

Abstract
Purpose
To develop an end-to-end convolutional neural network model for analyzing hematoxylin and eosin(H&E)-stained histological images, enhancing the performance and efficiency of nuclear segmentation and classification within the digital pathology workflow.
Methods
We propose a dual-mechanism feature pyramid fusion technique that integrates nuclear segmentation and classification tasks to construct the HistoNeXt network model. HistoNeXt utilizes an encoder-decoder architecture, where the encoder, based on the advanced ConvNeXt convolutional framework, efficiently and accurately extracts multi-level abstract features from tissue images. These features are subsequently shared with the decoder. The decoder employs a dual-mechanism architecture: The first branch of the mechanism splits into two parallel paths for nuclear segmentation, producing nuclear pixel (NP) and horizontal and vertical distance (HV) predictions, while the second mechanism branch focuses on type prediction (TP). The NP and HV branches leverage densely connected blocks to facilitate layer-by-layer feature transmission and reuse, while the TP branch employs channel attention to adaptively focus on critical features. Comprehensive data augmentation including morphology-preserving geometric transformations and adaptive H&E channel adjustments was applied. To address class imbalance, type-aware sampling was applied. The model was evaluated on public tissue image datasets including CONSEP, PanNuke, CPM17, and KUMAR. The performance in nuclear segmentation was evaluated using the Dice Similarity Coefficient (DICE), the Aggregated Jaccard Index (AJI) and Panoptic Quality (PQ), and the classification performance was evaluated using F1 scores and category-specific F1 scores. In addition, computational complexity, measured in Giga Floating Point Operations Per Second (GFLOPS), was used as an indicator of resource consumption.
Results
HistoNeXt demonstrated competitive performance across multiple datasets: achieving a DICE score of 0.874, an AJI of 0.722, and a PQ of 0.689 on the CPM17 dataset; a DICE score of 0.826, an AJI of 0.625, and a PQ of 0.565 on KUMAR; and performance comparable to Transformer-based models, such as CellViT-SAM-H, on PanNuke, with a binary PQ of 0.6794, a multi-class PQ of 0.4940, and an overall F1 score of 0.82. On the CONSEP dataset, it achieved a DICE score of 0.843, an AJI of 0.592, a PQ of 0.532, and an overall classification F1 score of 0.773. Specific F1 scores for various cell types were as follows: 0.653 for malignant or dysplastic epithelial cells, 0.516 for normal epithelial cells, 0.659 for inflammatory cells, and 0.587 for spindle cells. The tiny model’s complexity was 33.7 GFLOPS.
Conclusion
By integrating novel convolutional technology and employing a pyramid fusion of dual-mechanism characteristics, HistoNeXt enhances both the precision and efficiency of nuclear segmentation and classification. Its low computational complexity makes the model well suited for local deployment in resource-constrained environments, thereby supporting a broad spectrum of clinical and research applications. This represents a significant advance in the application of convolutional neural networks in digital pathology analysis.
Keywords: Nuclear segmentation, Nuclear classification, Feature pyramid, Convolutional neural network, Digital pathology
Introduction
In digital pathology, accurate segmentation and classification of cell nuclei are fundamental for effective diagnosis and the development of treatment strategies. Traditional pathological image diagnosis relies on manual analysis, which is inefficient and challenging, especially when dealing with large-scale and complex pathological images. With advances in digital scanning technology and the growing volume in pathological image data, there is an increasing demand for rapid and intelligent analysis techniques. As a result, the development of efficient automated techniques for the swift and precise segmentation and classification of cell nuclei has become an important research direction in this field.
In recent years, deep learning technology, particularly convolutional neural networks (CNNs), has demonstrated remarkable capabilities in medical image processing. These networks have excelled in tasks such as cell nucleus detection, segmentation, and classification. For example, the U-Net model improves feature extraction through a symmetric encoder-decoder structure and retains more contextual information through skip connections, substantially improving the accuracy of medical image segmentation and showing superior performance when dealing with detailed medical images [1]. The HoVer-Net model provides a high-performance solution for simultaneous cell nuclear segmentation and classification [2]. It employs three parallel decoding branches: the nuclear pixel (NP) branch to predict whether a pixel belongs to a cell nucleus, the horizontal and vertical distance (HV) branch to estimate the horizontal and vertical distances of nuclear pixels to their centroids, and the type prediction (TP) branch to classify the type of each cell nucleus pixel. This model has achieved state-of-the-art performance in cell nuclear segmentation and classification tasks on multiple public datasets.
Recent research has further expanded the potential of deep learning in medical image analysis. Srikantamurthy et al. proposed a hybrid CNN-LSTM model to classify benign and malignant subtypes in breast cancer histopathology images, achieving 99% precision in binary classification [3]. This highlights the promise of combining different neural network architectures to enhance performance. Additionally, Gudhe et al. introduced a deep learning method based on Bayesian dropout, achieving a mean F1 score of 0.893 on the PanNuke dataset [4], surpassing several existing models [5]. This approach not only improved segmentation accuracy, but also quantified prediction uncertainty, offering new directions for future research.
Despite these advancements, traditional CNNs face limitations when dealing with ultra-long contextual semantic images, and their performance can degrade when handling highly heterogeneous and complex pathological images. Furthermore, current methods often struggle with class imbalance, notably in distinguishing normal from malignant epithelial cells, and face challenges in maintaining consistent performance across varying image resolutions. These challenges underscore the need for more robust architectures that can effectively capture both local and global features while maintaining computational efficiency.
The Transformer architecture, originally designed for long-sequence natural language data, has gradually demonstrated its potential in image analysis tasks. For example, the CellViT model shows the potential advantages of transformers in pathological image segmentation tasks compared to traditional CNNs [6]. ConvNeXt, by redesigning convolutional blocks and simplifying the network structure based on Transformer principles, enhances performance and efficiency over conventional CNNs, particularly when dealing with images featuring complex textures and morphologies, thus demonstrating superior feature extraction capabilities [7]. Recent studies like CellViT have demonstrated the advantages of pure transformer architectures in nuclear segmentation tasks, achieving state-of-the-art performance, especially in handling complex tissue contexts. However, these approaches often require substantial computational resources and large-scale training data, which has led to the exploration of hybrid architectures that combine the strengths of both CNNs and Transformers. These developments suggest that further integration of CNNs with Transformers could optimize cell nuclear segmentation and classification performance.
To address these challenges and advance digital pathology analysis, we propose a computationally efficient approach that leverages the strengths of modern CNN architectures combined with dual-mechanism feature pyramid fusion. Drawing inspiration from the U-Net and HoVer-Net architectures and incorporating techniques such as ConvNeXt, dense connections, and attention mechanisms, we construct an end-to-end network model HistoNeXt for cell nuclear segmentation and classification. The performance of this model in both segmentation and classification is validated using multiple metrics including Dice Similarity Coefficient (DICE), Aggregated Jaccard Index (AJI), Detection Quality (DQ), Segmentation Quality (SQ), Panoptic Quality (PQ), and F1 scores on public datasets. The computational complexity is measured using Giga Floating Point Operations Per Second (GFLOPS) to evaluate the model resource consumption.
Materials and methods
Training Data
To effectively train and evaluate the HistoNeXt model, we utilize four publicly available histological image datasets: CONSEP [2], PanNuke [4],CPM17 [8] and KUMAR [9]. All datasets were meticulously annotated by professional pathologists, providing a detailed ground truth for nuclear segmentation and classification tasks.
The CONSEP dataset comprises 24,319 meticulously annotated nuclei from 41 hematoxylin and eosin (H&E)-stained colorectal cancer pathology slides, with each image measuring 10001000 pixels at 40x magnification. The dataset features detailed nuclear boundary annotations along with nuclear-type classifications, including inflammatory cells, normal epithelial cells, malignant or dysplastic epithelial cells, fibroblasts, muscle cells, endothelial cells, and other cell types. To align with clinical diagnostic priorities and improve model training efficiency, we maintain the critical distinction between normal and malignant epithelial cells while consolidating fibroblasts, muscle cells, and endothelial cells into a single category of spindle-shaped cells. This consolidation is justified because these stromal components often share similar morphological characteristics and their precise subclassification is less critical for cancer diagnosis compared to epithelial cell changes. The strength of the dataset lies in its diverse representation of nuclear morphologies and types within colorectal cancer tissue, as it includes samples from multiple patients demonstrating various stages of disease progression.
The PanNuke dataset represents one of the most extensive collections of histological nuclei annotations available, containing approximately 200,000 manually annotated nuclei from over 20,000 whole slide images spanning 19 different tissue types. The dataset consists of 7,904 image patches (256256 pixels, 40x magnification, 0.25m/px), with each nucleus annotated with instance segmentation masks and classification into five clinically relevant categories: neoplastic cells, non-neoplastic epithelial cells, inflammatory cells, connective/soft tissue cells, and dead cells. A distinguishing feature of PanNuke is its rigorous quality control by professional pathologists and its organization into three cross-validation folds, each of which contains a balanced distribution of tissue and cell types. This extensive coverage and careful annotation make it an exemplary benchmark for simultaneous nuclear segmentation and classification tasks.
The CPM17 (Computational Precision Medicine 2017) dataset is sourced from the 2017 Digital Pathology Challenge of the Medical Image Computing and Computer-Assisted Intervention Society (MICCAI). It includes tissue images from patients with various tumor types, specifically non-small cell lung cancer (NSCLC), head and neck squamous cell carcinoma (HNSCC), glioblastoma multiforme (GBM), and low-grade glioma (LGG). Despite various treatment strategies, patients often succumb to these complex and fatal cancers. The images were acquired from H&E-stained pathology slides using high-resolution scanners. There are 32 PNG images and the corresponding cell nucleus instance segmentation annotations for the training set, and another 32 PNG images and annotations for the test set, with resolutions of 500500 or 600600. The dataset contains 7570 nuclei in total, with each image containing dozens to hundreds of nuclei.
The KUMAR dataset is a multicenter collection of pathology images, including various tissue types from different organs such as breast, liver, kidney, prostate, bladder, colon, and stomach, from different patients. These images are cropped at a resolution of 10001000 from H&E-stained, 40x magnified Whole Slide Images (WSIs) enriched with nuclei regions of The Cancer Genomic Atlas (TCGA), formatted as TIF, and include 21,623 nuclei. Detailed nuclear boundary annotations are provided for each image. The dataset consists of 16 training cases and 14 test cases.
These four datasets, each with their unique characteristics and challenges, provide a diverse platform for training and validating the HistoNeXt model. Visual examples of the annotated images from these datasets are presented in Fig. 1.
Fig. 1.

Examples from CONSEP, PanNuke, CPM17 and KUMAR datasets
Data preprocessing and enhancement
The data preprocessing pipeline is specifically designed for H&E-stained histological images and optimized for HistoNeXt’s segmentation and classification tasks. Input images are normalized to the range [0,1] and center-cropped to 270270 pixels, providing broader contextual information compared to the network’s output size of 8080 pixels. The corresponding instance maps and type maps are extracted from annotations and processed with identical cropping parameters to maintain spatial alignment.
The data augmentation framework implements a nucleus-aware strategy with two distinct paths: shape augmentations that simultaneously transform both images and instance maps, and intensity augmentations that exclusively modify input images. This dual-path approach ensures the preservation of nuclear instance information while enhancing feature diversity.
Shape augmentations focus on preserving nuclear morphology while introducing spatial diversity. The framework employs three levels of geometric transformations: (1) basic affine transformations including scaling (0.9–1.1), rotation (±90°), and mild shearing (±5°) with nearest-neighbor interpolation to maintain nuclear boundaries; (2) Spatial enhancement through uniform random cropping to 270270 pixels combined with occasional elastic deformations (p = 0.3, : 0–20, : 5.0) to simulate tissue deformation; (3) Random horizontal and vertical flips with 0.5 probability each to enhance orientation invariance.
Intensity augmentations are hierarchically structured to enhance nuclear features while maintaining H&E staining characteristics. The primary components include: (1) Channel-specific adjustments, where both H-channel (nuclear staining) and E-channel (cytoplasmic staining) undergo independent intensity modifications (±8 intensity units, multiplication factor 0.95–1.05) with high probability (p=0.8); (2) Optical simulation through selective (p=0.3) application of Gaussian blur (: 0–0.5) or additive Gaussian noise (scale: 0–0.025255); (3) Local contrast enhancement (p=0.4) using a combination of CLAHE all channels (Contrast Limited Adaptive Histogram Equalization, clip limit: 1–2), gamma contrast adjustment (0.9–1.1) and edge sharpening (: 0–0.2).
During validation, only center cropping is applied to maintain consistent input dimensions. All augmentation operations are implemented using the imgaug library with deterministic execution to ensure consistency between images and their annotations. The effectiveness of this nucleus-aware augmentation strategy is thoroughly evaluated through ablation studies, with detailed results presented in Results section.
To address the class imbalance issue in nuclear type classification, we implemented a type-aware sampling strategy through a custom TypeBalanceSampler class. For each image patch, the sampler calculates the number of pixels belonging to each nuclear type and assigns the patch to the category of the most numerous type. Based on this categorization, different sampling rates are applied to patches containing different nuclear types.
For the CONSEP dataset, patches containing higher proportions of miscellaneous cells or normal epithelial cells are sampled 4.0 and 3.0 times more frequently, while maintaining original sampling rates for patches with other nuclear types. For the PanNuke dataset, patches with high proportions of dead cells are sampled 10 times more frequently. For CPM17 and KUMAR datasets, which contain only segmentation annotations without type information, no type-based sampling is applied. During training, patches are randomly sampled according to these rates, with replacement when oversampling is needed. This sampling strategy helps balance the training distribution of different nuclear types while preserving their natural spatial relationships within each patch, as demonstrated in Results section.
HistoNeXt design
The HistoNeXt model employs an encoder-decoder architecture combined with dual-mechanism feature pyramid fusion technology for cell nuclear segmentation and classification. To accommodate varying computational resources while maintaining performance, HistoNeXt offers four variants (Tiny, Base, Large, XLarge), with the Base variant serving as our reference implementation. The model accepts 270270 pixel inputs and produces 8080 pixel outputs, as illustrated in Fig. 2.
Fig. 2.

The framework of HistoNeXt using the Base variant as an example
Encoder
The encoder utilizes ConvNeXt architecture, incorporating Transformer design principles into convolutional networks for optimized feature extraction. The feature dimensions vary across variants: Tiny [96, 192, 384, 768], Base [128, 256, 512, 1024], Large [192, 384, 768, 1536], and XLarge [256, 512, 1024, 2048].
Taking the Base variant as reference, the encoder processes images through four progressive stages with 3, 3, 27, and 3 ConvNeXt blocks in each stage. Each block implements 77 convolutions for enhanced receptive fields, layer normalization for training stability, and inverted bottleneck design for efficient feature transformation. The encoder is initialized with parameters from the pre-trained ConvNeXt model (TIMM platform), initially trained on ImageNet-22k (22,000 classes, 384384 resolution) and fine-tuned on ImageNet-1k. This hierarchical architecture outputs multiscale features through four levels: low-level feature0 (128 channels), mid-level feature1 (256 channels), high-level feature2 (512 channels), and context feature3 (1024 channels).
Decoder
The decoder, inspired by HoVer-Net, integrates three specialized branches for simultaneous nuclear segmentation and classification through dual-mechanism feature pyramid fusion. Let denote encoder features at level with corresponding channel dimensions . The decoder employs two distinct fusion mechanisms: dense feature reuse for segmentation tasks (NP and HV branches) and channel attention for classification tasks (TP branch).
For the NP and HV branches, the core component is the Dense Connection Block (Fig. 3). Given input features , each layer i in an n-layer block performs:
| 1 |
where represents sequential operations:
| 2 |
with being a 33 convolution generating k new feature channels (growth rate).
Fig. 3.
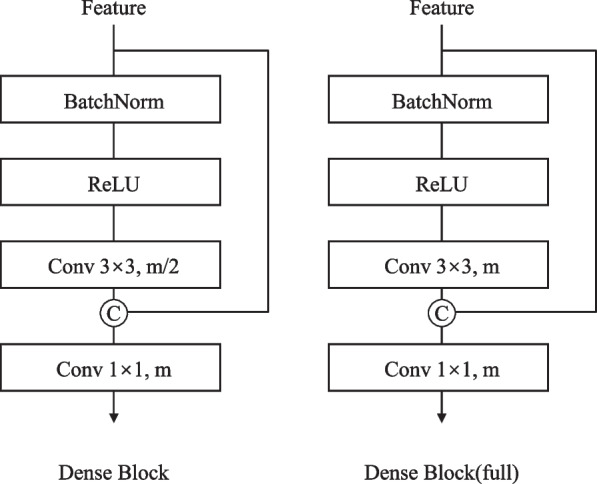
Design of the dense connection block
The forward propagation in NP / HV branches follows a bottom-up path with progressive feature fusion:
| 3 |
The process begins with Feature3, which is upsampled twofold through a 33 transposed convolution and center-cropped to match Feature2’s spatial dimensions. These features are concatenated channel-wise before passing through a Dense Connection Block. This pattern repeats across four stages with progressively deeper Dense Blocks: a 2-layer block with growth rate 512 in stage one, followed by 4-layer (growth rate 256), 6-layer (growth rate 128) and 4-layer (growth rate 64) blocks in subsequent stages. The final stage concludes with a 11 convolution to generate the output predictions.
The TP branch implements a distinct forward propagation path using the ConvCAM block (Fig. 4). The channel attention mechanism within each ConvCAM block computes attention weights as:
| 4 |
where and are 11 convolutions with reduction ratio 16.
Fig. 4.
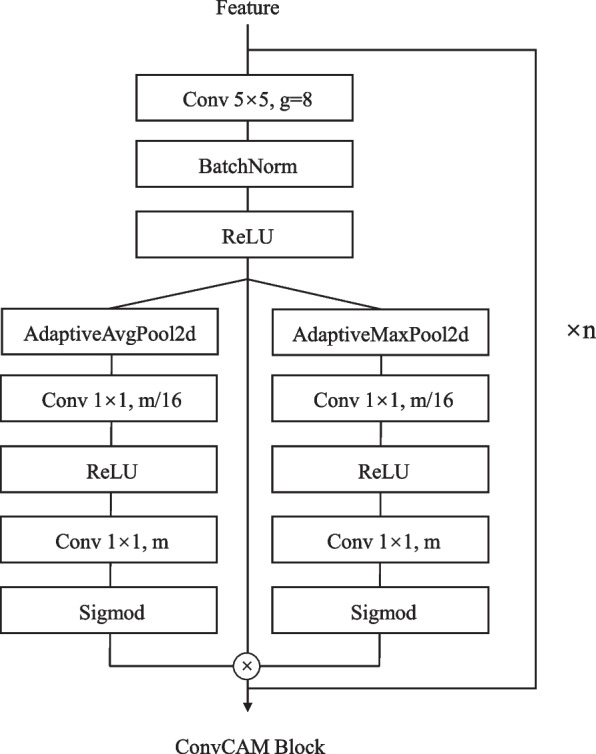
Design of the ConvCAM block
Unlike the NP / HV branches, the forward path of the TP branch begins by upsampling Feature3 through a 55 transposed convolution. The upsampled features first pass through a ConvCAM block before concatenation with feature2. This pattern continues through four stages, with the ConvCAM blocks increasing in depth from one to four layers. Each ConvCAM block incorporates 55 grouped convolutions (groups=12), batch normalization, ReLU activation, and the channel attention mechanism. The final outputs maintain 8080 spatial dimensions, with the NP / HV branches producing two-channel feature maps and the TP branch generating channels corresponding to nuclear types.
The decoder architecture maintains the identical structure across all variants, with channel dimensions scaled proportionally to the encoder. Compared to the Base variant described above, the Tiny variant reduces channel dimensions by half (starting from 768 channels at Feature3), while the Large and XLarge variants increase dimensions by 1.5 and 2 times, respectively (starting from 1536 and 2048 channels). Growth rates in Dense Connection Blocks and channel dimensions in ConvCAM blocks are scaled accordingly, ensuring consistent architectural design while accommodating different computational constraints. For example, in the Tiny variant, the first stage Dense Block operates with a growth rate of 256 rather than 512, while in the Large variant it increases to 768. This scaling strategy allows HistoNeXt to maintain feature representation capacity proportional to model size while preserving the dual-mechanism feature pyramid fusion approach.
Computational efficiency
The multi-variant design enables systematic performance-efficiency trade-offs. HistoNeXt-Tiny (33.7 GFLOPS, 82.6M parameters) targets resource-constrained environments, while HistoNeXt-Base (69.5 GFLOPS, 184.8M parameters) provides balanced performance. HistoNeXt-Large (155.7 GFLOPS, 414.0M parameters) and HistoNeXt-XLarge (276.0 GFLOPS, 734.4M parameters) offer enhanced capabilities for complex cases and specialized applications. This scalable architecture facilitates flexible deployment across varying computational environments while maintaining core functionality.
Training
Training strategy
The HistoNeXt training process employs a two-phase strategy to optimize the performance of the model while addressing the challenges of nuclear segmentation and classification tasks. In phase 1, we freeze the encoder parameters to stabilize feature extraction and focus on optimizing the decoder networks. In phase two, we unfreeze all the parameters for end-to-end fine-tuning, allowing the model to adapt its feature extraction capabilities to the specific characteristics of the histological images.
The model training utilizes the Adam optimizer with =0.9 and =0.999. The initial learning rate is set to , determined through extensive parameter studies. We implement a step decay learning rate schedule, reducing the rate by a factor of 0.1 every 25 epochs in phase one and also every 25 epochs in phase two. This schedule helps maintain stable training while allowing the model to converge to better local optima.
The batch size is set to 64 during phase one to provide stable gradient statistics and optimize GPU memory utilization. In phase two, we reduce the batch size to 16 to accommodate the increased memory requirements of full-model gradient computation while maintaining effective batch statistics. Training continues until the model meets the early stopping criteria, which typically occurs between 80–120 epochs based on validation performance. Early stopping is implemented with a patience of 20 epochs.
Model checkpoints are saved at 5-epoch intervals, with the best model selected based on validation metrics including the DICE, AJI, and type-specific F1 scores. This dual-phase training strategy with carefully tuned hyperparameters enables effective knowledge transfer from the pre-trained encoder while optimizing for the specific requirements of nuclear analysis in histological images.
Loss functions
To effectively train the three decoder branches, we employ specialized loss functions tailored to each branch’s objectives. The total loss combines the losses from individual branches:
| 5 |
For the NP branch, we implement an asymmetric loss to handle the foreground-background pixel imbalance:
| 6 |
where and represent predictions for positive and negative samples, with and as focusing parameters. Predictions are reduced to [0.05, 0.95] for numerical stability.
The HV branch utilizes a combination of mean squared error (MSE) and mean squared gradient error (MSGE):
| 7 |
where MSE is computed as:
| 8 |
and MSGE incorporates Sobel gradient computation:
| 9 |
where represents the gradients computed using 55 Sobel kernels, and the focus masks restrict the gradient loss to nuclear regions.
For the TP branch, we employ focal loss to address multi-class imbalance:
| 10 |
where is the predicted probability for the target class, is the balancing factor, and is the focusing parameter. The predictions are normalized across classes and clipped to ensure numerical stability.
This combination of loss functions was determined through extensive ablation studies. The asymmetric loss effectively handles the severe foreground-background imbalance in nuclear segmentation, while the focal loss improves the model’s performance on rare nuclear types. The MSE-MSGE combination in the HV branch ensures accurate coordinate prediction while maintaining sensitivity to nuclear boundaries.
Class imbalance handling
The nuclear type distribution in histological images typically exhibits a marked class imbalance, which poses challenges for training deep learning models. To address this issue, we implement a type-aware sampling strategy while maintaining spatial relationships within tissue contexts.
Our sampling strategy employs a dynamic multiplier mechanism for different types of nuclear. For an image patch P, let represent the set of nuclear types present in the patch, where n is the number of nuclei. The dominant type is determined by:
| 11 |
The sampling weight for each nuclear type is adjusted according to its frequency in the training set, with underrepresented classes receiving higher weights. During training, each patch is sampled with probability proportional to , where ranges from 1.0 to 20.0 depending on the frequency of the class. This approach facilitates several key advantages: It allows for dynamic adjustment of sampling rates based on class distributions, enhances the representation of minority classes through controlled oversampling, and preserves the natural spatial relationships between nuclei.
This sampling approach, combined with the asymmetric and focal loss functions described in Loss functions section, effectively improves the model’s performance on minority classes. Experimental results demonstrate substantial improvements in F1 scores for underrepresented classes while maintaining strong performance in majority classes. The method shows consistent effectiveness across different histological image datasets, demonstrating its generalizability to various nuclear segmentation and classification tasks.
Training monitoring and overfitting prevention
To ensure stable training and prevent overfitting, we implemented systematic monitoring and regularization strategies. The performance of the model was evaluated every five epochs using two primary metrics: AJI for segmentation quality and F1 scores for classification precision. These metrics were tracked in a separate validation set comprising 20% of the total data.
Model checkpoints were saved based on a weighted combination of AJI and class-wise F1 scores:
| 12 |
where represents individual F1 scores for each nuclear type category.
Early stopping was implemented with a patience of 20 epochs, terminating training if no improvement in the validation score was observed. This mechanism effectively prevented overfitting while ensuring sufficient model convergence. The effectiveness of our training strategy is demonstrated by the stable improvement in both segmentation and classification metrics throughout the training process.
Evaluation metrics
To comprehensively evaluate the performance of the HistoNeXt model, we use multiple complementary metrics for both segmentation and classification tasks following HoVer-Net [2].
| 13 |
where X and Y represent the predicted and ground-truth segmentation masks. The DICE ranges from 0 to 1, with higher values indicating better segmentation overlap.
| 14 |
where represents the i-th ground truth instance, is the predicted instance that has the maximal intersection over the union with , and U is the set of unmatched predicted instances. AJI provides a more stringent evaluation of instance segmentation quality, specifically for touching nuclei.
For detection and segmentation quality assessment:
| 15 |
where TP denotes matched pairs of segments (true positives), FP denotes unmatched predicted segments (false positives), and FN denotes unmatched ground truth segments (false negatives).
| 16 |
| 17 |
To provide a thorough evaluation of both instance segmentation and classification performance in the PanNuke dataset following Gamper et al. [4], we compute two variants of PQ:
The binary PQ(bPQ) evaluates instance segmentation performance by treating all nuclei as one class, regardless of their type:
| 18 |
where N is the total number of images and is calculated treating all nuclei in the image i as a single class.
The multi-class PQ(mPQ) extends the evaluation to consider classification performance by computing PQ separately for each nuclei class and averaging:
| 19 |
where C is the set of nuclei classes and is calculated only matching instances of class c in image i with an IoU threshold of 0.5. This metric is specifically designed for the evaluation of the PanNuke dataset as it is insensitive to class imbalance by equally weighting each nuclear type.
For classification performance evaluation:
| 20 |
where , , and denote true positives, false positives, and false negatives for detection.
For each nuclear class c, we further define the class-specific F1 score as:
| 21 |
where and represent correctly classified instances of class c and correctly classified instances of types other than c respectively, and denote false positives and false negatives of class c, and and are detection-level false positives and negatives. This formulation incorporates both classification accuracy and detection performance into the class-specific evaluation.
Results
Experimental setup
The experimental evaluation of HistoNeXt was performed on a heterogeneous computing platform equipped with an AMD EPYC 7663 56-core processor with 80GB system memory, primarily utilizing an NVIDIA RTX4090 GPU with 24GB VRAM. For experiments requiring larger memory capacity, such as batch size analysis with 128 samples or evaluations on the PanNuke dataset, an NVIDIA A6000 with 48GB of VRAM was used. The implementation was carried out using the PyTorch 1.10 framework with CUDA 11.3 acceleration. The system ran on Ubuntu 20.04.6 LTS and the software stack included CUDA 11.7, Python 3.8.19, PyTorch 1.10, NumPy 1.24.4, JupyterLab 4.2.3, and Scikit-learn 1.3.2.
To ensure rigorous validation while maintaining experimental efficiency, we performed ablation studies using the HistoNeXt-Tiny variant on the CONSEP dataset. These studies encompassed data augmentation strategies, loss function configurations, class imbalance handling techniques, and hyperparameter optimization. We trained different versions of HistoNeXt on multiple datasets, including CONSEP, PanNuke, CPM17, and KUMAR, to evaluate both segmentation and classification performance.
The performance of the model was evaluated using the metrics defined in Evaluation metrics section, including segmentation metrics (DICE, AJI, DQ, SQ, PQ), dataset-specific metrics for PanNuke (bPQ, mPQ) and classification metrics (overall and class-specific F1 scores).
Ablation studies
To rigorously evaluate the effectiveness of different components in HistoNeXt, we conducted extensive ablation studies on the CONSEP dataset using the HistoNeXt-Tiny variant. These experiments systematically assessed the impact of data augmentation strategies, loss function configurations, class imbalance handling techniques, and variations in hyperparameter.
Data augmentation strategy analysis
To systematically evaluate data augmentation strategies, we conducted experiments with five progressive augmentation configurations using HistoNeXt-Tiny on the CONSEP dataset. Each strategy was designed to address specific challenges in the segmentation and classification of nuclear instances. The quantitative results of this analysis are presented in Table 1.
Table 1.
Performance comparison of different augmentation strategies
| Strategy | DICE | AJI | DQ | SQ | PQ | F1 Score |
|---|---|---|---|---|---|---|
| Baseline | 0.792 | 0.471 | 0.545 | 0.730 | 0.400 | 0.706 |
| Basic | 0.832 | 0.566 | 0.653 | 0.765 | 0.501 | 0.751 |
| Moderate | 0.832 | 0.567 | 0.656 | 0.765 | 0.503 | 0.750 |
| Strong | 0.839 | 0.571 | 0.660 | 0.762 | 0.504 | 0.737 |
| Full | 0.840 | 0.576 | 0.665 | 0.767 | 0.511 | 0.761 |
The baseline strategy uses only center cropping. The basic strategy introduces geometric transformations within restricted ranges (scale: 0.9–1.1, rotation: ±90°, shear: ±5°) to preserve nuclear morphology. The moderate strategy further enhances augmentation by adding H&E staining-specific modifications, including channel-wise intensity adjustments (±8) and multiplication factors (0.95–1.05). The strong strategy incorporates optical simulation with Gaussian blur () and noise (scale ).
Our full strategy combines all previous augmentations with nucleus-aware enhancements: 1) Morphology-preserving geometric transformations. 2) Adaptive H&E channel adjustments with a probability of 0.8. 3) Local contrast enhancement using CLAHE (clip limit: 1–2). 4) Elastic deformation (, ) with a probability of 0.3.
This complete strategy achieves optimal performance by carefully balancing the preservation of nuclear morphology and the diversity of augmentation. Controlled geometric transformations maintain structural integrity while the adaptive H&E augmentations enhance robustness to staining variations. This integrated approach improves the DICE score by 4.8% (from 0.792 to 0.840) and AJI by 10.5% (from 0.471 to 0.576) compared to the baseline, while simultaneously increasing the overall classification score F1 by 5. 5% (from 0.706 to 0.761). Consistent improvements across all metrics demonstrate the effectiveness of our proposed augmentation framework.
Loss function configuration
Building upon previously defined loss functions, we conducted systematic ablation studies to identify the optimal loss combination for each branch.
The ablation results demonstrate the effectiveness of our proposed loss configuration. For the NP branch, using asymmetric loss alone, without DICE loss, enhances both segmentation metrics: the DICE score rises to 0.844, and the AJI score increases to 0.594. In the TP branch, focal loss alone yields improved classification performance, achieving an overall F1 score of 0.770. This optimal configuration, which combines MSE+MSGE for the HV branch, asymmetric loss for the NP branch, and focal loss for the TP branch, provides the best performance across all evaluation metrics. Detailed ablation results are presented in Table 2.
Table 2.
Ablation study of loss function configurations on CONSEP dataset
| Configuration | DICE | AJI | Overall F1 |
|---|---|---|---|
| NP: BCE+DICE, TP: BCE+DICE | 0.840 | 0.576 | 0.761 |
| NP: BCE+DICE, TP: Focal+DICE | 0.842 | 0.582 | 0.756 |
| NP: Asym+DICE, TP: BCE+DICE | 0.842 | 0.584 | 0.758 |
| NP: Asym+DICE, TP: Focal+DICE | 0.842 | 0.581 | 0.752 |
| NP: Asym, TP: Focal | 0.844 | 0.594 | 0.770 |
All configurations maintain MSE+MSGE for the HV branch. Asym Asymmetric loss, Focal Focal loss
Class imbalance handling
To address class imbalance in nuclear type classification, we evaluated three sampling strategies with increasing complexity. Table 3 presents the quantitative comparison of these approaches.
Table 3.
Performance comparison of different sampling strategies
| Strategy | Overall F1 | Malig | Normal | Inflam | Spindle | Misc |
|---|---|---|---|---|---|---|
| No resampling | 0.770 | 0.621 | 0.504 | 0.622 | 0.647 | 0.173 |
| Uniform | 0.765 | 0.596 | 0.541 | 0.623 | 0.566 | 0.150 |
| Type-aware | 0.767 | 0.620 | 0.497 | 0.645 | 0.576 | 0.335 |
Malig Malignant epithelial, Normal Normal epithelial, Inflam Inflammatory, Misc Miscellaneous
The type-aware sampling strategy employs differential sampling rates for underrepresented nuclear types. Resulting in a notable improvement in detecting minority classes, Specifically, the F1 score for these minority classes increased from 0.173 to 0.335, while maintaining competitive performance in other nuclear types. In contrast to uniform sampling, which resulted in degraded performance across most classes, our type-aware strategy effectively balances class representation without compromising the model’s ability to identify majority classes.
The strategy achieved optimal results when combined with the asymmetric and focal losses described in Loss functions section, highlighting the effectiveness of our integrated approach to address class imbalance in nuclear-type classification.
Hyperparameter variations
We conducted systematic experiments to analyze the impact of critical hyperparameters on model performance. The baseline configuration used a learning rate of , a batch size of 16, a focal loss gamma of 2.0, an asymmetric positive focusing factor of 1.0, and a combination of MSE-MSGE for coordinate prediction. Through extensive experimentation, we explored variations in the learning rate, batch size, and loss parameters, ultimately identifying an optimized configuration. Table 4 presents the detailed results of these experiments.
Table 4.
Systematic analysis of hyperparameter variations
| Configuration | Segmentation Metrics | Classification F1 Scores | |||||||
|---|---|---|---|---|---|---|---|---|---|
| DICE | AJI | PQ | Overall | Malig | Normal | Inflam | Spindle | Misc | |
| Baseline | 0.841 | 0.589 | 0.527 | 0.767 | 0.619 | 0.497 | 0.645 | 0.576 | 0.258 |
| LR=5e-5 | 0.842 | 0.587 | 0.531 | 0.767 | 0.638 | 0.502 | 0.650 | 0.582 | 0.315 |
| Batch=64 | 0.841 | 0.592 | 0.531 | 0.771 | 0.651 | 0.377 | 0.633 | 0.578 | 0.260 |
| Batch=128 | 0.841 | 0.587 | 0.526 | 0.758 | 0.626 | 0.389 | 0.628 | 0.559 | 0.076 |
| Asym =0.5 | 0.844 | 0.594 | 0.538 | 0.776 | 0.622 | 0.385 | 0.655 | 0.585 | 0.372 |
| Focal =3.0 | 0.844 | 0.591 | 0.532 | 0.770 | 0.626 | 0.466 | 0.651 | 0.581 | 0.356 |
| Final Config | 0.842 | 0.593 | 0.532 | 0.770 | 0.656 | 0.516 | 0.647 | 0.580 | 0.265 |
Malig Malignant epithelial, Normal Normal epithelial, Inflam Inflammatory, Misc Miscellaneous
The experimental results reveal several key findings. First, reducing the learning rate to 5e-5 improved the detection of a rare class, but had only a marginal impact on overall segmentation metrics. Increasing the batch size to 64 and 128 accelerated faster training convergence but led to noticeable degradation in classification performance, particularly for normal epithelial and miscellaneous cells. Although an asymmetric positive focusing factor of 0.5 yielded the highest segmentation metrics (DICE = 0.844, AJI = 0.594) and improved the detection of inflammatory and miscellaneous cells, it notably hindered the model’s ability to distinguish epithelial cells (malignant F1=0.622, normal F1=0.385). Adjusting the focal loss gamma to 3.0 provided better balance, but still showed some instability.
Our final configuration (learning rate=, batch size=64, focal gamma=2.0, asymmetric positive focusing factor=1.0) was chosen as it achieved the best balance between segmentation and classification performance. Notably, it significantly improved epithelial cell detection (malignant F1 = 0.656, normal F1 = 0.516) while maintaining strong performance across other metrics. This configuration highlights the importance of balancing the quality of overall segmentation with the accuracy of nuclear-type classification, especially for clinically relevant cell types such as malignant and normal epithelial cells.
Multi-dataset performance evaluation
CONSEP dataset evaluation
Table 5 presents the systematic comparison between HistoNeXt-Tiny and HoVer-Net on the CONSEP dataset, demonstrating both segmentation and classification performance.
Table 5.
Performance comparison on CONSEP dataset
| Model | Segmentation Metrics | Classification F1 Scores | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DICE | AJI | DQ | SQ | PQ | Overall | Malig | Normal | Inflam | Spindle | Misc | |
| HoVer-Neta | 0.839 | 0.554 | 0.640 | 0.762 | 0.489 | 0.729 | 0.509 | 0.091 | 0.495 | 0.494 | 0.266 |
| HistoNeXt-Tiny | 0.843 | 0.592 | 0.687 | 0.772 | 0.532 | 0.773 | 0.653 | 0.516 | 0.659 | 0.587 | 0.251 |
aModel re-trained by ourselves with modified nuclear type categories
Malig Malignant epithelial, Normal Normal epithelial, Inflam Inflammatory, Misc Miscellaneous
The results show that HistoNeXt-Tiny achieves superior performance across all metrics despite its lighter architecture. Specifically, it improves F1 scores for challenging categories such as malignant epithelial cells (0.653 vs. 0.509) and normal epithelial cells (0.516 vs. 0.091). Example results are shown in Fig. 5.
Fig. 5.

Example comparison of segmentation and classification results on the CONSEP dataset: HistoNeXt (bottom), HoVer-Net (middle), and Ground Truth (top)
PanNuke dataset evaluation
Table 6 presents a performance comparison on the PanNuke dataset. HistoNeXt-Tiny (33.7 GFLOPS) outperforms CellViT256 in segmentation metrics (mPQ: 0.4967 vs. 0.4846) while maintaining comparable classification performance (F1: 0.82). HistoNeXt-Large achieves competitive results comparable to CellViT-SAM-H, with a bPQ of 0.6794 and an overall F1 score of 0.82. The results across different HistoNeXt variants demonstrate that the classification performance remains stable while the segmentation metrics improved slightly with increasing model capacity.
Table 6.
Performance comparison on PanNuke dataset
| Model | Segmentation Metrics | Classification F1 Scores | ||||||
|---|---|---|---|---|---|---|---|---|
| mPQ | bPQ | Overall | Neop | Non-neop | Inflam | Connect | Dead | |
| HoVer-Net [10] | 0.4629 | 0.6596 | 0.80 | 0.62 | 0.56 | 0.54 | 0.49 | 0.31 |
| CellViT256 [6] | 0.4846 | 0.6696 | 0.82 | 0.69 | 0.70 | 0.58 | 0.52 | 0.37 |
| CellViT-SAM-H [6] | 0.4980 | 0.6793 | 0.83 | 0.71 | 0.73 | 0.58 | 0.53 | 0.36 |
| HistoNeXt-Tiny | 0.4967 | 0.6779 | 0.82 | 0.70 | 0.71 | 0.58 | 0.52 | 0.36 |
| HistoNeXt-Base | 0.4914 | 0.6765 | 0.82 | 0.70 | 0.71 | 0.58 | 0.52 | 0.35 |
| HistoNeXt-Large | 0.4940 | 0.6794 | 0.82 | 0.71 | 0.71 | 0.59 | 0.53 | 0.36 |
Results for all models are evaluated on the official three-fold splits of the PanNuke dataset. Neop Neoplastic, Non-neop Non-neoplastic epithelial, Inflam Inflammatory, Connect Connective tissue
Nuclear segmentation on CPM17 and KUMAR datasets
As shown in Table 7, HistoNeXt-Tiny demonstrates competitive performance on both datasets. To further assess the generalization capability of HistoNeXt, we compared the segmentation performance of HistoNeXt-Tiny with that of HoVer-Net on the CPM17 and KUMAR datasets. The sample results are shown in Figs. 6 and 7.
Table 7.
Segmentation performance on CPM17 and KUMAR datasets
| Model | CPM17 | KUMAR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DICE | AJI | DQ | SQ | PQ | DICE | AJI | DQ | SQ | PQ | |
| HoVer-Net [2] | 0.869 | 0.705 | 0.854 | 0.814 | 0.697 | 0.826 | 0.618 | 0.770 | 0.773 | 0.597 |
| HistoNeXt-Tiny | 0.874 | 0.722 | 0.855 | 0.804 | 0.689 | 0.826 | 0.625 | 0.737 | 0.763 | 0.565 |
Fig. 6.

Example comparison of segmentation results on the CPM17 dataset: HistoNeXt (bottom), HoVer-Net (middle), and Ground Truth (top)
Fig. 7.

Example comparison of segmentation results on the KUMAR dataset: HistoNeXt (bottom), HoVer-Net (middle), and Ground Truth (top)
Discussion
Cell nuclear segmentation and classification are fundamental tasks in digital pathology, serving as the primary steps in automated workflows that directly impact pathological diagnosis and subsequent treatment decisions. With the growing volume of pathological image data, rapid and accurate automated analysis techniques are crucial for enhacing pathologist efficiency, reducing human error, and supporting large-scale clinical research.
In recent years, deep learning technology has made remarkable progress in nuclear segmentation and classification tasks, with CNNs and Transformers as the two main architectures. CNNs, which are based on hierarchical convolutional computations, are especially suitable for handling data with fixed structures and exhibit strong feature extraction capabilities in medical image processing. This has led to the development of many models and techniques designed for image analysis tasks. Notable segmentation models include U-Net, SegNet [11], DeepLab [12], and MASK R-CNN [13], with techniques such as Encoder-Decoder Architecture, Skip Connections, and Feature Pyramid Networks (FPN) [14] having enhanced segmentation performance. For image classification tasks, models such as ResNet [15], DenseNet [16], and EfficientNet [17], have been widely adopted, with performance enhancing techniques such as Transfer Learning, Batch Normalization, and Regularization Techniques. However, the innate limitations of small and uneven receptive fields in CNNs restrict their performance in image segmentation and classification tasks [18].
Comparative analysis on public datasets provides insights into model specialization and adaptability. On the CONSEP dataset, our error analysis reveals specific challenges in nuclear classification tasks. The model occasionally misclassifies normal epithelial cells as malignant, particularly in borderline cases where normal cells exhibit slight nuclear enlargement or irregular morphology. This confusion contributes to the relatively low F1 score (0.516) for normal epithelial cells compared to malignant cells (0.653). Additionally, in regions with densely packed nuclei, while overall detection remains accurate, precise boundary delineation becomes more challenging.
On the PanNuke dataset, HistoNeXt demonstrates remarkable scalability and efficiency. The Tiny variant achieves impressive results with only 33.7 GFLOPS, surpassing CellViT256 in segmentation metrics while maintaining comparable classification performance. This suggests that our dual-mechanism feature pyramid fusion approach effectively captures essential features even in the lightweight configuration. HistoNeXt-Large demonstrates balanced performance across different cell types, matching CellViT-SAM-H in the detection of neoplastic (F1 = 0.71) and connective tissue (F1 = 0.53), while achieving slightly higher scores for inflammatory cells (F1 = 0.59), despite its markedly higher computational demands.
HistoNeXt excels in computational efficiency while maintaining competitive performance. The Tiny variant, which requires approximately one-sixth of the computational resources of HoVer-Net, achieves comparable or superior results across multiple metrics. This efficiency makes HistoNeXt highly valuable for practical clinical applications, especially in resource-constrained environments or for analyzing rare disease datasets where large-scale, high-quality data collection is often challenging.
Internal validation on whole slide images with 20002000 resolution tiles, conducted on proprietary datasets that cannot be publicly shared, indicates HistoNeXt’s potential scalability to higher-resolution applications. This capability is particularly relevant for clinical settings, where whole slide image analysis is becoming a standard practice. The model maintains performance comparable to standard resolution processing when using a sliding-window approach, demonstrating the effectiveness of our feature extraction mechanism and local-global feature fusion strategy.
The success of HistoNeXt’s dual-mechanism feature pyramid fusion approach, notably in the Tiny variant, suggests that thoughtful architectural design and efficient feature utilization can effectively compensate for reduced model capacity. The dense connection blocks in the NP and HV branches efficiently handle segmentation tasks, while the channel attention mechanism in the TP branch effectively captures classification-relevant features. This balanced design enables robust performance across varying computational constraints, making the model suitable for various clinical and research applications.
These findings indicate that, while HistoNeXt advances nuclear segmentation and classification capabilities, there are opportunities for further improvement. Future work could focus on enhancing the model’s ability to distinguish subtle morphological differences between normal and malignant cells, developing more sophisticated approaches for addressing class imbalance in nuclear-type classification, and optimizing performance on varying image resolutions. The integration of modern CNN architectures with carefully designed feature fusion mechanisms provides a foundation for future innovations in automated digital pathology analysis.
Conclusion
HistoNeXt advances the field of digital pathology through its novel dual-mechanism feature pyramid fusion approach and efficient architecture design. The model achieves superior performance across multiple public datasets, with notable strengths in computational efficiency and nuclear instance segmentation. Key achievements include improved AJI scores on the CONSEP dataset (0.592 vs 0.554), and on PanNuke, HistoNeXt-Tiny achieves remarkable efficiency at only 33.7 GFLOPS, surpassing CellViT256 in segmentation metrics (mPQ: 0.4967 vs 0.4846) and achieving classification performance comparable to state-of-the-art models (F1 score 0.82). These results establish HistoNeXt as a promising tool for automated nuclear analysis in clinical pathology, offering a balance between precision and efficiency that makes it particularly well suited for deployment in resource-constrained environments.
Acknowledgements
Not applicable.
Clinical trial number
Not applicable.
Code availability
The custom code is not currently publicly available due to intellectual property considerations but is available from the corresponding author upon reasonable request with appropriate confidentiality agreements.
Abbreviations
- AJI
Aggregated Jaccard Index
- BCE
Binary Cross-Entropy
- bPQ
binary Panoptic Quality
- CLAHE
Contrast Limited Adaptive Histogram Equalization
- CNN
Convolutional Neural Network
- DICE
Dice Similarity Coefficient
- DQ
Detection Quality
- FPN
Feature Pyramid Network
- GBM
Glioblastoma Multiforme
- GFLOPS
Giga Floating Point Operations Per Second
- H[NONSPACE
&E] Hematoxylin and Eosin
- HNSCC
Head and Neck Squamous Cell Carcinoma
- HV
Horizontal and Vertical Distance
- LGG
Low-Grade Glioma
- mPQ
multi-class Panoptic Quality
- MSE
Mean Squared Error
- MSGE
Mean Squared Gradient Error
- NP
Nuclear Pixel
- NSCLC
Non-Small Cell Lung Cancer
- PQ
Panoptic Quality
- SQ
Segmentation Quality
- TCGA
The Cancer Genomic Atlas
- TP
Type Prediction
- ViT
Vision Transformer
- WSI
Whole Slide Image
Authors’ contributions
JC conducted the experiments and drafted the manuscript. RW wrote the English version of the manuscript and contributed to data interpretation. WD contributed the medical aspects of the research and provided critical revision. HH organized the writing of the manuscript and provided overall supervision. SW organized the experiments and provided overall supervision. All authors have read and approved the submitted version of the manuscript.
Funding
No funding was received for conducting this study.
Data availability
The datasets used and analysed during the current study are publicly available. The CONSEP, CPM17 and KUMAR dataset can be found in the GitHub repository associated with that study:HoVer-Net. https://github.com/vqdang/hover_net (Accessed on [2024-08-13]). PanNuke dataset is available from: https://warwick.ac.uk/fac/cross_fac/tia/data/pannuke (Accessed on [2024-11-4]). Any additional data generated during our analysis are available from the corresponding author upon reasonable request.'.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hua He and Shiyong Wang contributed equally to this work.
Contributor Information
Hua He, Email: hehua1624@smmu.edu.cn.
Shiyong Wang, Email: luohong_wsy@smmu.edu.cn.
References
- 1.Ronneberger O, Fischer P, Brox T. U-Net: Convolutional Networks for Biomedical Image Segmentation. In: Navab, N., Hornegger, J., Wells, W., Frangi, A. (eds) Medical Image Computing and Computer-Assisted Intervention – MICCAI 2015. MICCAI 2015. Lecture Notes in Computer Science. Springer, Cham. 2015;9351. 10.1007/978-3-319-24574-4_28.
- 2.Graham S, Vu QD, Raza SEA, Azam A, Tsang YW, Kwak JT, et al. Hover-Net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med Image Anal. 2019;58:101563. 10.1016/j.media.2019.101563. [DOI] [PubMed] [Google Scholar]
- 3.Srikantamurthy MM, Rallabandi VS, Dudekula DB, Natarajan S, Park J. Classification of benign and malignant subtypes of breast cancer histopathology imaging using hybrid CNN-LSTM based transfer learning. BMC Med Imaging. 2023;23(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gamper J, Alemi Koohbanani N, Benet K, Khuram A, Rajpoot N. Pannuke: an open pan-cancer histology dataset for nuclei instance segmentation and classification. In: Digital Pathology: 15th European Congress, ECDP 2019, Warwick, UK, April 10–13, 2019, Proceedings 15. Springer; 2019. pp. 11–9.
- 5.Gudhe NR, Kosma VM, Behravan H, Mannermaa A. Nuclei instance segmentation from histopathology images using Bayesian dropout based deep learning. BMC Med Imaging. 2023;23(1):162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horst F, Rempe M, Heine L, Seibold C, Keyl J, Baldini G, et al. CellViT: Vision Transformers for precise cell segmentation and classification. Med Image Anal. 2024;94:103143. 10.1016/j.media.2024.103143. [DOI] [PubMed] [Google Scholar]
- 7.Liu Z, Mao H, Wu CY, Feichtenhofer C, Darrell T, Xie S. A convnet for the 2020s. In: Proceedings of the IEEE/CVF conference on computer vision and pattern recognition. IEEE, New Orleans, LA, USA; 2022. pp. 11976–86.
- 8.Vu QD, Graham S, Kurc T, To MNN, Shaban M, Qaiser T, et al. Methods for Segmentation and Classification of Digital Microscopy Tissue Images. Front Bioeng Biotechnol. 2019;7. 10.3389/fbioe.2019.00053. [DOI] [PMC free article] [PubMed]
- 9.Kumar N, Verma R, Sharma S, Bhargava S, Vahadane A, Sethi A. A Dataset and a Technique for Generalized Nuclear Segmentation for Computational Pathology. IEEE Trans Med Imaging. 2017;36(7):1550–60. 10.1109/TMI.2017.2677499. [DOI] [PubMed] [Google Scholar]
- 10.Gamper J, Koohbanani NA, Benes K, Graham S, Jahanifar M, Khurram SA, Azam A, Hewitt K, Rajpoot N. PanNuke Dataset Extension, Insights and Baselines. arXiv:2003.10778 [eess.IV]. 2020.
- 11.Badrinarayanan V, Kendall A, Cipolla R. SegNet: A Deep Convolutional Encoder-Decoder Architecture for Image Segmentation. IEEE Trans Pattern Anal Mach Intell. 2017;39(12):2481–95. 10.1109/TPAMI.2016.2644615. [DOI] [PubMed] [Google Scholar]
- 12.Chen LC, Papandreou G, Schroff F, Adam H. Rethinking atrous convolution for semantic image segmentation. arXiv preprint arXiv:170605587. 2017. 10.48550/arXiv.1706.05587.
- 13.He K, Gkioxari G, Dollar P, Girshick R. Mask R-CNN. IEEE Trans Pattern Anal Mach Intell. 2020;42(2):386–97. 10.1109/TPAMI.2018.2844175. [DOI] [PubMed] [Google Scholar]
- 14.Lin TY, Dollar P, Girshick R, He K, Hariharan B, Belongie S. Feature Pyramid Networks for Object Detection. IEEE Comput Soc. 2017. 10.1109/CVPR.2017.106.
- 15.He K, Zhang X, Ren S, Sun J. Deep Residual Learning for Image Recognition. IEEE; 2016. 10.1109/CVPR.2016.90.
- 16.Huang G, Liu Z, Laurens VDM, Weinberger KQ. Densely Connected Convolutional Networks. IEEE Comput Soc. 2016. 10.1109/CVPR.2017.243. [Google Scholar]
- 17.Tan M, Le Q. Efficientnet: Rethinking model scaling for convolutional neural networks. In: Proceedings of the 36th International Conference on Machine Learning. PMLR, Cambridge, MA, USA; 2019. pp. 6105–14.
- 18.Li J, Chen J, Tang Y, Wang C, Landman BA, Zhou SK. Transforming medical imaging with Transformers? A comparative review of key properties, current progresses, and future perspectives. Med Image Anal. 2023;85:102762. 10.1016/j.media.2023.102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analysed during the current study are publicly available. The CONSEP, CPM17 and KUMAR dataset can be found in the GitHub repository associated with that study:HoVer-Net. https://github.com/vqdang/hover_net (Accessed on [2024-08-13]). PanNuke dataset is available from: https://warwick.ac.uk/fac/cross_fac/tia/data/pannuke (Accessed on [2024-11-4]). Any additional data generated during our analysis are available from the corresponding author upon reasonable request.'.