

ABSTRACT
TAX1BP1 is a selective macroautophagy/autophagy receptor that inhibits NFKB and RIGI-like receptor (RLR) signaling to prevent excessive inflammation and maintain homeostasis. Selective autophagy receptors such as SQSTM1/p62 and OPTN are phosphorylated by the kinase TBK1 to stimulate their selective autophagy function. However, it is unknown if TAX1BP1 is regulated by TBK1 or other kinases under basal conditions or during RNA virus infection. Here, we found that TBK1 and IKBKE/IKKi function redundantly to phosphorylate TAX1BP1 and regulate its autophagic turnover through canonical macroautophagy. TAX1BP1 phosphorylation promotes its localization to lysosomes, resulting in its degradation. Additionally, we found that during vesicular stomatitis virus infection, TAX1BP1 is targeted to lysosomes in an ATG8-family protein-independent manner. Furthermore, TAX1BP1 plays a critical role in the clearance of MAVS aggregates, and phosphorylation of TAX1BP1 controls its MAVS aggrephagy function. Together, our data support a model whereby TBK1 and IKBKE license TAX1BP1-selective autophagy function to inhibit MAVS and RLR signaling.
Abbreviations: ATG: autophagy related; BafA1: bafilomycin A1; CALCOCO2: calcium binding and coiled-coil domain 2; GFP: green fluorescent protein; IFA: indirect immunofluorescence assay; IFN: interferon; IκB: inhibitor of nuclear factor kappa B; IKK: IκB kinase; IRF: interferon regulatory factor; KO: knockout; LAMP1: lysosomal associated membrane protein 1; LIR: LC3-interacting region; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MAVS: mitochondrial antiviral signaling protein; MEF: mouse embryonic fibroblast; MOI: multiplicity of infection; IKBKG/NEMO: inhibitor of nuclear factor kappa B kinase regulatory subunit gamma; NFKB: nuclear factor kappa B; OPTN: optineurin; Poly(I:C): polyinosinic-polycytidylic acid; RB1CC1/FIP200: RB1 inducible coiled-coil 1; RIGI: RNA sensor RIG-I; RLR: RIGI-like receptor; SDD-AGE: semi-denaturing detergent-agarose gel electrophoresis; SeV: Sendai virus; SLR: SQSTM1-like receptor; SQSTM1: sequestosome 1; TAX1BP1: Tax1 binding protein 1; TBK1: TANK binding kinase 1; TNF: tumor necrosis factor; TRAF: TNF receptor associated factor; VSV: vesicular stomatitis virus; ZnF: zinc finger.
KEYWORDS: Aggrephagy, autophagy, IKBKE/IKKi, MAVS, TAX1BP1, TBK1
Introduction
Pattern recognition receptors detect conserved molecular features of viruses and other pathogens known as pathogen-associated molecular patterns. In the RIGI-like receptor (RLR) pathway the cytoplasmic RNA helicases RIGI and IFIH1/MDA5 (interferon induced with helicase C domain 1) recognize nucleic acid derived from RNA viruses and trigger signaling pathways through the mitochondrial protein MAVS, leading to activation of NFKB and IRF3 transcription factors that upregulate expression of proinflammatory cytokines and type I IFNs respectively [1,2]. MAVS recruits E3 ubiquitin ligases such as TRAF2, TRAF3, TRAF5 and TRAF6 to conjugate lysine 63 (K63)-linked polyubiquitin chains that recruit the adaptor IKBKG/NEMO and kinases TBK1 and IKBKE/IKKi [3–8]. TBK1 and IKBKE/IKKi directly phosphorylate IRF3 and IRF7 to trigger their dimerization, nuclear localization and activation of type I IFN to restrict virus replication [9,10].
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved lysosomal degradation pathway critical for homeostasis. Autophagy is initiated by the recruitment of membranes to form a cup-shaped lipid structure known as a phagophore which sequesters substrates by expanding around them, resulting in an enclosed double-membrane vesicle termed an autophagosome. Following the sequestration of substrates, the autophagosome fuses with lysosomes generating autolysosomes to degrade its contents [11,12]. Concomitant with expansion, the phagophore is conjugated with ATG8-family member proteins, such as MAP1LC3/LC3 and GABARAP (GABA type A receptor-associated protein), that play key roles in the biogenesis and maturation of autophagosomes as well as cargo recruitment [13]. This cargo recruitment (i.e., selective autophagy) results in the recruitment of specific cargo to phagophores including protein aggregates/misfolded proteins (aggrephagy), damaged organelles such as mitochondria (mitophagy) or pathogenic microbes (xenophagy) [14]. Targeted degradation of specific cargo is mediated by specialized autophagy receptors that recognize and link cargo to phagophores. Cargo destined for phagophores are typically modified by post-translational modifications such as ubiquitination which can be detected by autophagy receptors containing ubiquitin binding domains [15]. Furthermore, autophagy receptors harbor LC3-interacting regions (LIRs) that link cargo to phagophore membranes [16]. The best characterized selective autophagy receptors consist of the SQSTM1/p62 (sequestosome 1)-like receptor (SLR) family including SQSTM1, OPTN (optineurin), NBR1, CALCOCO2 and TAX1BP1 [17]. Autophagy has been linked to the negative regulation of RLR signaling [18,19], yet the precise mechanisms remain unknown.
TAX1BP1 was originally identified in yeast two-hybrid screens as a binding protein of the human T lymphotropic virus 1 Tax protein, the ubiquitin-editing enzyme TNFAIP3/A20 and the E3 ubiquitin ligase TRAF6 [20–22]. TAX1BP1 inhibits canonical NFKB signaling, together with E3 ligases ITCH and RNF11 (ring finger protein 11), by acting as an adaptor for the ubiquitin-editing enzyme TNFAIP3 [23–26]. In addition to regulating NFKB signaling, TAX1BP1 also inhibits the RLR pathway and the induction of type I IFN triggered by RNA virus infection or transfection with the double-stranded RNA mimetic poly(I:C) [27]. Furthermore, TAX1BP1 blocks RLR-mediated apoptosis by interacting with and promoting MAVS degradation [28]. TAX1BP1 also suppresses the TLR3 (toll like receptor 3) and TLR4 pathways by targeting the adaptor TICAM1 (TIR domain containing adaptor molecule 1) for degradation [29,30]. TAX1BP1 contains two LIR motifs and functions as a selective autophagy receptor [31–33]. Furthermore, the second zinc finger domain (ZnF2) in the carboxyl terminus of TAX1BP1 can bind to K63-linked polyubiquitin chains [33,34]. Therefore, TAX1BP1 targets ubiquitinated cargo via ZnF2 and recruits cargo to developing autophagosomes via the LIR domains. TAX1BP1 also interacts with MYO6 (myosin VI), a cytoskeletal actin-based motor protein that regulates vesicular transport, to induce autophagosome maturation [33]. Therefore, TAX1BP1 exerts multiple roles in autophagy including cargo selection and autophagosome maturation [35]. TAX1BP1 can remove damaged mitochondria (mitophagy) together with OPTN and CALCOCO2 [36], and pathogenic bacteria including Salmonella typhimurium and Mycobacterium tuberculosis (xenophagy) [33,37]. A recent study has linked TAX1BP1 to the clearance of protein aggregates (i.e., polyQ HTT [huntingtin] fragments and TARDBP/TDP-43) in the brain thus implicating TAX1BP1 as an aggrephagy receptor [38].
Despite the important roles of TAX1BP1 in the inhibition of innate immune signaling pathways, it remains unclear how the selective autophagy function of TAX1BP1 is regulated and if TAX1BP1 functions as an aggrephagy receptor in the regulation of innate immunity. We previously reported that phosphorylation of TAX1BP1 by the kinase CHUK/IKKα plays a critical role in the termination of TNF and IL1B (interleukin 1 beta)-induced NFKB signaling [39]; however, it is unknown if phosphorylation of TAX1BP1 regulates its autophagy function. In this study, we have identified TBK1 and IKBKE/IKKi as key regulators of TAX1BP1 macroautophagic degradation that function redundantly, at steady state and during RLR stimulation with transfected poly(I:C). However, during vesicular stomatitis virus (VSV) infection, both TBK1 and IKBKE/IKKi, and to a lesser extent the ATG8-family protein conjugation machinery, are dispensable for TAX1BP1 degradation, whereas RB1CC1/FIP200 is essential for degradation of TAX1BP1, suggesting a role for noncanonical lysosomal delivery of TAX1BP1 during VSV infection. Furthermore, TAX1BP1 mediates the clearance of MAVS aggregates, both basally and during RNA virus infection, and phosphorylation of TAX1BP1 stimulates its MAVS aggrephagy function.
Results
TAX1BP1 is phosphorylated by IKBKE/IKKi and TBK1 kinases
We previously reported that the CHUK subunit of IKK phosphorylates TAX1BP1 to promote the termination of NFKB signaling [39]. During the course of our studies on TAX1BP1 regulation of RLR signaling, we found that the noncanonical IκB kinases TBK1 and IKBKE/IKKi also phosphorylated TAX1BP1. Overexpression of CHUK, IKBKE/IKKi and TBK1 all induced a slower migrating form of TAX1BP1 (Figure 1A). Interestingly, IKBKE/IKKi (and to a lesser extent TBK1) overexpression was associated with the loss of TAX1BP1 protein (Figure 1A). Treatment with lambda phosphatase converted the slower migrating band to a faster migrating form of TAX1BP1, thus confirming phosphorylation (Figure 1B). To investigate if phosphorylation by IKBKE/IKKi or TBK1 was required for the observed band shift of TAX1BP1, Flag TAX1BP1 was co-transfected with WT TBK1 or IKBKE/IKKi or their kinase dead mutants (TBK1K38A and IKBKE/IKKiK38A). As expected, WT TBK1 and IKBKE/IKKi both induced a band shift, while their kinase dead mutants Flag-TBK1K38A and Flag-IKBKE/IKKiK38A failed to do so (Figure 1C). In vitro kinase assays with purified recombinant proteins demonstrated that TBK1 and IKBKE/IKKi could both directly phosphorylate TAX1BP1, although IKBKE/IKKi induced a more obvious TAX1BP1 band shift (Figure 1D). To determine whether the loss of TAX1BP1 protein expression observed with overexpression of TBK1 or IKBKE/IKKi was due to lysosomal degradation, we transfected DLD-1 cells with these kinases in the presence or absence of bafilomycin A1 (BafA1) which inhibits acidification of lysosomes. BafA1 treatment stabilized TAX1BP1 expression (Figure 1E) suggesting that the expression/activation of these kinases induces the lysosomal degradation of TAX1BP1. To confirm that loss of TAX1BP1 protein associated with overexpression of the noncanonical IκB kinases was the result of macroautophagy, we overexpressed TBK1 and IKBKE/IKKi in cells deficient for the ATG8-conjugation protein ATG3. As expected, loss of ATG3 stabilized TAX1BP1 upon overexpression of TBK1 or IKBKE/IKKi (Figure 1F). To identify the IKBKE/IKKi-inducible TAX1BP1 phosphorylation sites in an unbiased manner, we used liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). As expected, IKBKE/IKKi induced a slower migrating band shift in TAX1BP1 (Figure 1G). A total of four bands were excised from the gel (both phosphorylated and unphosphorylated TAX1BP1 as controls) and subjected to LC-MS/MS analysis, which identified a total of 13 IKBKE/IKKi-inducible TAX1BP1 phosphorylation sites in TAX1BP1 (Figure 1H).
Figure 1.
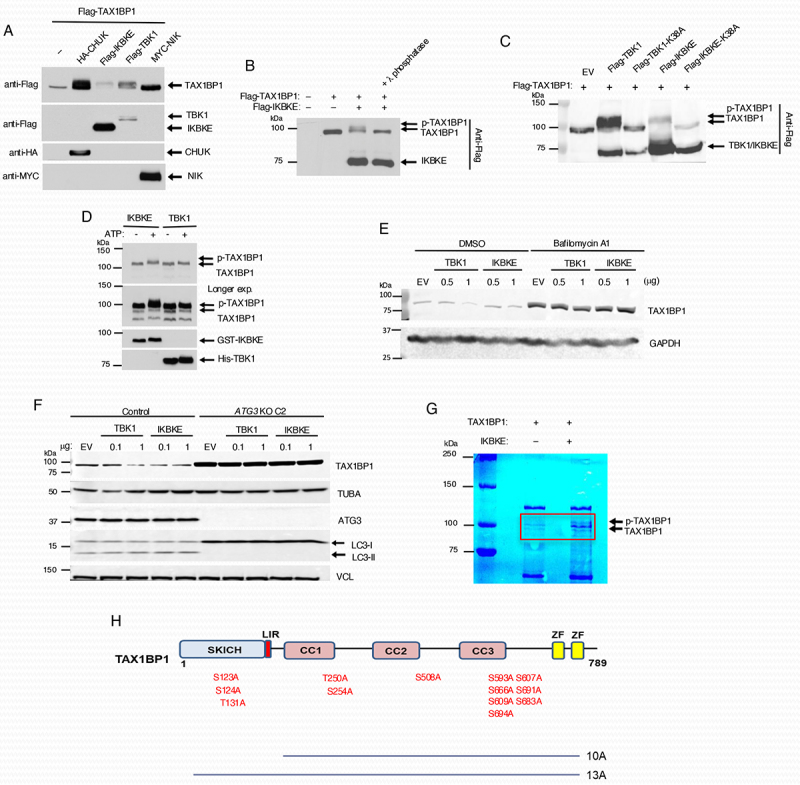
IκB kinases induce TAX1BP1 phosphorylation. (A) 293T cells were co-transfected with Flag-TAX1BP1 together with the indicated kinase plasmids and 24 h later lysed, and the cell extracts were immunoblotted with the indicated antibodies. (B) 293T cells were transfected with the indicated plasmids and lysates were incubated with λ-phosphatase for 30 min prior to immunoblotting with anti-Flag. (C) 293T cells were transfected with Flag-TAX1BP1 and either Flag-TBK1, Flag-IKBKE/IKKi or kinase dead mutants Flag-TBK1K38A or Flag-IKBKE/IKKiK38A, respectively. Lysates were subjected to immunoblotting with anti-Flag antibody. (D) in vitro kinase assays. Purified GST-tagged TAX1BP1 (300 ng) was incubated with 50 ng of purified recombinant GST-tagged IKBKE/IKKi or hexahistidine-tagged TBK1 in the presence or absence of ATP. The reaction mixtures were subjected to immunoblotting with antibodies to TAX1BP1, IKBKE/IKKi and TBK1. (E) DLD-1 cells were transfected with the indicated amount of plasmids for 24 h and then treated with BafA1 (20 nM) for 18 h. Cells were then lysed and subjected to immunoblotting analysis with the indicated antibodies. (F) Control and ATG3 KO DLD-1 cells were transfected with the indicated amount of plasmids for 24 h and then lysed and subjected to immunoblotting with the indicated antibodies. (G) Colloidal blue staining of in vitro kinase reaction mixtures containing TAX1BP1 with or without IKBKE/IKKi. Individual bands within the red rectangle were cut and gelextracted for mass spectrometry (MS) analysis. (H) Schematic diagram of TAX1BP1 domains. The indicated ten and thirteen predicted phosphorylation sites were substituted with alanine, generating the TAX1BP1 10A and 13A mutants, respectively. SKICH, the SKIP carboxy homology domain; LIR, LC3-interacting region; CC, coiled-coil domain; ZF, zinc finger domain.
Mapping of TAX1BP1 phosphorylation sites
To validate the putative TAX1BP1 phosphorylation sites, we generated a panel of TAX1BP1 point mutants with all 13 putative sites mutated to alanine (designated as 13A) as well as the 10 sites (designated as 10A) downstream of coiled coil domain 1 (CC1) (Figure 1H). IKBKE/IKKi overexpression induced the phosphorylation of wild-type (WT) TAX1BP1, but not of 13A or 10A mutants (Figure 2A). Since the TAX1BP1 10A mutant was indistinguishable from 13A with regard to the lack of phosphorylation and degradation we focused on this mutant for subsequent experiments. We next generated a new panel of rescue mutants where each of the potential phosphorylation sites was individually restored back to the original amino acid in the context of TAX1BP1 10A (Figure 2B). These are designated as TAX1BP1 9A/WT amino acid. This panel of TAX1BP1 mutants was transfected into cells and then infected with a model RNA virus, VSV-GFP (vesicular stomatitis virus [VSV] encoding a green fluorescent protein [GFP] reporter), followed by western blotting to assess phosphorylation. As expected, WT TAX1BP1 was phosphorylated and degraded upon VSV infection (Figure 2C). However, TAX1BP1 10A was resistant to VSV-induced phosphorylation and degradation (Figure 2C). Densitometric analysis of VSV-induced TAX1BP1 degradation from three independent experiments revealed that reconstitution of either serines 254 or 593 partially rescued the degradation of TAX1BP1 (Figure 2D). Similar results were observed with overexpression of IKBKE/IKKi, as well as a prominent band shift of TAX1BP1 observed with reconstitution of serine 666 (Figure S1). Since virus-induced TAX1BP1 degradation was not fully restored with single point mutations, we next generated a TAX1BP1 compound mutant with S254, S593 and S666 in the context of 10A (designated as 7A; Figure 2B). An additional mutant was generated with T250, S254, S593 and S666 in the context of 10A, as reconstitution of threonine 250 yielded slight degradation of TAX1BP1 by IKBKE/IKKi overexpression (designated as 6A; Figure 2B; Figure S1). Remarkably, VSV-induced TAX1BP1 degradation was fully restored by the 7A and 6A mutants (Figure 2E) suggesting that S254, S593 and possibly S666 act synergistically in promoting TAX1BP1 phosphorylation/degradation.
Figure 2.

Three phosphoserine residues are involved in TAX1BP1 degradation. (A) Immunoblotting analysis of the extracts derived from 293T cells transfected with WT Flag-TAX1BP1 and mutants (10A and 13A) together with, or without, Flag-IKBKE/IKKi. For better separation of phosphorylated TAX1BP1, a 6% gel was used. (B) Schematic of restored TAX1BP1 10A variants at single or multiple phosphorylation residue(s). (C) 293T cells transfected with WT Flag-TAX1BP1 and mutants together with HA-tagged TAX1BP1 10A for 24 h were infected with or without VSV-GFP for 6 h at an MOI of 1, and the cell extracts were subjected to immunoblotting with the indicated antibodies. (D) Densitometric analysis of TAX1BP1 band intensity from three independent experiments presented in (C). Densitometric analysis was performed with ImageJ. Unpaired Student’s t-test, **p < 0.01, *p < 0.05. (E) Immunoblotting analysis of the extracts derived from 293T cells transfected with WT Flag-TAX1BP1 and variants (10A, 7A, and 6A) and 24 h later infected with or without VSV-GFP as above. As shown in (B), 7A was generated by restoring the three potential phosphorylation sites, S254, S593, and S666 in 10A, and 6A was generated by restoring T250 in 7A.
Loss of either TBK1 or IKBKE/IKKi does not impair poly(I:C)- or VSV-induced TAX1BP1 degradation
We hypothesized that virus infection-mediated phosphorylation and autophagic degradation of TAX1BP1 could be mediated by IKBKE/IKKi based upon the enhanced degradation observed during IKBKE/IKKi overexpression. To test this notion, we generated IKBKE/IKKi knockout (KO) DLD-1 cell lines using CRISPR-Cas9 technology. In addition, we generated DLD-1 cell lines deficient in the closely related kinase TBK1 due to their functional redundancy. DLD-1 cells were used because of high basal levels of TAX1BP1 expression [28]. DLD-1 cells were transduced with recombinant lentiviruses expressing Cas9 and either IKBKE/IKKi or TBK1 gRNAs followed by limiting dilution and clonal analysis of TBK1 and IKBKE/IKKi KOs. Multiple clones of TBK1 and IKBKE/IKKi KOs were identified and two clones each were selected for further experimentation. Surprisingly, poly(I:C)-induced TAX1BP1 degradation remained intact in TBK1 or IKBKE/IKKi KO cells (Figures 3A,B). Similar to what was observed with poly(I:C) transfection, loss of either TBK1 or IKBKE/IKKi did not prevent degradation of TAX1BP1 in response to VSV infection (Figure 3C).
Figure 3.

TBK1 and IKBKE/IKKi redundantly promote TAX1BP1 degradation in response to poly(I:C) transfection but are dispensable for VSV-induced TAX1BP1 degradation. (A, B) Immunoblotting analyses of the extracts derived from the following cells: two different TBK1 KO DLD-1 cell lines (A) and two differentIKBKE/IKKi KO DLD-1 cell lines (B) transfected with 2.5 µg/ml poly(I:C) for 0, 4 and 6 h. (C) Immunoblotting analysis of control, TBK1 KO or IKBKE/IKKi KO DLD-1 cells infected with VSV-GFP at the indicated MOIs for 16 h. (D) Immunoblotting analysis of WT and TBK1 IKBKE/IKKi dKO DLD-1 cells transfected with the indicated concentration of poly(I:C) for 6 h. A lipofectamine only control is designated as “LO.” (E) Immunoblotting analysis of WT and TBK1 IKBKE/IKKi dKO DLD-1 cells infected with VSV-GFP at the indicated MOIs for 24 h.
TBK1 and IKBKE/IKKi are redundant for RLR-induced TAX1BP1 degradation but dispensable for TAX1BP1 degradation triggered by VSV infection
To address the possibility of functional compensation between TBK1 and IKBKE/IKKi, we generated TBK1 IKBKE/IKKi double-KO (TBK1 IKBKE/IKKi dKO) DLD-1 cells and infected these cells with VSV-GFP. Similarly, TAX1BP1 degradation was unimpaired in TBK1 IKBKE/IKKi dKO cells infected with VSV-GFP (Figure 3D). However, the degradation of TAX1BP1 was impaired in the TBK1 IKBKE/IKKi dKO cells when RLR signaling was induced by the transfection of poly(I:C) (Figure 3E).
RLR stimulation promotes the degradation of TAX1BP1 through macroautophagy
To confirm that poly(I:C) induced the autophagic degradation of TAX1BP1, we performed confocal microscopy after poly(I:C) transfection to assess TAX1BP1 colocalization with MAP1LC3B using DLD-1 cells stably expressing a GFP-MAP1LC3B fusion protein. TAX1BP1 was found to colocalize with MAP1LC3B following treatment with BafA1, which was significantly enhanced with poly(I:C) transfection (Figure 4A,B). Interestingly, TAX1BP1 was found to colocalize with MAP1LC3B in TBK1 IKBKE/IKKi dKO DLD-1 cells following BafA1 treatment in large punctate structures (Figure 4A,B). Upon further investigation, TAX1BP1 was found to basally colocalize with both ubiquitin (Figure S2A) and SQSTM1 (Figure S2B) in the absence of TBK1 and IKBKE/IKKi.
Figure 4.

TAX1BP1 is targeted to LC3-positive vesicles in response to poly(I:C) transfection. (A) Immunofluorescence assays of WT and TBK1 IKBKE/IKKi dKO DLD-1 cells transfected with 5 µg/ml poly(I:C) for 6 h in the presence of BafA1 (100 nM). Scale bar: 10 µm. (B) Colocalization analysis of TAX1BP1 and LC3 following poly(I:C) transfection. Manders coefficients were derived using ImageJ. Coefficients shown are representative of TAX1BP1 signal overlapping with LC3 signal from 50 cells chosen at random across five fields per condition. Unpaired Student’s t-test. ****p < 0.0001, *p < 0.05.
TBK1 and IKBKE/IKKi regulate the basal turnover of TAX1BP1
During the course of these studies, we noticed that the basal expression of TAX1BP1 protein was consistently increased in TBK1 IKBKE/IKKi dKO cells which was confirmed by quantification in several independent experiments (Figure 5A). Treatment of cells with Amlexanox, a specific inhibitor of TBK1 and IKBKE/IKKi, also increased basal expression of TAX1BP1 (Figure 5B). To provide further evidence that the basal phosphorylation of TAX1BP1 caused its degradation, we treated cells with the phosphatase inhibitor calyculin A. Indeed, calyculin A promoted TAX1BP1 degradation in WT DLD-1 cells, which was impaired in TBK1 IKBKE/IKKi dKO cells (Figure 5C). Therefore, it appears that dynamic regulation of TAX1BP1 phosphorylation and dephosphorylation controls its turnover.
Figure 5.
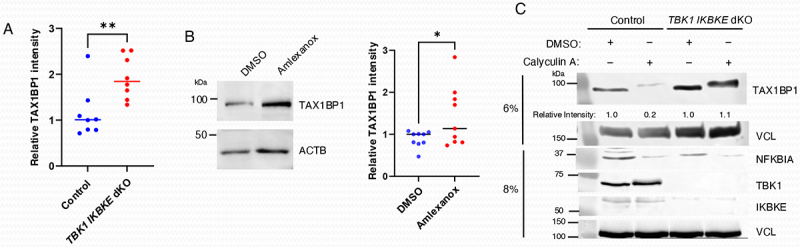
TBK1 and IKBKE/IKKi regulate the basal turnover of TAX1BP1. (A) TAX1BP1 protein expression was quantified by ImageJ using lysates from WT and TBK1 IKBKE/IKKi dKO DLD-1 cells by normalizing TAX1BP1 expression to loading controls. Data were derived from eight independent experiments. Unpaired Student’s t-test, **p < 0.01. (B) Immunoblot analysis of extracts from DLD-1 cells treated with DMSO or Amlexanox (100 µg/ml) for 17 h. TAX1BP1 expression was quantified by ImageJ using lysates from DMSO- or Amlexanox-treated DLD-1 cells. Data were derived from nine independent experiments. Unpaired Student’s t-test, *p < 0.05. (C) WT and TBK1 IKBKE/IKKi dKO DLD-1 cells were treated with calyculin A for 30 min, and lysates were immunoblotted with the indicated antibodies. TAX1BP1 expression was quantified by ImageJ.
TAX1BP1 is degraded by ATG8-family proteindependent autophagy and ATG8-family protein independent lysosomal delivery during VSV infection
The most prominent role of selective autophagy receptors is their ability to shuttle cargo to the developing phagophore, typically through an interaction between the LIR domain of autophagy receptors and ATG8-family member proteins (e.g., MAP1LC3B) that decorate the expanding phagophore. Based on the observed differences between TAX1BP1 degradation triggered by poly(I:C) transfection (TBK1 and IKBKE/IKKi-dependent) versus VSV infection (TBK1 and IKBKE/IKKi-independent) we sought to identify these distinct TAX1BP1 degradation pathways. TAX1BP1 has been shown to undergo lysosomal degradation in the absence of ATG8-family protein phagophore conjugation machinery factors [40,41]. Based on these findings and the observed colocalization of TAX1BP1 and MAP1LC3B after poly(I:C) transfection (Figure 4), we next sought to determine if the ATG8-family protein conjugation machinery was required for the degradation of TAX1BP1 during VSV infection. To this end we used CRISPR-Cas9 to generate multiple clones of ATG3 KO DLD-1 cells. ATG3 functions as an E2-like enzyme in the conjugation of phosphatidylethanolamine to MAP1LC3/LC3-I to yield MAP1LC3/LC3-II. Direct stimulation of RLR signaling by poly(I:C) transfection yielded TAX1BP1 degradation in control cells as expected, however a total impairment of TAX1BP1 degradation occurred in ATG3 KO cells (Figure 6A). Interestingly, we observed diminished but not complete impairment of TAX1BP1 degradation during VSV infection (Figure 6B), implicating multiple avenues for the degradation of TAX1BP1 during VSV infection.
Figure 6.

TAX1BP1 degradation induced by poly(I:C), but not VSV infection, requires canonical macroautophagy/ATG8-family protein conjugation machinery. (A) Immunoblot analysis of control or ATG3 KO DLD-1 cells transfected with poly(I:C) at the indicated concentrations for 6 h. A lipofectamine only vehicle control is indicated as “LO.” (B) Immunoblot analysis of control or ATG3 KO DLD-1 cells infected with VSV-GFP at the indicated MOIs for 16 h. Densitometric analysis was performed using ImageJ.
RB1CC1/FIP200 is required for the degradation of TAX1BP1 in response to VSV infection or RLR activation
Recent studies have identified pathways in which selective autophagy receptors can be delivered to lysosomes in the absence of ATG8-family member proteins in which RB1CC1/FIP200, a member of the ULK1 initiation complex that interacts with TAX1BP1, is required [40]. Remarkably, TAX1BP1 degradation in response to VSV infection was almost completely abrogated in rb1cc1–/– MEFs, but not in atg3–/– MEFs (Figure 7A). VSV-GFP infection in both atg3–/– and rb1cc1–/– MEFs was comparable to WT MEFs as examined by Incucyte S3 live-cell analysis (data not shown). To rule out cell-type/species specificity, we generated RB1CC1/FIP200 KO DLD-1 cells using CRISPR-Cas9 and as observed in MEFs, TAX1BP1 degradation was completely impaired in RB1CC1/FIP200 KO DLD-1 cells during VSV infection (Figure 7B), as well as in response to poly(I:C) transfection (Figure 7C). To further support these data, we generated DLD-1 cells stably expressing a TAX1BP1-eGFP fusion protein by transducing TAX1BP1 KO DLD-1 cells with a lentivirus expressing a gRNA-resistant form of TAX1BP1 fused with eGFP. eGFP-positive cells were sorted and a single clone was isolated, and then knocked out for RB1CC1/FIP200 (RB1CC1/FIP200 KO) using CRISPR-Cas9, of which a single clone was then isolated for further analysis. Flow cytometry was used to assess TAX1BP1 lysosomal degradation during VSV infection as eGFP is quenched in acidic environments such as the lysosome. As expected, the RB1CC1/FIP200 KO cells were resistant to TAX1BP1 lysosomal degradation during VSV infection compared to control cells (Figure 7D). Taken together, these data suggest that TAX1BP1 degradation induced by VSV infection likely occurs via both ATG8-family protein-dependent and RB1CC1/FIP200-dependent/ATG8-independent lysosomal pathways.
Figure 7.

RB1CC1/FIP200 is required for TAX1BP1 degradation in response to VSV infection and poly(I:C) transfection. (A) WT, rb1cc1–/– and atg3–/– MEFs were infected with VSV-GFP (MOI = 1) for 13 h and subjected to immunoblotting with anti-TAX1BP1 and anti-ACTB/β-actin antibodies. (B) Control and RB1CC1/FIP200 KO DLD-1 cells were infected with VSV-GFP at the indicated MOIs for 20 h and then subjected to immunoblotting analysis with the indicated antibodies. Densitometric analysis was performed using ImageJ. (C) Control and RB1CC1/FIP200 KO DLD-1 cells were transfected with the indicated concentration of poly(I:C) for 6 h and then subjected to immunoblotting analysis with the indicated antibodies. A lipofectamine only control is designated as “LO.” Densitometric analysis was performed using ImageJ. (D) Control and RB1CC1/FIP200 KO cells stably expressing a TAX1BP1-eGFP fusion protein were infected with VSV for 20 h (MOI = 1) and then analyzed by flow cytometric analysis of eGFP fluorescence intensity. Geometric mean fluorescence intensity (gMFI) was quantified using FlowJo 10 software. Data were derived from three independent experiments. Unpaired Student’s t-test. **p < 0.01.
TAX1BP1 clears MAVS aggregates in a phosphorylation-dependent manner
Thus far our experiments have established that TAX1BP1 undergoes ATG8-family protein-dependent and -independent degradation during RLR activation and VSV infection, and this process is dependent on its phosphorylation. It remains unclear what cargo is recruited to autophagosomes by TAX1BP1 for the homeostatic control of the RLR pathway. We previously demonstrated that TAX1BP1 interacts with the mitochondrial adaptor MAVS, and targets MAVS for degradation [28]. Basal expression of MAVS protein is elevated in TAX1BP1 KO cells and the half-life of MAVS is significantly increased in the absence of TAX1BP1 [28]. Dysregulated MAVS expression in TAX1BP1 KO cells results in increased type I IFN and apoptosis in response to RNA virus infection [28]. Upon activation of RLR signaling MAVS forms large aggregates with properties of amyloid fibers and prions including: 1) formation of fiber-like polymers, 2) ability to “infect” the endogenous protein and convert it to aggregate forms, 3) resistance to protease digestion and 4) resistance to detergent solubilization [42]. A common approach to analyze MAVS aggregates is SDD-AGE (semi-denaturing detergent agarose gel electrophoresis), which can detect large polymers between 200–4000 kDa [42,43]. Therefore, SDD-AGE was utilized to analyze MAVS aggregates in WT and tax1bp1–/– MEFs infected with Sendai virus (SeV). In WT MEFs, SeV infection triggered the formation of MAVS aggregates as expected (Figure 8A). Remarkably, MAVS aggregates were spontaneously produced in uninfected tax1bp1–/– MEFs, and SeV infection further increased MAVS aggregates in knockout cells (Figure 8A). Similar results were obtained with TAX1BP1 KO 293T cells generated by CRISPR-Cas9 and infected with VSV-GFP (Figure 8B). In agreement with our previous experiments, TAX1BP1 clearance of MAVS aggregates was dependent on the noncanonical IκB kinases TBK1 and IKBKE/IKKi, and in their absence MAVS aggregates were detected basally and further accumulated during VSV-GFP infection (Figure 8C). Similar to both TAX1BP1 KO and TBK1 IKBKE/IKKi dKO cells, MAVS aggregates accumulated both basally and during virus infection in cells lacking ATG3 or RB1CC1/FIP200 (Figure S3).
Figure 8.
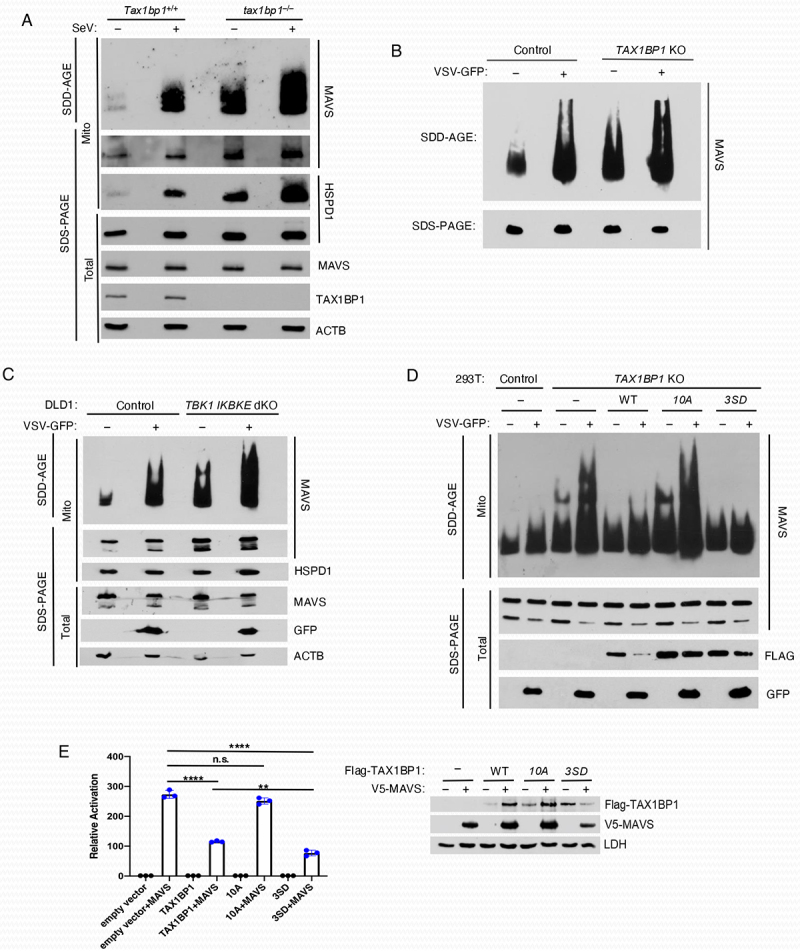
TAX1BP1 promotes MAVS degradation via aggrephagy. (A) SDD-AGE analysis of MAVS. Crude mitochondria were isolated from Tax1bp1+/+ and tax1bp1–/– MEFs infected with SeV (25 HA units/ml) for 6 h, and extracts separated on SDD-AGE and SDS-PAGE gels and immunoblotted with the indicated antibodies. (B) Control and TAX1BP1 KO 293T cells were infected with VSV-GFP and lysates subjected to SDD-AGE for MAVS aggregates and SDS-PAGE to examine expression of MAVS. (C) Control and TBK1 IKBKE/IKKi dKO DLD-1 cells were infected with VSV-GFP and crude mitochondrial extracts separated on SDD-AGE and SDS-PAGE gels followed by immunoblotting with the indicated antibodies. (D) TAX1BP1 KO 293T cells were transiently transfected with gRNA-resistant forms of WT TAX1BP1, phosphorylation mutant 10A TAX1BP1 and phosphomimetic mutant 3SD TAX1BP1 plasmids for 24 h and then infected with VSV-GFP. Lysates were subjected to SDD-AGE for MAVS aggregates and SDS-PAGE for the indicated proteins. (E) Dual luciferase reporter assays. 293T cells were co-transfected with V5-MAVS and Flag-TAX1BP1 WT, 10A or 3SD at a ratio of 1:8 along with IFNB promoter-driven firefly luciferase and TK (thymidine kinase) promoter-dependent Renilla luciferase reporter plasmids for 24 h. The data are presented as mean ± standard deviation of biological triplicates. The remaining cell lysates were subjected to immunoblotting with anti-Flag, anti-V5 and anti-LDH antibodies. One-way ANOVA with Dunnett’s post hoc test or unpaired Student’s t-test. ****p < 0.0001, **p < 0.01, n.s.=not significant.
To examine the direct effect of TAX1BP1 phosphorylation on its ability to clear MAVS aggregates we generated a phosphomimetic mutant (“3SD”) in which the three serines identified earlier (S254, S593, and S666) were mutated to aspartic acid to compare with the phosphorylation-null mutant, 10A. We then performed transient transfection reconstitution experiments in TAX1BP1 KO 293T cells with gRNA-resistant WT TAX1BP1, phosphorylation mutant 10A TAX1BP1 and phosphomimetic mutant 3SD TAX1BP1. SDD-AGE analysis revealed that WT TAX1BP1 inhibited MAVS aggregates and the 3SD phosphomimetic TAX1BP1 was more effective at clearing MAVS aggregates. However, the 10A mutant was impaired in clearing either basal or VSV-induced MAVS aggregates (Figure 8D). Furthermore, the phosphomimetic 3SD TAX1BP1 mutant was more effective than WT TAX1BP1 in suppressing MAVS-induced IFN activation (Figure 8E). However, the TAX1BP1 phosphorylation mutant 10A was impaired in the inhibition of MAVS-IFN induction (Figure 8E).
TAX1BP1 phosphorylation promotes its lysosomal localization
We next sought to determine mechanistically how phosphorylation of TAX1BP1 promotes its autophagy function. We first examined TAX1BP1 dimerization since dimerization of SLRs is required for their autophagy function. To determine if TAX1BP1 phosphorylation regulated its dimerization, we performed NanoBiT assays (Promega) with WT TAX1BP1, phosphorylation mutant 10A and phosphomimetic 3SD. However, the 10A and 3SD mutants exhibited comparable dimerization to WT TAX1BP1 (Figure S4). Therefore, TAX1BP1 phosphorylation does not appear to promote its dimerization. We next sought to determine if phosphorylation regulated the localization of TAX1BP1 to lysosomes. To this end, we examined the subcellular localization of WT TAX1BP1, the phosphorylation mutant 10A, and phosphomimetic 3SD in transfected TAX1BP1 KO HeLa cells. Cells were treated with the protease inhibitor leupeptin to inhibit autophagic flux and prevent TAX1BP1 degradation by poly(I:C) transfection. WT TAX1BP1 colocalization with LAMP1, a marker of lysosomes, was significantly increased by poly(I:C) transfection (Figure 9A,B). However, the phosphorylation-deficient 10A mutant was impaired in poly(I:C)-mediated colocalization with lysosomes (Figure 9A,B). Remarkably, the TAX1BP1 phosphomimetic 3SD colocalized persistently with lysosomes, which was not further increased by poly(I:C) (Figure 9A,B). To determine the role of IKBKE/IKKi in TAX1BP1 lysosomal targeting, we overexpressed IKBKE/IKKi with TAX1BP1 and stained for LAMP1. Overexpression of IKBKE/IKKi promoted TAX1BP1 colocalization with LAMP1 (Figure S5). Therefore, IKBKE/IKKi-mediated TAX1BP1 phosphorylation at serines 254, 593 and 666 directs its localization to lysosomes.
Figure 9.
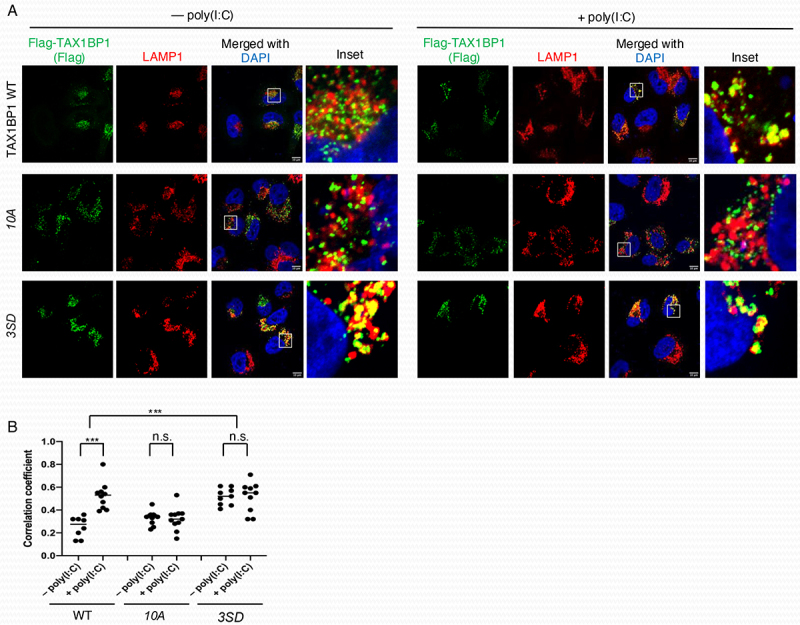
Phosphorylation of TAX1BP1 is required for localization to lysosomes. (A) Immunofluorescence assays of TAX1BP1 KO HeLa cells transfected with Flag-TAX1BP1 WT, 10A or 3SD (S254D, S593D and S666D) and 24 h later transfected with 2.5 µg/ml poly(I:C) for 6 h in the presence of 20 µm leupeptin. Scale bar: 10 µm. (B) Pearson’s correlation coefficient was calculated to measure colocalization between TAX1BP1 and LAMP1 in 8-12 cells randomly selected from each sample. Unpaired Student’s t-test, ***p < 0.001, n.s. = not significant.
Discussion
In this manuscript we have found that the selective autophagy receptor TAX1BP1 can be phosphorylated by both TBK1 and IKBKE/IKKi, with 13 putative IKBKE/IKKi-induced phosphorylation sites in TAX1BP1 identified by mass spectrometry analysis. TAX1BP1 phosphorylation triggers its lysosomal degradation with serines 254, 593 and 666 playing the most critical roles. IKBKE/IKKi and TBK1 regulate basal and RLR-induced TAX1BP1 phosphorylation and degradation through canonical (ATG8-family protein dependent) SLR-mediated macroautophagy; however, it appears that TAX1BP1 is also degraded during VSV infection through an ATG8-family protein-independent/RB1CC1/FIP200-dependent lysosomal delivery pathway. Finally, we found that TAX1BP1 serves as a phosphorylation-dependent aggrephagy receptor for MAVS, and TAX1BP1-deficient cells exhibit a spontaneous accumulation of MAVS aggregates.
Phosphorylation plays important functional roles in the regulation of selective autophagy receptors. TBK1 phosphorylates OPTN, CALCOCO2 and SQSTM1 during bacterial infection and mitophagy to enhance ubiquitin binding and cargo recruitment to autophagosomes [44–49]. Although TBK1 was shown to interact with TAX1BP1 in a previous study [50], it was not examined whether TBK1 played any role in TAX1BP1 autophagy function. Our data indicate that TBK1 and IKBKE/IKKi can phosphorylate TAX1BP1, but surprisingly both were dispensable for VSV-induced TAX1BP1 degradation (Figure 3). We previously found that CHUK interacts with TAX1BP1 and phosphorylates TAX1BP1 on serines 593 and 666 for the inhibition of NFKB signaling [39]; however, the effect of this phosphorylation on TAX1BP1 autophagy function was not examined in our previous study. TBK1 and IKBKE/IKKi share identical phosphorylation motifs which overlap with CHUK and IKBKB substrate specificities [51–53]. IKK phosphorylation sites usually have acidic or phosphorylated amino acids both amino-terminal and carboxyl-terminal to the phosphorylation site. Although S254 and S593 in TAX1BP1 are both putative IKK/TBK1 phosphorylation sites [39], S666 does not conform to an IKK consensus site and is likely phosphorylated by a kinase other than IKKs. S254 and S593 are positioned immediately downstream of coiled-coil domains 1 and 3 respectively, which overlap with self-oligomerization regions of TAX1BP1 [22,54]. However, TAX1BP1 phosphorylation at S254 or S593 did not enhance its dimerization (Figure S4). Rather, IKBKE/IKKi-mediated TAX1BP1 phosphorylation promotes its lysosomal localization (Figure 9 and Figure S5), possibly due to conformational changes in TAX1BP1 that may facilitate its trafficking and/or binding to other factors needed for autophagosome/autolysosome targeting.
Two recent studies have described lysosomal degradation pathways involving TAX1BP1 that are independent of ATG7 and MAP1LC3/LC3 lipidation [40,41]. In these studies, TAX1BP1 and RB1CC1/FIP200 were both shown to be critical for ATG8-family protein-independent autophagy through TAX1BP1-dependent recruitment and clustering of RB1CC1/FIP200 around substrates. Here, we identify a physiological setting (i.e., VSV infection) in which ATG8-family protein-independent lysosomal delivery of SLRs appears to play a critical role. Interestingly, TAX1BP1 degradation by VSV infection and poly(I:C) transfection is completely blocked in rb1cc1–/– MEFs and RB1CC1/FIP200 KO DLD-1 cells (Figure 7). Future studies should determine if TAX1BP1 and RB1CC1/FIP200 coordinate ATG8-family protein-independent lysosomal targeting pathways of substrates during infection with VSV as well as other RNA viruses.
MAVS forms large prion-like aggregates during RNA virus infection that propagate downstream signaling for the activation of TBK1 and IRF3, and type I IFN. However, it remains poorly understood how MAVS aggregates are resolved once viral infections are cleared to suppress inflammation and autoimmunity. Indeed, MAVS aggregates can be detected in peripheral blood mononuclear cells of systemic lupus erythematosus patients and are associated with increased levels of type I IFN [55]. The E3 ligase MARCH5 inhibits RNA virus-induced MAVS aggregates by ubiquitinating MAVS to trigger its proteasomal degradation [56]. The deubiquitinase YOD1 also antagonizes MAVS aggregation by cleaving K63-linked poly-Ub chains on MAVS [57]. MAVS is also negatively regulated by autophagic degradation although the mechanisms remain poorly understood. The E3 ligases RNF34 and MARCH8 have been shown to ubiquitinate MAVS to promote CALCOCO2-dependent autophagic degradation of MAVS [18,19]. Our results indicate that TAX1BP1 serves a nonredundant role in preventing the spontaneous formation of MAVS aggregates and also inhibits MAVS aggregates formed during RNA virus infections (Figure 8). In addition to TAX1BP1-deficient cells, MAVS accumulates in cells lacking TBK1 and IKBKE/IKKi (Figure 8C), ATG3 (Figure S3A) and RB1CC1/FIP200 (Figure S3B). Taken together, these observations suggest that TAX1BP1 functions as an aggrephagy receptor for MAVS through canonical ATG8-family protein-dependent, SLR-mediated autophagy. This MAVS-aggrephagy function for TAX1BP1 is congruent with recent studies that described TAX1BP1 as an aggrephagy receptor in the brain [38] and in regulating TICAM1/TRIF aggregates that form in response to LPS stimulation [29].
In summary, we describe a novel regulatory role for phosphorylation in the regulation of TAX1BP1 autophagosomal degradation and aggrephagy function. Since several viral proteins such as human T lymphotropic virus 1 Tax [54], human papillomavirus E2 [58] and measles virus nucleoprotein [59] interact with TAX1BP1 or target TAX1BP1 for cleavage (coxsackievirus B3 proteinase 2A) [60], it will be interesting in future studies to examine if viruses exploit TAX1BP1 phosphorylation to inhibit MAVS and RLR signaling.
Materials and methods
Cell culture
Human embryonic kidney 293T (HEK293T; CRL-3216) and DLD-1 cells (CCL-221) were purchased from the American Type Culture Collection. TAX1BP1 KO HeLa cells were provided by Dr. Richard Youle (National Institute of Neurological Disorders and Stroke) [36]. tax1bp1–/– MEFs were described previously [23]. atg3–/– [61] and rb1cc1–/– [62] MEFs were obtained from the indicated sources. Cell lines and MEFs were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Corning, 10–013-CV) or RPMI-1640 (Corning, 10–040-CV) supplemented with 10% fetal bovine serum, streptomycin and penicillin at 37°C and 5% CO2. The cell lines were tested for mycoplasma contamination using MycoAlert® Mycoplasma Detection kit (Lonza, LT07–703) and if necessary cultured in the presence of Plasmocin™ treatment (InvivoGen, ant-mpt-1) or Plasmocin™ prophylactic (InvivoGen, ant-mpp). Transient transfection with plasmids was performed using GenJet Plus (SignaGen Laboratories, SL100499), and transfection with poly(I:C) was performed using Lipofectamine 2000 (ThermoFisher Scientific, 11668019) or Lipofectamine 3000 (ThermoFisher Scientific, L300015) following the manufacturer’s instructions.
Reagents
Poly(I:C) (P1530), bafilomycin A1 (SML1661) and leupeptin (SAE0153) were purchased from MilliporeSigma. λ-phosphatase was from New England Biolabs (P0753S). Calyculin A was from Cell Signaling Technology (9902). Amlexanox was from InvivoGen (inh-amx). SeV (Cantrell strain) was purchased from Charles River Laboratories (10100816).
Virus infections
Cells were starved for 1 h in serum-free DMEM or RPMI and inoculated with VSV-GFP or SeV for 1 h at the indicated multiplicity of infection (MOI) in serum-free DMEM or RPMI, and further incubated in complete DMEM or RPMI for the indicated times.
Western blotting
Antibodies used in western blotting were anti-LDH (Santa Cruz Biotechnology, sc-33781), anti-MYC (Santa Cruz Biotechnology, sc-40), anti-MAVS (Santa Cruz Biotechnology, sc-166583), anti-HSPD1/HSP60 (Santa Cruz Biotechnology, sc-271215), anti-V5 (ThermoFisher Scientific, R960–25), anti-TAX1BP1 (Abcam, ab176572), anti-TAX1BP1 (Proteintech 14424–1-AP), anti-SQSTM1/p62 (Cell Signaling Technology, 8025), anti-NFKBIA/IκBα (Cell Signaling Technology, 4812), anti-MAVS (Cell Signaling Technology, 4983), anti-TBK1 (Cell Signaling Technology, 3504), anti-IKBKE/IKKi (Cell Signaling Technology, 2905), anti-GFP (Santa Cruz Biotechnology, sc-9996), anti-ACTB/β-Actin (Santa Cruz Biotechnology, sc-47778), anti-GAPDH (Santa Cruz Biotechnology, sc-32233), anti-MAP1LC3/LC3 (Proteintech 14600–1-AP), anti-ATG3 (Proteintech 11262–2-AP), anti-RB1CC1 (Proteintech 17250–1-AP), anti-VCL/vinculin (Santa Cruz Biotechnology, sc-73614), anti-TUBA/α-Tubulin (Santa Cruz Biotechnology, sc-5286), anti-Flag (MilliporeSigma, F3165), anti-V5 agarose (MilliporeSigma, A7345), anti-ACTB/β-Actin (MilliporeSigma, A1978), anti-DYKDDDDK (L5) gel (BioLegend, 651502), anti-HA (MilliporeSigma, 11583816001), rabbit IgG HRP-linked whole Ab (Cytiva, NA934) and mouse IgG HRP-linked whole Ab (Cytiva, NA931). Cells were lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% IGEPAL CA-630 [MilliporeSigma, I8896], and 0.25% deoxycholic acid [Fisher Scientific, BP349–100]) containing a protease inhibitor cocktail (Fisher Scientific, PI78443) and protein phosphatase inhibitors, including 10 mM NaF and 5 mM Na3VO4. Cell lysates were separated on SDS-PAGE, transferred to nitrocellulose or polyvinylidene difluoride membranes, and immunoblotted with appropriate primary antibodies diluted in SuperBlock™ (PBS) blocking buffer (ThermoFisher Scientific, 37515). Following incubation with horseradish peroxidase-labeled appropriate secondary antibody, immunoreactive bands were visualized by an enhanced chemiluminescence (ECL) reagent on an X-ray film or Azure 600 Western Blot Imaging System. ImageJ software (NIH) or Image Lab software (Bio-Rad) was used to quantify the intensities of bands.
Immunofluorescence microscopy
Antibodies used in immunofluorescence experiments were anti-TAX1BP1 (Cell Signaling Technology, 5105S), anti-SQSTM1/p62 (MBL Life Science, PM045), anti-ubiquitin (Cayman, 14220), anti-LAMP1 (Sino Biological, 11215-R107), Alexa Fluor 488 goat anti-mouse IgG (ThermoFisher Scientific, A11029) and Alexa Fluor 594 goat anti-rabbit IgG (ThermoFisher Scientific, A11037). Cells grown on a coverslip (and transfected) were fixed in Image-iT™ fixative (ThermoFisher Scientific, R37814) and permeabilized in 0.5% Triton X-100 (MilliporeSigma, T8787) prepared in PBS (Corning, 21–040-CV). Following incubation with SuperBlock™ PBS blocking buffer for 1 h at room temperature, coverslips were incubated with primary antibodies, washed with PBS, and then incubated with appropriate fluorescent dye-conjugated secondary antibodies. Coverslips were mounted in ProLong™ Gold Antifade Mounting medium containing 4’, 6-diamidino-2-phenylindole (DAPI; ThermoFisher Scientific, P36965) on glass slides and cells were imaged on a Zeiss 700 or Leica SP8 Falcon confocal laser scanning microscope with a 63× oil-immersion objective and Zen software. Manders correlation coefficient was calculated to measure colocalization between TAX1BP1 and MAP1LC3/LC3, ubiquitin, and SQSTM1 using Coloc2 (ImageJ).
Flow cytometry assays
Cells were harvested following trypsinization and washed three times with PBS. Following a PBS wash, cells were resuspended in Flow Cytometry Staining Buffer (ThermoFisher, 00-4222-26) and assayed using a BD Symphony 17 cytometer. Flow cytometry analysis was performed using FlowJo 10 software. Cell sorting of eGFP-TAX1BP1 DLD-1 cells was performed with a BD FACSMelody cell sorter.
Plasmids and nucleic acid manipulation
All polymerase chain reaction (PCR) amplification and site-directed mutagenesis were performed using Platinum™ Pfx, SuperFi™ DNA polymerase (ThermoFisher Scientific, 1235–1010) or the QuikChange II mutagenesis kit (Agilent, 200523). Subcloning of open reading frames (ORFs) and their derivatives into pICE [63] and other expression plasmids was conducted using appropriate restriction enzyme sites (Table 1). The gRNAs for each gene were synthesized by Integrated DNA Technologies, and cloned into plentiCRISPR v2-puro [64] or plentiCRISPR v2-blasticidin (a gift from Mohan Babu [Addgene 83480; http://n2t.net/addgene:83480; RRID:Addgene_83480]) using BsmBI. pHAGE-eGFP-TAX1BP1 was a gift from Wade Harper (Harvard Medical School) [67]. Oligonucleotides are listed in Table 2.
Table 1.
Plasmids used in the study.
| Name | Source/Cloning or Mutagenesis | Identifier |
|---|---|---|
| lentiCRISPR v2-puro | [64] | Addgene 52961; deposited by Feng Zhang |
| lentiCRISPR v2-puro_gRNAs | Cloning with BsmBI | N/A |
| lentiCRISPR v2-blasticidin | A gift from Mohan Babu (University of Regina) | Addgene 83480 |
| lentiCRISPR v2-blasticidin_gRNAs | Cloning with BsmBI | N/A |
| lentiCRISPR v2-puro TBK1 gRNA | [65] | N/A |
| lentiCRISPR v2-puro IKBKE/IKKi gRNA | [65] | N/A |
| pHAGE-eGFP-TAX1BP1 | A gift from Wade Harper (Harvard Medical School) | N/A |
| pICE | [63] | Addgene 46960; deposited by Steve Jackson |
| pICE_V5 | [66] | N/A |
| pICE_V5-MAVS | Cloning with BamHI and MluI | N/A |
| Flag-MAVS | [27] | N/A |
| HA-CHUK/IKKα | [39] | N/A |
| Flag-IKBKE/IKKi | [27] | N/A |
| Flag-IKBKE/IKKiK38A | A gift from Tom Maniatis (Columbia University) | Addgene 26202 |
| Flag-TBK1 | [27] | N/A |
| Flag-TBK1K38A | Mutagenesis | N/A |
| MYC-MAP3K14/NIK | A gift from Shao-Cong Sun (MD Anderson Cancer Center) | N/A |
| pCLXSN_Flag-TAX1BP1 | [39] | N/A |
| pBABEpuro GFP-MAP1LC3/LC3 | A gift from Jayanta Debnath (University of California, San Francisco) | Addgene 22405 |
| pICE_Flag-TAX1BP1 WT and variants | Cloning with BamHI and MluI | N/A |
| plenti.puro_TAX1BP1 WT and variants | Cloning with AgeI and SalI | N/A |
| plenti.puro | A gift from Melina Fan (Addgene) | Addgene 7218 |
| pRK5_HA-TAX1BP1 10A | Cloning with SalI and NotI | N/A |
| pBiT 1.1N_TAX1BP1 WT and variants | Cloning with XhoI and SacI | N/A |
| pBiT 2.1N_TAX1BP1 WT and variants | Cloning with XhoI and SacI | N/A |
| HaloTag-SmB (negative control) | Promega | 2014 |
| psPAX2 | A gift from Didier Trono (École Polytechnique Fédérale de Lausanne) | Addgene 12260 |
| VSV-G | [39] | N/A |
| pCL-Ampho | A gift from Shao-Cong Sun (MD Anderson Cancer Center) | N/A |
| IFN-β-Luc | [27] | N/A |
| pRL-TK | Promega | E2231 |
Table 2.
Oligonucleotides used in the study.
| Name | Forward or Reverse | Sequences (5’ to 3’) |
|---|---|---|
| RB1CC1/FIP200 gRNA | Forward | CACCGTATGTATTTCTGGTTAACAC |
| Reverse | AAACGTGTTAACCAGAAATACATAC | |
| ATG3 gRNA | Forward | CACCGAGGTGTAATTACCCCAGAAG |
| Reverse | AAACCTTCTGGGGTAATTACACCTC | |
| TAX1BP1 gRNA | Forward | CACCGAATTGTGTACTAGCATTCCA |
| Reverse | AAACTGGAATGCTAGTACACAATTC | |
| TAX1BP1 resistant to gRNA | Forward | GTCAACTGCGTCCTGGCTTTTCAAGGATATTACCTTCCAAATGAT |
| Reverse | TCCTTGAAAAGCCAGGACGCAGTTGACTGTTGATCCTTCCACATA | |
| TAX1BP1 S123A/S124A mutagenesis | Forward | CCTTTCCAGTTTCGAGCTGCTGCTCCAGTTGAAGAGCTGCTT |
| Reverse | AAGCAGCTCTTCAACTGGAGCAGCAGCTCGAAACTGGAAAGG | |
| TAX1BP1 T131A mutagenesis | Forward | GTTGAAGAGCTGCTTGCTATGGAAGATGAAGGA |
| Reverse | TCCTTCATCTTCCATAGCAAGCAGCTCTTCAAC | |
| TAX1BP1 T250A mutagenesis | Forward | GCAATTGAAAAAGAAGCCGAATTAGACAGT |
| Reverse | ACTGTCTAATTCGGCTTCTTTTTCAATTGC | |
| TAX1BP1 S254A mutagenesis | Forward | GAAACCGAATTAGACGCTTTAAAGGACAAACTC |
| Reverse | GAGTTTGTCCTTTAAAGCGTCTAATTCGGTTTC | |
| TAX1BP1 S508A mutagenesis | Forward | GTAGATGTAAAGCCAGCACCTTCTGCAGCAGAG |
| Reverse | CTCTGCTGCAGAAGGTGCTGGCTTTACATCTAC | |
| TAX1BP1 S593A mutagenesis | Forward | TATAAAGAACTTAAAAGGGCTCTAGAAAATCCAGCAGAA |
| Reverse | TTCTGCTGGATTTTCTAGAGCCCTTTTAAGTTCTTTATA | |
| TAX1BP1 S607A/S609A mutagenesis | Forward | AAAATGGAAGGTCAGAATGCCCAGGCTCCTCAATGTTTCAAAACA |
| Reverse | TGTTTTGAAACATTGAGGAGCCTGGGCATTCTGACCTTCCATTTT | |
| TAX1BP1 S666A mutagenesis | Forward | CCTGTCAGAGTCCCCGCTTGGGGACTGGAAGAC |
| Reverse | GTCTTCCAGTCCCCAAGCGGGGACTCTGACAGG | |
| TAX1BP1 S683A mutagenesis | Forward | CAGCCTGCTCGAAACTTTGCTCGGCCTGATGGCTTAGAG |
| Reverse | CTCTAAGCCATCAGGCCGAGCAAAGTTTCGAGCAGGCTG | |
| TAX1BP1 T250 A->T mutagenesis | Forward | GCAATTGAAAAAGAAACCGAATTAGACGCTTTA |
| Reverse | TAAAGCGTCTAATTCGGTTTCTTTTTCAATTGC | |
| TAX1BP1 S254 A->S mutagenesis | Forward | GAAGCCGAATTAGACAGTTTAAAGGACAAACTC |
| Reverse | GAGTTTGTCCTTTAAACTGTCTAATTCGGCTTC | |
| TAX1BP1 S508 A->S mutagenesis | Forward | GTAGATGTAAAGCCATCACCTTCTGCAGCAGAG |
| Reverse | CTCTGCTGCAGAAGGTGATGGCTTTACATCTAC | |
| TAX1BP1 S593 A->S mutagenesis | Forward | AAAGAACTTAAAAGGAGTCTAGAAAATCCAGCA |
| Reverse | TGCTGGATTTTCTAGACTCCTTTTAAGTTCTTT | |
| TAX1BP1 S607 A->S mutagenesis | Forward | ATGGAAGGTCAGAATTCCCAGGCTCCTCAATGT |
| Reverse | ACATTGAGGAGCCTGGGAATTCTGACCTTCCAT | |
| TAX1BP1 S609 A->S mutagenesis | Forward | GGTCAGAATGCCCAGAGTCCTCAATGTTTCAAA |
| Reverse | TTTGAAACATTGAGGACTCTGGGCATTCTGACC | |
| TAX1BP1 S666 A->S mutagenesis | Forward | CCTGTCAGAGTCCCCTCTTGGGGACTGGAAGAC |
| Reverse | GTCTTCCAGTCCCCAAGAGGGGACTCTGACAGG | |
| TAX1BP1 S683 A->S mutagenesis | Forward | CAGCCTGCTCGAAACTTTAGTCGGCCTGATGGCTTAGAG |
| Reverse | CTCTAAGCCATCAGGCCGACTAAAGTTTCGAGCAGGCTG | |
| TAX1BP1 S691 A->S mutagenesis | Forward | GATGGCTTAGAGGACTCTGAGGATGCCAAAGAA |
| Reverse | TTCTTTGGCATCCTCAGAGTCCTCTAAGCCATC | |
| TAX1BP1 S694 A->S mutagenesis | Forward | GAGGACGCTGAGGATAGCAAAGAAGATGAGAAT |
| Reverse | ATTCTCATCTTCTTTGCTATCCTCAGCGTCCTC | |
| TAX1BP1 S254D mutagenesis | Forward | GAAACCGAATTAGACGATTTAAAGGACAAACTC |
| Reverse | GAGTTTGTCCTTTAAATCGTCTAATTCGGTTTC | |
| TAX1BP1 S593D mutagenesis | Forward | AAAGAACTTAAAAGGGATCTAGAAAATCCAGCA |
| Reverse | TGCTGGATTTTCTAGATCCCTTTTAAGTTCTTT | |
| TAX1BP1 S666D mutagenesis | Forward | CCTGTCAGAGTCCCCGATTGGGGACTGGAAGAC |
| Reverse | GTCTTCCAGTCCCCAATCGGGGACTCTGACAGG | |
| TBK1 K38A mutagenesis | Forward | GATTTATTTGCTATCGCAGTATTTAATAACATA |
| Reverse | TATGTTATTAAATACTGCGATAGCAAATAAATC |
CRISPR-Cas9-mediated gene knockout
CRISPR-Cas9-mediated genetic ablation of the indicated genes in DLD-1 and 293T cells was performed as previously described [68]. Antibiotic-resistant individual clones were isolated by limiting dilution. The ATG3 [41] and RB1CC1 [69] gRNAs were previously described. TBK1 and IKBKE/IKKi gRNAs cloned in pLentiCRISPRv2 were kindly provided by Dr. Fangfang Zhou (Soochow University, Suzhou, Jiangsu, China) [65].
Retroviral infections
Retroviral infections were performed as previously described [70]. Briefly, 293T cells were transfected with pCL-Ampho, VSV-G and pBABEpuro GFP-MAP1LC3/LC3 (a gift from Jayanta Debnath [Addgene 22405; http://n2t.net/addgene:22405; RRID:Addgene_22405]). The supernatants were filtered and used to infect DLD-1 cells in the presence of polybrene/hexadimethrine bromide (10 μg/ml; MilliporeSigma, H9268). GFP+ cells were obtained by cell sorting to obtain stable, bulk DLD-1 GFP-MAP1LC3/LC3 cells.
In vitro kinase and phosphatase assays
Purified GST-tagged recombinant full-length human TAX1BP1 protein (Novus Biologicals, H00008887-P01) was incubated with purified GST-tagged IKBKE/IKKi (Thermo Fisher Scientific, PV4875) in buffer A containing 20 mM Tris, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 1 mM Na3VO4, 5 mM β-glycerophosphate (MilliporeSigma, G9422), 2 mM DTT, 0.02% Triton X-100 and 200 µM ATP (Fisher Scientific, 11–915-GM) for 10 min at 30°C or with purified hexahistidine-tagged TBK1 (ThermoFisher Scientific, PV3504) in buffer B containing 50 mM Tris, pH 7.5, 10 mM MgCl2, 2 mM DTT, 0.025% Triton X-100 and 200 µM ATP for 20 min at 30°C. The reaction was terminated by boiling in 1× SDS-sample buffer for 5 min. For dephosphorylation of TAX1BP1, lysates from 293T transfected cells were incubated with 800 U λ-phosphatase at 30°C for 30 min and then subjected to SDS-PAGE analysis.
Mass spectrometry (MS) analysis
Coomassie Brilliant Blue-stained gel pieces were de-stained and subjected to reduction (5 mM DTT for 45 min at 60) and alkylation (20 mM iodoacetamide [Promega, VB1010] for 20 min at room temperature in the dark). Samples were subsequently proteolyzed with 10 ng/µl trypsin (Promega, V5280) overnight at 37°C. Dry extracted peptides after clean-up were re-suspended in 8 µl 0.1% formic acid. Titanium dioxide was used for phosphopeptide enrichment. Protein identification by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of peptides was performed using an LTQ Orbitrap Velos MS (ThermoFisher Scientific) interfaced with a nanoAcquity LC system (Waters, Corp.). Peptides were fractionated by reverse-phase HPLC on a 75 µm x 15 cm PicoFrit column with a 15 µm emitter (New Objective) in-house packed with Magic C18AQ (Michrom BioResources, Inc.) using a 0–60% acetonitrile:0.1% formic acid gradient over 70 min at 300 nl/min. Eluting peptides were sprayed directly into an LTQ Orbitrap Velos at 2.0 kV. Survey scans were acquired from 350–1,800 m/z with up to 10 peptide masses individually isolated with a 1.9 Da window and fragmented (MS/MS) using a collision energy of 40 and 30s dynamic exclusion. Precursor and fragment ions were analyzed at 30,000 and 7500 resolution, respectively. Peptide sequences were identified from isotopically resolved masses in MS and MS/MS spectra extracted with and without deconvolution using Thermo Scientific MS2 processor and Xtract software. Data was searched for in the human RefSeq database, with oxidation on methionine (variable), deamidation NQ (variable), phosphoSTY (variable) and carbamidomethyl on cysteine as (fixed) modifications, using Proteome Discoverer 1.3 software.
Semi-denaturing detergent agarose gel electrophoresis (SDD-AGE)
SDD-AGE was performed as previously described [66]. Briefly, crude mitochondria isolated by differential centrifugation were resuspended in 1× sample buffer (0.5× Tris-borate-EDTA [TBE], 10% glycerol, 2% SDS, and 0.0025% bromophenol blue) and loaded onto a 1.5% agarose gel. After electrophoresis in running buffer (1× TBE and 0.1% SDS) for 1 h with a constant voltage of 100 V at 4°C, proteins were transferred to a nitrocellulose membrane for western blotting. SDD-AGE with transfected MAVS and TAX1BP1 was performed similarly but with whole cell lysates.
Dual luciferase reporter assays
Cells were transfected with the desired plasmids together with IFNB-Luc [27] and the Renilla reporter pRL-TK-Luc (Promega, E2241) as an internal control. After 24 h, cells were lysed in passive lysis buffer (Promega, E1941) and subjected to dual-luciferase assays as recommended by the manufacturer (Promega, E1910). Luminescence was measured using a GloMax® Explorer luminometer (Promega, GM3500). Results are presented as the relative firefly luciferase activity over the Renilla luciferase activity.
NanoBiT assays
NanoBiT assays (Promega, N2014) were performed as previously described [66]. Briefly, 293T cells were seeded in a 6-well plate and transfected with pairs of NanoBiT constructs. After 24 h, cells were collected, washed two times in PBS (pH 7.4), and resuspended in 1 ml of Opti-MEM I Reduced Serum Media (ThermoFisher Scientific, 11058021). The cell suspension (100 µl) was transferred to a white-walled 96-well plate in triplicate, and 20 µl of Nano-Glo® Luciferase substrate furimazine (Promega, N205A) diluted in PBS at a ratio of 1:100 was added to each well. After incubation for 5 min at room temperature, luminescence was measured using a GloMax luminometer.
Statistical analysis
Data are presented as mean ± standard deviation from a representative experiment with triplicate samples. Statistical analysis was performed in GraphPad Prism 8 and indicated in the Figure legends. Statistical significance was set at a p value of < 0.05.
Supplementary Material
Acknowledgements
We thank Drs. Hong-Gang Wang (Penn State College of Medicine) and Shengkan (Victor) Jin (Rutgers University, Robert Wood Johnson Medical School) for atg3–/– MEFs, Dr. Jun-Lin Guan (University of Cincinnati College of Medicine) for rb1cc1–/– MEFs, Dr. Richard Youle for TAX1BP1 KO HeLa cells, Dr. Fangfang Zhou for TBK1 and IKBKE/IKKi CRISPR-Cas9 plasmids, Dr. Wade Harper for pHAGE-eGFP-TAX1BP1 plasmid and Drs. Siddharth Balachandran (Fox Chase Cancer Center) and Glen Barber (University of Miami, Miller School of Medicine) for VSV-GFP. We also thank Dr. Robert Cole of Johns Hopkins School of Medicine for assistance with mass spectrometry. The Advanced Light Microscopy Core (RRID: SCR_022526) and Flow Cytometry Core (RRID:SCR_021134), and services and instruments used in this project were funded, in part, by the Pennsylvania State University College of Medicine via the Office of the Vice Dean of Research and Graduate Students and the Pennsylvania Department of Health using Tobacco Settlement Funds (CURE). The content is solely the responsibility of the authors and does not necessarily represent the official views of the University or College of Medicine. The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions. This work was supported, in part, by NIH grant R01 AI162815 (to EWH).
Funding Statement
The work was supported by the National Institutes of Health [R01AI162815].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2024.2394306
References
- [1].Hornung V, Ellegast J, Kim S, et al. 5’-triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- [2].Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi: 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- [3].Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–988. doi: 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- [4].Seth RB, Sun L, Ea CK, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates nf-kappaB and IRF 3. Cell. 2005;122(5):669–682. doi: 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- [5].Oganesyan G, Saha SK, Guo B, et al. Critical role of TRAF3 in the toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439(7073):208–211. doi: 10.1038/nature04374 [DOI] [PubMed] [Google Scholar]
- [6].Zhao T, Yang L, Sun Q, et al. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8(6):592–600. doi: 10.1038/ni1465 [DOI] [PubMed] [Google Scholar]
- [7].Guo B, Cheng G.. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem. 2007;282(16):11817–11826. doi: 10.1074/jbc.M700017200 [DOI] [PubMed] [Google Scholar]
- [8].Liu S, Chen J, Cai X, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2:e00785. doi: 10.7554/eLife.00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fitzgerald KA, McWhirter SM, Faia KL, et al. Ikkepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4(5):491–496. doi: 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- [10].Sharma S, tenOever BR, Grandvaux N, et al. Triggering the interferon antiviral response through an ikk-related pathway. Science. 2003;300(5622):1148–1151. doi: 10.1126/science.1081315 [DOI] [PubMed] [Google Scholar]
- [11].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. doi: 10.1038/s41580-018-0003-4 [DOI] [PubMed] [Google Scholar]
- [12].Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42. doi: 10.1016/j.cell.2018.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19(5):2092–2100. doi: 10.1091/mbc.e07-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marshall RS, Vierstra RD. Autophagy: the master of bulk and selective recycling. Annu Rev Plant Biol. 2018;69(1):173–208. doi: 10.1146/annurev-arplant-042817-040606 [DOI] [PubMed] [Google Scholar]
- [15].Grumati P, Dikic I. Ubiquitin signaling and autophagy. J Biol Chem. 2018;293(15):5404–5413. doi: 10.1074/jbc.TM117.000117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020;432(1):80–103. doi: 10.1016/j.jmb.2019.07.016 [DOI] [PubMed] [Google Scholar]
- [17].Lamark T, Svenning S, Johansen T, et al. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61(6):609–624. doi: 10.1042/EBC20170035 [DOI] [PubMed] [Google Scholar]
- [18].Jin S, Tian S, Luo M, et al. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective Autophagic degradation in human cells. Mol Cell. 2017;68(2):308–322 e4. doi: 10.1016/j.molcel.2017.09.005 [DOI] [PubMed] [Google Scholar]
- [19].He X, Zhu Y, Zhang Y, et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. Embo J. 2019;38(14):e100978. doi: 10.15252/embj.2018100978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gachon F, Peleraux A, Thebault S, et al. CREB-2, a cellular cre-dependent transcription repressor, functions in association with tax as an activator of the human T-cell leukemia virus type 1 promoter. J Virol. 1998;72(10):8332–8337. doi: 10.1128/JVI.72.10.8332-8337.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].De Valck D, Jin DY, Heyninck K, et al. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene. 1999;18(29):4182–4190. doi: 10.1038/sj.onc.1202787 [DOI] [PubMed] [Google Scholar]
- [22].Ling L, Goeddel DV. T6BP, a TRAF6-interacting protein involved in IL-1 signaling. Proc Natl Acad Sci USA. 2000;97(17):9567–9572. doi: 10.1073/pnas.170279097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shembade N, Harhaj NS, Liebl DJ, et al. Essential role for TAX1BP1 in the termination of tnf-alpha-, IL-1- and lps-mediated nf-kappaB and JNK signaling. Embo J. 2007;26(17):3910–3922. doi: 10.1038/sj.emboj.7601823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Harhaj EW, Dixit VM. Regulation of nf-kappaB by deubiquitinases. Immunol Rev. 2012;246(1):107–124. doi: 10.1111/j.1600-065X.2012.01100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shembade N, Harhaj NS, Parvatiyar K, et al. The E3 ligase itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008;9(3):254–262. doi: 10.1038/ni1563 [DOI] [PubMed] [Google Scholar]
- [26].Shembade N, Parvatiyar K, Harhaj NS, et al. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate nf-kappaB signalling. Embo J. 2009;28(5):513–522. doi: 10.1038/emboj.2008.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 2010;285(20):14999–15009. doi: 10.1074/jbc.M110.109819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Choi YB, Shembade N, Parvatiyar K, et al. TAX1BP1 restrains virus-induced apoptosis by facilitating itch-mediated degradation of the mitochondrial adaptor MAVS. Mol Cell Biol. 2017;37(1). doi: 10.1128/MCB.00422-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang Q, Liu TT, Lin H, et al. TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLOS Pathog. 2017;13(9):e1006600. doi: 10.1371/journal.ppat.1006600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Samie M, Lim J, Verschueren E, et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat Immunol. 2018;19(3):246–254. doi: 10.1038/s41590-017-0042-6 [DOI] [PubMed] [Google Scholar]
- [31].Newman AC, Scholefield CL, Kemp AJ, et al. TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical nf-kappaB signalling. PLOS ONE. 2012;7(11):e50672. doi: 10.1371/journal.pone.0050672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tumbarello DA, Waxse BJ, Arden SD, et al. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012;14(10):1024–1035. doi: 10.1038/ncb2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tumbarello DA, Manna PT, Allen M, et al. The autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of salmonella typhimurium by autophagy. PLOS Pathog. 2015;11(10):e1005174. doi: 10.1371/journal.ppat.1005174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Iha H, Peloponese JM, Verstrepen L, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective nf-kappaB activation. Embo J. 2008;27(4):629–641. doi: 10.1038/emboj.2008.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].White J, Suklabaidya S, Vo MT, et al. Multifaceted roles of TAX1BP1 in autophagy. Autophagy. 2022;19(1):44–53. doi: 10.1080/15548627.2022.2070331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi: 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Budzik JM, Swaney DL, Jimenez-Morales D, et al. Dynamic post-translational modification profiling of mycobacterium tuberculosis-infected primary macrophages. Elife. 2020;9. doi: 10.7554/eLife.51461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sarraf SA, Shah HV, Kanfer G, et al. Loss of TAX1BP1-directed autophagy results in protein aggregate accumulation in the brain. Mol Cell. 2020;80(5):779–795 e10. doi: 10.1016/j.molcel.2020.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shembade N, Pujari R, Harhaj NS, et al. The kinase IKKalpha inhibits activation of the transcription factor nf-kappaB by phosphorylating the regulatory molecule TAX1BP1. Nat Immunol. 2011;12(9):834–843. doi: 10.1038/ni.2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ohnstad AE, Delgado JM, North BJ, et al. Receptor-mediated clustering of FIP200 bypasses the role of LC3 lipidation in autophagy. Embo J. 2020;39(24):e104948. doi: 10.15252/embj.2020104948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Goodwin JM, Dowdle WE, DeJesus R, et al. Autophagy-independent lysosomal targeting regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017;20(10):2341–2356. doi: 10.1016/j.celrep.2017.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hou F, Sun L, Zheng H, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–461. doi: 10.1016/j.cell.2011.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zamorano Cuervo N, Osseman Q, Grandvaux N. Virus infection triggers MAVS polymers of distinct molecular weight. Viruses. 2018;10(2):56. doi: 10.3390/v10020056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Thurston TL, Ryzhakov G, Bloor S, et al. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10(11):1215–1221. doi: 10.1038/ni.1800 [DOI] [PubMed] [Google Scholar]
- [45].Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts salmonella growth. Science. 2011;333(6039):228–233. doi: 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA. 2016;113(15):4039–4044. doi: 10.1073/pnas.1523926113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Matsumoto G, Wada K, Okuno M, et al. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44(2):279–289. doi: 10.1016/j.molcel.2011.07.039 [DOI] [PubMed] [Google Scholar]
- [48].Pilli M, Arko-Mensah J, Ponpuak M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37(2):223–234. doi: 10.1016/j.immuni.2012.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60(1):7–20. doi: 10.1016/j.molcel.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fu T, Liu J, Wang Y, et al. Mechanistic insights into the interactions of NAP1 with the SKICH domains of NDP52 and TAX1BP1. Proc Natl Acad Sci USA. 2018;115(50):E11651–E11660. doi: 10.1073/pnas.1811421115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hutti JE, Turk BE, Asara JM, et al. I{kappa}B kinase {beta} phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the nf-{kappa}b pathway. Mol Cell Biol. 2007;27(21):7451–7461. doi: 10.1128/MCB.01101-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hutti JE, Porter MA, Cheely AW, et al. Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKepsilon. PLOS ONE. 2012;7(7):e41494. doi: 10.1371/journal.pone.0041494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Marinis JM, Hutti JE, Homer CR, et al. IkappaB kinase alpha phosphorylation of TRAF4 downregulates innate immune signaling. Mol Cell Biol. 2012;32(13):2479–2489. doi: 10.1128/MCB.00106-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chin KT, Chun AC, Ching YP, et al. Human T-Cell leukemia virus oncoprotein tax represses nuclear receptor-dependent transcription by targeting coactivator TAX1BP1. Cancer Res. 2007;67(3):1072–1081. doi: 10.1158/0008-5472.CAN-06-3053 [DOI] [PubMed] [Google Scholar]
- [55].Shao WH, Shu DH, Zhen Y, et al. Prion-like aggregation of mitochondrial antiviral signaling protein in lupus patients is associated with increased levels of type I interferon. Arthritis Rheumatol. 2016;68(11):2697–2707. doi: 10.1002/art.39733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yoo YS, Park YY, Kim JH, et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat Commun. 2015;6(1):7910. doi: 10.1038/ncomms8910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu C, Huang S, Wang X, et al. The otubain YOD1 suppresses aggregation and activation of the signaling adaptor MAVS through Lys63-linked deubiquitination. J Immunol. 2019;202(10):2957–2970. doi: 10.4049/jimmunol.1800656 [DOI] [PubMed] [Google Scholar]
- [58].Wang X, Naidu SR, Sverdrup F, et al. Tax1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. J Virol. 2009;83(5):2274–2284. doi: 10.1128/JVI.01791-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Petkova DS, Verlhac P, Rozieres A, et al. Distinct contributions of autophagy receptors in measles virus replication. Viruses. 2017;9(5):123. doi: 10.3390/v9050123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mohamud Y, Xue YC, Liu H, et al. Autophagy receptor protein tax1-binding protein 1/traf6-binding protein is acellular substrate of enteroviral proteinase. Front Microbiol. 2021;12:647410. doi: 10.3389/fmicb.2021.647410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sou YS, Waguri S, Iwata J, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19(11):4762–4775. doi: 10.1091/mbc.e08-03-0309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gan B, Peng X, Nagy T, et al. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and tsc-mTOR signaling pathways. J Cell Biol. 2006;175(1):121–133. doi: 10.1083/jcb.200604129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Britton S, Coates J, Jackson SP. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol. 2013;202(3):579–595. doi: 10.1083/jcb.201303073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–784. doi: 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang S, Xie F, Chu F, et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKvarepsilon-mediated phosphorylation. Nat Immunol. 2017;18(7):733–743. doi: 10.1038/ni.3744 [DOI] [PubMed] [Google Scholar]
- [66].Vo MT, Smith BJ, Nicholas J, et al. Activation of nix-mediated mitophagy by an interferon regulatory factor homologue of human herpesvirus. Nat Commun. 2019;10(1):3203. doi: 10.1038/s41467-019-11164-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Eapen VV, Swarup S, Hoyer MJ, et al. Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. Elife. 2021;10. doi: 10.7554/eLife.72328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Choi YB, Choi Y, Harhaj EW, et al. Peroxisomes support human herpesvirus 8 latency by stabilizing the viral oncogenic protein vFLIP via the MAVS-TRAF complex. PLOS Pathog. 2018;14(5):e1007058. doi: 10.1371/journal.ppat.1007058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Assar EA, Tumbarello DA. Loss of the essential autophagy regulators FIP200 or Atg5 leads to distinct effects on focal adhesion composition and organization. Front Cell Dev Biol. 2020;8:733. doi: 10.3389/fcell.2020.00733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shembade N, Harhaj NS, Yamamoto M, et al. The human T-cell leukemia virus type 1 tax oncoprotein requires the ubiquitin-conjugating enzyme Ubc13 for nf-kappaB activation. J Virol. 2007;81(24):13735–13742. doi: 10.1128/JVI.01790-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.