

Abstract
CRISPR-Cas9 genome editing is a powerful tool for assessing the functional role of candidate genes. In vitro CRISPR/Cas9 screens have been used to rapidly assess the role of thousands of genes in the differentiation and function of immune populations. However, the physiological relevance of a gene is often dependent on signals received in the tissue microenvironment, such as exposure to growth factors, chemokines, cytokines, and cell contact-dependent signals, which may not be recapitulated in an in vitro setting. Additionally, in vitro approaches are not sufficient to induce the differentiation of all cell populations limiting the cell types that can be screened. This has posed a major barrier to understanding the genes regulating the differentiation of germinal center B cells. Here, we describe an approach to perform an in vivo Crispr-Cas9 screen to specifically ablate genes in activated B cells. Using this approach, we have been able to reveal novel transcriptional regulators of germinal center B cell differentiation following viral infection.
Keywords: B cells, CRISPR-Cas9 screen, Single-guide RNAs, Retroviral transduction, Bone marrow chimeras
1. Introduction
CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated protein 9) is a powerful genome editing approach that has been widely used to understand the genes underlying key biological processes. This method was developed based on a defense mechanism that operates in bacteria and archaea to protect against infection by viruses, such as bacteriophages [1]. Injection of phage DNA into CRISPR-containing bacterial cells results in cleavage of the DNA into fragments, which are then incorporated into the CRISPR loci. The CRISPR loci are then transcribed with the resulting pre CRISPR RNA (crRNA) processed by a Cas9/RNaseIII complex to generate mature crRNA. Mature crRNAs are then coupled to transactivating CRISPR RNA (tracrRNA), which are bound by Cas protein effectors. This allows for the Cas protein to be targeted to and cleave the phage DNA upon subsequent infection [2, 3].
CRISPR/Cas systems are classified into class 1 and class 2 systems. Class I systems have effector modules composed of multiple Cas proteins that form a crRNA-binding complex. By contrast, class 2 systems have a single, multidomain crRNA-binding Cas protein [4]. CRISPR/Cas9 is a class 2 system and involves a single-guide RNA (sgRNA)-targeted Cas9 endonuclease derived from Streptococcus pyogenes [5]. The Cas9 protein contains two nuclease domains that cleave the complementary and noncomplementary DNA strands of the target sequence [6]. The cleavage efficiency of the Cas9 protein is dependent on the presence of a valid protospacer adjacent motif (PAM) sequence at the non-target strand downstream of the cleavage site. The sgRNA contains a crRNA sequence that is fused to the scaffold of the tracrRNA sequence.
The application of Crispr-Cas9 mediated gene ablation in vivo was enabled through the development of a Cre-dependent Cas9 knockin mouse [7]. We generated Rosa26-Cas9flox/+ Cγ1Cre mice to specifically express Cas9 protein in B cells that express the immunoglobulin heavy constant gamma 1 locus, thereby enriching for Cas9 expression in germinal center-derived B cells [8–10]. We then retrovirally transduced bone marrow from these mice with sgRNAs targeting candidate genes to reveal a novel role for the transcriptional factors Hhex and Tle3 in memory B cell development following viral infection [9]. Here, we describe how to perform an in vivo Crispr/Cas9 screen to assess the role of candidate genes in activated B cells. Specifically, we detail the approach for designing sgRNAs for in vivo target gene ablation, cloning of the retroviral constructs encoding the sgRNAs, and generation of bone marrow chimeras for in vivo screening of target genes.
2. Materials
All the materials and conditions are kept sterile throughout the experiment and used according to manufacturer’s instructions.
2.1. Retroviral Packaging
Platinum-E (Plat-E) retroviral packaging cell line (Cell Biolabs).
Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum, 1% L-Glutamine, 1% antibiotics: 10,000 IU Penicillin and 10,000 μg/mL Streptomycin, and 1% HEPES 10 mM pH 7.2.
BsgI restriction enzyme.
Nuclease-free water.
PCR thermal cycler.
Gel extraction kit.
NEBuilder HiFi DNA Assembly Kit.
DH5α competent cells.
LB agar-Amp plates.
Ampicillin.
Plasmid DNA miniprep kit.
Lipofectamine 3000.
pTR-MSCV-IRES-Thy1.1 plasmid DNA.
pTR-MSCV-IRES-BFP plasmid DNA.
sgRNA primers specific for gene of interest.
Opti-MEM Reduced-Serum Medium.
Sequencing Primer: 5′-CCCCTTGAACCTCCTCGTT-3′.
Cell culture dishes: 10 cm, 6-well plate, 24-well plate.
Centrifuge tubes: 15 mL and 50 mL.
Round-bottom tubes: 5 mL and 14 mL.
Microcentrifuge tubes: 1.5 mL.
Incubator shaker.
Inverted microscope.
2.2. Bone Marrow Chimeras
Donor mice: Rosa26-Cas9flox/+ Cre+ (see Note 1).
Recipient mice.
5-Fluorouracil.
Ethanol (70%).
Phosphate-buffered saline (pH 7.2–7.4).
Biosafety cabinet.
CO2 incubator.
RPMI (2% FBS, 1% L-Glutamine, 1% P/S, 1% HEPES).
Bone marrow cell culturing media: DMEM containing 15% fetal bovine serum, 1% L-Glutamine, antibiotics (10,000 IU) Penicillin and (10,000 μg/mL) Streptomycin in a 100-fold working concentration (MT30001CI), and 10 mM HEPES (pH 7.2) supplemented with IL-3, IL-6, and stem cell factor (at concentrations of 20, 50, and 100 ng/m, respectively).
Cell strainer (70 μm).
Needle (27-gauge).
Syringe (5 mL).
Scissors and forceps.
Syringe plunger (3 mL).
24-well culture plates.
Syringe filters (0.22 μm pore size).
Polybrene transfection reagent.
HEPES buffer.
Sulfamethoxazole/Trimethoprim.
X-ray irradiator.
Mice irradiation pies.
3. Methods
3.1. Designing sgRNA Sequences
After selecting a gene of interest, search for the mouse transcript ID using the Ensembl genome database (Fig. 1). The transcript ID that is flagged as Ensembl Canonical is the most conserved and highly expressed transcript and will generally be the transcript used to design sgRNAs (see Note 2).
Use the CRISPR Guide RNA Design Tool on Benchling.com to search for the gene of interest. Select the mouse genome and the transcript that corresponds to the ID found on Ensembl. Then input the information about design type (single guide), guide length (20), genome (mus musculus), and PAM (NGG). Now go to the sequence map and highlight the first 300 bp of exon 1. In the Design Crispr tab, press the Create button to generate sgRNA sequences for the target sequence. If the total sequence of exon 1 is less than 300 bp, you should highlight additional exons until you have a total of 300 bp. Then sort the sequences based on on-target score and export the results (see Note 3).
Use the CRISPick program from the Broad Institute to independently determine the highest scoring sgRNA sequences. Select the appropriate reference genome (mouse), mechanism (CRISPRko), and enzyme (SpyoCas9 (NGG)). Then paste in the 300 bp sequence that was used to generate the sgRNA sequences on Benchling, and generate a list of the top ten guide RNA sequences based on raw ranking, cut position, and mutual spacing.
Cross-reference the results of the Benchling and CRISPick analysis to identify sgRNA sequences that are overlapping. Among the overlapping sequences, select two sequences that have the highest on target efficacy score and efficiency score. Avoid choosing two sequences that target overlapping region in their target sequence. Use Nucelotide BLAST to verify that the chosen sgRNA sequences are specific for the gene of interest (see Note 4).
If the selected sgRNA sequence does not have guanine as first base, add G as the first base. Then add 5′ and 3′ sequences that overlap with the pTR-MSCV-IRES-Thy1.1 and pTR-MSCV-IRES-BFP plasmids (plasmid available upon request). The final primer sequence will be 5′-GTGGAAAGGACGAAAcacc-(sgRNA sequence)-gttttagagctaGAAAtag-3′ (see Note 5). The two sgRNA sequences are cloned into plasmids containing different reporters (Thy1.1 and blue fluorescent protein (BFP)) so that the transduced cells can be detected based on expression of reporter corresponding to the specific sgRNA.
Fig. 1.
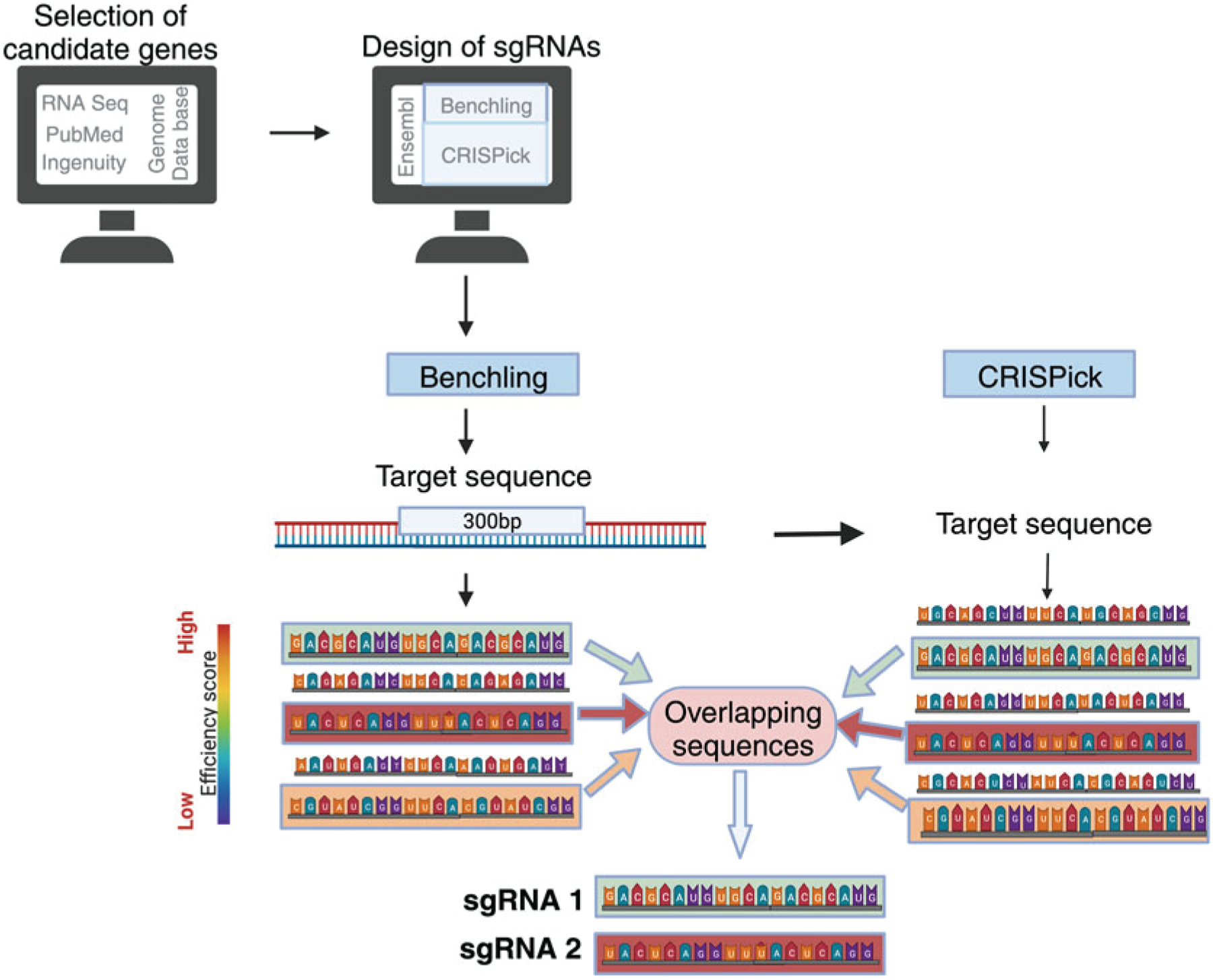
Schematic for method used to design sgRNA sequence
3.2. Clone sgRNAs into pTR-MSCV-IRES Plasmid
Perform a restriction digest to generate a cut pTR-MSCV-IRES-Thy1.1 and pTR-MSCV-IRES-BFP plasmid. Incubate 4 μg uncut plasmid with 4ul BsgI (New England Biolabs) for 2–3 h at 37 °C. Then perform a gel extraction to purify the band around 6 kb (see Note 6). There will also be a 2 kb band corresponding to the stuffer region.
Use Gibson cloning to clone the sgRNA-containing primers into the cut pTR-MSCV-IRES-Thy1.1 and pTR-MSCV-IRES-BFP plasmids using the NEBuilder HiFi DNA Assembly Master Mix (Fig. 2). We generally cloned sgRNA 1 into pTR-MSCV-IRES-Thy1.1 and sgRNA 2 into pTR-MSCV-IRES-BFP. Cloning is performed using NEBuilder HiFi DNA Assembly Cloning Kit. Add 50 ng of the appropriate cut pTR-MSCV-IRES plasmid, 5 ng sgRNA primer, and 5 μL NEB-uilder HiFi DNA Assembly Master Mix to nuclease-free water for a total reaction volume of 10 μL. Then incubate at 50 °C for 15 min using a PCR machine.
Transform the cloned products into DH5α competent cells, and spread the transformed bacterial cell culture on LB agar ampicillin plates for antibiotic selection of transformed cells. Incubate agar plates at 37 °C for 16–18 h.
Next day, pick three to four colonies from each plate, and use to inoculate to 3 mL LB media containing 100 μg/mL ampicillin in 14 mL round-bottom tubes. Allow the culture to grow for 16–18 h with aeration 37 °C for 16–18 h. Then purify the plasmids using a plasmid DNA miniprep kit, and submit the plasmid for sequencing using a primer with the sequence CCCCTTGAACCTCCTCGTT (see Note 7).
Fig. 2.

Schematic for method used generate sgRNA-containing retroviruses
3.3. Retroviral Packaging of Vectors and Generation of Bone Marrow Chimera
Day 1
-
1
Inject donor mice (Rosa26-Cas9flox/+ Cre+) intraperitoneally with 150 mg/kg 5-FU (see Note 8) (Fig. 3). Bone marrow from one adult donor mouse is typically sufficient for transfer to three to four recipients.
Fig. 3.
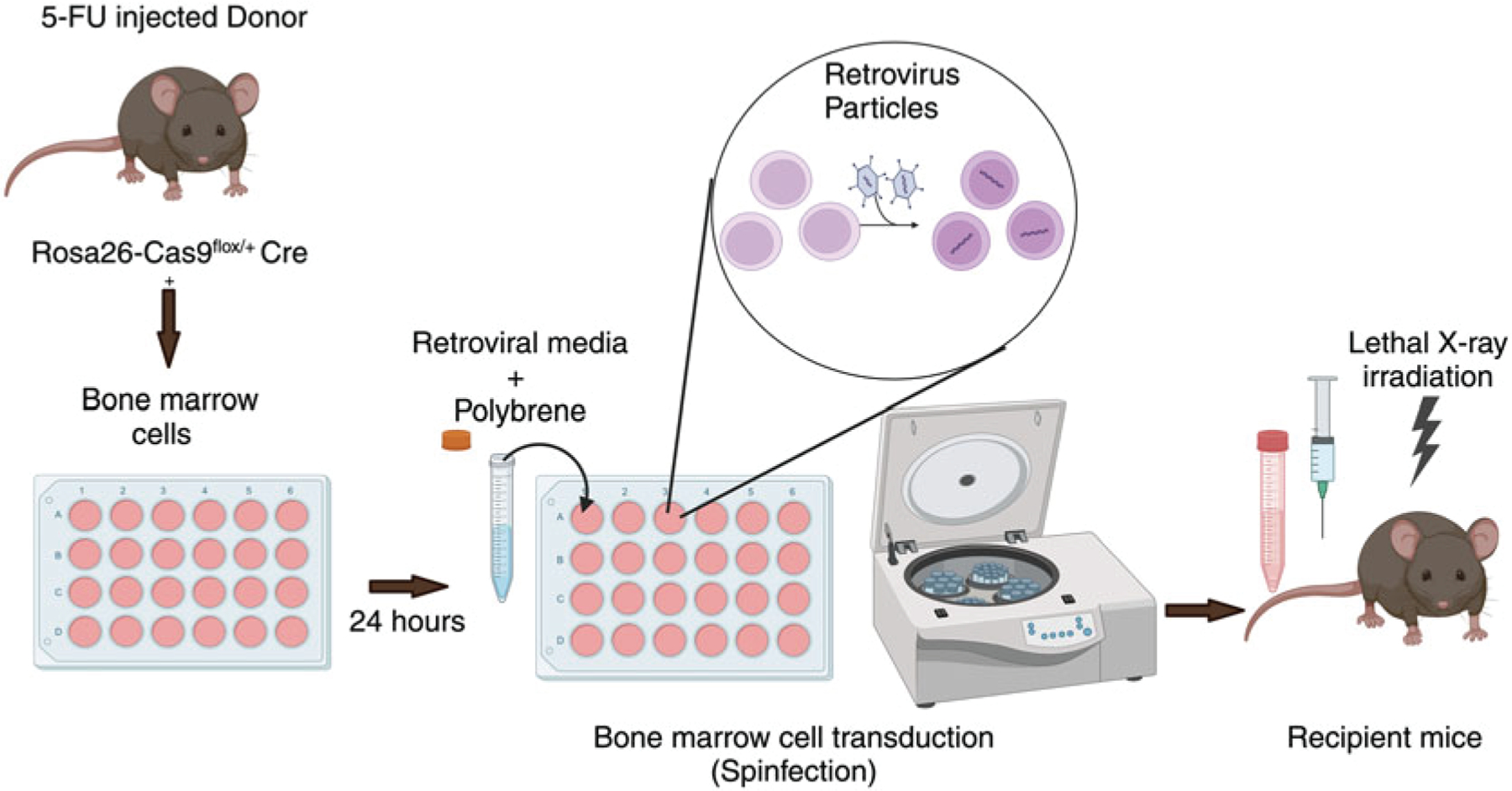
Schematic for method used to generate bone marrow chimeras
Day 4
-
2
Seed Plat-E cells in a six-well plate with a seeding density that is sufficient for achieving 75% confluency the next day. Use DMEM (10% FBS, 1% L-Glutamine, 1% P/S, 1% HEPES) for culturing Plat-E cells (see Note 9). One well of a six-well plate is needed to generate sufficient retrovirus for one recipient mouse (i.e., if you have ten recipient mice, you should split ten wells for transfection)
Day 5
-
3
Check Plat-E cells to confirm that cells are about 75% confluent (see Note 10). Replace media with pre-warmed complete DMEM without antibiotics without disturbing the cells, and return plate to CO2 incubator for 1 h (see Note 11).
-
4
Prepare lipid mix by adding 5 μL lipofectamine 3000 to 125 μL Opti-MEM per well in 1.5 mL microcentrifuge tubes or 5 mL tubes (i.e., add 50 μL lipofectamine to 1250uL Opti-MEM if you have ten wells total). Mix well and incubate for 5 min at room temperature (see Note 12).
-
5
For each well, add 2 μg total plasmid (1 μg of plasmid containing sgRNA 1 and 1 μg of plasmid containing sgRNA 2) to 125 μL Opti-MEM, and then add 5 μL P3000 reagent, and mix well in 1.5 mL microcentrifuge tubes or 5 mL tubes.
-
6
Add lipid mix and plasmid DNA mixtures at a 1:1 ratio. Mix well by gently vortexing or pipetting up and down. Incubate for 20 min at room temperature to allow lipofectamine to form complexes with the plasmid DNA.
-
7
Add 250 μL of the DNA–lipid complex to appropriate well dropwise to distribute evenly. Let plates sit undisturbed in biosafety hood for 5 min to settle before moving to CO2 incubator.
-
8
After 6 h, change the media to complete DMEM with antibiotics, and return the plate to the incubator (see Note 13). Avoid disturbing the cells while changing the media.
Day 6
-
9
Collect the tibia and femurs of the 5-FU injected donor mice as follows: Euthanize the donor mice and spray with 70% ethanol. Remove the abdominal skin all the way to the hind legs. Then cut along the spine to separate the legs from the body taking care to avoid cutting the bones. Collect intact legs in 2% RPMI in 50 mL centrifuge tube on ice.
-
10
Sterilize the legs by submerging in 50 mL tube containing 70% ethanol. Then transfer the legs to 50 mL tube containing cold PBS for 2 min, 2 times to wash. Clean the leg bones of any muscle tissue, and then separate the tibia, femur, and hip joint without breaking the individual segments. Cut the bones from one end, and flush the bone marrow with RPMI (2% FBS, 1% L-Glutamine, 1% P/S, 1% HEPES) onto a 70 μm cell strainer using a 27-gauge needle attached to a 5 mL syringe into a petri dish. Then cut the bone from the other end and flush again. Repeat the same process with other bones (see Note 14). Then mash the flushed bone marrow cells using a syringe plunger to dissociate any cell clumps, and collect the cells in 15 mL tube. Spin down the bone marrow cells by centrifugation at 526× g for 5 min at 4 °C.
-
11
Resuspend the pellet in an appropriate volume of DMEM containing 15% FBS, 20 ng/mL IL-3, 50 ng/mL IL-6, 100 ng/mL SCF, 1% P/S, and 1% HEPES, and add to a 24-well plate. Then place the plate in a CO2 incubator. Culture media is supplemented with cytokines to induce bone marrow cell proliferation. The number of wells in which bone marrow cells are seeded should correspond to the number of wells that were transfected on day 5. Total volume should be about 700 μL per well.
Day 7
-
12
Inspect the bone marrow cells under a microscope. Wells should be confluent with bone marrow cells and show signs that they are actively proliferating. Spin down the plate by centrifugation at 1460× g for 10 min at 30 °C to adhere cells to the bottom of the plate.
-
13
Harvest the retrovirus containing media from Plat-E cells transfected in six-well plates on day 5. Filter the retroviral media using 0.45-micron syringe filter to get rid of cell debris. Add polybrene (1:1000—4 μg/mL), and HEPES buffer (1: 100) to retroviral media (see Note 15). Mix gently without vortexing (see Note 16). Replace the media on the Plat-E cells with 1.3 mL fresh pre-warmed complete DMEM, and return to CO2 incubator. Retrovirus will be harvested from these cells on day 8 for the second spinfection (see Note 17).
-
14
Aspirate cytokine-containing media from the bone marrow cells gently without disturbing the bone marrow cells. Collect the media in a 15 mL tube, and incubate in 37 °C water bath. This media will be re-used to resuspend bone marrow cells after first spinfection.
-
15
Add 1 mL of retroviral media to each well of 24-well plate, and mix the bone marrow cells by pipetting up down gently (see Note 18). Avoid the formation of bubbles. Resuspension of bone marrow cells should be done in stages to minimize the time for which the cells are sitting without media (see Note 19).
-
16
Perform spinfection by centrifuging plate with bone marrow cells at 1460× g at 30 °C for 90–120 min with brake off. Then discard the retroviral media by careful aspiration, and add 500 μL of pre-warmed cytokine-containing media (collected earlier) per well to bone marrow cells. Return plate to CO2 incubator.
Day 8
-
17
Irradiate recipient mice using an X-ray irradiator with a total dose of 950–1100 rad (see Note 20). This dose is generally split into two doses administered approximately between 3 and 4 h apart to minimize damage to the gut and lung cells (see Note 21). Mice should be maintained on antibiotic containing water (i.e., Sulfamethoxazole/Trimethoprim; 5.3 mL stock in 400 mL autoclaved drinking water) to prevent opportunistic infection for 2 weeks following irradiation as mice are at higher risk of infection for that duration.
-
18
Repeat spinfection procedure from day 7. After the completion of second spinfection, carefully aspirate retroviral-containing media from the bone marrow cells. Then dislodge cells from plate using PBS by pipetting up and down and collecting the cell suspension in a 15 mL centrifuge tube. Repeat this step 2–3 times, and then check the 24-well plate under the microscope to confirm that cells are no longer adhered to plate.
-
19
Spin down the transduced bone marrow cells down at 526× g for 5 min at 4 °C. Then resuspend cells in an appropriate volume of PBS based on the number of recipient mice, and place on ice until time of transfer. We generally will resuspend cells in about 200 μL PBS per recipient.
-
20
Inject 200 μL of cell suspension per mouse intravenously to lethally irradiated mice using a 1 mL insulin syringe. Recipient mice can be analyzed at 6–8 weeks post reconstitution to check the transduction efficiency in peripheral blood mononuclear cells. Mice are now ready to be infected or immunized as desired to assess the role of candidate genes.
Acknowledgments
This work was supported by grants from the NIH (DP2AI169978 and K22AI153015 [all to B.J.L.]). All figures were created with Biorender.com.
Notes
Donor mice were obtained by crossing Rosa26-LSL-Cas9 mice with Cγ1Cre (010611) or AidCre (007770) to generate mice in which the Cas9 protein is specifically expressed in activated B cells [8, 11]. It is important to only use Rosa26-Cas9flox/+ not Rosa26-Cas9flox/flox mice as bone marrow donors to avoid potential toxicity in Cre-expressing cells [12]. Do not cross the Rosa26-LSL-Cas9 to a tamoxifen inducible Cre (i.e., S1pr2-ERT2cre mice) as this will result in the death of Cas9-expressing cells due to excess toxicity [12, 13].
Differences in the version of the mouse genome that are being used by Ensembl, Benchling, and Nucelotide BLAST can result in some sgRNA sequences not matching the targeted gene after BLAST analysis.
The first few exons of gene are generally targeted to increase the probability that a premature stop codon will be generated resulting in no protein product being translated. An alternative approach is to target specific functional domains of the protein. To do this, search the cDNA sequence of the same gene on Interpro (InterProScan—InterPro (ebi.ac.uk)). Locate a domain that is reported to be important for the protein function. Then determine based on the amino acid sequence in which exon encodes that domain. Then create sgRNA sequences that are specific for the target sequence.
If a gene has multiple splicing variants, then it’s important to target exons present in all splice variants.
We generally order a single strand primer for the Gibson cloning reaction. However, ordering a double stranded primer may increase cloning efficiency.
Gel extraction should be performed using a gel imager that does not use UV light to avoid generating mutations. If this is not possible, 1 mmol/L guanosine or cytidine can be added to the electrophoresis buffer to protect against UV-induced damage [14].
A control guide RNA sequence should be cloned using a sequence that is not specific to any genes in the mouse gene. We typically use the sequence GCGAGGTATTCGGCTCCGCG.
Dissolve 5-FU by incubating on a rotator for about 1 h at 37 °C or by vortexing for 5–10 min. 5-FU inhibits DNA synthesis and will result in the death of rapidly dividing cells [15].
Plat-E cells are based on the 293T cell line and contain gag, pol, and env genes, thus allowing retroviral packaging with a single plasmid transfection.
Performing transfection using cells that are too sparse or dense can result in lower transfection efficiency. Target should be confluency between 65% and 75% prior to transfection.
Cationic lipid reagents increase cell permeability and may increase the amount of antibiotics delivered to the cells, resulting in cytotoxicity and reduced transfection efficiency [16].
It is good practice to make enough lipid mix for one extra well to ensure you have sufficient volume for all wells.
The media on the transfected cells can also be replaced the following morning rather than after 6 h. However, this may result in a slight decrease in the amount of retrovirus present in the media at the time of transduction.
Bones typically turn white in color after they are adequately flushed. If bones do not turn white, you may want to perform an additional flushing step.
Polybrene transfection reagent is a cationic polymer that acts by neutralizing the repulsion between retrovirus and cell surface, thus enabling cell contact for infection.
Retroviral particles are sensitive to shearing, air, physical stress, and temperature fluctuation [17]. Therefore, it is necessary to maintain gentle handling techniques for retroviruses especially avoiding formation of bubbles while handling retroviral media.
Retroviral titer generally peaks 60–72 h after transfection [18]. However, we have observed that there is a slight decrease in the retroviral titer of the media used for the second spinfection. Therefore, you may have an increased transduction efficiency if you transfect a second set of Plat-E cells to generate retrovirus for the second spinfection rather than simply replacing the media on the Plat-E cells used for the first spinfection.
Bone marrow cells are mixed up and down after adding retrovirus to ensure that all cells have the same exposure to retroviral particles.
Do not let the bone marrow cells sit without media as it will dry the cells and can impact viability. Hence, the removal of cytokine-containing media from wells and addition of retroviral media should be done in sets of five to six wells at once then moving to the next set of wells.
You can also irradiate the mice the day before you transfer the cells. However, irradiation should be performed within 24 h of when bone marrow cells will be transferred.
Waiting longer than 4 h between radiation doses can result in a decreased engraftment efficiency [19].
References
- 1.Hille F, Richter H, Wong SP, Bratovič M, Ressel S, Charpentier E (2018) The biology of CRISPR-Cas: backward and forward. Cell 172:1239–1259. 10.1016/j.cell.2017.11.032 [DOI] [PubMed] [Google Scholar]
- 2.Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, van der Oost J, Koonin EV (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9:467–477. 10.1038/nrmicro2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, Moineau S, Mojica FJM, Scott D, Shah SA, Siksnys V, Terns MP, Venclovas Č, White MF, Yakunin AF, Yan W, Zhang F, Garrett RA, Backofen R, van der Oost J, Barrangou R, Koonin EV (2020) Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 18:67–83. 10.1038/s41579-019-0299-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koonin EV, Makarova KS, Zhang F (2017) Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37:67–78. 10.1016/j.mib.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156:935–949. 10.1016/j.cell.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F (2014) CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159:440–455. 10.1016/j.cell.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, Waisman A, Egert A, Ghitza D, Rajewsky K (2006) Tracking germinal center B cells expressing germ-line immunoglobulin γ1 transcripts by conditional gene targeting. Proc Natl Acad Sci U S A 103:7396–7401. 10.1073/pnas.0602353103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG (2020) The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat Immunol 21:1082–1093. 10.1038/s41590-020-0713-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanson CH, Henry B, Andhare P, Lin FJ, Pak H, Turner JS, Adams LJ, Liu T, Fremont DH, Ellebedy AH, Laidlaw BJ (2023) CD62L expression marks a functionally distinct subset of memory B cells. Cell Rep 42:113542. 10.1016/j.celrep.2023.113542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, Nussenzweig MC (2008) AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell 135:1028–1038. 10.1016/j.cell.2008.09.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurachi M, Ngiow SF, Kurachi J, Chen Z, Wherry EJ (2019) Hidden caveat of inducible Cre recombinase. Immunity 51:591–592. 10.1016/j.immuni.2019.09.010 [DOI] [PubMed] [Google Scholar]
- 13.Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, Kurosaki T (2016) Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17:861–869. 10.1038/ni.3460 [DOI] [PubMed] [Google Scholar]
- 14.Grndemann D, Schmig E (1996) Protection of DNA during preparative agarose gel electrophoresis against damage induced by ultraviolet light. BioTechniques 21:898–903. 10.2144/96215rr02 [DOI] [PubMed] [Google Scholar]
- 15.Focaccetti C, Bruno A, Magnani E, Bartolini D, Principi E, Dallaglio K, Bucci EO, Finzi G, Sessa F, Noonan DM, Albini A (2015) Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS One 10:e0115686. 10.1371/journal.pone.0115686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P, Nagarajan A, Uchil PD (2019) Lipofection. Cold Spring Harb Protoc 2019:pdb.top096248. 10.1101/pdb.top096248 [DOI] [PubMed] [Google Scholar]
- 17.Beer C, Meyer A, Müller K, Wirth M (2003) The temperature stability of mouse retroviruses depends on the cholesterol levels of viral lipid shell and cellular plasma membrane. Virology 308:137–146. 10.1016/s0042-6822(02)00087-9 [DOI] [PubMed] [Google Scholar]
- 18.Liao J, Wei Q, Fan J, Zou Y, Song D, Liu J, Liu F, Ma C, Hu X, Li L, Yu Y, Qu X, Chen L, Yu X, Zhang Z, Zhao C, Zeng Z, Zhang R, Yan S, Wu T, Wu X, Shu Y, Lei J, Li Y, Zhang W, Wang J, Reid RR, Lee MJ, Huang W, Wolf JM, He T-C, Wang J (2017) Characterization of retroviral infectivity and superinfection resistance during retrovirus-mediated transduction of mammalian cells. Gene Ther 24:333–341. 10.1038/gt.2017.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui Y-Z, Hisha H, Yang G-X, Fan T-X, Jin T, Li Q, Lian Z, Ikehara S (2002) Optimal protocol for total body irradiation for allogeneic bone marrow transplantation in mice. Bone Marrow Transplant 30:843–849. 10.1038/sj.bmt.1703766 [DOI] [PubMed] [Google Scholar]