


Abstract
Rad54 plays key roles in homologous recombination (HR) and double-strand break (DSB) repair in yeast, along with Rad51, Rad52, Rad55 and Rad57. Rad54 belongs to the Swi2/Snf2 family of DNA-stimulated ATPases. Rad51 nucleoprotein filaments catalyze DNA strand exchange and Rad54 augments this activity of Rad51. Mutations in the Rad54 ATPase domain (ATPase–) impair Rad54 function in vitro, sensitize yeast to killing by methylmethane sulfonate and reduce spontaneous gene conversion. We found that overexpression of ATPase– Rad54 reduced spontaneous direct repeat gene conversion and increased both spontaneous direct repeat deletion and spontaneous allelic conversion. Overexpression of ATPase– Rad54 decreased DSB-induced allelic conversion, but increased chromosome loss and DSB-dependent lethality. Thus, ATP hydrolysis by Rad54 contributes to genome stability by promoting high-fidelity DSB repair and suppressing spontaneous deletions. Overexpression of wild-type Rad54 did not alter DSB-induced HR levels, but conversion tract lengths were reduced. Interestingly, ATPase– Rad54 decreased overall HR levels and increased tract lengths. These tract length changes provide new in vivo evidence that Rad54 functions in the post-synaptic phase during recombinational repair of DSBs.
INTRODUCTION
In the yeast Saccharomyces cerevisiae, DNA double-strand breaks (DSBs) are repaired by homologous recombination (HR) and either precise or imprecise non-homologous end-joining (NHEJ). Imprecise NHEJ repairs <1% of DSBs, and precise NHEJ repairs 20–30% of nuclease-induced DSBs (1). Thus, most DSBs in yeast are repaired by HR, which includes conservative events such as gene conversion and crossing over and, in direct repeats, the non-conservative process termed single-strand annealing (SSA) (2). Gene conversion involves initiation, end processing, heteroduplex DNA (hDNA) formation and resolution of intermediates. Conversion can be initiated by DSBs that are processed to 3′-single-stranded DNA tails by a poorly understood mechanism involving MRE11, RAD50 and XRS2 (2). hDNA formation is thought to involve DNA strand invasion (synapsis) by Rad51, a homolog of Escherichia coli RecA, and possibly branch migration of Holliday junctions. Initially, single-stranded 3′ tails are thought to be coated with the heterotrimeric single-strand DNA binding protein RPA. Rad52 and the Rad55/Rad57 heterodimer then facilitate replacement of RPA with Rad51, forming a nucleoprotein filament that invades a homologous duplex with the assistance of Rad54 (3,4). Rad54 was also shown to enhance hDNA extension in the postsynaptic phase of DNA strand exchange in vitro (5). Inactivation of RAD51, RAD52, RAD54, RAD55 or RAD57 severely impairs HR and confers extreme radiosensitivity (reviewed in 2). Once hDNA is formed, the resulting intermediate is resolved into recombination products by mismatch repair and/or resolution of Holliday junctions (6–8). Thus, conversion tracts reflect both hDNA formation and mismatch repair of hDNA, yet little is known about factors that regulate hDNA formation and gene conversion tract lengths.
Rad54 is a member of the Swi2/Snf2 family of DNA-stimulated ATPases that modulate protein:duplex DNA interactions in disparate molecular processes (3,5,9–12). It has been proposed that Rad54 is important in the synaptic phase (homology search and strand invasion) (3,9,10,13,14) and in the postsynaptic phase (hDNA extension) (5). Rad51, Rad55 and Rad57 have conserved Walker ATPase domains and display DNA-dependent ATPase activity in vitro. ATP binding probably induces conformational changes that are essential for the activity of these proteins. By extension of studies of the Rad3 ATPase (15), mutation of the conserved lysine residue in the Walker domain to alanine (K→A) in Rad54 is thought to prevent ATP binding, whereas mutation of lysine to arginine (K→R) is expected to prevent ATP hydrolysis but not ATP binding. The latter mutation often shows milder phenotypes. For example, yeast overexpressing rad51-K191A are as sensitive to methylmethane sulfonate (MMS) and show the marked DSB repair defects characteristic of rad51Δ strains, whereas overexpression of rad51-K191R fully complements the MMS sensitivity of rad51Δ strains (16). Expression of hRAD51-K133R complements the lethal defect of RAD51-defective chicken DT40 cells (17), but this mutant protein does not fully complement defects in gene targeting in DT40 cells or DSB-induced HR in mouse cells (17,18). The Rad54 ATPase is much more potent than that of the Rad51 and Rad55/Rad57 proteins (3,9–11,14), and catalytic ATP hydrolysis is essential for Rad54 function during in vitro DNA strand exchange (3,5,9,10,13,14). However, only limited information is available about the in vivo roles of the Rad54 ATPase. Yeast carrying rad54-K341R or rad54-K341A (hereafter rad54-KR, rad54-KA) are hypersensitive to the lethal effects of MMS (9,19), and show reduced levels of spontaneous gene conversion, sporulation and spore viability (9). Human RAD54-K189R partially complements the sensitivity of mRAD54 knockout ES cells to γ-rays and mitomycin C (20). MMS and mitomycin C do not directly produce DSBs, but such damage is repaired primarily by HR in yeast. Although DSBs are directly induced by γ-rays, many other forms of DNA damage are produced by γ-rays, including base damage and single-strand breaks.
Given the key role of Rad54 in HR and DSB repair, we were interested in determining the effects of overexpressed wild-type and ATPase-defective (ATPase–) Rad54 on spontaneous HR and HR induced by a defined (nuclease) DSB. We found that excess wild-type Rad54 had no effect on spontaneous direct repeat or allelic HR, but ATPase– Rad54 enhanced both spontaneous deletions between direct repeats and spontaneous allelic gene conversion. Excess wild-type Rad54 had little effect on DSB-dependent killing or the efficiency of DSB-induced gene conversion, but ATPase– Rad54 decreased DSB-induced gene conversion, and increased both DSB-dependent cell killing and loss of broken chromosomes. Increased levels of wild-type Rad54 were found to decrease gene conversion tract lengths, whereas increased levels of ATPase-defective Rad54 increased tract lengths. These alterations in tract lengths provide in vivo evidence that Rad54 functions late in recombinational repair of DSBs.
MATERIALS AND METHODS
Plasmid DNAs
Plasmids were prepared as described previously (21). Plasmid pSE271 is a pUC19 derivative with TRP1, ARS1 and CEN4. Plasmid ADH-R54 is a pBR322 derivative with a 2-micron circle origin, TRP1 and RAD54 controlled by the ADH1 promoter. ADH-R54Arg and ADH-R54Ala are derivatives of ADH-R54 with rad54-KR or rad54-KA mutations, respectively (13). The Rad54 vectors were kindly provided by P. Sung (University of Texas, Austin).
Yeast strains and analysis of Rad54 protein levels
Yeast culture and chromosome modification were described earlier (8,22). Yeast strains are listed in Table 1, and structures of chromosomal recombination substrates are shown in Figure 1. The haploid strain JW3082 (22) has ura3 direct repeats, and an integrated copy of a GAL1-promoter-controlled HO nuclease gene (GALHO). One ura3 gene is inactivated by insertion of an HO site at position 432 (HO432), and the second by a +1 frameshift mutation at position 764 (X764). Strain JW3084 is identical to JW3082 except it lacks GALHO (22). The MATa-inc/MATα diploid strain JC3517-13 has these same ura3 alleles at allelic loci and GALHO; JC3520 is identical to JC3517-13 except it lacks GALHO (8). JC3545 is a MATa-inc/MATa-inc derivative of JC3517-13 created by brief GALHO induction as described previously (1). Strains with direct repeat or allelic recombination substrates were transformed with TRP1 plasmids (empty control vector pSE271, ADH-R54, ADH-R54Arg or ADH-R54Ala) by electroporation, and Trp+ transformants were maintained on tryptophan omission medium to retain plasmids. Rad54 protein levels were measured by immunoblotting as described previously (19).
Table 1. Yeast strains.
| Name | Genotype | Source or reference |
|---|---|---|
| JW3082 | MATa-inc, ade2-101, his3-200, lys2-801::pHSSGALHO::LYS2, trp1-Δ1, leu2-Δ1, ura3-X764-LEU2-ura3R-HO432 | (22) |
| JW3084 | Same as JW3082 except lacks GALHO | (22) |
| DY3427 | MATα, ade2-101, his3-200, lys2-801, trp1-Δ1, leu2-Δ1, RscBam-ura3-X764-LEU2 | (8) |
| JC3441 | MATa-inc, ade2-101, his3-200:HIS3:telV, lys2-801::pHSSGALHO::LYS2, trp1-Δ1, leu2-Δ1, RscRI-ura3R-HO432-LEU2 | (8) |
| JC3517-13 | Diploid product of DY3427 × JC3441 | (8) |
| JC3545 | Same as JC3517-13 except MATa-inc/MATa-inc | This study |
| JC3520-13 | Same as JC3517-13 except lacks GALHO | (8) |
Figure 1.

Recombination substrates and products. (A) In haploid strains, 1.2 kb ura3 direct repeats flank pUC19 and LEU2. The left copy is inactivated by X764, and the right copy (shaded) is inactivated by insertion of an HO site at NcoI (HO432). The four principal HR products and phenotypes are shown below. Gene conversions (GC) retain LEU2 (Leu+); short tract GC does not convert the X764 frameshift mutation; long tract GC includes X764, giving Ura– products. (B) The same ura3 genes present at allelic positions in diploids. The chromosome carrying the HO site is marked with HIS3 near the chromosome V telomere ∼100 kb from ura3. Conversions not associated with a crossover retain HIS3 (top two products); BIR and 50% of crossovers result in HIS3 loss. Loss of the broken chromosome (not shown) also leads to HIS3 loss.
Recombination assays and cell viability
Spontaneous HR rates were determined in strains lacking GALHO by using fluctuation analysis as described previously (8). DSB-induced recombination frequencies were measured in strains with GALHO by using non-selective assays (8,22). Briefly, cells from 2-day-old patches of parent strains were inoculated into 1.5 ml of YPGly medium and incubated for 24 h. Cultures were divided, cells were harvested by centrifugation, suspended in 1.5 ml of YPD (uninduced control) or YPGal (with 2% galactose; HO nuclease-induced), grown for 6 h and plated on YPD and tryptophan omission media. Cell viability was assessed for glucose- and galactose-grown cultures by measuring plating efficiency (PE), calculated as the ratio of YPD colonies to the number of cells plated (determined with a Coulter Counter). Plasmid loss was assessed by replica-plating YPD colonies to tryptophan omission medium. Under these experimental conditions, ∼25% of cells lose ARS1/CEN4 plasmids, and 5–10% of cells lose 2-micron circle plasmids; plasmid loss rates were not affected by RAD54 overexpression, nor by DSB induction (data not shown). DSB-induced HR was measured in haploid strains as follows. Colonies arising on tryptophan omission medium were replica-plated to medium lacking uracil and/or leucine to identify Ura+ Leu+ (gene conversion plus rare unequal exchanges), Ura+ Leu– (deletion) and Ura– Leu– (deletion) products. Nearly all Ura– Leu+ recombinants result from long-tract gene conversion. These were distinguished from parental cells by re-induction of GALHO on YPGal plates and subsequent replica-plating to uracil omission medium (22,23). Re-induction produces many Ura+ papillae in each parental colony whereas Ura– Leu+ recombinants, which are homozygous X764, do not yield Ura+ papillae. Less than 5% of Ura– Leu+ products (∼0.1% of total events) result from imprecise NHEJ (1). HR frequencies were calculated as the ratio of recombinants per colony arising on tryptophan omission medium; this procedure ensures that all products that were scored retained RAD54 or control plasmids. HR in diploid strains produces Ura+ and Ura– products that were identified using uracil omission media and re-induction assays, as above. Diploid strains also carried a telomere-proximal HIS3 gene (HIS3:telV) linked to ura3-HO432. Sectored Ura+/– colonies were very rare in haploid strains but common in diploid strains; these may result from segregation of X764 or from independent events in G2. Cell viability of MMS-treated cells was calculated as the PE after 6 days growth on tryptophan omission medium containing 0.01% MMS. Statistical analyses were performed with t tests.
RESULTS
Experimental design
We assayed spontaneous and DSB-induced HR in cells overexpressing wild-type Rad54 protein, or mutant Rad54 proteins presumed to be defective in ATP binding (rad54-KA) or proficient in ATP binding but defective in ATP hydrolysis (rad54-KR); we refer to both mutant proteins as ATPase– Rad54. Wild-type or ATPase– Rad54 were overexpressed in haploid and diploid strains carrying direct repeat and allelic recombination substrates. Haploid strain JW3082 (22) carries ura3 alleles arranged as direct repeats flanking pUC19 and LEU2 (Fig. 1A). One copy of ura3 was inactivated by a +1 frameshift mutation (X764); the second copy was inactivated by an HO site insertion (HO432). JW3082 has a MATa-inc mutation to prevent HO cleavage of MAT and subsequent mating-type interconversion and diploidization. The single-base MATa-inc mutation does not affect MAT coding potential; in this report MATa-inc and ‘a’ are equivalent. Direct repeat recombination yields products with one of four phenotypes. Ura+ Leu+ products typically reflect short tract gene conversion, which conserves the gross structure of the direct repeat; Ura+ Leu+ products may also result from unequal sister chromatid exchange and yield a ura3 triplication, but these events are rare in JW3082 and related strains (22,24). Essentially all Ura– Leu+ products arise by long tract gene conversion involving co-conversion of HO432 and X764. Ura+ Leu– and Ura– Leu– products reflect loss of pUC19, LEU2 and one copy of ura3 by intrachromosomal crossover, SSA or unequal sister chromatid exchange (24–26). Leu– events are collectively described as ‘deletions’.
Diploid strain JC3517-13 (MATa-inc/MATα) (8) has these same ura3 genes at allelic positions (Fig. 1B). Allelic recombination yields Ura+ and Ura– products (short and long tract gene conversion, respectively) and sectored Ura+/– products which probably reflect independent events in G2 cells. Diploids also carry a telomere-proximal HIS3 gene (HIS3:telV) on the same chromosome arm as ura3-HO432. HIS3:telV is useful for monitoring G2 crossovers (half of which lead to HIS3:telV loss and half to HIS3:telV gain), chromosome loss and break-induced replication (BIR). In wild-type cells, nearly all HIS3:telV loss results from G2 crossovers (8). We also examined DSB-induced HR in a MAT homozygous derivative of JC3517-13 (MATa-inc/MATa-inc) called JC3545 because HR efficiency and cell survival following DNA damage are lower in MAT homozygous versus heterozygous cells (1, reviewed in 27). In addition, RAD54 mRNA is reportedly increased in MAT heterozygous cells (28,29), although MAT status apparently does not influence Rad54 protein levels (19). GALHO induction in these strains increases HR by >100-fold (8,22), and similar results were obtained in the present study (data not shown). Spontaneous HR rates were measured in haploid and a/α diploid cells carrying these recombination substrates but lacking GALHO. Wild-type and mutant Rad54 were overexpressed from the strong ADH1 promoter in 2-micron circle vectors with TRP1 (kindly provided by P. Sung). Control cells carried a TRP1/ARS1/CEN4 vector to allow parallel plating strategies for all experiments. Rad54 is normally present at moderate levels (19) and cells with Rad54 expression vectors had greatly increased Rad54 protein levels (Fig. 2). Although it appeared that there was ∼2-fold higher levels of wild-type versus ATPase– Rad54, this difference does not affect the conclusions of this study.
Figure 2.
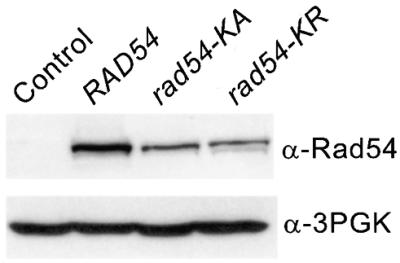
Immunoblot analysis of wild-type and ATPase– Rad54. Protein extracts were prepared from JW3082 expressing wild-type levels of Rad54 (control), and from JW3082 overexpressing RAD54, rad54-KA or rad54-KR. In each lane 50 µg of protein was loaded, the proteins were separated by SDS–PAGE, transferred to PVDF membranes and detected with antibodies to Rad54 (α-Rad54) using ECL reagents (Amersham Pharmacia Biotech, Piscataway, NJ). α-Rad54 detects both wild-type and ATPase– Rad54. Parallel lanes were probed with antibodies to 3-phosphoglycerate kinase (α-3PGK). A brief exposure is shown to compare overexpression levels; with this exposure Rad54 expressed from the native chromosomal locus cannot be detected.
Excess ATPase– Rad54 enhances spontaneous direct repeat deletion and allelic gene conversion
Spontaneous HR and/or resistance to ionizing radiation is enhanced by overexpression of wild-type HR proteins, including human RAD52 in monkey cells (30), Chinese hamster RAD51 in Chinese hamster ovary cells (31,32), human RAD51 and human RAD52 in human cells (33) and yeast RAD52 in human cells (34). Spontaneous HR rates in haploid and diploid cells overexpressing wild-type or ATPase– Rad54 are shown in Figure 3. Excess wild-type Rad54 had no effect on spontaneous direct repeat or allelic HR. Petukhova et al. (9) showed that spontaneous direct repeat gene conversion is reduced in cells expressing ATPase– Rad54. Similarly, we observed 2–3-fold lower rates of spontaneous direct repeat conversion with overexpression of ATPase– Rad54. Interestingly, ATPase– Rad54 had the opposite effect on spontaneous allelic gene conversion and direct repeat deletion, with rates enhanced by 3–10-fold. The distinct effects of ATPase– Rad54 on spontaneous direct repeat and allelic gene conversion may reflect differences in the roles of Rad54 and the Rad54 homolog, Tid1 (also called Rdh54), in sister chromatid versus homolog interactions, or perhaps different effects on replication-associated HR (see Discussion).
Figure 3.

ATPase– Rad54 enhances rates of spontaneous direct repeat deletion and allelic gene conversion. Spontaneous HR yielding Ura+ recombinants was assayed by fluctuation analysis of 11 independent parental colonies of JW3084 (Direct Repeats) or JC3520 (Allelic). For direct repeats, Ura+ products were replica plated to leucine omission medium to determine rates for Ura+ Leu+ (gene conversion) and Ura+ Leu– (deletion). Cells carried an empty (TRP1) vector as control, or they overexpressed wild-type (RAD54) or ATPase– Rad54 (rad54-KR and rad54-KA).
Effect of overexpressed wild-type and ATPase– Rad54 on DSB- and MMS-dependent cell killing
Clever et al. (19) found that overexpression of wild-type Rad54 caused significant cell killing in untreated and MMS-treated haploid cells, with milder effects in diploid cells. Given the importance of Rad54 in recombinational repair of DSBs, and the importance of HR in conferring resistance to DNA damaging agents (2,27), we tested whether excess Rad54 would increase cell killing after induction of one to two DSBs per cell by HO nuclease (Fig. 4A). Excess wild-type Rad54 had no effect on DSB-dependent killing in haploid or a/α diploid cells, whereas cell killing in a/a diploid cells was increased by ∼20% (P = 0.046). Overexpression of ATPase– Rad54 led to similar modest increases in DSB-dependent cell killing in haploid and a/a diploids, but there was no significant effect in a/α diploids. These results are in sharp contrast to the 50–95% killing of MMS-treated cells expressing ATPase– Rad54 (Fig. 4B) (19). Similar to the results of Clever et al. (19), we found that rad54-KR caused more MMS-dependent cell death than rad54-KA, but this was not the case for DSB-dependent cell killing. Although MMS induces Rad54 ∼10-fold (19), this increase in wild-type protein probably has little effect since constitutive overexpression from high-copy plasmids gave wild-type or ATPase– Rad54 levels that appeared to be at least another order of magnitude higher.
Figure 4.

Dominant negative effects of excess wild-type or ATPase– Rad54 on cell survival following DNA damage. (A) DSB-dependent cell killing. PEs were determined following 24 h growth in glycerol medium and 6 h growth in either glucose or galactose medium. DSB-dependent cell killing was determined by dividing the galactose PE by the glucose PE for each of three to four determinations per strain. The ratios were converted to percentages, and the averages ± SEM were plotted after normalizing control values to 100%. Values <100% indicate greater DSB-dependent cell killing relative to control; *, P < 0.05; **, P = 0.012. (B) MMS-dependent cell killing. PEs were determined for colonies grown in the presence or absence of 0.01% MMS for 6 days. MMS-dependent cell killing was determined as the ratio of PEs for MMS-treated to untreated cells for each of four determinations per strain. The ratios were converted to percentages, and the averages ± SEM are plotted after normalizing control values to 100%. Values <100% indicate greater MMS-dependent cell killing relative to control; **, P ≤ 0.01.
Excess wild-type and ATPase– Rad54 differentially affect DSB-induced direct repeat gene conversion and deletion
Overexpression of wild-type RAD51 or RAD52 in mammalian cells has been shown to have stimulatory or inhibitory effects on DSB-induced HR (33,35). As shown in Figure 5, excess wild-type Rad54 had no effect on DSB-induced direct repeat gene conversion, but decreased deletions ∼2-fold. In contrast, excess ATPase– Rad54 strongly reduced DSB-induced direct repeat gene conversion, but had relatively mild effects on deletions. These effects tended to balance such that total direct repeat HR frequencies were reduced by ∼2-fold with excess wild-type or ATPase– Rad54. In haploid cells overexpressing ATPase– Rad54, there was a correlation between reduced gene conversion and increased DSB-dependent cell killing, but a similar correlation was not observed with excess wild-type Rad54 (Figs 4A and 5).
Figure 5.

Dominant negative effects of excess wild-type or ATPase– Rad54 on DSB-induced direct repeat recombination. Frequencies of each of the four phenotypic classes, plus totals of all classes are shown for JW3082 carrying a control vector or overexpressing RAD54, rad54-KR or rad54-KA. Data are averages ± SD for four determinations per strain; *, P < 0.02; **, P < 0.003.
Excess ATPase– Rad54 reduces DSB-induced allelic gene conversion and increases chromosome loss
Direct repeat and allelic HR differ in several respects. In direct repeats, HR can occur by non-conservative (SSA) or conservative (gene conversion) mechanisms; sister chromatid interactions are thought to predominate; and homology is limited to the repeat length (1.2 kb in the present study). For allelic interactions, HR cannot occur by SSA, detected HR events result from homolog but not sister chromatid interactions and homology extends the entire length of the chromosome. The contribution of Rad54 may differ in sister chromatid versus homolog interactions (36–38). Thus, we were interested in whether excess wild-type or ATPase– Rad54 would differentially affect DSB-induced allelic and direct repeat HR. We were also interested in the effects of MAT status in diploid cells since MAT heterozygosity enhances HR efficiency (1,27), and diploid cells have 2-fold higher levels of RAD54 mRNA and protein than haploid cells (19,28). In a/α cells, excess wild-type Rad54 had little effect on the total frequency of DSB-induced gene conversion, whereas these events were reduced 2–3-fold by ATPase– Rad54 (Fig. 6A); qualitatively similar, but somewhat stronger effects were seen in a/a cells (Fig. 6B). ATPase– Rad54 greatly increased the relative fraction of Ura– His– products, which can arise by long-tract gene conversion associated with a crossover, BIR or chromosome loss (Fig. 1B). Quantitative Southern hybridization (data not shown) indicated that increases in Ura– His– products were due to chromosome loss (Fig. 6). When product spectra were adjusted for chromosome loss, there was little difference in fractions of His+ and His– products among cells expressing normal or high levels of wild-type Rad54 or ATPase– Rad54 (Table 2). Thus, these changes in Rad54 expression do not alter the rates of crossing over or BIR.
Figure 6.

Dominant negative effects of excess wild-type or ATPase– Rad54 on DSB-induced allelic recombination. (A) Frequencies of Ura+, Ura– and sectored Ura+/– products, HR totals and chromosome loss are shown for JC3517-13 (MATa/α) carrying a control vector or overexpressing RAD54, rad54-KR or rad54-KA (see Table 2 for distributions of His+ and His– phenotypes). Data are averages ± SD for four determinations per strain; *, P < 0.05; **, P < 0.01. (B) Data for JC3545 (MATa/a) as above.
Table 2. Excess wild-type or ATPase– Rad54 does not affect rates of crossing over or BIR.
| Percentage of allelic HR productsa | |||||
|---|---|---|---|---|---|
| Overexpression | MAT | Ura+ His+ | Ura+ His– | Ura– His+ | Ura– His– |
| None |
a/α |
36.5 |
0.9 |
57.6 |
5.0 |
|
RAD54 |
a/α |
44.0 |
1.2 |
50.6 |
4.1 |
|
rad54-KR |
a/α |
25.4 |
1.4 |
64.2 |
9.0 |
|
rad54-KA |
a/α |
18.2 |
1.8 |
67.2 |
12.9 |
| None |
a/a |
35.4 |
0.6 |
60.8 |
3.1 |
|
RAD54 |
a/a |
45.6 |
0.6 |
48.1 |
5.6 |
|
rad54-KR |
a/a |
27.9 |
0.6 |
66.4 |
5.0 |
| rad54-KA | a/a | 31.9 | 1.3 | 59.4 | 7.3 |
aValues are based on averages of four determinations; excludes sectored products (Ura+/– and/or His+/–).
Gene conversion tract lengths are reduced by excess wild-type Rad54 and increased by ATPase– Rad54
Since Rad54 augments Rad51 function in vitro (9), we expected that excess wild-type Rad54 would enhance strand invasion in vivo, promote hDNA extension and thereby increase gene conversion tract lengths. Conversely, we predicted that ATPase– Rad54 would reduce conversion tract lengths. We focused on allelic conversion tract lengths since crossing over is associated with long gene conversion tracts (39), and crossovers between direct repeats produce deletions that cannot be distinguished from SSA or unequal sister chromatid exchange. We estimated tract lengths by determining the fraction of DSB-induced HR products with tracts that extend to X764 (Ura–). Note that in strains expressing ATPase– Rad54, many Ura– His– products result from chromosome loss (Fig. 6). Since chromosome loss frequencies were estimated by analyzing relatively few Ura– His– products (10–25 per strain), it was possible that inaccuracies in chromosome loss measurements would compromise our tract length measurements, but this was not the case: tract lengths calculated from the full data set (Fig. 7A) are essentially the same as those calculated from His+ products, which do not reflect chromosome loss (Fig. 7B). Surprisingly, tract lengths were reduced by overexpression of wild-type Rad54 in both a/α and a/a cells. Conversely, tract lengths were increased significantly by rad54-KR in both cell types, and by rad54-KA in a/α cells. These changes in tract lengths support a role for Rad54 in postsynapsis in vivo.
Figure 7.

Effects of excess wild-type or ATPase– Rad54 on gene conversion tract lengths. (A) Allelic tract lengths for JC3517-13 (MATa/α) and JC3545 (MATa/a) were calculated as the ratio of long tracts (sum of Ura– + half of Ura+/–) divided by total recombinants. Data are averages ± SD for four determinations per strain; *, P ≤ 0.05; **, P < 0.01. (B) Allelic tract lengths calculated from His+ data.
DISCUSSION
Rad54 suppression of spontaneous direct repeat deletion and allelic gene conversion
Cells with null mutations in rad51, rad54, rad55 or rad57 display enhanced rates of spontaneous deletions between direct repeats (40). There is increasing evidence that spontaneous recombination is replication dependent, reflecting recombinational bypass of DNA lesions, formation of DSBs when a replication fork encounters a single-stranded break, or processing of an arrested replication fork into a double-stranded end by fork reversal or direct breakage (reviewed in 41,42). Defects in HR proteins may increase spontaneous deletions because spontaneous DSBs are shunted to other repair pathways. In direct repeats, such DSBs will likely be shunted to SSA leading to repeat deletion. Alternatively, HR defects may abort recombination causing cell death, chromosome loss or shunting of HR intermediates to alternative pathways. We found that spontaneous direct repeat deletion is enhanced by overexpression of rad54-KR and rad54-KA (Fig. 3), consistent with ATPase– Rad54 acting in a dominant negative manner. Thus, spontaneous direct repeat deletions are enhanced in rad54Δ or by ATPase– Rad54, indicating that Rad54 ATP hydrolysis is required to suppress this type of genetic instability.
We found that overexpression of ATPase– Rad54 reduced spontaneous gene conversion rates between direct repeats by ∼2-fold. A stronger (>40-fold) reduction was seen when RAD54 was replaced by rad54-KR or rad54-KA (9), indicating that overexpression gives a partial dominant negative phenotype. Interestingly, spontaneous allelic conversion rates were increased by 3–4-fold by ATPase– Rad54 (Fig. 3). These opposite effects may reflect differences in mechanisms and/or differential roles of HR proteins in direct repeat versus allelic conversion. Direct repeat conversion probably involves sister chromatid interactions (43), whereas allelic conversion must involve homolog interactions. In addition, homolog and sister chromatid interactions are differentially affected by mutations in Rad54 and Tid1 (36–38), and ATPase– Rad54 may enhance spontaneous allelic conversion by interfering with the competing sister chromatid pathway. Note that ATPase– Rad54 enhances spontaneous allelic conversion, but reduces DSB-induced allelic conversion. Given the key role of Rad54 in DSB repair, it is not surprising that ATPase– Rad54 has dominant negative effects on DSB-induced conversion (discussed further below). In contrast, enhanced spontaneous allelic conversion may reflect inhibition of sister chromatid interactions (in which Rad54 plays an important role) (37), shifting some events toward allelic interactions mediated by Tid1 (36,38). Studies with tid1 mutants are in progress to test this hypothesis.
Effects of wild-type and ATPase– Rad54 on DSB-induced recombination
Excess wild-type or ATPase– Rad54 has distinct effects on DSB-induced direct repeat conversions and deletions. Excess ATPase– Rad54 reduces conversion but has little effect on deletions, while excess wild-type Rad54 has little effect on conversion but reduces deletions (Fig. 5). Reduced conversion probably reflects dominant negative effects on this conservative DSB repair pathway. The minimal effect of ATPase– Rad54 on deletions can be explained since most deletions probably arise by SSA which requires Rad52, but not Rad51, Rad54, Rad55 or Rad57 (44). Defects in Rad51-dependent HR increase DNA damage-induced cell killing (reviewed in 27). With direct repeats, reduced conversion did not correlate with increased DSB-dependent cell killing (Figs 4 and 5); this can similarly be explained by shunting DSBs to Rad51- (and Rad54-)independent SSA. It is not clear why overexpression of wild-type Rad54 preferentially reduces direct repeat deletions.
Overexpression of wild-type Rad54 suppresses meiotic defects of dmc1 mutants, and it was proposed that this reflects activation of a Rad54/Rad51-dependent DSB-repair pathway normally suppressed in meiosis (i.e., by the synaptonemal complex) (45). We found that overexpression of wild-type Rad54 does not affect the efficiency of DSB-induced allelic conversion. Excess Rad54 does, however, influence product spectra (see below). ATPase– Rad54 reduced DSB-induced gene conversion and increased chromosome loss and, in a/a diploid cells, increased cell killing. It is possible that ATPase– Rad54 interferes with HR at a late stage in a way that prevents DSBs from being shunted toward NHEJ, frequently leading to loss of broken chromosomes.
Rad54 regulation of gene conversion tract lengths
Rad54 has proposed roles in the synaptic and post-synaptic phases of HR facilitating homology search, strand invasion and hDNA extension (3,5,9,10,13,20). The homology of Rad54 with chromatin remodeling factors like Swi2/Snf2 led to the proposal that Rad54 is involved in chromatin remodeling during HR (46). However, not all Swi2/Snf2-like proteins remodel chromatin, and the common function of this class of proteins may be more general in modulating protein:duplex DNA interactions (47); for example, Mot1 removes the TATA-binding protein from the TATA box (48). Rad54 stimulates in vitro DNA strand exchange by Rad51 with nucleosome-free DNA substrates, resulting in increased formation of joint molecules and faster extension of hDNA (3,5,9,10,13,14,20). We tested whether overexpression of wild-type Rad54 would promote hDNA formation and increase gene conversion tract lengths in vivo. Instead, tract lengths were reduced by excess wild-type Rad54, and increased by excess ATPase– Rad54 (Fig. 7). These changes in tract lengths provide in vivo evidence that Rad54 acts during late HR stages, consistent with studies of in vitro DNA strand exchange (5), meiotic focus formation (49) and genetic suppression/epistasis (50–52). One model to account for the observed changes in tract lengths suggests that Rad54 plays a role in stabilizing hDNA. In this view, excess Rad54 would stabilize shorter hDNA regions enough to allow these to progress to mature recombination products. Conversely, defective Rad54 would destabilize intermediates with limited hDNA and these would fail to mature to products, whereas intermediates with longer hDNA may be stable enough to mature to products in the absence of functional Rad54. However, this model cannot account for the observation that the total number of events is unchanged when wild-type Rad54 is overexpressed. Alternatively, excess Rad54 may limit hDNA formation and thus tract length by stimulating Rad51 turnover, while excess ATPase– Rad54 slows Rad51 turnover, promoting hDNA extension and increasing tract lengths. Biochemical studies show that stoichiometry is of critical importance for recombination proteins. While the overexpression results strongly suggest a role of Rad54 in postsynapsis (conversion tract length), it is difficult to infer the precise in vivo role of Rad54 in this process from studies in which protein levels are not in stoichiometric balance.
Rad54 ATP hydrolysis and maintenance of genomic stability
Studies of several ATPases involved in HR, including RecA, Rad51 and Rad57, raised questions about the importance of ATP hydrolysis in their functions. For example, RecA and yeast Rad51 require ATP binding, but not hydrolysis, to catalyze in vitro homologous strand exchange (16,53,54); overexpression of rad51KR complements the MMS sensitivity of yeast rad51 mutants (16); the radiosensitivity of null rad57 yeast is fully complemented by rad57KR (55); and human RAD51KR complements the replication defect of chicken rad51 mutants (17). It has been suggested that Swi/Snf-type proteins use the energy from ATP hydrolysis to modulate protein–DNA interactions directly (47) or indirectly by altering DNA conformation (3,9–11). ATP hydrolysis is important for Rad54 function in vitro in DNA strand exchange (3,5,9,10,13,14), and in vivo to confer MMS resistance (9,19) and to prevent spontaneous direct repeat deletions (9). We found that rad54-KR and rad54-KA share several phenotypes, including increased spontaneous direct repeat deletions and allelic conversion, reduced efficiency of conservative DSB repair, increased loss of broken chromosomes and increased gene conversion tract lengths. These results suggest that, unlike Rad51, Rad55 or Rad57, catalytic ATP hydrolysis is required for Rad54 function(s) that promote genome stability. A role for Rad54 in maintaining genome stability is consistent with mammalian RAD54 acting as a tumor suppressor, along with two other HR proteins, BRCA1 and BRCA2 (56–58), and with the identification of human RAD54 mutations in tumors (59,60).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Patrick Sung for providing Rad54 expression vectors, and Mark Brenneman, Chris Allen, Jennifer Clikeman, Patrick Sung and Mary Ann Osley for helpful comments. This work was supported by grant CA55302 to J.A.N. from the National Cancer Institute of the NIH, and by grant GM58015 from the Institute of General Medical Sciences of the NIH to W.-D.H.
REFERENCES
- 1.Clikeman J.A., Khalsa,G.J., Barton,S.L. and Nickoloff,J.A. (2001) Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKu-dependent and -independent mechanisms. Genetics, 157, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paques F. and Haber,J.E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazin A.V., Bornarth,C.J., Solinger,J.A., Heyer,W.D. and Kowalczykowski,S.C. (2000) Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell, 6, 583–592. [DOI] [PubMed] [Google Scholar]
- 4.Sung P. (1997) Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev., 11, 1111–1121. [DOI] [PubMed] [Google Scholar]
- 5.Solinger J.A. and Heyer,W.D. (2001) Rad54 protein stimulates the postsynaptic phase of Rad51 protein-mediated DNA strand exchange. Proc. Natl Acad. Sci. USA, 98, 8447–8453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nickoloff J.A. and Hoekstra,M.F. (1998) Double-strand break and recombinational repair in Saccharomyces cerevisiae. In Nickoloff,J.A. and Hoekstra,M.F. (eds), DNA Damage and Repair, Vol. 1: DNA Repair in Prokaryotes and Lower Eukaryotes. Humana Press, Totowa, NJ, pp. 335–362. [Google Scholar]
- 7.Weng Y.-s. and Nickoloff,J.A. (1998) Evidence for independent mismatch repair processing on opposite sides of a double-strand break in Saccharomyces cerevisiae. Genetics, 148, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nickoloff J.A., Sweetser,D.B., Clikeman,J.A., Khalsa,G.J. and Wheeler,S.L. (1999) Multiple heterologies increase mitotic double-strand break-induced allelic gene conversion tract lengths in yeast. Genetics, 153, 665–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petukhova G., Van Komen,S., Vergano,S., Klein,H. and Sung,P. (1999) Yeast Rad54 promotes Rad51-dependent homologous DNA pairing via ATP hydrolysis-driven change in DNA double helix conformation. J. Biol. Chem., 274, 29453–29462. [DOI] [PubMed] [Google Scholar]
- 10.van Komen S., Petukhova,G., Sigurdsson,S., Stratton,S. and Sung,P. (2000) Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell, 6, 563–572. [DOI] [PubMed] [Google Scholar]
- 11.Tan T.L.R., Essers,J., Citterio,E., Swagemakers,S.M.A., de Wit,J., Benson,F.E., Hoeijmakers,J.H.J. and Kanaar,R. (1999) Mouse Rad54 affects DNA conformation and DNA-damage-induced Rad51 foci formation. Curr. Biol., 9, 325–328. [DOI] [PubMed] [Google Scholar]
- 12.Fyodorov D.V. and Kadonaga,J.T. (2001) The many faces of chromatin remodeling: SWItching beyond transcription. Cell, 106, 523–525. [DOI] [PubMed] [Google Scholar]
- 13.Petukhova G., Stratton,S. and Sung,P. (1998) Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature, 393, 91–94. [DOI] [PubMed] [Google Scholar]
- 14.Solinger J.A., Lutz,G., Sugiyama,T., Kowalczykowski,S.C. and Heyer,W.D. (2001) Rad54 protein stimulates heteroduplex DNA formation in the synaptic phase of DNA strand exchange via specific interactions with the presynaptic Rad51 nucleoprotein filament. J. Mol. Biol., 307, 1207–1221. [DOI] [PubMed] [Google Scholar]
- 15.Sung P., Higgins,D., Prakash,L. and Prakash,S. (1988) Mutation of lysine-48 to arginine in the yeast Rad3 protein abolishes its ATPase and DNA helicase activities but not the ability to bind ATP. EMBO J., 7, 3263–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sung P. and Stratton,S.A. (1996) Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. J. Biol. Chem., 271, 27983–27986. [DOI] [PubMed] [Google Scholar]
- 17.Morrison C., Shinohara,A., Sonoda,E., Yamaguchi-Iwai,Y., Takata,M., Weichselbaum,R.R. and Takeda,S. (1999) The essential functions of human Rad51 are independent of ATP hydrolysis. Mol. Cell. Biol., 19, 6891–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stark J.M., Hu,P., Pierce,A.J., Moynahan,M.E., Ellis,N. and Jasin,M. (2002) ATP-hydrolysis by mammalian Rad51 has a key role during homology-directed DNA repair. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 19.Clever B., Schmuckli-Maurer,J., Sigrist,M., Glassner,B.J. and Heyer,W.D. (1999) Specific negative effects resulting from elevated levels of the recombinational repair protein Rad54p in Saccharomyces cerevisiae. Yeast, 15, 721–740. [DOI] [PubMed] [Google Scholar]
- 20.Swagemakers S.M.A., Essers,J., de Wit,J., Hoeijmakers,J.H.J. and Kanaar,R. (1998) The human Rad54 recombinational DNA repair protein is a double-stranded DNA-dependent ATPase. J. Biol. Chem., 273, 28292–28297. [DOI] [PubMed] [Google Scholar]
- 21.Taghian D.G. and Nickoloff,J.A. (1996) Subcloning strategies and protocols. In Harwood,A. (ed.), Basic DNA and RNA Protocols. Humana Press, Totowa, NJ, Vol. 58, pp. 221–235. [DOI] [PubMed]
- 22.Cho J.W., Khalsa,G.J. and Nickoloff,J.A. (1998) Gene conversion tract directionality is influenced by the chromosome environment. Curr. Genet., 34, 269–279. [DOI] [PubMed] [Google Scholar]
- 23.Weng Y.-s., Whelden,J., Gunn,L. and Nickoloff,J.A. (1996) Double-strand break-induced gene conversion: examination of tract polarity and products of multiple recombinational repair events. Curr. Genet., 29, 335–343. [DOI] [PubMed] [Google Scholar]
- 24.Nickoloff J.A., Singer,J.D., Hoekstra,M.F. and Heffron,F. (1989) Double-strand breaks stimulate alternative mechanisms of recombination repair. J. Mol. Biol., 207, 527–541. [DOI] [PubMed] [Google Scholar]
- 25.Ray A., Siddiqi,I., Kolodkin,A.L. and Stahl,F.W. (1988) Intrachromosomal gene conversion induced by a DNA double-strand break in Saccharomyces cerevisiae. J. Mol. Biol., 201, 247–260. [DOI] [PubMed] [Google Scholar]
- 26.Fishman-Lobell J., Rudin,N. and Haber,J.E. (1992) Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell. Biol., 12, 1292–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nickoloff J.A. and Haber,J.E. (2001) Mating-type control of DNA repair and recombination in Saccharomyces cerevisiae. In Nickoloff,J.A. and Hoekstra,M.F. (eds), DNA Damage and Repair, Vol. 3: Advances from Phage to Humans. Humana Press, Totowa, NJ, pp. 107–124. [Google Scholar]
- 28.Cole G.M., Schild,D., Lovett,S.T. and Mortimer,R.K. (1987) Regulation of RAD54- and RAD52-lacZ gene fusion in Saccharomyces cerevisiae in response to DNA damage. Mol. Cell. Biol., 7, 1078–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galitski T., Saldanha,A.J., Styles,C.A., Lander,E.S. and Fink,G.R. (1997) Ploidy regulation of gene expression. Science, 285, 251–254. [DOI] [PubMed] [Google Scholar]
- 30.Park M.S. (1995) Expression of human RAD52 confers resistance to ionizing radiation in mammalian cells. J. Biol. Chem., 270, 15467–15470. [DOI] [PubMed] [Google Scholar]
- 31.Vispe S., Cazaux,C., Lesca,C. and Defais,M. (1998) Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res., 26, 2859–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnaudeau C., Helleday,T. and Jenssen,D. (1999) The RAD51 protein supports homologous recombination by an exchange mechanism in mammalian cells. J. Mol. Biol., 289, 1231–1238. [DOI] [PubMed] [Google Scholar]
- 33.Kim P.M., Allen,C., Wagener,B.M., Shen,Z. and Nickoloff,J.A. (2001) Overexpression of human RAD51 and RAD52 reduces double-strand break-induced homologous recombination in mammalian cells. Nucleic Acids Res., 29, 4352–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson B.L., Thyagarajan,B., Krueger,L., Hirsch,B. and Campbell,C. (1996) Elevated levels of recombinational DNA repair in human somatic cells expressing the Saccharomyces cerevisiae RAD52 gene. Mutat. Res., 363, 179–189. [DOI] [PubMed] [Google Scholar]
- 35.Lambert S. and Lopez,B.S. (2000) Characterization of mammalian RAD51 double strand break repair using non-lethal dominant-negative forms. EMBO J., 19, 3090–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klein H.L. (1997) RDH54, a RAD54 homolog in Saccharomyces cerevisiae, is required for mitotic diploid-specific recombination and repair and for meiosis. Genetics, 147, 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arbel A., Zenvirth,D. and Simchen,G. (1999) Sister chromatid-based DNA repair is mediated by RAD54, not by DMC1 or TID1. EMBO J., 18, 2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinohara M., Shitayamaguchi,E., Buerstedde,J.M., Shinagawa,H., Ogawa,H. and Shinohara,A. (1997) Characterization of the roles of the Saccharomyces cerevisiae RAD54 gene and a homologue of RAD54, RDH54/TID1, in mitosis and meiosis. Genetics, 147, 1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguilera A. and Klein,H.L. (1989) Yeast intrachromosomal recombination: long gene conversion tracts are preferentially associated with reciprocal exchange and require the RAD1 and RAD3 gene products. Genetics, 123, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDonald J.P. and Rothstein,R. (1994) Unrepaired heteroduplex DNA in Saccharomyces cerevisiae is decreased in RAD1 RAD52-independent recombination. Genetics, 137, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothstein R., Michel,B. and Gangloff,S. (2000) Replication fork pausing and recombination or “gimme a break”. Genes Dev., 14, 1–10. [PubMed] [Google Scholar]
- 42.Cox M.M. (2000) Recombinational DNA repair in bacteria and the RecA protein. Prog. Nucleic Acid Res. Mol. Biol., 63, 311–366. [DOI] [PubMed] [Google Scholar]
- 43.Kadyk L.C. and Hartwell,L.H. (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics, 132, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ivanov E.L., Sugawara,N., Fishman-Lobell,J. and Haber,J.E. (1996) Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics, 142, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bishop D.K., Nikolski,Y., Oshiro,J., Chon,J., Shinohara,M. and Chen,X. (1999) High copy number suppression of the meiotic arrest caused by a dmc1 mutation: REC114 imposes an early recombination block and RAD54 promotes a DMC1-independent DSB repair pathway. Genes Cells, 4, 425–443. [DOI] [PubMed] [Google Scholar]
- 46.Sugawara N., Ivanov,E.L., Fishman-Lobell,J., Ray,B.L., Wu,X. and Haber,J.E. (1995) DNA structure-dependent requirements for yeast RAD genes in gene conversion. Nature, 373, 84–86. [DOI] [PubMed] [Google Scholar]
- 47.Pazin M.J. and Kadonaga,J.T. (1997) SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions? Cell, 88, 737–740. [DOI] [PubMed] [Google Scholar]
- 48.Auble D.T., Wang,D.Y., Post,K.W. and Hahn,S. (1997) Molecular analysis of the SNF2/SWI2 protein family member MOT1, an ATP-driven enzyme that dissociates TATA-binding protein from DNA. Mol. Cell. Biol., 17, 4842–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shinohara M., Gasior,S.L., Bishop,D.K. and Shinohara,A. (2000) Tid1/Rdh54 promotes colocalization of Rad51 and Dmc1 during meiotic recombination. Proc. Natl Acad. Sci. USA, 97, 10814–10819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palladino F. and Klein,H.L. (1992) Analysis of mitotic and meiotic defects in Saccharomyces cerevisiae SRS2 DNA helicase mutants. Genetics, 132, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schild D. (1995) Suppression of a new allele of yeast RAD52 by overexpression of RAD51, mutations in srs2 and ccr4, or mating-type heterozygosity. Genetics, 140, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rattray A.J. and Symington,L.S. (1995) Multiple pathways for homologous recombination in Saccharomyces cerevisiae. Genetics, 139, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J.-I., Cox,M.M. and Inman,R.B. (1992) On the role of ATP hydrolysis in RecA protein-mediated DNA strand exchange. I. Bypassing a short heterologous insert in one DNA substrate. J. Biol. Chem., 267, 16438–16443. [PubMed] [Google Scholar]
- 54.Rosselli W. and Stasiak,A. (1991) The ATPase activity of RecA is needed to push the DNA strand exchange through heterologous regions. EMBO J., 10, 4391–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson R.D. and Symington,L.S. (1995) Functional differences and interactions among the putative RecA homologues Rad51, Rad55 and Rad57. Mol. Cell. Biol., 15, 4843–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moynahan M.E., Chiu,J.W., Koller,B.H. and Jasin,M. (1999) Brca1 controls homology-directed DNA repair. Mol. Cell, 4, 511–518. [DOI] [PubMed] [Google Scholar]
- 57.Moynahan M.E., Pierce,A.J. and Jasin,M. (2001) BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell, 7, 263–272. [DOI] [PubMed] [Google Scholar]
- 58.Xia F., Taghian,D.G., DeFrank,J.S., Zeng,Z.C., Willers,H., Iliakis,G. and Powell,S.N. (2001) Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc. Natl Acad. Sci. USA, 98, 8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuda M., Miyagawa,K., Takahashi,M., Fukuda,T., Kataoka,T., Asahara,T., Inui,H., Watatani,M., Yasutomi,M., Kamada,N. et al. (1999) Mutations in the RAD54 recombination gene in primary cancers. Oncogene, 18, 3427–3430. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez R., Silva,J.M., Dominguez,G., Garcia,J.M., Martinez,G., Vargas,J., Provencio,M., Espana,P. and Bonilla,F. (1999) Detection of loss of heterozygosity at RAD51, RAD52, RAD54 and BRCA1 and BRCA2 loci in breast cancer: pathological correlations. Br. J. Cancer, 81, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]