‐5 cells was evaluated through the CCK‐8 assay and EdU incorporation assay. As presented in Figure 2A, TGF‐β1 significantly stimulated the proliferation of MRC‐5 cells, which was inhibited by SAHA in a concentration‐dependent manner. Similarly, the percentage of EdU‐positive cells decreased in a dose‐dependent manner in groups treated with SAHA compared to TGF‐β1 (Figure 2B,C). Next, wound healing assays were used to test the effects of SAHA on the migration of MRC‐5 cells. The results showed that SAHA markedly and dose‐dependently inhibited the migration of MRC‐5 cells (Figure 2D,F). Together, these results indicate that SAHA can inhibit the proliferation and migration of TGF‐β1‐stimulated MRC‐5 cells.
FIGURE 2.
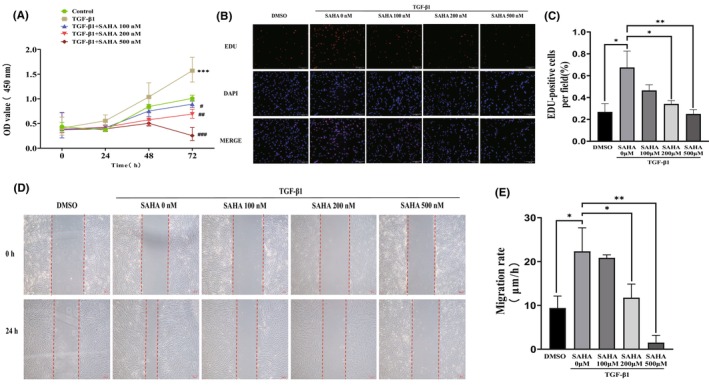
SAHA inhibits TGF‐β1‐induced proliferation and migration of MRC‐5 cells. (A) The CCK‐8 assay of MRC‐5 cells exposed to different concentrations of SAHA for 24, 48, and 72 h in the presence of TGF‐β1 (n = 3). (B, C) Fluorescence images and quantification of EdU incorporation assay in MRC‐5 cells co‐cultured with TGF‐β1 and different concentrations of SAHA for 24 h (n = 3). (D, E) Images and quantification of wound healing assay in MRC‐5 cells treated with TGF‐β1 and different concentrations of SAHA for 24 h (n = 3). The bars represent the mean ± SD of three separate experiments. * indicates p < 0.050, ** indicates p < 0.010, *** indicates p < 0.001, **** indicates p < 0.0001. # indicates p < 0.050, ## indicates p < 0.010, ### indicates p < 0.001.
3.2. SAHA increases mitochondrial ROS and the expression of p66Shc
Pulmonary fibrosis has been demonstrated to be associated with oxidative stress. To assess the effect of SAHA on mitochondrial ROS generation of MRC‐5 cells induced by TGF‐β1, we loaded MRC‐5 cells with mito‐SOX, a fluorescent probe targeting mitochondria ROS in viable cells. We found that SAHA dose dependently increased mitochondrial ROS induced by TGF‐β1 (Figure 3A,B). p66Shc, a master regulator of mitochondrial ROS, is a crucial mediator of oxidative stress. It has been demonstrated that p66Shc plays an important role in oxidative stress by regulating the generation of mitochondrial ROS. Therefore, we aimed to test whether SAHA influences the expression of p66Shc in MRC‐5 cells induced by TGF‐β1 and found that both whole‐cell and mitochondrial p66Shc expression (Figure 4A,B,D) and p66shc mRNA (Figure 4C) levels were stimulated by different concentrations of SAHA.
FIGURE 3.
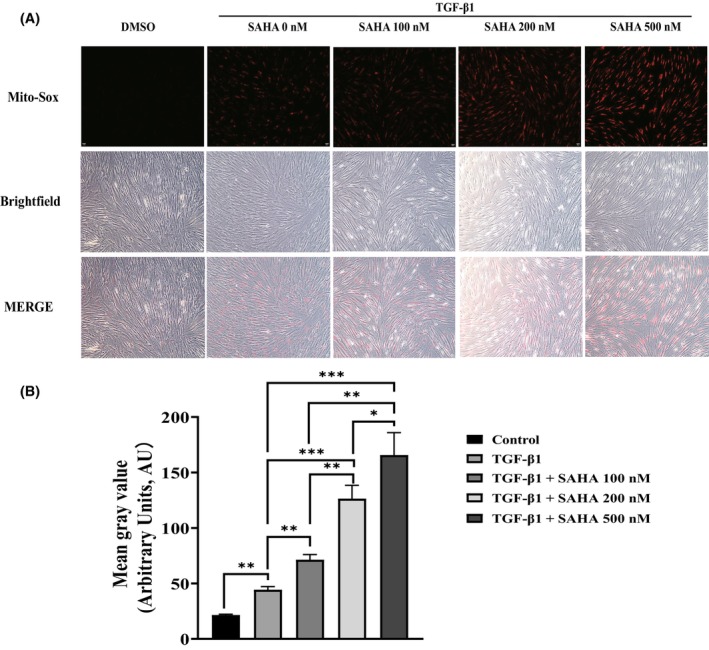
SAHA increases mitochondrial ROS in MRC‐5 cells. (A) Fluorescence and bright‐field images of MRC‐5 cells treated with mito‐SOX red after exposure to TGF‐β1 and different concentrations of SAHA (n = 3). (B) Quantification of mean gray value of mito‐SOX (n = 3). The bars represent the mean ± SD of three separate experiments. * indicates p < 0.050, ** indicates p < 0.010, *** indicates p < 0.001.
FIGURE 4.
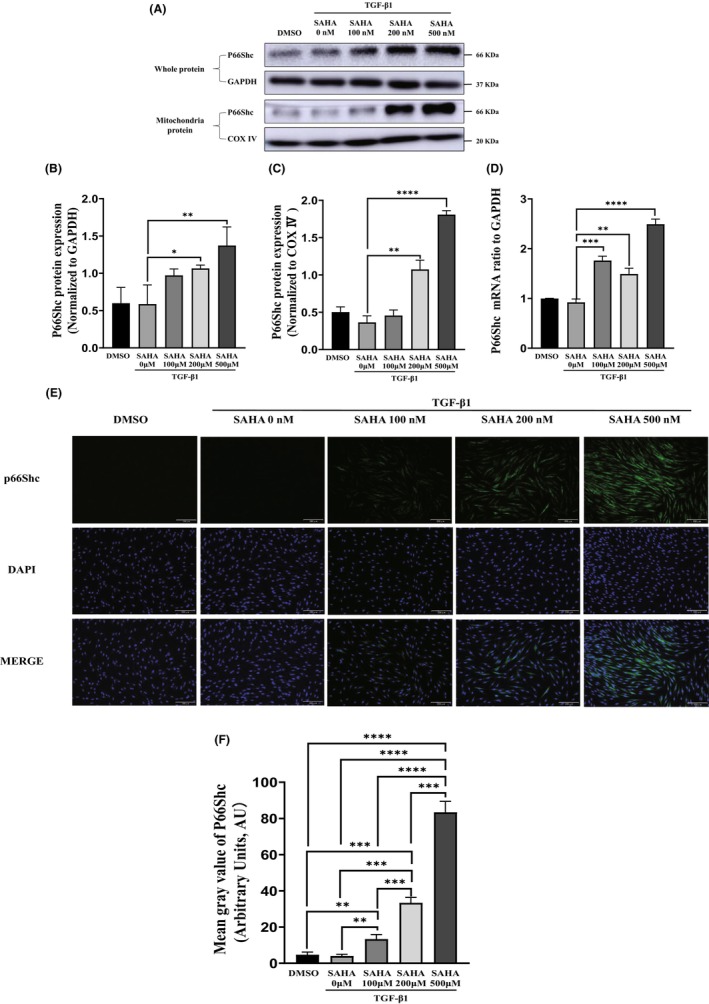
SAHA increases the expression of p66Shc. (A, B, C, and D) Protein expression (both whole‐cell protein and mitochondrial protein) and mRNA expression of p66Shc in MRC‐5 cells treated with TGF‐β1 and different concentrations of SAHA (n = 3). (E, F) Immunofluorescence images and quantification of p66Shc (green) in MRC‐5 cells treated with TGF‐β1 and different concentrations of SAHA (n = 3). The bars represent the mean ± SD of three separate experiments. * indicates p < 0.050, ** indicates p < 0.010, *** indicates p < 0.001, **** indicates p < 0.0001.
3.3. p66Shc silencing reverses the effects of SAHA
To further confirm that p66Shc mediates the effects of SAHA on lung fibroblasts induced by TGF‐β, MRC‐5 cells were transfected with three p66Shc siRNAs, and knockdown efficiency was evaluated via Western blot and qRT‐PCR. All the siRNAs effectively silenced p66Shc, and we selected one siRNA for subsequent experiments (Figure 5A–C).
FIGURE 5.
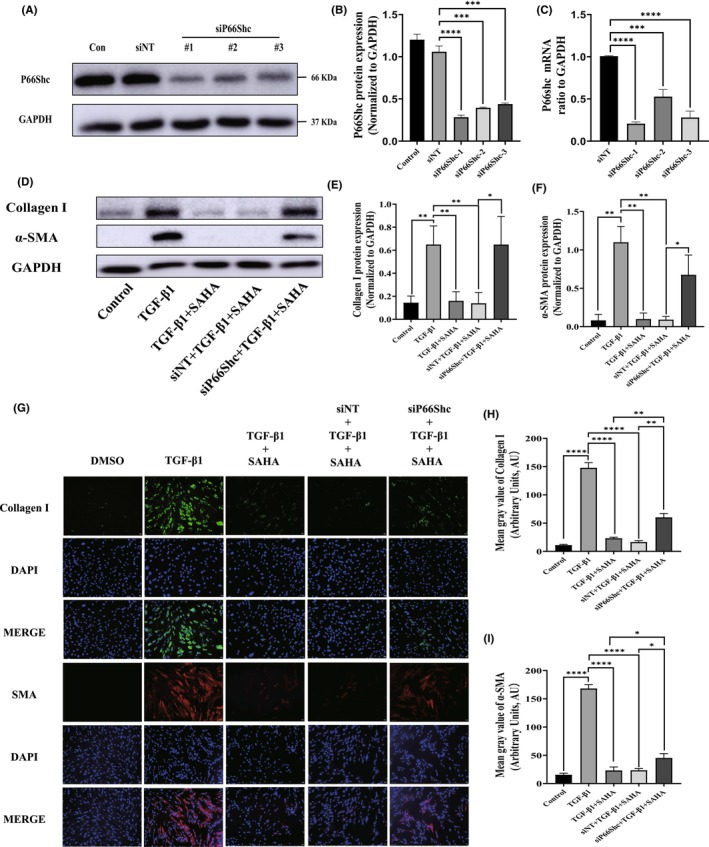
Silencing p66Shc inhibits the differentiation of lung fibroblasts. (A, B, and C) Protein and mRNA expression of p66Shc in MRC‐5 cells transfected with either NC siRNA or p66Shc siRNA for 24 h (n = 3). (D, E, and F) Protein and mRNA expression of collagen I and α‐SMA in MRC‐5 cells transfected with either NC siRNA or p66Shc siRNA for 24 h in the presence of TGF‐β1 and SAHA (n = 3). (G, H, and I) Immunofluorescence images and quantification of collagen I (green) and α‐SMA (red) in MRC‐5 cells (n = 3). The bars represent the mean ± SD of three separate experiments. * indicates p < 0.050, ** indicates p < 0.010, *** indicates p < 0.001, **** indicates p < 0.0001.
After transfection with p66Shc siRNA, the effects of SAHA on expression of collagen I and α‐SMA protein and mRNA were significantly reversed compared to SAHA and TGF‐β1 co‐treatment group (Figure 5D–G). The p66Shc knockdown also significantly promoted the proliferation of MRC‐5 cells that had been inhibited by SAHA (Figure 6A,B). Next, wound healing assays were used to test the effects of p66Shc on the migration of MRC‐5 cells treated by SAHA and TGF‐β1. After p66Shc siRNA treatment, the migratory ability dramatically increased compared to the SAHA and TGF‐β1 co‐treatment group (Figure 6C). These results indicated that p66Shc mediates the effects of SAHA on lung fibroblasts.
FIGURE 6.
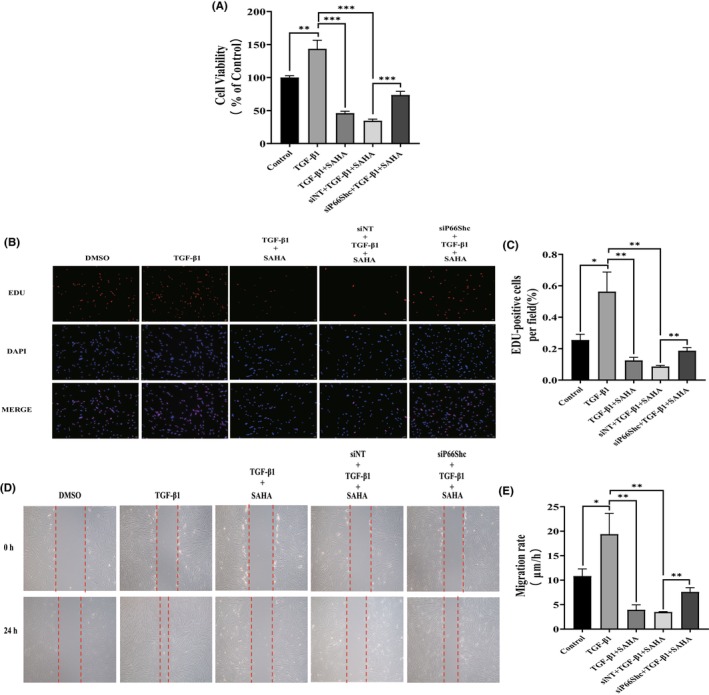
Silencing p66Shc inhibits the proliferation and migration of lung fibroblasts. (A) Cell viability of MRC‐5 cells exposed to SAHA and TGF‐β1, and transfected with either NC siRNA or p66Shc siRNA for 24 h (n = 3). (B and C) Immunofluorescence images and statistical analysis of EdU incorporation assay in MRC‐5 cells transfected with either NC siRNA or p66Shc siRNA and co‐cultured with TGF‐β1 and SAHA (n = 3). (D and E) Images and statistical analysis of the wound healing assay in MRC‐5 cells transfected with either NC siRNA or p66Shc siRNA and treated with TGF‐β1 and SAHA (n = 3). The bars represent the mean ± SD of three separate experiments. * indicates p < 0.050, ** indicates p < 0.010, *** indicates p < 0.001, **** indicates p < 0.0001.
3.4. Histone modifications mediate the upregulation of p66Shc expression by SAHA
Histone modification, including acetylation and methylation, is essential for the gene regulation of most biological processes. 31 It has been found that prolonged SAHA treatment regulates modification of histone acetylation, which is associated with the expression of apoptosis‐related genes in lung fibroblasts isolated from patients with IPF24. In this study, results from Western blot also showed that TGF‐β1 and SAHA could alter the levels of histone acetylation and methylation in lung fibroblasts (Figure 7A–D). The expression of p66Shc has been found to be regulated by histone acetylation and methylation. 29 , 32 To explore the underlying mechanism by which p66Shc is upregulated upon SAHA treatment, ChIP‐qPCR was performed. The results showed that SAHA treatment resulted in an enrichment of H3K9Ac and H3K4Me3 in the region of the p66Shc gene (Figure 7E). Previous studies have demonstrated both H3K9Ac and H3K4Me3 are markers of transcription activation. 33 , 34 , 35 Therefore, we speculate that SAHA increases p66shc expression through histone modifications.
FIGURE 7.
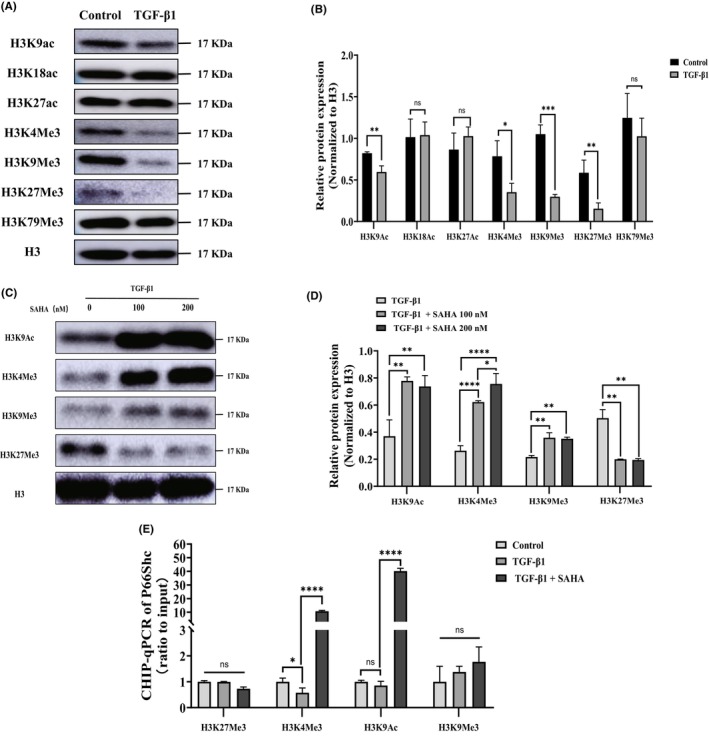
Histone modifications mediate the upregulation of p66Shc expression by SAHA. (A, B, C, and D) Acetylation and methylation modifications of histone H3 in lung fibroblasts after treatment with TGF‐β1 and SAHA (n = 3). (E) Modification of histone H3 in the region of the p66Shc gene after TGF‐β1 and SAHA treatment (n = 3). The bars represent the mean ± SD of three separate experiments. * indicates p < 0.050, ** indicates p < 0.010, *** indicates p < 0.001, **** indicates p < 0.0001.
4. DISCUSSION
In the present study, we demonstrated that SAHA inhibits lung fibroblast activation and migration by increasing the expression of p66Shc. More specifically, SAHA promotes mitochondrial ROS generation and augments p66Shc expression by elevating levels of H3K9Ac and H3K4Me3—active histone markers—in the region of the p66Shc gene in lung fibroblast. However, silencing p66Shc significantly reversed the aforementioned effects of SAHA on lung fibroblasts. Taken together, our study suggests that SAHA plays a central role in pulmonary fibrosis, and pharmacological targeting of p66Shc may offer a novel approach to the treatment of lung fibrosis.
Pulmonary fibrosis is characterized by fibroblast activation and ECM deposition, accompanied by the destruction of tissue structure. 36 Currently, the pathogenesis of pulmonary fibrosis remains unclear, and there are limited therapeutic targets and drugs available. It is urgent to find effective treatment strategies for this fatal disease. HDACs, as an important class of epigenetic‐modifying enzymes, are involved in the onset and progression of aging and fibrosis in various tissues and organs. 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 HDAC inhibitors, including SAHA, have been shown to have anti‐aging and anti‐fibrotic effects in various animal models. 11 , 12 , 13 SAHA has been approved by the FDA for anti‐cancer treatment. Interestingly, a published study found that SAHA can suppress TGF‐β‐induced cell proliferation and differentiation, as well as ECM production, by inhibiting both Smad and non‐Smad signal pathways in human conjunctival fibroblast cells. 37 Additionally, it has been reported that SAHA can rescue inflammatory cystic fibrosis by modulating immune responses. 38 Treatment with 5 μM SAHA for 24 h significantly decreased collagen production in lung fibroblasts derived from both normal human subjects and patients with IPF, without pro‐apoptotic effects, 23 which aligns with our mitochondrial membrane potential assays (data not shown). However, treatment with SAHA for an extended period (more than 60 h) significantly induced apoptosis of lung fibroblasts derived from IPF patients through histone modifications and DNA methylation, while also attenuating bleomycin‐induced pulmonary fibrosis. 24 As a result, SAHA appears to have anti‐fibrotic potential for IPF treatment.
It has been demonstrated that low levels of oxygen free radicals can stimulate the proliferation of human fibroblasts. 39 Lung fibroblasts isolated from patients with IPF also showed increased production of mitochondrial ROS, which contributed to the senescence of these pathological cells and may be associated with their resistance to apoptosis. 40 Our results also suggest that TGF‐β can induce a slight increase in mitochondrial ROS. However, excessive accumulation of mitochondrial ROS can damage fibroblasts and induce apoptosis. 41 , 42 , 43 SAHA promotes apoptosis in cancer cells by increasing ROS production. 44 In this study, we showed that SAHA stimulates a dramatic increase in mitochondrial ROS in lung fibroblasts, which was negatively correlated with decreased cell viability and the abilities of proliferation, differentiation, and migration. P66Shc, a protein located in mitochondria, is involved in ROS production. Once p66Shc translocates into the mitochondria, it affects mitochondrial cristae structure and function by oxidizing cytochrome C and catalyzing the reduction of oxygen to hydrogen peroxide. 45 Meanwhile, p66Shc has a fascinating impact on lifespan and aging. Numerous studies have confirmed that p66Shc promotes aging in various tissues and organs by regulating oxidative stress and inducing apoptosis. 26 Interestingly, some studies have found that knockout of the p66Shc gene can significantly increase lifespan in model organisms and enhance their resilience to various pathological damages. 26 However, other studies have shown that p66Shc knockout does not extend lifespan but only alleviates organ atrophy, infertility, and weight loss. 26 Additionally, one study found that p66Shc knockout in mice accelerates lung aging. 46 Furthermore, the experiment did not investigate mice older than 24 months, so the possibility of further deterioration in lung function in p66Shc knockout mice cannot be ruled out. The underlying biological mechanisms remain unclear and may be related to increased levels of lung inflammation. 46 In contrast, in vitro studies have shown that microRNA let‐7a can delay replicative senescence of human diploid fibroblasts by inhibiting the expression of p66Shc. 47 Therefore, we further explored the effect of SAHA on the expression of p66Shc and found that SAHA increases the expression of p66Shc in both whole cell and mitochondria. Silencing p66Shc reversed the inhibition of SAHA on the proliferation, differentiation, and migration of lung fibroblasts. However, previous studies have suggested that p66Shc mediates the activation of hepatic stellate cells and liver fibrosis through mitochondrial ROS and NOD‐like receptor protein 3 inflammasome activation. 27 The discrepancy in p66Shc function regarding organ fibrosis between these two studies may stem from the different disease models used, which need validation in future research.
Gene expression can be governed by epigenetic mechanisms. Several HDAC inhibitors have been identified as effective anti‐fibrotic agents in various forms of organ fibrosis, such as givinostat and trichostatin A. Givinostat effectively attenuates liver fibrosis by regulating lipid metabolism‐related genes, 48 while trichostatin A has been shown to ameliorate renal tubulointerstitial fibrosis through the JNK‐dependent Notch‐2 signaling pathway. 49 Additionally, trichostatin A was found to increase the expression of p66Shc by upregulating the activity of p66Shc promoter. 29 Previous reports indicate that SAHA regulates gene expression by modulating histone acetylation and methylation, thereby influencing chromatin accessibility. 50 In our work, SAHA was shown to increase the active histone markers H3K9Ac35 and H3K4Me3 33 , 34 in the region of the p66Shc gene.
Collectively, this study demonstrated that SAHA ameliorates pulmonary fibrosis through increasing expression of p66Shc and production of ROS. The mechanism by which p66Shc expression was modulated involved histone modification. Since SAHA has already been approved for cancer treatment, it might serve as a potential therapeutic drug for treatment of pulmonary fibrosis.
AUTHOR CONTRIBUTIONS
Qiong Wang and Xing Lyu proposed the idea. Yiheng Dong and Jieting Peng conducted experiments. Yiheng Dong, Jieting Peng, and Xiangyu Zhang analyzed data and performed statistical analysis. Yiheng Dong and Jieting Peng wrote the manuscript. Qiong Wang and Xing Lyu reviewed and revised the manuscript. All authors have given consent to the publication of this study.
FUNDING INFORMATION
This paper was funded by the Natural Science Foundation of Hunan Province [General Program, No. 2024JJ5489].
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
The authors have nothing to report.
Dong Y, Peng J, Zhang X, Wang Q, Lyu X. SAHA inhibits lung fibroblast activation by increasing p66Shc expression epigenetically. Aging Med. 2024;7:790‐801. doi: 10.1002/agm2.12385
Contributor Information
Qiong Wang, Email: wangqiong4324@csu.edu.cn.
Xing Lyu, Email: xinglyu@csu.edu.cn.
REFERENCES
- 1. Spagnolo P, Kropski JA, Jones MG, et al. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798. doi: 10.1016/j.pharmthera.2020.107798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cho SJ, Stout‐Delgado HW. Aging and Lung Disease. Annu Rev Physiol. 2020;82:433‐459. doi: 10.1146/annurev-physiol-021119-034610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol. 2022;17:515‐546. doi: 10.1146/annurev-pathol-042320-030240 [DOI] [PubMed] [Google Scholar]
- 4. Alder JK, Armanios M. Telomere‐mediated lung disease. Physiol Rev. 2022;102:1703‐1720. doi: 10.1152/physrev.00046.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spencer LG, Loughenbury M, Chaudhuri N, Spiteri M, Parfrey H. Idiopathic pulmonary fibrosis in the UK: analysis of the British Thoracic Society electronic registry between 2013 and 2019. ERJ Open Res. 2021;7:00187‐2020. doi: 10.1183/23120541.00187-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ong CH, Tham CL, Harith HH, Firdaus N, Israf DA. TGF‐β‐induced fibrosis: a review on the underlying mechanism and potential therapeutic strategies. Eur J Pharmacol. 2021;911:174510. doi: 10.1016/j.ejphar.2021.174510 [DOI] [PubMed] [Google Scholar]
- 7. Tsukui T, Wolters PJ, Sheppard D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature. 2024;631:627‐634. doi: 10.1038/s41586-024-07660-1 [DOI] [PubMed] [Google Scholar]
- 8. Zhang X, Liu H, Zhou JQ, et al. Modulation of H4K16Ac levels reduces pro‐fibrotic gene expression and mitigates lung fibrosis in aged mice. Theranostics. 2022;12:530‐541. doi: 10.7150/thno.62760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sivashanmugam K, Kandasamy M, Subbiah R, Ravikumar V. Repurposing of histone deacetylase inhibitors: a promising strategy to combat pulmonary fibrosis promoted by TGF‐β signalling in COVID‐19 survivors. Life Sci. 2021;266:118883. doi: 10.1016/j.lfs.2020.118883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramzan F, Vickers MH, Mithen RF. Epigenetics, microRNA and metabolic syndrome: a comprehensive review. Int J Mol Sci. 2021;22:5047. doi: 10.3390/ijms22095047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pasyukova EG, Vaiserman AM. HDAC inhibitors: a new promising drug class in anti‐aging research. Mech Ageing Dev. 2017;166:6‐15. doi: 10.1016/j.mad.2017.08.008 [DOI] [PubMed] [Google Scholar]
- 12. Pasyukova EG, Symonenko AV, Rybina OY, Vaiserman AM. Epigenetic enzymes: a role in aging and prospects for pharmacological targeting. Ageing Res Rev. 2021;67:101312. doi: 10.1016/j.arr.2021.101312 [DOI] [PubMed] [Google Scholar]
- 13. McIntyre RL, Daniels EG, Molenaars M, Houtkooper RH, Janssens GE. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol Med. 2019;11:e9854. doi: 10.15252/emmm.201809854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen F, Gao Q, Wei A, et al. Histone deacetylase 3 aberration inhibits klotho transcription and promotes renal fibrosis. Cell Death Differ. 2021;28:1001‐1012. doi: 10.1038/s41418-020-00631-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, Yang Y, Yang F, et al. HDAC9‐mediated epithelial cell cycle arrest in G2/M contributes to kidney fibrosis in male mice. Nat Commun. 2023;14:3007. doi: 10.1038/s41467-023-38771-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Travers JG, Wennersten SA, Peña B, et al. HDAC inhibition reverses preexisting diastolic dysfunction and blocks covert extracellular matrix remodeling. Circulation. 2021;143:1874‐1890. doi: 10.1161/circulationaha.120.046462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weng L, Ye J, Yang F, et al. TGF‐β1/SMAD3 regulates programmed cell death 5 that suppresses cardiac fibrosis post‐myocardial infarction by inhibiting HDAC3. Circ Res. 2023;133:237‐251. doi: 10.1161/circresaha.123.322596 [DOI] [PubMed] [Google Scholar]
- 18. Korfei M, Skwarna S, Henneke I, et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax. 2015;70:1022‐1032. doi: 10.1136/thoraxjnl-2014-206411 [DOI] [PubMed] [Google Scholar]
- 19. Huang SK, Scruggs AM, Donaghy J, et al. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013;4:e621. doi: 10.1038/cddis.2013.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li M, Zheng Y, Yuan H, Liu Y, Wen X. Effects of dynamic changes in histone acetylation and deacetylase activity on pulmonary fibrosis. Int Immunopharmacol. 2017;52:272‐280. doi: 10.1016/j.intimp.2017.09.020 [DOI] [PubMed] [Google Scholar]
- 21. Chen F, Gao Q, Zhang L, Ding Y, Wang H, Cao W. Inhibiting HDAC3 (histone deacetylase 3) aberration and the resultant Nrf2 (nuclear factor erythroid‐derived 2‐related Factor‐2) repression mitigates pulmonary fibrosis. Hypertension. 2021;78:e15‐e25. doi: 10.1161/hypertensionaha.121.17471 [DOI] [PubMed] [Google Scholar]
- 22. Yoon S, Kang G, Eom GH. HDAC inhibitors: therapeutic potential in fibrosis‐associated human diseases. Int J Mol Sci. 2019;20:1329. doi: 10.3390/ijms20061329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Z, Chen C, Finger SN, et al. Suberoylanilide hydroxamic acid: a potential epigenetic therapeutic agent for lung fibrosis? Eur Respir J. 2009;34:145‐155. doi: 10.1183/09031936.00084808 [DOI] [PubMed] [Google Scholar]
- 24. Sanders YY, Hagood JS, Liu H, Zhang W, Ambalavanan N, Thannickal VJ. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J. 2014;43:1448‐1458. doi: 10.1183/09031936.00095113 [DOI] [PubMed] [Google Scholar]
- 25. Zhang X, Liu H, Hock T, Thannickal VJ, Sanders YY. Histone deacetylase inhibition downregulates collagen 3A1 in fibrotic lung fibroblasts. Int J Mol Sci. 2013;14:19605‐19617. doi: 10.3390/ijms141019605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mir HA, Ali R, Mushtaq U, Khanday FA. Structure‐functional implications of longevity protein p66Shc in health and disease. Ageing Res Rev. 2020;63:101139. doi: 10.1016/j.arr.2020.101139 [DOI] [PubMed] [Google Scholar]
- 27. Zhao Y, Wang Z, Feng D, et al. p66Shc contributes to liver fibrosis through the regulation of mitochondrial reactive oxygen species. Theranostics. 2019;9:1510‐1522. doi: 10.7150/thno.29620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang S, Zhao L, Han Y, et al. Probucol ameliorates renal injury in diabetic nephropathy by inhibiting the expression of the redox enzyme p66Shc. Redox Biol. 2017;13:482‐497. doi: 10.1016/j.redox.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ventura A, Luzi L, Pacini S, Baldari CT, Pelicci PG. The p66Shc longevity gene is silenced through epigenetic modifications of an alternative promoter. J Biol Chem. 2002;277:22370‐22376. doi: 10.1074/jbc.M200280200 [DOI] [PubMed] [Google Scholar]
- 30. Shou J, Chen PJ, Xiao WH. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol Metab Syndr. 2020;12:14. doi: 10.1186/s13098-020-0523-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grosicki GJ, Fielding RA, Lustgarten MS. Gut microbiota contribute to age‐related changes in skeletal muscle size, composition, and function: biological basis for a gut‐muscle Axis. Calcif Tissue Int. 2018;102:433‐442. doi: 10.1007/s00223-017-0345-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim YR, Kim CS, Naqvi A, et al. Epigenetic upregulation of p66shc mediates low‐density lipoprotein cholesterol‐induced endothelial cell dysfunction. Am J Physiol Heart Circ Physiol. 2012;303:H189‐H196. doi: 10.1152/ajpheart.01218.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yancoskie MN, Maritz C, van Eijk P, Reed SH, Naegeli H. To incise or not and where: SET‐domain methyltransferases know. Trends Biochem Sci. 2023;48:321‐330. doi: 10.1016/j.tibs.2022.10.003 [DOI] [PubMed] [Google Scholar]
- 34. Wang H, Helin K. Roles of H3K4 methylation in biology and disease. Trends Cell Biol. 2024. doi: 10.1016/j.tcb.2024.06.001. [DOI] [PubMed] [Google Scholar]
- 35. Xu J, Ma H, Jin J, et al. Super‐resolution imaging of higher‐order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep. 2018;24:873‐882. doi: 10.1016/j.celrep.2018.06.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meyer KC. Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis. Expert Rev Respir Med. 2017;11:343‐359. doi: 10.1080/17476348.2017.1312346 [DOI] [PubMed] [Google Scholar]
- 37. Futakuchi A, Inoue T, Fujimoto T, et al. Molecular mechanisms underlying the filtration bleb‐maintaining effects of Suberoylanilide hydroxamic acid (SAHA). Invest Ophthalmol Vis Sci. 2017;58:2421‐2429. doi: 10.1167/iovs.16-21403 [DOI] [PubMed] [Google Scholar]
- 38. Bodas M, Mazur S, Min T, Vij N. Inhibition of histone‐deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respir Res. 2018;19:2. doi: 10.1186/s12931-017-0705-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murrell GA, Francis MJ, Bromley L. Modulation of fibroblast proliferation by oxygen free radicals. Biochem J. 1990;265:659‐665. doi: 10.1042/bj2650659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schuliga M, Pechkovsky DV, Read J, et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J Cell Mol Med. 2018;22:5847‐5861. doi: 10.1111/jcmm.13855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3:350‐356. doi: 10.1513/pats.200601-001TK [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi YH. Trans‐cinnamaldehyde prevents oxidative stress‐induced apoptosis in V79‐4 Chinese hamster lung fibroblasts through the Nrf2‐mediated HO‐1 activation. Biol Pharm Bull. 2020;43:1707‐1714. doi: 10.1248/bpb.b20-00407 [DOI] [PubMed] [Google Scholar]
- 43. Wu Q, Zhou Y, Feng FC, Jin YH, Wang ZC, Zhou XM. Probing into the mechanism of alkaline citrus extract promoted apoptosis in pulmonary fibroblasts of bleomycin‐induced pulmonary fibrosis mice. Evid Based Complement Alternat Med. 2018;2018:9658950. doi: 10.1155/2018/9658950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brodská B, Holoubek A. Generation of reactive oxygen species during apoptosis induced by DNA‐damaging agents and/or histone deacetylase inhibitors. Oxidative Med Cell Longev. 2011;2011:253529. doi: 10.1155/2011/253529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Boengler K, Bornbaum J, Schlüter KD, Schulz R. P66shc and its role in ischemic cardiovascular diseases. Basic Res Cardiol. 2019;114:29. doi: 10.1007/s00395-019-0738-x [DOI] [PubMed] [Google Scholar]
- 46. Castro CFG, Nardiello C, Hadzic S, et al. The role of the redox enzyme p66Shc in biological aging of the lung. Aging Dis. 2024;15:911‐926. doi: 10.14336/ad.2023.0715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu F, Pang L, Cai X, et al. Let‐7‐repressesed Shc translation delays replicative senescence. Aging Cell. 2014;13:185‐192. doi: 10.1111/acel.12176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang HM, Fan SJ, Zhou XR, et al. Histone deacetylase inhibitor givinostat attenuates nonalcoholic steatohepatitis and liver fibrosis. Acta Pharmacol Sin. 2022;43:941‐953. doi: 10.1038/s41401-021-00725-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tung CW, Hsu YC, Cai CJ, et al. Trichostatin a ameliorates renal tubulointerstitial fibrosis through modulation of the JNK‐dependent Notch‐2 signaling pathway. Sci Rep. 2017;7:14495. doi: 10.1038/s41598-017-15162-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pun MD, Wu HH, Olatunji FP, Kesic BN, Peters JW, Berkman CE. Phosphorus containing analogues of SAHA as inhibitors of HDACs. J Enzyme Inhib Med Chem. 2022;37:1315‐1319. doi: 10.1080/14756366.2022.2063281 [DOI] [PMC free article] [PubMed] [Google Scholar]