

Summary
We apply a single-molecule chromatin fiber sequencing (Fiber-seq) protocol designed for amplification-free cell-type-specific mapping of the regulatory architecture at nucleosome resolution along extended ∼10-kb chromatin fibers to neuronal and non-neuronal nuclei sorted from human brain tissue. Specifically, application of this method enables the resolution of cell-selective promoter and enhancer architectures on single fibers, including transcription factor footprinting and position mapping, with sequence-specific fixation of nucleosome arrays flanking transcription start sites and regulatory motifs. We uncover haplotype-specific chromatin patterns, multiple regulatory elements cis-aligned on individual fibers, and accessible chromatin at 20,000 unique sites encompassing retrotransposons and other repeat sequences hitherto “unmappable” by short-read epigenomic sequencing. Overall, we show that Fiber-seq is applicable to human brain tissue, offering sharp demarcation of nucleosome-depleted regions at sites of open chromatin in conjunction with multi-kilobase nucleosomal positioning at single-fiber resolution on a genome-wide scale.
Keywords: nucleosomal array, nucleosomal offset, transcription factor footprint, postmortem brain, long-read sequencing, adenine methylation, cytosine methylation, Hia5 methyltransferase
Graphical abstract
Highlights
-
•
Genome-scale long-read nucleosomal positioning mapping in human brain
-
•
Single-molecule transcription factor footprinting in FACS-sorted brain nuclei
-
•
Fiber-seq reproduces chromatin accessibility maps at superior resolution
Motivation
Nucleosomes are the basic unit of chromatin, but genome-scale maps of nucleosome positioning along chromatin fibers do not exist for brain and many other complex tissues. Conventional chromatin accessibility assays designed to map nucleosome-depleted regions via nucleolytic digestion face major limitations such as limited resolution and sequence bias, with additional shortcomings from PCR-generated short-read libraries including poor annotation for an estimated 50% of the human genome. To address this, we tested a brain-adapted single-molecule chromatin fiber sequencing (Fiber-seq) protocol designed for amplification-free adenine-methyltransferase tagging of extranucleosomal DNA in neuronal and, separately, non-neuronal nuclei in situ.
Peter et al. introduce a brain-adapted version of the Fiber-seq protocol for amplification-free genome-scale profiling of nucleosomal positions along ∼10-kb chromatin fibers in FACS-sorted brain nuclei in situ. Fiber-seq resolves transcription factor footprints and sequence-specific fixation of nucleosome arrays at single-molecule resolution, with sharp demarcation of nucleosome-depleted regions.
Introduction
Genome function is tightly linked to the 3D organization of nuclear DNA into chromatin structures across multiple scales. The nucleosome is the basic unit of chromatin fibers comprising a core histone octamer and approximately 147 bp of DNA wrapped around it.1 Neighboring nucleosomes are interconnected by linker DNA into nucleosomal arrays, assembling as heterogeneous 8- to 24-nm-wide chromatin fibers, with nucleosome positioning along the fiber thought to reflect a complex regulome driven by genomic sequence, ATP-dependent chromatin remodeling, and transcriptional activity2,3,4 and to serve as a strong determinant for chromosomal organization into functional domains.5,6
To date, no brain-related genome-scale maps exist for nucleosome positioning and occupancy (the latter being defined as the fraction of cells carrying a nucleosome at a specific genomic region2). However, genomic sites of low nucleosomal density, commonly referred to as “accessible chromatin,” have been mapped in brain with DNase- and transposase-based assays or related nucleolytic approaches7 and are thought to include sequences where transcription factors (TFs) and other chromatin regulatory proteins bind directly to genomic DNA. This includes ∼79,000 active enhancers8 that could regulate gene expression relevant for human-specific cognition and neuropsychiatric disease9,10,11,12 and brain-specific neoplasms.12 Unfortunately, conventional chromatin accessibility mappings face major limitations that impede deeper understanding of the brain’s epigenomic landscape and fall short of charting nucleosome positioning in the brain’s neuronal and glial genomes. This is because DNase- and transposase-based assays, while intended to map nucleosome-depleted sequences, show a strong bias toward genomic sites with overall loose chromatin, and therefore, these techniques suffer from decreased resolution and inflated size calculation of nucleosome-depleted sequences.7 Additional shortcomings of conventional techniques for genome-scale chromatin accessibility mapping, such as the DNase I digestion-based DNase-sequencing (DNase-seq) method and the, by now more widely used, assay for transposase-accessible chromatin (ATAC)-seq, include requirements for titration experiments, and their intrinsic cleavage preferences necessitate bias corrections.13 These techniques therefore offer only a crude approximation of the regulatory architecture of chromatin fibers. Furthermore, short-read sequencing in general is conducted with 100- to 150-bp fragments of exponentially amplified DNA, resulting in a significant loss of resolution at genomic regions that are not uniquely spanned by overlapping short reads. Thus, genome-scale chromatin accessibility mapping by short-read sequencing is suboptimal at low-complexity repetitive loci, duplicated regions, tandem arrays, and complex structural variants.14 As these types of sequences constitute far more than 50% of the human genome,15 the epigenomic composition of the majority of the nuclear genome is left unexplored.
Here, we bypass these limitations by adapting single-molecule chromatin fiber sequencing (Fiber-seq),16 a novel technology hitherto applied only to peripheral cells, which was designed to map the open chromatin landscape and nucleosomal positions at the resolution of single chromatin fibers, each ∼10 kb in length. Specifically, we demonstrate the application of Fiber-seq to sorted intact adult human brain nuclei, amplification free, and on a genome-wide scale. We present initial reference maps for nucleosomal positioning specific to neuronal and non-neuronal nuclei and demarcate nucleosome-depleted regulatory sequences with sharper resolution compared to conventional nucleolytic short-read chromatin assays. We uncover tens of thousands of nucleosome-depleted regions (NDRs) and actively regulated genomic “dark matter,” including repetitive sequences that hitherto had remained epigenomically unmappable. We highlight the ability of this approach to uncover haplotype-specific nucleosome positioning and occupancy using recently introduced Fiber-seq analytical pipelines,17 and co-regulation of cis-bound promoter and enhancer elements at single-fiber levels. We confirm for the human brain long-standing principles previously shown only in simple eukaryote models and cell lines, such as the invariance in sequence-specific occupancy of nucleosomes bordering transcription start sites (TSSs) at active promoters.
Results
Cell-type-specific Fiber-seq in brain nuclei in situ
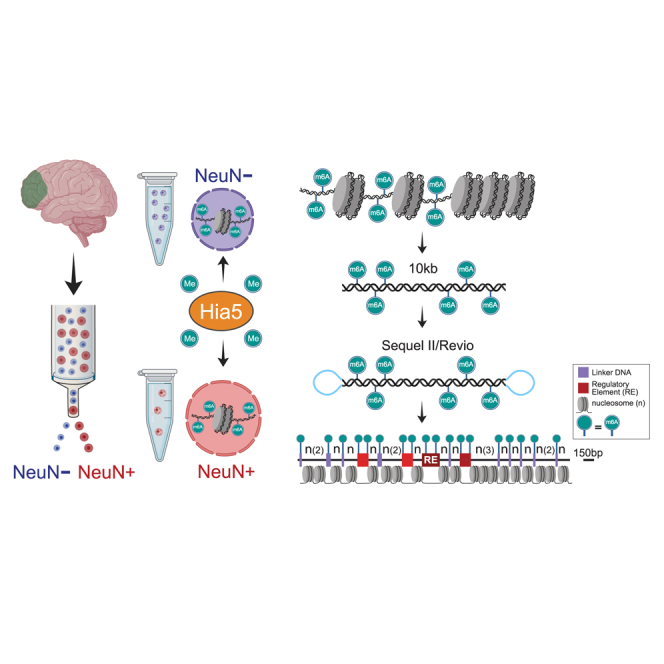
Epigenomic regulation is cell-type specific.18 To more comprehensively test the potential of Fiber-seq in brain, we separated nuclei from human prefrontal cortex (PFC) by immunotagging and fluorescence-activated sorting into neuronal (NeuN+) and non-neuronal (NeuN−) fractions of nuclei (Figure 1A; Table S1). Quality control checks, including single-nucleus RNA-seq from sorted NeuN+ and NeuN− nuclei (n = 22,844 and n = 13,977 nuclei, respectively), confirmed complete separation of the neuronal and non-neuronal fractions (Figures S1A–S1C). As expected from transcriptomics-based cell typing of the adult human cortex,19 the NeuN+ fraction included multiple subtypes of glutamatergic and GABAergic neurons, while the largest share of the NeuN− fraction comprised oligodendrocytes and their precursors, followed by astrocytic and microglial populations (Figure S1B). Sorted NeuN+ and NeuN− nuclei then served as input material for the generation of Fiber-seq libraries (Figures 1B–1D). For each sample, 0.4–2.5 × 106 sorted nuclei (Figure 1E) were incubated in situ with recombinant Hia5 m6A adenine methyltransferase (m6A-MTase) (Figures S2A and S2B) and processed for PacBio Sequel II sequencing, without any polymerase chain reaction (PCR). Thus, in each Fiber-seq library, each sequenced DNA molecule (Figures 1C and 1D) directly originates from a sorted PFC nucleus and represents a single chromatin fiber.
Figure 1.

Single chromatin fiber long-read epigenomic profiling by cell type in human brain
(A–D) Work flow. (A) Fluorescence-activated cell sorting (FACS) of NeuN immunostained neuronal and non-neuronal nuclei from the region of interest (PFC) and in situ incubation of the intact nuclei with adenine methyltransferase Hia5 for m6A tagging of extranucleosomal genomic DNA, followed by (B) amplification-free PacBio (Sequel II or Revio) long-read sequencing of ∼10-kb single DNA molecules derived directly from the brain nuclei. (C and D) Early step computational analyses include annotation to the telomere-to-telomere T2T-CHM13v2.0/hs1 genome, mapping of Hia5 methylation-sensitive patches (MSPs)/nucleosome-depleted regions, and calling of linker DNA (purple) and regulatory elements (red, brown), such as promoters and enhancers, at single-fiber resolution. (C) Example fiber visualizations were taken from the NeuN− Fiber-seq reference set, showing the promoter region of the chr18 SEH1L gene locus. Portions of fibers shown (fiber 1–4) are marked by green bars in (D). Fiber 4 in (C) is shown at enlarged scale, highlighting Fiber-seq-generated nucleosomal position map at single-nucleosome resolution. (D) hs1-T2T genome browser shots for PFC non-neuronal chromatin profiles at the SEH1L locus, including (top to bottom) “peak” landscapes for (blue) ATAC, (green) H3K4me3, (red) H3K27ac-seq, and (orange) CTCF ENCODE (Enc.) neural cell line (CTCF short-read-based peak landscapes). Fiber-seq long-read sequencing: (brown) m5CpG tracks, (black) Fiber-seq FIRE scores of MSPs. NeuN− single DNA molecules = single chromatin fibers as indicated; green-shaded single DNA molecules/chromatin fibers are highlighted as representative fiber examples shown in (C), including the fourth fiber at 150 bp resolution. Purple vertical rectangles mark linker DNA. Red/brown MSPs scoring as regulatory elements (P, promoter, and other REs, regulatory elements) on fiber (FIRE false discovery rate [FDR], orange, 0.10 ≥ q > 0.05; bright red, 0.05 ≥ q > 0.01; dark red-brown, q ≤ 0.01).
(E) Representative FACS showing efficient separation of non-neuronal (NeuN−) from neuronal (NeuN+) nuclei.
(F) Genome-scale proportion of methyladenines (m6A/total A; y axis) in 46 Fiber-seq-processed samples of human brain nuclei. Each sample/aliquot was sequenced at low depth (see text), and the m6A/A was computed as an average across all DNA molecules of a sample. The x axis marks the number of nuclei/nU Hia5 enzyme for each sample of nuclei (log scale, x axis). Blue dot, NeuN− reference sample sequenced with 11,143,795 reads/median of 30-fold genomic coverage; red dots, NeuN+ samples (4 aliquots × 2 brains) picked for sequencing to a total of 4,749,556 reads/median of 20-fold genomic coverage.
Accessibility mapping methods are sensitive to enzyme concentrations and various other factors.20 We explored whether variations in nucleus-to-Hia5 enzyme ratio (N/E) could impact the overall proportion of methyladenines. We conducted a pilot study on n = 46 Fiber-seq assays (n = 23 brains × 2 cell types, NeuN+ and NeuN−) by systematically varying the N/E and keeping all other conditions unaltered across experiments. We titrated the N/E from 1 × 103 to 1 × 107 nuclei/nU Hia5 (we defined via an m6A ELISA the relative Hia5 enzyme activity as 16.6 nmol/min/mL) (Figures S2C–S2E), followed by shallow sequencing (N reads/Fiber-seq library; range ∼400–600,000, median ∼500,000). As expected, there was overall an inverse correlation between the genome-wide extent of Hia5 methylation and the N/E ratio (Figure 1F). Fiber-seq samples with an N/E in the range of 1 × 104 to 2 × 105 nuclei/nU Hia5 were associated with the target m6A/A of 0.03–0.09 (median from all single DNA molecules). Furthermore, control experiments with samples processed with the Fiber-seq protocol but without Hia5 enzyme in the methylation reaction displayed m6A levels effectively at zero (Figure S2F). Importantly, this is consistent with recent studies reporting extremely low, or non-detectable, levels of endogenous m6A in genomic DNA from normal brain-derived cells, which reportedly do not exceed 16 ppm.21 Therefore, levels of endogenous m6A, if the mark is present at all, remain several thousand-fold below the m6A levels observed in our Hia5-treated brain nuclei and could not impact Hia5-based epigenomic mapping. Furthermore, we wanted to address whether tissue conditions and cell type or experimental conditions, including sequencing parameters, affect the quality of the Fiber-seq output, so we analyzed these in our PFC Fiber-seq samples (Table S1). We did this using a linear mixed-effects model with m6A proportion as the dependent variable and PMI and NeuN as the independent variables, adjusting for the covariates Hia5 concentration (nuclei per Hia5 unit) and circular consensus sequence (CCS) coverage, including a random intercept across BrainID to account for repeated measures. The dependent variable m6A proportion was transformed, using the square-root function, to ensure normality, which was confirmed using a Shapiro-Wilk test (p = 0.30). Results show that N nuclei per Hia5 unit (p < 0.001) with R −0.78 (p = 0) and CCS coverage (p < 0.05) with R −0.48 (p = 0.002), but not PMI or cell type, were significantly affecting the m6A proportion in our samples (Table S2A; Figure S3A).
To assess the impact of genome-level adenine methylation (m6A/A proportion) on the calling of methylation-sensitive patches (MSPs), nucleosomes, and Fiber-seq inferred regulatory elements (FIREs), we first calculated the correlation of these values using our PFC Fiber-seq samples (Table S1). As expected, m6A proportion showed strong correlations with the average sizes of MSPs and nucleosomes (Figure S3B). This was expected because MSPs are called based on the m6A distribution, and the nucleosomes are inversely called from the MSP distribution in the fibertools pipeline.22 Similarly, the number of FIREs showed a correlation with m6A proportion level and the read numbers as well. This was also expected, for FIREs are called from MSPs using a machine learning classifier,17 and low read numbers and inappropriate adenine methylation levels would fail to call FIREs. Since both m6A proportion and read numbers could affect the FIRE calling, we next tested the association using a linear mixed-effects model with FIRE as the dependent variable, m6A proportion as the independent variable, read numbers and cell type (NeuN) as covariates, a random intercept across BrainID to account for repeated measures, and an interaction between the m6A proportion and the read numbers. This analysis found that the interaction between the m6A proportion and the read numbers was significantly associated with the number of FIREs being called in our samples (Table S2B; Figure S3C).
We then generated a PFC NeuN− reference set from a 32-year-old female donor, with a total of 11,143,795 reads (fibers) and a median of 30-fold genomic coverage (Table S1, sample SA9B, NeuN−). To assess cell-type-specific regulation, we generated an additional PFC NeuN+ Fiber-seq reference set merged from four libraries of two 24-year-old brain donors (one female/one male), with a combined total reads (fibers) count of 4,749,556 and a median of 20-fold genomic coverage (Table S1, samples SA21A–24A, NeuN+).The final NeuN− and NeuN+ samples showed passing levels of CCS coverage, read length, m6A/A proportion, and MSP and nucleosome size distribution, with median mononucleosome/end-to-end linker DNA base pair lengths of 150/52 bp for NeuN+ and 142/46 bp for NeuN− (Figures 2A–2D, S4A, and S4B).
Figure 2.

Hia5 m6A tagging of accessible chromatin and internucleosomal DNA in neurons and non-neurons at single-molecule resolution
(A–D) Quality controls for PFC NeuN+ (red) and NeuN− (blue) Fiber-seq, including (A) proportional representation of DNA molecules by circular consensus sequence (CCS) coverage, (B) read length, (C) m6A/A per read, and (D) genome-wide counts of (Hia5) methylation sensitive patches (MSPs) (y axis) by base pair length (x axis).
(E and F) Heatmap for 6-kb sequences centered on 45,992 ATAC-seq peaks each for (E) NeuN+ and (F) NeuN−, split by k-means (k = 2) into promoter prominent (NeuN+) ATAC cluster 1 and (NeuN−) cluster 3 and enhancer prominent (NeuN+) ATAC cluster 2 and (NeuN−) cluster 4, as indicated. Numbers in parentheses indicate number of peaks by cluster. Pie charts display the genomic annotation of each cluster. Cell-type-specific alignments (NeuN+, NeuN−) are shown for (left to right) ATAC-seq, H3K4me3 and H3K27ac ChIP-seq, and m6A signal in the (by cell type) corresponding Fiber-seq libraries (see text). Notice strong cell-type-specific NeuN+ versus NeuN− differential regulation for enhancer-dominated clusters 2 and 4 compared to promoter-dominated clusters 1 and 3.
(G) Signal difference by cell type (NeuN+ versus NeuN−) for each of the 39,807 neuronal ATAC-seq peaks from cluster 2 (E) and for each of the 39,988 non-neuronal ATAC-seq peaks from cluster 4 (F), using the formula shown on the y axis of each plot. For each set of enhancer peaks, the difference in ATAC counts and percentage accessibility was computed for every peak at each position and then divided by the total number of peaks in the cluster to determine the average difference in ATAC-seq counts and Fiber-seq percentage accessibility at each position relative to the center of the ATAC-seq peaks. For NeuN+ enhancers, NeuN− signal was subtracted from NeuN+ signal, while for NeuN− enhancers, NeuN+ signal was subtracted from NeuN− signal. The exact equation used to calculate the per-position difference in ATAC-seq counts and Fiber-seq percentage accessibility is noted in the y axis label of each plot. The ATAC-seq counts for NeuN+ and NeuN− are denoted as ANeuN+ and ANeuN−, respectively, while for Fiber-seq percentage accessibility they are referred to as FNeuN+ and FNeuN−. The NeuN+ and NeuN− enhancer ATAC peaks are denoted as Ncluster2 and Ncluster4.
(H) Representative browser shots, ∼15 kb wide, comparing hs1-T2T genome annotated neuronal and non-neuronal chromatin from PFC (top) short-read epigenomic landscapes with (bottom) Fiber-seq signals for (left) GRIN1 NMDA receptor gene locus (chromosome 9q34.3) and (right) long non-coding RNA MALAT1 locus (chromosome 11q13.1) (right). Top to bottom, as indicated, GRIN1 gene map, NeuN+- and, separately, NeuN−- specific “peak” landscapes for (blue) ATAC, (green) H3K4m3, (red) H3K27ac-seq, and (orange) H1 neural cell line (ENCODE) CCCTC-binding factor (CTCF) peak landscape. Fiber-seq long-read sequencing: (brown) m5CpG tracks by cell type, (black) NeuN+ Fiber-seq FIRE scores of methylation-sensitive patches (MSPs); NeuN+ and NeuN− single DNA molecules are as indicated; blue-shaded single DNA molecules/chromatin fibers are highlighted as representative examples and shown at the bottom again at a higher (150 bp) resolution, with purple-colored vertical rectangles marking linker DNA (examples marked by “L” on fiber), with single-nucleosome (“n” on fiber) resolution, and red/brown-colored MSPs scoring as regulatory elements (P, promoter, and RE, regulatory element on fiber) by FIRE analyses (orange, FDR 0.10 ≥ q > 0.05; bright red, FDR 0.05 ≥ q > 0.01; dark red-brown FDR q ≤ 0.01). Notice prominence of FIRE nucleosome-depleted regions (NDRs) for GRIN1 in neuronal and for MALAT1 in non-neuronal fibers.
Using these non-neuronal and neuronal Fiber-seq reference datasets, we first examined whether, on a genome-wide scale, “open” chromatin peaks mappable by the short-read sequencing-based ATAC-seq, which is the most widely applied chromatin accessibility assay in brain, are tracked by a corresponding Hia5 m6A signal in Fiber-seq reads. To this end, we applied k-means (n = 2) clustering to the top scoring 45,992 cell-type-specific ATAC-seq peaks for each cell type, NeuN+ and NeuN−,23 together with chromatin immunoprecipitation-sequencing (ChIP-seq) data for H3K4me3 and H3K27ac.24 We identified clusters corresponding to gene-proximal promoters (clusters 1 [NeuN+] and 3 [NeuN−]) and enhancers and other distal regulatory elements (clusters 2 [NeuN+] and 4 [NeuN−]) (Figures 2E and 2F). Promoter clusters (1 and 3) showed enrichment of H3K4me3 and H3K27ac signal in NeuN+ and NeuN−, while enhancer clusters (2 and 4) showed enrichment of only H3K27ac (Figures S4C–S4F). To further confirm that Fiber-seq accessibility is detectable and comparable to ATAC-seq within our ATAC-seq peak clusters, we used the fibertools pipeline to identify single-molecule nucleosome footprints and MSPs22, and then used the FIRE pipeline to identify MSPs with features consistent with accessible regulatory elements (i.e., FIREs) along each molecule, as well as peaks of significantly enriched Fiber-seq chromatin accessibility.17 Notably, each of the peaks’ total FIRE score in the corresponding cell-type-specific Fiber-seq libraries sequencing signal correlated with the ATAC peak levels in the neuronal and non-neuronal ATAC-seq libraries, respectively (promoters, clusters 1 and 3: NeuN+, r = 0.574; NeuN−, r = 0.683, p < 0.00001; enhancers, clusters 2 and 4: NeuN+, r = 0.41; NeuN−, r = 0.55, p < 0.00001) (Figures S4G and S4H). Therefore, genomic sites with transposase-defined accessible chromatin as defined in conventional, neuronal and non-neuronal ATAC-seq match to sequences that are called as MSP/nucleosome-depleted regulatory elements in the (by cell type) corresponding Fiber-seq libraries. Furthermore, on a genome-wide scale, cell-type specificity is primarily defined by distal tissue-specific enhancer sequences (compared with a much lesser cell-specific degree at promoters).25 We therefore examined cell-type-specific regulation in our Fiber-seq datasets. Distal regulatory elements of NeuN+ cluster 2 and NeuN− cluster 4, both of which are dominated by enhancers, showed strongest cell-type-specific Fiber-seq and ATAC-seq and histone H3K27ac signals, compared to moderate differences in promoter-dominated clusters 1 and 3 (Figures S4C–S4F). Robust cell-type-specific NeuN+ versus NeuN− Fiber-seq effects were further confirmed with peak-by-peak differential analyses (Figure 2G), with the Fiber-seq differentials at both promoter and enhancer tightly matching the corresponding sequence-matched NeuN+ versus NeuN− differentials in the ATAC-seq datasets (Figure S5). Furthermore, three of the five highest scoring Gene Ontology categories for NeuN+ Fiber-seq peaks were dominated by neuron-specific biological processes, including neuron ion channel regulation and dendritic spine plasticity, while none of the top scoring NeuN−-specific Fiber-seq peaks included neuron-specific functions but instead were reflective of immune functions, chemotaxis, and metabolism (Tables S3A and S3B). These findings, taken together, strongly suggest that regulation of Fiber-seq-defined NDRs is highly specific for neurons compared to non-neurons.
Furthermore, NeuN−- and NeuN+-specific signals for transcriptional histone marks, including H3K4me3 and H3K27ac,24 sharply flanked the m6A signal in the corresponding cell-type-specific Fiber-seq libraries (Figures 2E, 2F, and S4C–S4F). Representative genome browser shots highlight cell-type-specific short- and long-read neuronal and non-neuronal chromatin profiles for the NMDA receptor GRIN1 and non-coding MALAT1 gene loci, with FIREs showed prominence for GRIN1 in neuronal and for MALAT1 in non-neuronal fibers within their respective promoter regions (Figure 2H). While the short-read libraries generally demarcate active chromatin at the corresponding promoter sequences, Fiber-seq shows much sharper boundaries of regulatory elements at single nucleosome resolution on the individual fibers, with fiber-to-fiber variability in sequence-specific positioning of nucleosomes and NDRs, which at that level cannot be assessed by conventional ATAC- or histone ChIP-seq (Figure 2H). Overall, our Fiber-seq data detected 43,457 peaks in the PFC NeuN− Fiber-seq peak reference set, with 34,861 peaks meeting a minimum coverage of 10 (Figure S6A; Table S4; see STAR Methods). Peak-specific detection limits in our NeuN− Fiber-seq library showed a minimum of 35.7% of fibers required to be in an open/accessible state for us to call a peak (Figure S6A; Table S4; see STAR Methods) Note that, to control for cell-type specificity, we also generated a PFC NeuN+ peak set from our NeuN+ Fiber-seq reference library, but due to sample material limitations we did not reach sufficient sequencing depth to call a sufficient number of peaks for a deeper genomic analysis (Figure S6B).
We applied k-means clustering (n = 4) to these Fiber-seq peaks and integrated frontal lobe NeuN−-specific ATAC-seq26 and histone H3K4me3 and H3K27ac24 ChIP-seq datasets into the analysis pipeline, together with a CTCF (CCCTC binding factor) ChIP-seq dataset for the H1 neural cell line (ENCODE dataset ENCSR822CE). Of note, Fiber-seq (F) peak-defined cluster 1 matched overwhelmingly to promoters and promoter-proximal sequences; cluster 2 was predominantly defined by CTCF peaks at a broader range of regulatory elements, based on ChIP-seq signal enrichment and genomic annotation (Figure 3A). Furthermore, NeuN− Fiber-seq peaks of clusters 1–3 together comprised ∼40% of all Fiber-seq peaks and were closely matched by corresponding signals of NeuN− ATAC- and histone ChIP-seq datasets (Figure 3A). However, the 19,978 peaks of Fiber-seq cluster 4, which account for the majority, or ∼60%, of the entire population of Fiber-seq peaks (across all clusters 1–4, all peaks with coverage >10), while showing some ATAC-seq alignments (Figure S6C), lacked a corresponding site-specific enrichment in ATAC- and ChIP-seq datasets (Figure 3A). Interestingly, 60% of cluster 4 sequences are repetitive DNA elements, which is a much larger share compared to the 2%–25% repeat DNA contribution to clusters 1–3 (Figure S6D). This observation would suggest that our long-read single chromatin fiber sequencing, which exceeds the base pair length of conventional short-read libraries by two orders of magnitude, could capture many actively regulated repeat elements at the site of specific gene loci that otherwise would remain unmappable by conventional ATAC-, histone-, and CTCF-ChIP-seq. Overall, across fiber clusters 1–4, cluster 4 peaks showed significant enrichment for repeat peaks relative to the other peak clusters, with up to ∼69-fold difference in residual (chi-squared test, df = 3, p < 2.2e16 (Figure 3B). LINE (long interspersed nuclear element), satellite, and simple repetitive sequences account for the majority of cluster 4 peaks, followed by SINEs (short interspersed nuclear elements), LTRs (long terminal repeats), and DNA transposons (Figure 3C). Representative genome browser shots highlight the examples for FIRE peaks corresponding to the four identified clusters (Figure 3H). These include, at single-nucleosome resolution, active chromatin at the site of short retroelements, including mammalian interspersed repeats (MIRs), an ancient SINE species that comprises 2.5% of the human genome and maximally spans 250 bp (Figure 3H).
Figure 3.

Fiber-seq FIRE peak-based analysis of nucleosome-depleted regions (NDRs)
(A) Fiber-seq FIRE-defined peak heatmap of PFC NeuN− nuclei population, with k-means Fiber-seq clusters 1–4 and their alignments by corresponding sequences from PFC NeuN− open chromatin ATAC-seq, transcriptional histone marks H3K4me3 and H3K27ac, and Encode H1 neural cell line CTCF ChIP-seq conventional short-read sequencing.
(B) Cluster-specific enrichment scores for repeat and non-repeat sequences; note that repeat enrichment is specific for cluster 4 (chi-squared, df = 3, p = 2.2 × 10−16).
(C) Total peak count by type of DNA repeat and Fiber-seq cluster, as indicated. Notice that retroelements, including LINEs, SINEs, and LTR-associated fiber peaks (NDRs), are overwhelmingly locating to fiber cluster 4 with only very minimal contributions by clusters 1–3.
(D) PFC NeuN− Fiber-seq and ATAC-seq peak size distribution profiles; notice several-fold sharper peak size in former compared to latter.
(E) Top scoring HOMER regulatory motifs to each for the four Fiber-seq peak clusters, 1–4 (p, Kolmogorov-Smirnov).
(F) Fine-grained regulatory motif footprint for NFY TF with fiber cluster-specific binding scores as indicated (∗∗∗p < 1e−16, Kruskal-Wallis); note sharp footprints centered in NDRs.
(G) Plots depicting the average proportion of adenines that are methylated in fibers centered on n =1,855 specific CTCF binding sites (see text), with fibers with (top) MSP harboring a CTCF footprint and (bottom) MSP with no footprint. Arrows point to local minima adjacent to MSP, indicating strong phasing with well-positioned nucleosomes and cis actuation of flanking nucleosomal array. See also, as additional control, Figure S6F for fibers with a CTCF motif but no MSP.
(H) Representative hs1-T2T genome browser shots of cluster-specific genomic loci, with single-fiber resolution by cluster. (Left to right) Cluster 1 OLIG2, cluster 2 intergenic, cluster 3 DPF3, and cluster 4 ATOC6 gene locus. (Top to bottom) PFC NeuN−-specific “peak” landscapes for (blue) ATAC, (green) H3K4me3, (red) H3K27ac-seq, and (orange) CTCF ENCODE neural cell line CTCF short-read-based peak landscapes. Fiber-seq long-read sequencing: (brown) m5CpG tracks by cell type, (black) NeuN+ Fiber-seq FIRE scores of MSPs; NeuN+ and NeuN− single DNA molecules are as indicated; green-shaded single DNA molecules/chromatin fibers are highlighted as representative examples and shown at the bottom again at a higher (150 bp) resolution, with purple vertical rectangles marking linker DNA (examples marked by “L” on fiber), and single-nucleosome (“n” on fiber) resolution. Red/brown-colored MSPs scoring as regulatory elements (P, promoter, and other RE, regulatory element, on fiber) (FIRE FDR, orange, 0.10 ≥ q > 0.05; bright red, 0.05 ≥ q > 0.01; dark red-brown, q ≤ 0.01).
Motif analysis and transcription factor footprinting
Importantly, MTase-based accessibility mappings generally tend to provide far better resolution of NDRs and regulatory elements compared to hyperactive transposase, DNase digest, and other types of nucleolytic approaches,7 which typically require an artificial sliding-window computational step to limit peak size to (for example) 500 bp.27 We examined this in our Hia5-treated brain nuclei. Indeed, PFC NeuN− Fiber-seq versus ATAC peak sizes revealed, across all peak clusters, 1–4 (Figure 3A), a consistent multi-fold decrease in Fiber-seq peak length (median 231 bp, range 63–1,048 bp) compared to ATAC-seq (median 1,140 bp, range 136–2,972 bp) (p < 0.00001 via Wilcoxon rank-sum test) (Figure 3D). We then examined whether the FIRE-called Fiber-seq MSPs marking NDRs carry TF DNA binding motifs. We called cluster-specific Fiber-seq motifs using the HOMER known motif pipeline with the FIRE-called peaks as input and calculated the size of TF footprints (Table S5). Promoter-dominated (>95% of peaks) cluster 1 showed strongest enrichment for NFY and its jointly operating co-factor SP1 (Figure 3F), both critically important for oligodendrocyte survival28 and defined by a classical CCAAT and related ∼12-bp DNA binding motifs. Likewise, clusters 2 and 3 (Figure 3A), with a more mixed enhancer and promoter composition, showed strong enrichments for the pleiotropic transcriptional regulator and chromosomal loop organizer CTCF and respectively for SOX9, a prototype TF for neural stem cells differentiating into astrocytic lineages,29 and NEUROD1 as a regulator of microglial survival30 (Figure 3E; Table S5).
Of note, classical DNase I hypersensitive site mapping, and Fiber-seq on peripheral cell lines, reveals the physical occupancy of a DNA-bound TF as discreet “punctuation” surrounded by cleaved sequence (DNase I)31 or as a short m6A gap within an MSP (Fiber-seq),16 reflecting nucleotide-precise binding of the TF protein. We searched for TF binding footprints in our brain cluster 1–4 Fiber-seq peaks (Figure 3A). We counted the average proportion of m6A/A at respective TF motifs and flanking sequence aggregated across each peak cluster and then computed an aggregate binding score that represented the strength of binding at the TF binding across each cluster of peaks (see STAR Methods). As expected from our cluster-specific TF motif enrichments (Figure 3E), compared to all other clusters, significantly higher binding scores were observed for the NFY motif in cluster 1 (Figure 3F) and for CTCF in cluster 2 (Figure S6E). These types of sharp motif demarcation in the center of the cluster-specific FIRE Fiber-seq peaks, as shown for NPY and CTCF (Figures 3F and S6E), were representative of the broader group of top scoring TF in clusters 1 and 2 (Table S5).
Next, we asked whether the activity status of a regulatory sequence is coupled to nucleosome positioning effects in the surrounding chromatin. Specifically, we studied N = 1,855 cluster 2 FIRE peaks, each harboring a canonical 35-bp CTCF binding motif that had been further validated in a CTCF ChIP-seq dataset (ENCODE H1-derived neural cells)32 (Figure 3A). Of note, CTCF binding is strongly linked to specific motifs and typically lacks additional co-bound TFs.33 Each of the 1,855 peaks (minimum of 20 single fibers/locus), resulting in a total of n = 48,489 individual chromatin fibers, was assigned to one of the following three categories: (1) fiber with MSP harboring a CTCF footprint in the MSP, (2) fiber with an MSP but no footprint, and (3) fibers with a nucleosome occupying the CTCF binding motif. Remarkably, the CTCF footprint precisely centered to the CTCF motif within the MSP (Figures 3G and S7). However, all fibers with an MSP, regardless of the physical presence or absence of CTCF, showed adjacent to the MSP and motif center extremely well-positioned nucleosomes (Figure 3G). This type of nucleosomal phasing then became successively weaker with increasing nucleosome-to-CTCFMSP distance within the 2 kb of sequence surrounding the CTCFMSP (Figure 3G), a finding that is highly consistent with related studies in dividing cell lines.34 In sharp contrast, fibers with a nucleosome engulfing the CTCF sequence motif essentially lacked nucleosomal phasing (Figure S6F). Therefore, MSPs at the site of regulatory DNA carrying a CTCF motif are associated with an “actuation”16 of nucleosomal positioning in the surrounding portions of the chromatin fiber, regardless of the physical presence of CTCF protein. This actuation is lacking in fibers with closed (nucleosome-bound) regulatory DNA.
Enrichment of brain-related variants and haplotype phasing of single fibers from human brain
To gain first insights into potential disease-relevance of each cluster-specific set of FIRE-defined Fiber-seq peaks/NDRs in the non-neuronal nuclei, we computed the linkage-disequilibrium (LD)-score-partitioned heritability scores35 to examine the enrichment of common genetic variants identified by genome-wide association studies (GWASs) on 55 different brain- and non-brain-related traits (Table S6). Interestingly, non-brain-related autoimmune and metabolic disorders and traits such as height did not associate with any cluster. However, we observed strong enrichment in our enhancer-dominated cluster 3 peak set for neuropsychiatric disorders such as major depression, consistent with genetic36 and epigenomic evidence37,38,39 for a disease-relevant role of multiple types of glia (Figure 4A).
Figure 4.

Nucleosomes show sequence-specific fixation proximal to accessible TSS
(A) Enrichment of brain-specific traits based on the identity of GWAS-locus-specific SNPs in each cluster of Fiber-seq-called peaks, excluding peaks located at repetitive regions. Significant traits are labeled with ∗ (p < 0.05) and # (p < 0.01).
(B) Volcano plot of Fiber-seq peaks with haplotype-specific differences (N = 3,708), with the difference in accessibility on the x axis and the −log10 p value on the y axis. Peaks with nominal p < 0.05 by a Fisher test are colored red (H1 high) and blue (H2 high).
(C) UCSC genome browser visualization of fiber-to-fiber variability including a significantly haplotype differential Fiber-seq peak and Fiber-seq reads phased for haplotype at the ZNF343 promoter (H1, n.s. as indicated; H2, p < 0.05, Fisher test).
(D) Violin plots showing the distribution of haplotype-specific FIRE score at significant (top) H1-high and (bottom) H2-high peaks. The p value comparing the score was calculated using a paired t test (p < 1.42e−50 for both).
(E) Schematic showing the calculation of nucleosome offsets for a single NDR at a TSS region. Each row represents a fiber, with the accessible patches colored red (FIRE) and purple (linker) and the nucleosomes positioned between them, in stylized fashion. The offset was computed by subtracting the center of the nucleosome at a randomly selected reference fiber from all other nucleosomes at that position relative to the location of the NDR.
(F) Boxplots showing the distribution of nucleosome offsets for the first five nucleosomes upstream and downstream TSSs with a FIRE peak. Peak-adjacent nucleosomes are colored purple, while all others are shown in gray. The p values were computed across all distributions comparing the most proximal nucleosomes to all others using Wilcoxon rank sum (p < 0.0001).
(G) Boxplots showing the distribution of nucleosome offsets for the first five nucleosomes upstream and downstream centered at randomly selected internucleosomal linker regions; linker-adjacent nucleosomes are shown in purple, while all others are shown in gray. In the randomly picked regions, no nucleosomes showed significant differences in offsets compared to the rest, via Wilcoxon rank sum.
(H) Histogram showing the absolute genomic distance between each two peaks within pairs of significant (red, N = 436, p < 0.05) and not significant (gray, N = 1,472) co-actuated pairs of peaks.
(I) Representative UCSC genome browser capture of the PLEKHG3 promoter, which is detected to have a significant co-actuation event with a nearby peak (p < 0.05).
(J) Violin plot showing the proportion of co-actuated fibers versus total fibers across all significant (red) and non-significant (gray) co-actuated pairs. Significant pairs have a median co-actuation proportion of 0.33 with an interquartile range (IQR) of 0.15. Non-significant pairs have a median co-actuation proportion of 0.18 with an IQR of 0.11.
Haplotypes are the combination of specific alleles along a multi-kilobase stretch of sequence on a chromosome as a reflection of shared ancestry and LD.40 Allele-specific epigenomic differences could impact regulatory non-coding DNA and gene expression at these sites.41 However, haplotype-defining single-nucleotide polymorphisms and other structural DNA variants typically are spaced apart many hundreds of base pairs,40,42 and haplotype resolved (epi)genomic maps are difficult to construct from short-read libraries.43 Here, by taking advantage of long-read DNA molecules aligned to the T2T genome CHM13v2.0 assembly, we were able to phase 94.8% of our long reads into diploid haplotypes, de novo, without pre-existing parental variant information, by combining an established AI-powered variant calling information and k-mer counting pipeline (see STAR Methods). Using the FIRE pipeline, we identified peaks in our NeuN− fibers that showed allele-specific differences in percentage accessibility (Fisher’s exact test, nominal p < 0.05) and found 124, or 3.3%, of a total of 3,708 peaks called with detectable haplotype differences (Figures 4B and 4C). As a group, haplotype-specific FIRE scores for these 124 peaks carried highly significant differences between their corresponding haplotypes (paired t test, p < 2 × 10−50, Figure 4D). This is likely a conservative estimate, since we are limited by the per-haplotype sequencing depth of 14–15 reads, which dampens our statistical power. A representative example would be the promoter of ZNF343, a primate-specific zinc finger gene robustly expressed in human brain and estimated to bind to 200 promoters in the genome44 (Figure 4C).
Preferential positioning of nucleosomes at regulatory elements
Active promoters in human peripheral cells are defined by NDRs just upstream of and 10–50 bp into the TSS, with flanking nucleosomes often occupying near-identical sequences across many individual cells at the site of active and poised/paused, promoters.45 The “offset,” defined as variability in nucleosome positioning around a unique genomic sequence, sequentially increases for nucleosomes that are further removed from the TSS NDR2,3,46 (Figure 4E). We examined this for our active promoters (n = 4,544 promoters, range 133–439 bp, median 237 bp) and, separately as a control, randomly selected internucleosomal linker regions (n = 20,000, range 1–823 bp, median 53 bp) (Figures 4F and 4G), since nucleosomes should be randomly distributed at non-regulatory regions of the genome. Indeed, the offset score, or the variability in sequence-specific nucleosomal positioning across fibers for promoters of active regulatory elements, was lowest for the −1 nucleosome, with sequentially higher offsets of nucleosomes farther up- and downstream of the NDR (p < 0.0001, Wilcoxon rank sum). When fibers with nucleosomal occupancy at TSSs were excluded, offset was significant both for the −1 and the +1 nucleosome (Figures 4F and S8A). However, nucleosomes surrounding the random regions did not show preferential positioning at −1 and +1 nucleosomes and no significant differences exist in offset for any of the nucleosome positions relative to the random regions (p = 0.28, Wilcoxon rank sum) (Figures 4G and S8B).
In addition to these sequence-specific fixations and offset of −1 and +1 nucleosomes, MSPs at TSSs or FIRE peaks showed strong nucleosomal phasing and then became successively weaker with increasing nucleosome-to-TSSMSP distance (Figure S8C) and increasing nucleosome-to-FIREMSP distance (Figures S8C and S8D), very similar to the type of nucleosomal phasing observed at MSPs at the site of CTCF motifs (Figure 3G). We noted distinct phasing within the first 1,000 bp upstream of annotated TSSs (regardless of presence or absence of a FIRE at the TSS MSP), but this was less evident for sequence-guided nucleosomal TSS phasing at the downstream +1 nucleosome. This could reflect fiber-to-fiber variation in the base-pair length of TSS-associated NDRs at the 3′ (compared to 5′) end of TSS NDRs, reflecting differential RNA polymerase II and chromatin remodeler activity. Consistent with this, when we re-computed TSS-associated nucleosomal phasing by limiting the analysis to fibers with a FIRE at the TSS, strong positioning effects were observed across the first 1,000 bp both up- and downstream of TSS-bound MSPs, with equally strong phasing at the −1 and +1 nucleosomes in particular (Figure S8D). In contrast, phasing was completely absent in fibers with an inactive promoter TSS due to nucleosomal occupancy (gray curves in Figures S8C and S8D). To further validate that nucleosomes in linker regions are more dynamic, while nucleosomes flanking accessible promoters are held in place, we quantified nucleosome sliding by plotting the average encroachment of nucleosomes into accessible patches across single fibers, with greater encroachment implying more dynamic positioning of nucleosomes across individual cells for a given region (Figures S9A and S9B).
Co-actuation of regulatory elements on the single-fiber level
Enhancers and repressors and related regulatory elements are short, ∼100- to 1,000-bp stretches of sequence densely populated by TF binding sites, which could functionally interact with the target gene promoter located on the same chromosome.47 According to “activity-by-contact” (ABC) or chromatin accessibility with chromosome conformation capture mappings, a single human bio sample could harbor 48,000 enhancer-gene connections, with a median genomic distance of 13–16 kb between enhancer and target promoter.48,49 Because at any given time point only a small proportion of cells in a cell population or tissue is thought to undergo active transcription at a particular gene,50 we would expect that a small fraction of chromatin fibers of our brain nuclei could display signs of co-actuation, or the coordinated nucleosomal depletion, of a promoter and a regulatory element residing on the same fiber. Indeed, after stringent statistical filtering (false discovery rate [FDR] Fisher’s p < 0.05, see STAR Methods), we counted 386 significant co-actuation events, out of 1,842 same-fiber pairwise regulatory element interactions (Figures 4H–4J). To determine if these significant interactions were occurring at a rate greater than what we expect based on random chance, we computed the difference between the actual proportion of co-actuated fibers and the expected proportion for significant and non-significant peak pairs. The average proportion of co-actuated fibers in significant peaks comparing actual versus expected is 0.149, while in the non-significant peaks the difference was 0.036 (p = 1.64 × 10−227, one-tailed t test) (Figures S9C and S9D).
Discussion
Here, we label NDRs and extranucleosomal linker DNA from intact brain nuclei in situ via m6A methylation by a prokaryotic N(6)-adenine MTase. In conjunction with amplification-free ∼10-kb long-read sequencing of single DNA molecules directly derived from the nuclei, we can map individual chromatin fibers of the PFC on a genome-wide scale. Our study advances the field by providing the field with a brain adapted step-by-step protocol for the cell-type-specific Fiber-seq technique, as exemplified by the NeuN+ and NeuN− populations. We present an initial nucleosome positioning map for the human brain at single-fiber resolution and highlight some of the advances and insights from this novel neuroepigenomic approach.
The Fiber-seq technique on brain nuclei, as presented here, offers critical advances on scales of 0.1 to 10 kb compared to PCR-generated short-read libraries from ensembles of DNA molecules generated from conventional transposase-based and other nucleolytic accessibility assays. For example, on the 100 bp scale, Fiber-seq allows resolution of individual nucleosomes (147 bp) and internucleosomal linker DNA on the single chromatin fibers, which enabled us to confirm principles of nucleosomal organization hitherto elusive to assess in complex tissues and established only in simple eukaryotes and cells in culture.2,3,46 For example, we confirmed the strong inverse relation between offset (variability in nucleosome occupancy on individual fibers) and proximity of −1 and +1 nucleosomes to the NDR at active promoters.2,3,46 Future studies will assess dynamic alterations in nucleosome positioning at regulatory elements in normal and diseased human brain. We expect to quantify expansions and shrinkages of specific promoter NDRs at the single-fiber level with precise base-pair distance measurements of the +1 nucleosome to the TSS as a control point for transcription, as reported for simple eukaryotes.51,52
The increased resolution for Fiber-seq-defined NDRs at neuronal and non-neuronal regulatory elements compared to ATAC-seq (Figure 3D) confirms that conventional transposase-based nucleolytic approaches tend to be imprecise while inflating base-pair length estimation of nucleosome-depleted sequences.7 An illustrative example is provided by the glial nucleoporin-encoding gene SEH1L,53 which, according to our Fiber-seq mappings, is sharply compartmentalized on many non-neuronal fibers into two neighboring NDRs, flanked by two back-to-back nucleosomes. In contrast, in ATAC-seq, the same genomic site produces a much broader peak with incomplete resolution of NDRs (Figure 1D). We note that our findings presented here are in line with other recently developed techniques, including the nucleosome positioning mapping in the single-molecule adenine methylated oligonucleosome assay (SAMOSA)54 and single DNA molecule nucleosomal patterning in yeast55,56 and other approaches with enzyme-mediated chemical tagging of non-nucleosomal DNA.16,55,56,57,58
We show that on the 10 kb scale, Fiber-seq allows for haplotype-specific resolution and assessment of co-actuation and co-regulation of regulatory elements positioned in cis on the same chromatin fiber. In addition, our long-read epigenomic profiling produced site-specific localization of ∼20,000 NDRs that hitherto could not be anchored to their unique location by previous short-read sequencing-based accessibility mapping. Among these, ∼60% were uniquely assigned to retrotransposons, including LINEs, SINE/ALUs, and LTR/ERVs. Of note, earlier DNase I hypersensitivity mappings in human peripheral cell lines had estimated similar magnitudes for retrotransposon-associated open chromatin sites in the primate including human genome, but could not proceed to locus-specific annotation.59 Fiber-seq allows accurate annotation of comparatively short retroelements, including MIRs, an ancient SINE species that comprises 2.5% of the human genome and maximally spans 250 bp (Figure 3H). Because MIRs serve as potential docking sites for RNA polymerases with high relevance for neurodegenerative disease,60,61 it should now be possible to conduct genome-wide surveys of MIR genomic activation in cell types at risk.
Furthermore, single-molecule long-read sequencing, with detection of methylated CpGs together with nucleosome position mapping (Figures 2H and 3H) could capture 89% of structural variation that had been missed by short-read sequencing62,63 and inform about epigenetic regulation, including the potential variability at sites associated with disease risk.
Limitations of the study and unresolved questions
One of the most remarkable findings emerging from early Fiber-seq studies in simple eukaryotes, including yeast, is a surprising degree of fiber-to-fiber heterogeneity, with many fibers showing pronounced deviations from the expected stereotypic nucleosomal organization, together with abundant nucleosome-free gaps in gene bodies and elsewhere in the genome.56 These observations are consistent with some of the fiber-to-fiber variabilities in nucleosome positioning reported here for the human brain, and furthermore, because each study used very different species/input materials and different fiber labeling techniques, technical factors are an unlikely explanation. While some of the interfiber heterogeneities in brain may be partially resolved by cell type, gene locus, and haplotype (Figures 2H, 3H, and 4C), additional work will be required to gain deeper understanding of the nucleosomal variabilities between chromatin fibers.56 This may also necessitate further optimization of the technique specifically at sites of adenine-depleted genomic sequences and further improvement of computational analyses, including m6A calling.22,64 Of note, brain Fiber-seq libraries with an m6A/A fraction in the range of 0.03–0.09 lead to computationally predicted average nucleosomal base-pair sizes (150 and 142 bp, respectively, for the two reference libraries presented here; Figure 4B), which are most closely aligned to the 147-bp “fixed” model.65 Alternative m6A calling tools exist.66 Future studies should directly compare these alternative computational tools for m6A calling efficiencies and nucleosomal and linker DNA size distributions.
Furthermore, the amplification-free approach for whole-genome Fiber-seq requires, in practical terms, a significant amount of input material (106 nuclei per assay in the present study). Together with the fact that Fiber-seq provides single-chromatin-fiber but not single-cell resolution, this poses a limitation in case the cell type of interest is rare or if tissue is scarce. The latter may be addressed, as shown here, by pooling multiple samples, albeit it needs to be clarified in future investigations how interindividual variability could increase the noise level when working with Fiber-seq libraries generated from pooled samples, including biological replicates.67
Resource availability
Lead contact
Requests for further information may be directed to and will be fulfilled by the lead contact, Schahram Akbarian (schahram.akbarian@mssm.edu).
Materials availability
Materials are available from the lead contact upon request. The study did not generate any unique reagents.
Data and code availability
-
•
Fiber-seq and single-nuclei sequencing data are available in the database for Genotypes and Phenotypes (dbGAP) under accession no. phs003771.v1.p1.
-
•
This study does not report original code.
-
•
Any additional information required to reproduce the data reported in this study is available from the lead contact upon request.
Acknowledgments
This work was supported by United States National Institute of Drug Abuse (NIDA) DP1 DA056018, National Institute of Mental Health (NIMH) R01 MH106056 (S.A.), and in part by the Bioinformatics for Next Generation Sequencing (BiNGS) shared resource facility within the Tisch Cancer Institute at the Icahn School of Medicine at Mount Sinai, which is partially supported by NIH grant P30CA196521. This work was also supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai and supported by the Clinical and Translational Science Awards (CTSA) grant UL1TR004419 from the National Center for Advancing Translational Sciences. Research reported in this paper was supported by the Office of Research Infrastructure of the National Institutes of Health under award nos. S10OD026880 and S10OD030463 (A.B.S.). The authors thank Dr. Jordi Ochando and personnel of the Mount Sinai flow cytometry core for resources and support. M.R.V. was supported by a training grant (T32) from the NIH (2T32GM007454-46). A.B.S. was supported by National Institutes of Health (NIH) grant 1DP5OD029630.
Author contributions
Conceptualization and research design, C.J.P. (wet lab) and A.A. (computational analyses). C.J.P. and A.A. contributed equally as first authors who performed experiments in collaboration with R.W., B.S.K., X.W., T.Y.L., V.E., T.D., M.F., K.G., and S.K.N. Resources, A.B.S., M.R.V., N.M.T., R.P.S., D.H., and P.R.; writing, C.J.P., A.A., B.J., D.H., and S.A., with input from all co-authors; supervision, D.H. and S.A. S.A. conceived the project and led the funding acquisition.
Declaration of interests
A.B.S. is a co-inventor on a patent relating to the Fiber-seq method (US17/995,058).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-N6-methyladenosine (m6A) | EMD Millipore | ABE572-I, RRID:AB_2892214 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell signaling technology | 7074, RRID:AB_2099233 |
| Chemicals, peptides, and recombinant proteins | ||
| D5000 ScreenTape | Agilent | 5067-5588 |
| D5000 Reagents | Agilent | 5067-5589 |
| SAM (32-mM) | NEB | B9003S |
| Water (nuclease free) | Ambion | AM9932 |
| 1 M Tris, pH 8.0 (nuclease free) | Invitrogen | AM9855G |
| 5 M NaCl (nuclease free) | Invitrogen | AM9760G |
| 2 M KCl (nuclease free) | Invitrogen | AM9640 |
| 0.5 M EDTA pH 8.0 | Invitrogen | AM9260G |
| 0.5 M EGTA, pH 8.0 | Alfa Aesar | J60767-AE |
| Spermidine, 5 M | Sigma | 85558-5G |
| 10% Triton X-100 (nuclease free) | Sigma | 93443-100ml |
| rBSA | EMD Millipore | 126609-10GM |
| Genomic DNA ScreenTape | Agilent Technologies | 5067-5365 |
| HisTrap HP | Cytiva | 17524802 |
| Streptavidin Coated 96-well ELISA plate | Thermo scientific | 15124 |
| Ultra TMP-ELISA substrate | Thermo scientific | 34028 |
| Critical commercial assays | ||
| SMRTBELL prep kit 3.0 | Pacific Biosciences | 102-141-700 |
| SMRTBELL barcoded adapter plate | Pacific Biosciences | 102-009-200 |
| AMPure PB beads | Pacific Biosciences | 100-265-900 |
| Chromium GEM-X Single Cell 3' Kit v4 16 rxns | 10x Genomics | 1000691 |
| Chromium GEM-X Single Cell 3' Chip Kit v4 4 chips | 10x Genomics | 1000690 |
| Dual Index Kit TT Set A 96 rxns | 10x Genomics | 1000215 |
| KAPA Library Quantification Kit Illumina® Platforms | Roche | KK4828 – 07960166001 |
| Megaruptor 3 DNAFluid kit | Diagenode | E07020001 |
| Megaruptor 3 Shearing kit | Diagenode | E07010003 |
| Monarch Genomic DNA kit | New England Biolabs | T3010L |
| Software and algorithms | ||
| Python | Python Software Foundation | https://www.python.org/ |
| Visual Studio Code | Microsoft | https://visualstudio.microsoft.com/ |
| Adobe Illustrator | Adobe Inc. | https://www.adobe.com/products/illustrator.html |
| Fibertools | Stergachis Lab | https://github.com/fiberseq/fibertools-rs |
| R | R Core Team | https://www.r-project.org/ |
| Numpy | Numpy Team | https://numpy.org/ |
| Pandas | NumFOCUS Inc. | https://pandas.pydata.org/ |
| Matplotlib | Matplotlib Development Team | https://matplotlib.org/ |
| Seaborn | Michael Waskom | https://seaborn.pydata.org/ |
| Scipy | Scipy Steering Council | https://scipy.org/ |
| Pyft | Stergachis Lab | https://py-ft.readthedocs.io/en/latest/ |
| Deposited data | ||
| Fiber-seq and single nuclei sequencing data | This paper | dbGAP: phs003771.v1.p1 |
Experimental model and study participant details
Specimens
For Fiber-seq, we used postmortem frontal cortex, including prefrontal Area 46 and fronto-motor Area 4 using samples provided by the Department of Pathology at Icahn School of Medicine at Mount Sinai and University of Maryland Brain and Tissue Bank. The demographic variables are summarized in Table S1. The tissue collections, including their genomic assessment, were approved by the ethics committees of the participating institutes.
Method details
Production of Hia5 enzyme
The complete coding sequence of Hia5 open reading frame (Haemophilus influenzae sequence GenBank file JF268249.1), with 843 bases corresponding to entire 281 amino acids of Hia5 enzyme, was E.coli codon optimized and synthesized as gene block (IDT technologies) in pUC-IDT vector and subsequently cloned into NdeI and XhoI sites of pET30(+) expression vector (Millipore Sigma, #69909) in frame with a C-terminal His.Tag (Figure S2A).
We empirically determined the optimal expression and production of Hia5 in E.coli T7 express (NEB, #C3013I), OD600 0.8 at 18oC, with 0.5 mM IPTG (Invitrogen, AM9464) for induction, and repeated 6-hour spins at 250-rpm, 2XYT medium (RPI, X15640-1000). Each Hia5 batch was purified in AKTA-Start HPLC using 5 liters of culture and 5ml HisTrap HP columns (Cytiva, 17524701). (Figure S2A). We verified the m6A methylation by indirect DpnI restriction digestion assay and direct anti-m6A antibody dot blot using an independent longer PCR generated 1kb Hia5 DNA substrate (Figure S2B).
Quantitative determination of Hia5 enzyme activity
We designed a 200bp DNA Hia5 substrate that includes 28% adenine bases and is 5’ (“one sided”) biotinylated to promote linear substrate presentation and avoid steric hindrance upon adherence to wells. 10 picomole of this substrate dissolved in 100μl of TBS-T (10mM Tris, pH 7.5; 180mM sodium chloride; 0.1% Tween-20) was adhered to each streptavidin coated well (96-wells, clear, preblocked by the manufacturer with SuperBlockTM Blocking Buffer; Thermo scientific #15124) for 60min at room temperature on RoTo Mix, speed-6, followed by washing with 150μl of TBS-T for 5min. Blocking was done with 100μl/well of 5% non-fat dry milk in TBST-T for 30min at room temperature, on RoTo Mix speed-6, followed by one wash of 150μl of TBS-T for 5min. Final wash was done with 150μl of Hia5 activation buffer (15mM, pH-8.0; 15mM NaCl; 60KCL, 1mM EDTA, pH8; 0.5mM EGTA, 0.5mM, Spermidine; 0.002% Triton-X100; rBSA 0.1%; 0.8mM SAM). Substrate methylation was done at 37oC for 60 minutes in a 100μl of activation buffer, followed by 3 washes with 1x TBS-T buffer for 5min. Substrate was then incubated with rabbit anti-m6A antibody (1:1000, EMD Millipore, Cat# ABE572-I-100UG) for 30min, followed by 3 washes with sodium phosphate buffer (100mM, pH 7.2, 0.1% Tween) (SPB-T) for 10min. Incubation with secondary anti-rabbit HRP (1:2500; Cell Signaling technology, 7074S,) was done for 30min, and followed by 3 washes with SPB-T for 10 min each. Assay detection was done with 1-Step Ultra TMBL substrate (Thermo Scientific, 34028) for 15min followed by stopping the reaction with 2M sulfuric acid. Assay was measured at 450nm (Figure S2C).
Cell type-specific Fiber-seq from human cerebral cortex
The protocol below is divided in 3 consecutive steps, starting with Hia5 adenine methylation of immunotagged and sorted nuclei, followed by purification and size selection of high molecular weight (HMW) genomic DNA, and preparation of PacBio HiFi Whole Genome libraries.
Adenine methylation: Nuclei were extracted from postmortem cerebral cortex gray matter, immunotagged with NeuN (neuronal nuclear marker) for separate collection of neuronal and non-neuronal nuclei by fluorescence-activated nuclei sorting as described68,69 (Figure 1E).
For each sample, 0.4 - 2.5 x 106 sorted nuclei (Table S1) were methylated using 230μl of Hia5 activation buffer supplemented with the methyl-donor S-Adenosylmethionine (SAM) (0.8mM), and Triton X-100 at submicellar concentration70,71, or 0.002% (0.03mM) for 20 min at 37oC on a rotating platform (1500 rounds per minute,RPM). The Hia5 enzyme activity was stopped by adding Sodium-dodechyl-sulfate (SDS) to 1% final SDS concentration (Figure S2D).
DNA Purification and size selection: Next, high molecular weight gDNA was extracted from methylated nuclei using New England biolabs Monarch Genomic DNA Purification kit (#T3010L) according to the manufacturer’s instructions. We fragmented high molecular genomic DNA to 8-15 kb using the Megaruptor DNA fluid kit and shearing kit (Diagenode # E07030001) at the Megaraptor instrument speed setting 40, 31 and 32. Next, the fragmented DNA was assessed for purity with a NanoDrop ND-1000 spectrophotometer using A260/280=1.8; A260/230>2.0 as cutoff criteria. Additional quality controls included the Qubit fluorometer together with high sensitivity double stranded DNA assay kits, and additional size distribution checks by TapeStation, Genomic DNA ScreenTape, Agilent# 5067-5365.
PacBio library generation: Next, up to 5 μg input material was processed using SMRTbell kit v3.0 including repairing and A-tailing for 60 min at 37oC, 65oC for 5 min, followed by adaptor ligation at 20oC for 30 min and nuclease treatment for 15 min at 37oC, and size selected again on 2.9x AMPure PB beads. Library quality was assessed for concentration (Qubit fluorometer; high sensitivity double stranded DNA assay kit), purity (Nanodrop), size (TapeStation, Genomic DNA ScreenTape 5067-5365 and Femto pulse) (Table S1). Methylation status was validated with m6A slot blot assay before sequencing on the Sequel-II(e) platform, followed by deeper sequencing on Sequel II and Revio PacBio platforms for samples that passed quality controls and had sufficient material left for the additional sequencing.
SMRT-sequencing
Sequel II: SMRTbell libraries were quantified using the Qubit 1X dsDNA HS assay and the Femto Pulse. Libraries within ∼15% of each other by length were pooled for preliminary sequencing of 2-4 plex SMRTcells to check methylation levels. Each pool was separately annealed to PacBio sequencing primer v4 and bound to polymerase 2.0 for sequencing on 8M SMRTcells on the Sequel II or Sequel IIe system, each with a 30-hour movie. After preliminary interpretation of results from the pools, chosen libraries were separately annealed to PacBio sequencing primer v4 and bound to polymerase 2.0 for sequencing on individual 8M SMRTcells on the Sequel II or Sequel IIe system, each with a 30-hour movie, at loading concentrations between 60-80pM.
Revio: Individual SMRTbell libraries that had previously been sequenced on Sequel II/IIe were separately annealed to the PacBio Revio sequencing primer and bound to Revio polymerase for sequencing on individual 25M SMRTcells, each with a 24-hour movie, at loading concentrations between 225-250pM. Read length (as determined from SMRT-seq) was not significantly different between the N=41 NeuN+ and NeuN- samples shown in Table S1 (mean ± S.D. kilobases); NeuN+, 11.46 ± 2.49; NeuN-, 10.97 ± 2.26, with Welch’s t = 0.50.
HiFi data generation and preprocessing
Sequel II: data were imported to PacBio’s SMRTLink 11.1 bioinformatics tool suite for HiFi data generation. Intramolecular error correcting was performed using the circular consensus sequencing (CCS) algorithm to produce highly accurate (>Q20) CCS reads, each requiring a minimum of 3 polymerase passes. Pooled samples were afterwards demultiplexed using the PacBio lima tool in SMRTLink 11.1 with default parameters.
Methylated (m5) CpGs are called using PacBio jasmine on the raw HiFi BAM file output (https://github.com/pacificbiosciences/jasmine/). Methylated CpG positions must have an associated ML score above 200 to be considered a true methylation.
Revio: HiFi data were generated automatically using Revio’s on-board CCS. Methylated CpGs are automatically identified using Revio’s on-board jasmine. Barcodes were trimmed by Revio before the data were exported to SMRTLink.
Fiber-seq data processing
The BAM file with CpG methylation and HiFi kinetics information is processed using fibertools-rs v0.4.2 to predict-m6a to call positions with methylated adenines with an false positive rate of 0.23% at the precision level of >95%22,64, and then stitch regions depleted in methylation together to predict the size and location of nucleosomes on individual fibers. Methylated adenines must have an ML score greater than 200 to be called. Nucleosome footprints are called on a per read basis using fibertools -add-nucleosome22,64 with a minimum size set to 75 bp. Methyltransferase sensitive patches (MSPs) are similarly identified using fibertools -add-nucleosome, and are defined as regions along each fiber that are not occupied by a nucleosome footprint. Next, reads with per-fiber chromatin information are aligned to the recently published T2TCHM13 v2.0 genome72, assembly hs1 using pbmm2 v1.13.0. Using the recently introduced Fiber-seq quality control pipeline22, we visualized the sequencing quality and methylation level, distributions of the per-read CCS coverage, read length, m6A vs total A proportion, and MSP size were generated using python v3.8.18 and seaborn (v0.13.2, RRID:SCR_018132). The positions of m6As, nucleosomes, MSPs, and mCpGs are extracted into a BED12 format file using fibertools-rs extract, converted to bigBed format using UCSC-Utils bedToBigBed v2.10, and visualized on the UCSC Genome Browser. Since PacBio Hifi reads have a median read quality >Q30, they allow for highly accurate variant calling and phasing of reads into maternal and paternal haplotypes. Variants are called using three variant callers, DeepVariant v1.5.0, sniffles (v2.2, RRID:SCR_017619), and pbsv v2.9.0. We use a combination of variants, phased using hiphase v1.2.1, and k-mers, counted and phased using meryl and merqury v1.373, in order to phase our reads into maternal and paternal haplotypes. If haplotype information is available, the variant calling step is not necessary. The haplotype information is encoded in the BAM file in the haplotag field. The variant calling pipeline that was used can be found publicly available (https://github.com/mrvollger/k-mer-variant-phasing).
Linear mixed effects model
Linear mixed effects model was analyzed in R v.4.1.0. Impact of genome-level adenine methylation on Fiber-seq computational outcomes (Figure S3). To test for normality of m6A variable, we computed Shapiro-Wilk’s test using the shapiro.test function of the stats v4.1.0. m6A profile were assessed using linear mixed-effect models with post-mortem interval (PMI), cell-type (NeuN), Hia5 concentration (nuclei per Hia5 unit), and CCS coverage as the fixed factors, and the brain ID as a random effect. A model was fit using lmer function of the lme4 v1.1.27. and was analyzed using lmerTest v3.1.3. Confidence interval was calculated using confint function of the stats v4.1.0. N nuclei per Hia5 unit was log-transformed to be closer to a normal distribution (p<0.001, Shapiro-Wilk’s test). m6A profile was assessed using linear mixed-effect models with post-mortem interval (PMI), cell-type (NeuN), Hia5 concentration (nuclei per Hia5 unit), and CCS coverage as the fixed factors, and the brain ID as a random effect. A model was fit using lmer function of the lme4 v1.1.27. and was analyzed using lmerTest v3.1.3. Pearson’s correlation coefficient was calculated using cor function of stats v4.1.0 package. Scatterplots and histograms were drawn using functions pairs, plot, and hist in R v4.1.0. Confidence interval was calculated using confint function of the stats v4.1.0. We chose the m6A / A proportion as the dependent variable because m6A is fundamental for Fiber-seq pipeline’s nucleosome calling and the regulatory element annotation22.
Identification of Fiber-seq inferred regulatory element (FIRE)
We used the recently introduced FIRE analysis pipeline, (https://github.com/fiberseq/FIRE/blob/main/docs/README.md). Briefly, the FIRE pipeline uses a semi-supervised machine learning classification framework to classify MSPs as to whether they likely originate from accessible chromatin gene regulatory elements, or internucleosomal linker regions17. Specifically, FIRE provides a precision value for each MSP, with higher precision values (i.e. ≥0.9) corresponding to MSPs that have features similar to accessible chromatin gene regulatory elements. In addition, the FIRE pipeline calculates aggregate FIRE scores across all reads mapping to a given genomic position, and also does this based on haplotype if haplotype phasing information is included within the BAM file. The final BAM file with the FIRE elements and precision scores encoded is read into python for downstream data analysis using pyft v0.4.0.
Fiber-seq peak calling
In addition, the FIRE pipeline also calculates peaks of chromatin accessibility directly from the underlying FIRE score data at a 5% false discovery rate (FDR). Specifically, using a control dataset where each read’s mapping position has been shuffled, and null distribution of aggregate FIRE scores are calculated, which is used to compute real aggregate FIRE scores that are higher than would be expected by chance. Peaks of accessibility are then identified around regions that have aggregate FIRE scores that exceed an FDR cutoff of 5%. Peaks with fewer than 10 fibers overlapping the region were filtered out from the analysis to increase statistical power at loci that are considered while keeping a critical mass of peaks to perform genome-wide analysis. The pseudo-bulk Fiber-seq and short-read sequencing peaks were clustered (k-means, k=4) and visualized using deepTools (v3.5.1, RRID:SCR_016366). The four clusters of peaks were annotated to genomic regions using R v4.1.0 and ChIPseeker v1.30.3 and visualized using ggplot2 v3.3.5. Peaks were annotated to repeats by overlapping each cluster using bedtools v2.31.0; peaks with a minimum of 25% of the peak region overlapping a repeat are called repetitive.
Motif analysis
Motifs were identified using HOMER (v4.10, RRID:SCR_010881) Known motif calling in each cluster of fiber-seq NeuN- peaks that were called from FIRE analysis and clustered using H3K4me3 and H3K27ac ChIP-seq data (specific to NeuN-). Motifs are visualized (seaborn v0.13.2) by plotting the average m6A per position at all motif sites across all fibers within each cluster of peaks. We calculate a score to represent the size of the footprint by subtracting the total m6A proportion across the motif site of size (S) from the sum of the maximum total score of the left and the right side across S/2 base pairs. We control for GC rich motifs, such as SP1, by considering the accessibility in the flanking sequence and normalizing the signal in the motif region to one another. To find the p-value when comparing groups, we calculate this score for each site that makes up the total set of regions for both sets and then compute a Kolmogorov–Smirnov test.
Fiber-seq footprinting
Regions of accessibility signal depletion within larger MSP accessible patches (footprints) are determined in Fiber-seq by searching for regions within MSPs that are completely devoid of methylated adenines on a single fiber-level within the footprinted region. Footprinting was done on canonical 35bp CTCF motif locations that are found within FIRE peaks that have CTCF signal by ChIP-seq, cluster together by k-means clustering, and have at least 20 fibers overlapping them using fibertools-rs ft footprint (v0.4.2). The CTCF ChIP-seq data were obtained from ENCODE H1-derived neural cells. Fibers were then classified into three categories: (i) having a methyltransferase sensitive patch (MSP) with a footprint at the motif site, (ii) having a methyltransferase sensitive patch (MSP) without a footprint, or (iii) having a nucleosome overlapping the locus. Fibers of each category were grouped together and the average proportion of adenines that had an m6A was calculated at each base and plotted centered at the motif, with a flank of 1000bp upstream and downstream. The reading of the fiber-seq data was done with pyft (v0.0.6), the data manipulation and calculations were performed with pandas (v2.0.3) and numpy (v1.24.4), and the plotting was done with matplotlib (v3.7.3) and seaborn (v0.13.2).
Fiber-seq Enrichr analysis
The fiber-seq peaks from NeuN+ and NeuN- were filtered to only include those with coverage greater than 20 fibers at the peak, since that is the lower of the two respective medians. Peaks were then annotated to gene-based genomic regions using ChIPseeker. The peaks that were not annotated to promoters (the non-promoter peaks) were then ranked by their percent accessibility, with the highest percent accessibility corresponding to the highest rank. The top 1000 genes by percent accessibility annotated to non-promoter NeuN+ and NeuN- peaks were selected for Enrichr term enrichment analysis using the GO Biological Processes database. A p-value of 0.05 was selected for 95% confidence in the enrichment of terms.
Fiber-seq signal difference in ATAC enhancer peaks analysis
After clustering, the peaks that make up clusters 2 and 4 from the ATAC-seq analysis are annotated as enhancer peaks. To quantify the average difference in signal between NeuN+ and NeuN- per position in the enhancer peaks in both ATAC-seq and Fiber-seq, first the difference in NeuN+ and NeuN- signal at each base was calculated for each peak. For ATAC-seq the RPKM normalized signal was used and for Fiber-seq the percent accessibility, the proportion of fibers that are called as accessible at any given base. Additionally, to maintain the correct directionality of the signal difference, in the NeuN+ enhancers, negative signal was subtracted from positive signal, while in the NeuN- enhancers, the opposite operation was performed. After computing the difference in signal for every peak, the average difference at each position was calculated and plotted as a line plot, relative to the center of the peak. The array generation and manipulation was performed with python (v3.8.18) and numpy (v1.24.4) and the plots were generated using matplotlib (v3.7.3).
The correlation of the difference in signal in Fiber-seq and ATAC-seq relative to the center of the ATAC peak was done by plotting the average percent accessibility from Fiber-seq as a function of the average ATAC-seq counts at each position. A linear regression was applied to compute the line of best fit and a p-value using scipy (v1.10.1). The graphs were plotted with matplotlib (v3.7.3, same as above).
Linkage disequilibrium (LD) enrichment
We conducted LD score enrichment analysis74 to assess the enrichment of both brain-related and non-brain-related GWAS for all peaks within four clusters after removal of peaks that overlapped with repeat elements. We limited the summary statistics for GWAS traits to European-only version whenever it was possible. Additionally, the broad major histocompatibility complex region (hg19: chr.6:25–35 Mb) was removed from every GWAS due to its intricate and extensive LD structure. LDSc was ran using default parameters.
Haplotyping
Variants were called with the tools as described above in the section ‘Fiber-seq data processing’. Haplotype-specific differences were computed at peaks that had a minimum of 10 fibers overlapping the peak in each haplotype. The difference was calculated by subtracting the percent accessibility in haplotype 2 (H2) from haplotype (H1). In order to determine if the distribution of FIRE elements across the two haplotypes was due to random chance, a Fisher test was used with the number of accessible fibers (fibers with a FIRE element overlapping the peak) in H1 and H2 vs the number of inaccessible fibers in H1 and H2. A p-value cutoff of 0.05 was selected for 95% confidence that the distribution of FIRE elements is not due to random chance. The calculation was done with numpy (v1.24.4) and pandas (v2.0.3), and the plot was generated using matplotlib (v3.7.3).
Co-actuation
A co-actuated site is defined as a site that shows a FIRE element with a precision value ≥0.9 at two separate genomic loci but on the same fiber, across multiple fibers. For a given set of peaks, pairs of peaks with a fiber-seq coverage (C) greater than 10 over both regions were chosen as candidates for co-actuation. At each pair, we calculate the number of actuated elements on both sites (B), neither site (N), the first site (A1) and the second site (A2). The proportion of co-actuated fibers at the locus is calculated by dividing B by C. The p-value is computed using a Fisher’s exact test using the number of fibers with FIRE elements at site 1 and 2, the number of fibers with a FIRE element at site 1 or site 2, and the number of fibers with no FIRE elements at either site.
Next, for each peak pair, significant and non-significant, the actual proportion of co-actuated fibers was calculated by dividing the number of co-actuated fibers by the total number of fibers that overlap both peaks in the pair. The expected proportion represents the probability that both loci are open, given that the two loci are independent of one another. Therefore, the expected proportion of co-actuated fibers for a given pair of peaks A and B, can be calculated by multiplying the proportion of accessible fibers in A by the proportion of accessible fibers in B. The expected proportion is subtracted from the actual proportion to calculate the difference per peak for each set of peaks, significant and non-significant. Then the average difference in the actual vs expected proportions is calculated and a one-tailed T-test is used to determine if the significant peaks have a greater average difference than the non-significant peaks. The actual (A), expected (E), and difference (D) value calculations are summarized by the following equations: A = (Aopen ⋂ Bopen)/ Ftotal , E = (Aopen ∝ Ftotal) × (Bopen ∝ Ftotal), D = A – E. The actual and expected values for each peak are plotted separately in two swarm plots, one per peak set, using matplotlib (v3.7.3) and seaborn (v0.13.2), with data cleaning and shaping using pandas (v2.0.3) and numpy (v1.24.4), and computed using scipy (v1.10.1).
Nucleosome positioning
To investigate the preferential positioning of nucleosomes proximal to promoters, single-fiber nucleosome positions for fibers overlapping the feature are centered at the start of the region of interest. The upstream and downstream 5 nucleosomes are obtained, and their centers are calculated. A reference fiber is chosen at random; the reference fiber nucleosome centers are subtracted from all other centers and the position-based average is calculated, like previously described.16 These values represent the average offset of nucleosomes as a function of the relative nucleosome position to the regulatory elements across a set of cells for a single locus. Then we plot a boxplot (seaborn v0.13.2) of the average offset for each nucleosome across the set of regions. The p-value is generated by comparing the distributions of the nucleosome offsets using a Wilcoxon test pairwise between the -1 and +1 nucleosome versus all other nucleosome positions.
Single nuclei RNA-seq data processing
A tissue block of Area 4 frontal cortex gray matter (brain ID ‘SN2a’, see Table S1) was sorted into NeuN+ and NeuN- fractions and processed for single nuclei RNA-seq on a 10x chromium platform, then sequenced as recently described75. Sequenced fastq files were aligned, filtered, barcoded and UMI counted using Cell Ranger Chromium Single Cell RNA-seq version 8.0.1, by 10X Genomics with CHM13 T2T v2.0 as the human genome reference (RRID:SCR_017344)76. Each sample was filtered to retain cells with ≥ 1000 UMIs, ≥400 genes expressed, and <10% of the reads mapping to the mitochondrial genome. NeuNneg and NeuNpos samples were merged using Seurat package (version 4.0.3, RRID:SCR_016341)77. UMI counts were then normalized so that each cell had a total of 10,000 UMIs across all genes and these normalized counts were log-transformed with a pseudocount of 1 using the “LogNormalize” function in the Seurat package. The top 2000 most highly variable genes were identified using the “vst” selection method of “FindVariableFeatures” function and counts were scaled using the “ScaleData” function.
Principal component analysis was performed using the top 2000 highly variable features (“RunPCA” function) and the top 30 principal components were used in the downstream analysis. K-Nearest Neighbor graphs were obtained by using the “FindNeighbors” function whereas the UMAPs were obtained by the “RunUMAP” function. The Louvain algorithm was used to cluster cells based on expression similarity. The resolution was set at 0.2 for optimal clustering. Cell clusters were annotated based on expression of canonical marker genes including MOG (oligodendrocytes), PDGFRA (oligodendrocyte progenitor cells), AQP4 (astrocytes), PTPRC (microglia), SYP (neuronal cells), SLC17A7 (Excitatory Neurons), and GAD1 (Inhibitory Neurons).
Short-read sequencing pre-processing and analysis
FASTQ files for 3 replicates each of NeuN+ and NeuN- ATAC-seq, and ChIP-seq of H3K27ac, and H3K4me3 were downloaded from psychENCODE. Raw data for 2 replicates of CTCF ChIP-seq were downloaded from ENCODE. Reads were first evaluated for their quality using FastQC (v0.11.8, RRID:SCR_014583) (Andrews 2010). Reads were then trimmed for adaptor sequences using Trim Galore! (v0.6.6, RRID:SCR_011847) and aligned to the T2T-CHM13v2.0 (T2T) genome, assembly hs1, using Bowtie2 (v2.1.0, RRID:SCR_016368) with default parameters or –X 2000 for ChIP-seq and ATAC-Seq respectively78. ATAC-seq reads aligned to mtDN A were removed. Reads mapping to multiple genomic locations (excluding chrM) were not excluded from the alignment and were randomly assigned to one of their highest mapping quality alignments. Picard (v2.2.4, RRID:SCR_006525) was used to remove duplicated reads (Picard Toolkit 2019). Post-filtering bam file for samples from the same assay and condition were merged using SAMtools v1.17.0 merge function. Coverage tracks (bigWig) were generated from Bam files using deepTools (v3.2.1, RRID:SCR_016366) bamCoverage with parameters --normalizeUsingRPKM --binsize 1079.
Peaks obtained from psychENCODE were lifted over to the T2T genome from hg38 using UCSC Utilities LiftOver (version 2023-10-17) and clustered into promoters and enhancers with deeptools k-means clustering, k=2, on the ATAC-seq and H3K4me3 and H3K27ac ChIP-seq for each condition, NeuN+ and NeuN-. Clustered peaks were annotated to genomic regions based on gene information using R v4.1.0 and ChIPseeker v1.30.3 and visualized using ggplot2 v3.3.5. Coverage tracks in bigWig format are visualized on the UCSC Genome Browser. ATAC peaks are ranked based on the average signal across the peak region; we used the top n=45,992 peaks each for NeuN+ and NeuN- (91,984 peaks total).
Quantification and statistical analysis
Statistical tests used in this study are described separately for each portion of the analysis in the sections above and indicated in the figure legends.
Published: December 3, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2024.100911.
Supplemental information
References
- 1.Cutter A.R., Hayes J.J. A brief review of nucleosome structure. FEBS Lett. 2015;589:2914–2922. doi: 10.1016/j.febslet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Struhl K., Segal E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michael A.K., Thomä N.H. Reading the chromatinized genome. Cell. 2021;184:3599–3611. doi: 10.1016/j.cell.2021.05.029. [DOI] [PubMed] [Google Scholar]
- 4.Gourisankar S., Krokhotin A., Wenderski W., Crabtree G.R. Context-specific functions of chromatin remodellers in development and disease. Nat. Rev. Genet. 2024;25:340–361. doi: 10.1038/s41576-023-00666-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oberbeckmann E., Oudelaar A.M. Genome organization across scales: mechanistic insights from in vitro reconstitution studies. Biochem. Soc. Trans. 2024;52:793–802. doi: 10.1042/BST20230883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oberbeckmann E., Quililan K., Cramer P., Oudelaar A.M. In vitro reconstitution of chromatin domains shows a role for nucleosome positioning in 3D genome organization. Nat. Genet. 2024;56:483–492. doi: 10.1038/s41588-023-01649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhie S.K., Schreiner S., Farnham P.J. Defining Regulatory Elements in the Human Genome Using Nucleosome Occupancy and Methylome Sequencing (NOMe-Seq) Methods Mol. Biol. 2018;1766:209–229. doi: 10.1007/978-1-4939-7768-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D., Liu S., Warrell J., Won H., Shi X., Navarro F.C.P., Clarke D., Gu M., Emani P., Yang Y.T., et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362 doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whalen S., Inoue F., Ryu H., Fair T., Markenscoff-Papadimitriou E., Keough K., Kircher M., Martin B., Alvarado B., Elor O., et al. Machine learning dissection of human accelerated regions in primate neurodevelopment. Neuron. 2023;111:857–873.e8. doi: 10.1016/j.neuron.2022.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de la Torre-Ubieta L., Stein J.L., Won H., Opland C.K., Liang D., Lu D., Geschwind D.H. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell. 2018;172:289–304.e18. doi: 10.1016/j.cell.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girskis K.M., Stergachis A.B., DeGennaro E.M., Doan R.N., Qian X., Johnson M.B., Wang P.P., Sejourne G.M., Nagy M.A., Pollina E.A., et al. Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron. 2021;109:3239–3251.e7. doi: 10.1016/j.neuron.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stepniak K., Machnicka M.A., Mieczkowski J., Macioszek A., Wojtas B., Gielniewski B., Poleszak K., Perycz M., Krol S.K., Guzik R., et al. Mapping chromatin accessibility and active regulatory elements reveals pathological mechanisms in human gliomas. Nat. Commun. 2021;12:3621. doi: 10.1038/s41467-021-23922-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minnoye L., Marinov G.K., Krausgruber T., Pan L., Marand A.P., Secchia S., Greenleaf W.J., Furlong E.E.M., Zhao K., Schmitz R.J., et al. Chromatin accessibility profiling methods. Nat. Rev. Methods Primers. 2021;1 doi: 10.1038/s43586-020-00008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warburton P.E., Sebra R.P. Long-Read DNA Sequencing: Recent Advances and Remaining Challenges. Annu. Rev. Genomics Hum. Genet. 2023;24:109–132. doi: 10.1146/annurev-genom-101722-103045. [DOI] [PubMed] [Google Scholar]
- 15.Treangen T.J., Salzberg S.L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stergachis A.B., Debo B.M., Haugen E., Churchman L.S., Stamatoyannopoulos J.A. Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science. 2020;368:1449–1454. doi: 10.1126/science.aaz1646. [DOI] [PubMed] [Google Scholar]
- 17.Vollger M.R., Swanson E.G., Neph S.J., Ranchalis J., Munson K.M., Ho C.-H., Sedeño-Cortés A.E., Fondrie W.E., Bohaczuk S.C., Mao Y., et al. A haplotype-resolved view of human gene regulation. bioRxiv. 2024 doi: 10.1101/2024.06.14.599122. Preprint at. [DOI] [Google Scholar]
- 18.Cheung I., Shulha H.P., Jiang Y., Matevossian A., Wang J., Weng Z., Akbarian S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2010;107:8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jorstad N.L., Close J., Johansen N., Yanny A.M., Barkan E.R., Travaglini K.J., Bertagnolli D., Campos J., Casper T., Crichton K., et al. Transcriptomic cytoarchitecture reveals principles of human neocortex organization. Science. 2023;382 doi: 10.1126/science.adf6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minnoye L., Marinov G.K., Krausgruber T., Pan L., Marand A.P., Secchia S., Greenleaf W.J., Furlong E.E.M., Zhao K., Schmitz R.J., et al. Chromatin accessibility profiling methods. Nat. Rev. Methods Primers. 2021;1:10. doi: 10.1038/s43586-020-00008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong Y., Cao L., Deikus G., Fan Y., Mead E.A., Lai W., Zhang Y., Yong R., Sebra R., Wang H., et al. Critical assessment of DNA adenine methylation in eukaryotes using quantitative deconvolution. Science. 2022;375:515–522. doi: 10.1126/science.abe7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jha A., Bohaczuk S.C., Mao Y., Ranchalis J., Mallory B.J., Min A.T., Hamm M.O., Swanson E., Dubocanin D., Finkbeiner C., et al. DNA-m6A calling and integrated long-read epigenetic and genetic analysis with fibertools. Genome Res. 2024;34:1976–1986. doi: 10.1101/gr.279095.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fullard J.F., Giambartolomei C., Hauberg M.E., Xu K., Voloudakis G., Shao Z., Bare C., Dudley J.T., Mattheisen M., Robakis N.K., et al. Open chromatin profiling of human postmortem brain infers functional roles for non-coding schizophrenia loci. Hum. Mol. Genet. 2017;26:1942–1951. doi: 10.1093/hmg/ddx103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Girdhar K., Hoffman G.E., Jiang Y., Brown L., Kundakovic M., Hauberg M.E., Francoeur N.J., Wang Y.C., Shah H., Kavanagh D.H., et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci. 2018;21:1126–1136. doi: 10.1038/s41593-018-0187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinz S., Romanoski C.E., Benner C., Glass C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fullard J.F., Hauberg M.E., Bendl J., Egervari G., Cirnaru M.D., Reach S.M., Motl J., Ehrlich M.E., Hurd Y.L., Roussos P. An atlas of chromatin accessibility in the adult human brain. Genome Res. 2018;28:1243–1252. doi: 10.1101/gr.232488.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grandi F.C., Modi H., Kampman L., Corces M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022;17:1518–1552. doi: 10.1038/s41596-022-00692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Begum G., Otsu M., Ahmed U., Ahmed Z., Stevens A., Fulton D. NF-Y-dependent regulation of glutamate receptor 4 expression and cell survival in cells of the oligodendrocyte lineage. Glia. 2018;66:1896–1914. doi: 10.1002/glia.23446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun W., Cornwell A., Li J., Peng S., Osorio M.J., Aalling N., Wang S., Benraiss A., Lou N., Goldman S.A., Nedergaard M. SOX9 Is an Astrocyte-Specific Nuclear Marker in the Adult Brain Outside the Neurogenic Regions. J. Neurosci. 2017;37:4493–4507. doi: 10.1523/JNEUROSCI.3199-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao Y., Du S., Yang B., Wang Y., Li Y., Li R., Zhou T., Du X., He Y., Wang Y., et al. NeuroD1 induces microglial apoptosis and cannot induce microglia-to-neuron cross-lineage reprogramming. Neuron. 2021;109:4094–4108.e5. doi: 10.1016/j.neuron.2021.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Hesselberth J.R., Chen X., Zhang Z., Sabo P.J., Sandstrom R., Reynolds A.P., Thurman R.E., Neph S., Kuehn M.S., Noble W.S., et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neph S., Vierstra J., Stergachis A.B., Reynolds A.P., Haugen E., Vernot B., Thurman R.E., John S., Sandstrom R., Johnson A.K., et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly T.K., Liu Y., Lay F.D., Liang G., Berman B.P., Jones P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22:2497–2506. doi: 10.1101/gr.143008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Day F.R., Ruth K.S., Thompson D.J., Lunetta K.L., Pervjakova N., Chasman D.I., Stolk L., Finucane H.K., Sulem P., Bulik-Sullivan B., et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 2015;47:1294–1303. doi: 10.1038/ng.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Als T.D., Kurki M.I., Grove J., Voloudakis G., Therrien K., Tasanko E., Nielsen T.T., Naamanka J., Veerapen K., Levey D.F., et al. Depression pathophysiology, risk prediction of recurrence and comorbid psychiatric disorders using genome-wide analyses. Nat. Med. 2023;29:1832–1844. doi: 10.1038/s41591-023-02352-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lutz P.E., Tanti A., Gasecka A., Barnett-Burns S., Kim J.J., Zhou Y., Chen G.G., Wakid M., Shaw M., Almeida D., et al. Association of a History of Child Abuse With Impaired Myelination in the Anterior Cingulate Cortex: Convergent Epigenetic, Transcriptional, and Morphological Evidence. Am. J. Psychiatry. 2017;174:1185–1194. doi: 10.1176/appi.ajp.2017.16111286. [DOI] [PubMed] [Google Scholar]
- 38.Aberg K.A., Dean B., Shabalin A.A., Chan R.F., Han L.K.M., Zhao M., van Grootheest G., Xie L.Y., Milaneschi Y., Clark S.L., et al. Methylome-wide association findings for major depressive disorder overlap in blood and brain and replicate in independent brain samples. Mol. Psychiatry. 2020;25:1344–1354. doi: 10.1038/s41380-018-0247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Q.S., Morrison R.L., Turecki G., Drevets W.C. Meta-analysis of epigenome-wide association studies of major depressive disorder. Sci. Rep. 2022;12 doi: 10.1038/s41598-022-22744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Do C., Lang C.F., Lin J., Darbary H., Krupska I., Gaba A., Petukhova L., Vonsattel J.P., Gallagher M.P., Goland R.S., et al. Mechanisms and Disease Associations of Haplotype-Dependent Allele-Specific DNA Methylation. Am. J. Hum. Genet. 2016;98:934–955. doi: 10.1016/j.ajhg.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.Y., et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snyder M.W., Adey A., Kitzman J.O., Shendure J. Haplotype-resolved genome sequencing: experimental methods and applications. Nat. Rev. Genet. 2015;16:344–358. doi: 10.1038/nrg3903. [DOI] [PubMed] [Google Scholar]
- 44.Farmiloe G., Lodewijk G.A., Robben S.F., van Bree E.J., Jacobs F.M.J. Widespread correlation of KRAB zinc finger protein binding with brain-developmental gene expression patterns. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020;375 doi: 10.1098/rstb.2019.0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schones D.E., Cui K., Cuddapah S., Roh T.Y., Barski A., Wang Z., Wei G., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radman-Livaja M., Rando O.J. Nucleosome positioning: how is it established, and why does it matter? Dev. Biol. 2010;339:258–266. doi: 10.1016/j.ydbio.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panigrahi A., O'Malley B.W. Mechanisms of enhancer action: the known and the unknown. Genome Biol. 2021;22:108. doi: 10.1186/s13059-021-02322-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nasser J., Bergman D.T., Fulco C.P., Guckelberger P., Doughty B.R., Patwardhan T.A., Jones T.R., Nguyen T.H., Ulirsch J.C., Lekschas F., et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. doi: 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He B., Chen C., Teng L., Tan K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA. 2014;111:E2191–E2199. doi: 10.1073/pnas.1320308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lionnet T., Czaplinski K., Darzacq X., Shav-Tal Y., Wells A.L., Chao J.A., Park H.Y., de Turris V., Lopez-Jones M., Singer R.H. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat. Methods. 2011;8:165–170. doi: 10.1038/nmeth.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abril-Garrido J., Dienemann C., Grabbe F., Velychko T., Lidschreiber M., Wang H., Cramer P. Structural basis of transcription reduction by a promoter-proximal +1 nucleosome. Mol. Cell. 2023;83:1798–1809.e7. doi: 10.1016/j.molcel.2023.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen H., Kharerin H., Dhasarathy A., Kladde M., Bai L. Partitioned usage of chromatin remodelers by nucleosome-displacing factors. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu M., Li M., Liu W., Yan M., Li L., Ding W., Nian X., Dai W., Sun D., Zhu Y., et al. Nucleoporin Seh1 maintains Schwann cell homeostasis by regulating genome stability and necroptosis. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.112802. [DOI] [PubMed] [Google Scholar]
- 54.Abdulhay N.J., McNally C.P., Hsieh L.J., Kasinathan S., Keith A., Estes L.S., Karimzadeh M., Underwood J.G., Goodarzi H., Narlikar G.J., Ramani V. Massively multiplex single-molecule oligonucleosome footprinting. Elife. 2020;9 doi: 10.7554/eLife.59404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y., Wang A., Liu Z., Thurman A.L., Powers L.S., Zou M., Zhao Y., Hefel A., Li Y., Zabner J., Au K.F. Single-molecule long-read sequencing reveals the chromatin basis of gene expression. Genome Res. 2019;29:1329–1342. doi: 10.1101/gr.251116.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boltengagen M., Verhagen D., Wolff M.R., Oberbeckmann E., Hanke M., Gerland U., Korber P., Mueller-Planitz F. A single fiber view of the nucleosome organization in eukaryotic chromatin. Nucleic Acids Res. 2024;52:166–185. doi: 10.1093/nar/gkad1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee I., Razaghi R., Gilpatrick T., Molnar M., Gershman A., Sadowski N., Sedlazeck F.J., Hansen K.D., Simpson J.T., Timp W. Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. Nat. Methods. 2020;17:1191–1199. doi: 10.1038/s41592-020-01000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shipony Z., Marinov G.K., Swaffer M.P., Sinnott-Armstrong N.A., Skotheim J.M., Kundaje A., Greenleaf W.J. Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat. Methods. 2020;17:319–327. doi: 10.1038/s41592-019-0730-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacques P.É., Jeyakani J., Bourque G. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simone R., Javad F., Emmett W., Wilkins O.G., Almeida F.L., Barahona-Torres N., Zareba-Paslawska J., Ehteramyan M., Zuccotti P., Modelska A., et al. MIR-NATs repress MAPT translation and aid proteostasis in neurodegeneration. Nature. 2021;594:117–123. doi: 10.1038/s41586-021-03556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carnevali D., Conti A., Pellegrini M., Dieci G. Whole-genome expression analysis of mammalian-wide interspersed repeat elements in human cell lines. DNA Res. 2017;24:59–69. doi: 10.1093/dnares/dsw048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mantere T., Kersten S., Hoischen A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019;10:426. doi: 10.3389/fgene.2019.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huddleston J., Chaisson M.J.P., Steinberg K.M., Warren W., Hoekzema K., Gordon D., Graves-Lindsay T.A., Munson K.M., Kronenberg Z.N., Vives L., et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017;27:677–685. doi: 10.1101/gr.214007.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jha A., Bohaczuk S.C., Mao Y., Ranchalis J., Mallory B.J., Min A.T., Hamm M.O., Swanson E., Dubocanin D., Finkbeiner C., et al. DNA-m6A calling and integrated long-read epigenetic and genetic analysis with fibertools. bioRxiv. 2023 doi: 10.1101/2023.04.20.537673. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zlatanova J., Bishop T.C., Victor J.M., Jackson V., van Holde K. The nucleosome family: dynamic and growing. Structure. 2009;17:160–171. doi: 10.1016/j.str.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 66.Dennis A.F., Xu Z., Clark D.J. Examining chromatin heterogeneity through PacBio long-read sequencing of M.EcoGII methylated genomes: an m6A detection efficiency and calling bias correcting pipeline. Nucleic Acids Res. 2024;52 doi: 10.1093/nar/gkae288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blainey P., Krzywinski M., Altman N. Points of significance: replication. Nat. Methods. 2014;11:879–880. doi: 10.1038/nmeth.3091. [DOI] [PubMed] [Google Scholar]
- 68.Jiang Y., Matevossian A., Huang H.S., Straubhaar J., Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kundakovic M., Jiang Y., Kavanagh D.H., Dincer A., Brown L., Pothula V., Zharovsky E., Park R., Jacobov R., Magro I., et al. Practical Guidelines for High-Resolution Epigenomic Profiling of Nucleosomal Histones in Postmortem Human Brain Tissue. Biol. Psychiatry. 2017;81:162–170. doi: 10.1016/j.biopsych.2016.03.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang L., Chai X., Sun P., Yuan B., Jiang B., Zhang X., Liu M. The Study of the Aggregated Pattern of TX100 Micelle by Using Solvent Paramagnetic Relaxation Enhancements. Molecules. 2019;24 doi: 10.3390/molecules24091649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tiller G.E., Mueller T.J., Dockter M.E., Struve W.G. Hydrogenation of triton X-100 eliminates its fluorescence and ultraviolet light absorption while preserving its detergent properties. Anal. Biochem. 1984;141:262–266. doi: 10.1016/0003-2697(84)90455-x. [DOI] [PubMed] [Google Scholar]
- 72.Rhie A., Nurk S., Cechova M., Hoyt S.J., Taylor D.J., Altemose N., Hook P.W., Koren S., Rautiainen M., Alexandrov I.A., et al. The complete sequence of a human Y chromosome. Nature. 2023;621:344–354. doi: 10.1038/s41586-023-06457-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rhie A., Walenz B.P., Koren S., Phillippy A.M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020;21:245. doi: 10.1186/s13059-020-02134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K., et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei J., Lambert T.Y., Valada A., Patel N., Walker K., Lenders J., Schmidt C.J., Iskhakova M., Alazizi A., Mair-Meijers H., et al. Single nucleus transcriptomics of ventral midbrain identifies glial activation associated with chronic opioid use disorder. Nat. Commun. 2023;14:5610. doi: 10.1038/s41467-023-41455-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Fiber-seq and single-nuclei sequencing data are available in the database for Genotypes and Phenotypes (dbGAP) under accession no. phs003771.v1.p1.
-
•
This study does not report original code.
-
•
Any additional information required to reproduce the data reported in this study is available from the lead contact upon request.