


Abstract
Cross-species single-cell RNA-seq data hold immense potential for unraveling cell type evolution and transferring knowledge between well-explored and less-studied species. However, challenges arise from interspecific genetic variation, batch effects stemming from experimental discrepancies and inherent individual biological differences. Here, we benchmarked nine data-integration methods across 20 species, encompassing 4.7 million cells, spanning eight phyla and the entire animal taxonomic hierarchy. Our evaluation reveals notable differences between the methods in removing batch effects and preserving biological variance across taxonomic distances. Methods that effectively leverage gene sequence information capture underlying biological variances, while generative model-based approaches excel in batch effect removal. SATURN demonstrates robust performance across diverse taxonomic levels, from cross-genus to cross-phylum, emphasizing its versatility. SAMap excels in integrating species beyond the cross-family level, especially for atlas-level cross-species integration, while scGen shines within or below the cross-class hierarchy. As a result, our analysis offers recommendations and guidelines for selecting suitable integration methods, enhancing cross-species single-cell RNA-seq analyses and advancing algorithm development.
Graphical Abstract
Graphical Abstract.

Introduction
Single-cell RNA-sequencing (scRNA-seq) has emerged as a key technology to understand the conservation and divergence of cell types across species (1–14). The increasing availability of scRNA-seq datasets across diverse species presents a valuable opportunity for exploring and describing evolutionary relationships of cell types from different species (15). Key to our understanding of cell type diversity and their evolutionary history is the cross-species integration of scRNA-seq data (13). Cross-species scRNA-seq data integration aims to harmonize high-throughput datasets from various sources into a coherent format for subsequent downstream comparative analysis. Such an approach would facilitate the exploration of evolutionary trajectories among cell types across the tree of life. For example, a comparative study based on the integration of scRNA-seq data from salamanders, lizards, turtles and mice led to the identification of conserved neurons shared amongst these species and sauropsids specific neurons (16). Moreover, cross-species scRNA-seq data integration stands out as a powerful tool, paving the path for transferring knowledge from well-studied species to those less explored ones, which often possess genomes that are either inadequately annotated or devoid of validated marker genes for distinct cell types. This provides critical reference points for cell type annotation and offers a deeper dive into their cellular intricacies (4,17–23). More than pure basic research, these integration-centric methodologies illuminate venues for further research topics, most crucially in the conservation efforts for environmentally significant but understudied species, such as reef building corals or the honeybee Apis mellifera, among others.
However, cross-species data integration presents a unique set of challenges. First, cross-species datasets often include many samples generated from various conditions and laboratories, which may vary in terms of sequencing platform, sequencing depth, quality, and experimental conditions. These undesirable technical variations, often referred to as batch effects, have the potential to induce shifts in data distributions and bias the results (14,24–27). Distinguishing between batch effects and biological heterogeneity becomes particularly challenging, especially when these factors overlap. Besides, the dataset scale becomes a severe problem in cross-species integration projects. For example, the Human Cell Atlas (HCA) stores data from 626 single-cell projects encompassing trillions of cells obtained from different tissues and conditions (28,29). Yet, the current cell atlas of the sea anemone Nematostella vectensis, a non-bilaterian animal, only contains 14 000 cells covering two developmental stages (30). These huge differences in dataset scale may lead to the masking of specific cell types in the smaller datasets. Further, the complex evolutionary history of genes poses a significant hurdle, particularly when dealing with datasets of varying sequencing quality. Paralogs, which arise from gene duplication events within a genome, can evolve novel functions compared with their ancestral genes, considerably complicating the integration process (12).
At present, there are at least 50 published integration methods for scRNA-seq data, of which six emphasize their effectiveness in cross-species integration. Previous benchmark studies primarily concentrated on addressing the broader challenge of batch effect removal problem (14). Notably, two distinct benchmark studies attempted to explore the intricacies of the cross-species integration problem, providing valuable guidelines on selecting appropriate methods and strategies (13,31). However, their scope was confined to a limited species spectrum coverage. Specifically, one study centered around integrating human and mouse immune datasets (13) while the other involved a constrained selection of only five vertebrate species (31). This narrow species spectrum coverage falls short of representing the diverse scenarios encountered in cross-species integration tasks, thereby lacking comprehensive guidance for such endeavors. Yet, resolving the evolutionary trajectories of cell types across large phylogenetic distances, a prerequisite to building a cell type tree of life requires a comprehensive approach using a broad species spectrum. So far, a systematic benchmark for integrating diverse species across the entire animal taxonomic hierarchy is still lacking.
Here, we present a pioneering benchmark study of cross-species single-cell RNA-seq data integration methods across various settings, spanning the entire animal tree of life hierarchy and including genus, family, order, class and phylum species integrations (Figure 1). Specifically, we examined the challenges associated with developmental time trajectory preservation, dataset scale differences, species-specific cell type identification, nested batch effects and varying data quality. In total, we created 36 cross-species integration tasks, consisting of over 4.7 million cells and 20 species. We benchmarked nine most widely used data integration techniques, each with its unique underlying algorithmic assumptions, that generally performed well in various data integration tasks (13): mutual nearest neighbors (fastMNN (32)) and its variations (Scanorama (33) and BBKNN (34)), linear correlation (Seurat v4 CCA (35)), the k-means algorithm (Harmony (36)), deep generative models (scVI (19) and scGen (37)) and gene sequence alignment (SAMap (12) and SATURN (38)). These methods require different input formats—such as one-to-one orthologous genes or homologous genes—and produce varying outputs. We have summarized their input requirements, output formats and usability in a table to facilitate comparison (Supplementary Table S1). Moreover, to evaluate their performance, we used 13 metrics focusing on two key aspects: batch effect removal and biological variance conservation. Batch effect removal metrics assess an integration method’s ability to eliminate technical biases between datasets, i.e. unintended differences due to technical variations rather than actual biological differences between the samples. High batch effect removal scores indicate effective mixing of cells from different species, with minimized batch-specific biases. This is essential for tasks such as cross-species reference mapping (39), where the goal is to align analogous cell types across species. In contrast, biological variance conservation metrics evaluate how well an integration method preserves meaningful biological differences inherent in the data. High biological conservation scores suggest that important biological signals—such as cell types and cellular states clusters—are retained post-integration, which is crucial for accurate downstream analysis. By balancing these two objectives, an ideal integration method effectively removes unwanted technical variations while preserving the true biological information necessary for meaningful insights. Our comprehensive benchmarking study provides a reference for researchers interested in performing scRNA-seq cross species data integration studies. Furthermore, we presented the first attempt of constructing a cell type tree of life using the best performing data integration method.
Figure 1.

Schematic diagram of the benchmarking workflow. Here, nine data integration methods were tested using 36 cross-species integration tasks. Integration results were evaluated using 13 metrics that assess batch effect removal (species mixing), nested batch effect removal and conservation of biological variance. The influence of imbalanced dataset and data sequencing depth on the methods were also assessed.
Materials and methods
Datasets and preprocessing
We conducted a comprehensive benchmarking of cross-species integration methods using 23 published datasets (Supplementary Tables S2 and S3). To ensure consistency, all datasets underwent quality control (minimum 200 genes per cell, minimum 10 cells per gene) and normalization (scanpy.pp.normalize_in_total, scanpy.pp.log1p) using the Scanpy v1.9.1 (40).
Certain methods (Seurat v4 CCA, fastMNN, BBKNN, scVI, scGen, Scanorama and Harmony) require the cross-species datasets to have the same variable gene names, as they only accept one-to-one orthologous genes as inputs. To address this, we used OrthoFinder v2.5.5 (41) to identify one-to-one orthologous genes between species (Supplementary Table S4). We then extracted subsets of the input datasets containing these identified one-to-one orthologous genes and used them as input for the integration methods. The remaining two methods, SAMap and SATURN, require gene sequence similarity information, the related sequences involved are provided in Supplementary Table S4. These two methods were provided with datasets containing original gene names and gene size after preprocessing.
We utilized the cell type annotations provided by the original authors of each published dataset, serving as the ground truth for our evaluations. To align cell types across species, we matched cell types based on their annotated names. For closely related species, when cell types had identical names in different species, we considered them directly comparable. For phylogenetically distant species, we did not attempt to match specific cell types directly. Instead, we used coarse-grained annotations, aligning cells based on broader cell lineages. This approach relied solely on published annotations to ensure reproducibility and accuracy.
Integration methods
Seurat v4 (CCA)
Seurat is one of the most famous tools used in single cell transcriptomic data analysis. In this study, we ran Seurat (version 4.3.0) (35) which set canonical correction analysis (CCA) as default for dimensionality reduction following the official tutorial https://satijalab.org/seurat/articles/integration_introduction.html. The algorithm identifies the ‘anchor pairs’ between two datasets which are the MNN on the shared subspace constructed by CCA. Based on the anchor pairs, the method can further integrate and project the original two datasets to a common hyperplane. As a result, Seurat returns a projected gene expression matrix. In our study, we first normalized the dataset, selected 2000 highly variable genes (HVGs) for anchors finding. For those datasets, whose genes amount are <2000 after filtering only to the one-to-one orthologous genes before integration and renaming the gene names accordingly, we selected all genes for anchors finding. After integration, we scaled the output and transformed it into principal component (PC) space. Finally, we extracted the top 30 PCs for further evaluation.
Harmony
Harmony (36) uses iterative clustering algorithm to remove all the batch effects from datasets. It firstly clusters cells by maximizing clustering diversity. For each cluster, Harmony subsequently calculates the cluster specific centroids and cell specific correction factors, followed by adjusting each cell by the correction factors. These processes are repeated until convergence. As a result, Harmony returns a corrected embedding. We ran Harmony (version 0.1.1) within R language by following its tutorial https://portals.broadinstitute.org/harmony/articles/quickstart.html. We first normalized and scaled datasets and selected top 2000 HVGs. For the datasets with <2000 genes after filtering based on the one-to-one orthologous genes, we used all the genes. We ran Harmony by setting the maximal number of clusters as 50 and the maximal number of iterations as 100 while leaving the other parameters as default settings. The corrected embedding outputs were used for further evaluation.
fastMNN
fastMNN (32), a method based on MNN, was employed to mitigate batch effects between datasets by leveraging MNN pairs identified on the PC space. To execute fastMNN, we utilized the SeuratWrapper package (version 0.3.1) and followed the tutorial available at https://github.com/satijalab/seurat-wrappers/blob/master/docs/fast_mnn.md. Prior to applying fastMNN, we performed data normalization, scaling and selection of top 2000 HVGs for each dataset. For the datasets with <2000 genes after the one-to-one orthologous genes filtering, we selected all genes. We ran fastMNN with its default settings, and then extracted the resulting embedding output for subsequent evaluation and analysis.
Batch-balanced k-nearest neighbors (BBKNN)
BBKNN (34), a widely used data integration method, leverages the k-nearest neighbor (kNN) graph approach to capture cell similarities within and across batches. In our analysis, we applied BBKNN using the Python library bbknn. To construct the kNN graph, BBKNN calculates the pairwise distances between cells within each batch. It then identifies the k nearest neighbors for each cell based on these distances. The parameter ‘neighbors_within_batch = 5″ was set to define the number of neighbors to consider within each batch during the graph construction step. This parameter choice determines the level of local connectivity within individual batches. After constructing the kNN graph, BBKNN proceeds to integrate the data by merging the graphs from different batches. It aligns the cells across batches by matching their shared nearest neighbors, facilitating the integration of cell populations that exhibit similar gene expression patterns. The parameter ‘trim = 50″ was set to trim the edges in the merged graph, removing low-confidence connections that may arise from batch effects or noisy data. Following integration, BBKNN employs an algorithm similar to the Uniform Manifold Approximation and Projection (UMAP) algorithm to compute the connectivity scores for each cell based on the neighborhood graph structure, revealing the underlying cellular relationships. The output weighted neighborhood graph is used for the evaluation.
scVI
scVI (19), a hierarchical Bayesian model based neural network, was employed to model the expression of each cell with the negative binomial distribution. The model incorporates non-linear transformations to capture and account for the dataset-specific random effects. To utilize scVI, we followed the tutorial available at https://docs.scvi-tools.org/en/latest/tutorials/notebooks/scrna/harmonization.html. In this analysis, we projected the input data into a 30-dimensional latent space using scVI (version 0.19.0), while leaving other parameters at their default values.
scGen
scGen (37), a neural network-based method, leverages variational autoencoders and latent space vector arithmetics to model and predict the gene expression changes in different cell types under perturbations. In our analysis, we utilized scGen (version 2.1.0) by following the tutorial provided at https://scgen.readthedocs.io/en/stable/tutorials/scgen_batch_removal.html. We set the maximum number of epochs to 100 and the batch size to 32, while leaving other parameters at their default values. The return corrected latent was used for downstream evaluation.
Scanorama
Scanorama (33) is a method specifically designed for integrating single-cell transcriptomic datasets by leveraging the concept of mutually nearest neighbors across different datasets. In our analysis, we utilized Scanorama (version 1.7) by following the tutorial provided at https://github.com/brianhie/scanorama#api-example-usage. We set the batch size parameter to 30, the kNN parameter to 10, and the return_dimred parameter to True. Scanorama outputs corrected gene expression matrices as integration results, which were further evaluated to assess the performance in integrating the cross-species transcriptomic datasets.
SATURN
SATURN (38) is a deep learning method designed specifically for integrating cross-species single-cell transcriptomic datasets. It employs a unique approach by leveraging both single-cell RNA expression data and protein embeddings derived from protein language models to map the datasets into a low-dimensional space. This is achieved through a pretrained conditional autoencoder structure. In our analysis, we followed the tutorial provided by SATURN at https://github.com/snap-stanford/SATURN/tree/main/Vignettes to run the method. We set the num_macrogenes and hv_genes as default. By utilizing SATURN, we aimed to capture the functional relationships between genes rather than relying solely on genomic similarity for integration. The return embedding was used for further evaluation.
SAMap
SAMap (12) is a comprehensive integration method that leverages both protein sequence similarity and gene expression correlation to facilitate the integration of cross-species single-cell transcriptomic datasets. First, SAMap calculates MNN between species by considering the similarity of protein sequences and the correlation of gene expression profiles. This step enables the identification of biologically relevant cell-to-cell correspondences across species. Second, SAMap utilizes the cross-species MNN to transform the feature space from different species into a unified, stitch joint space. To run SAMap in our analysis, we followed the tutorial provided at https://github.com/atarashansky/SAMap/blob/main/SAMap_vignette.ipynb. The output of SAMap is a cell-to-cell neighborhood graph, which captures the relationships and similarities between cells from different species. The neighborhood graph is used for further evaluation.
Evaluated metrics
Similar to scIB, we treated the species labels as batch effects. We considered the evaluation metrics from two aspects: the batch effect removal and biological variance conservation. For evaluating batch effect removal, we utilized the following metrics: Adjusted Rand Index (ARI) for batch labels, Adjusted Silhouette Width (ASW) for batch labels, integrated Local Inverse Simpson’s Index (iLISI), Normalized Mutual Information (NMI) for batch labels, kBET statistic, Principal Component Regression for batch labels and graph connectivity. These metrics collectively captured the ability of the methods to mitigate batch effects and integrate the data from different species. All the metrics were computed based on the method outputs.
To assess the conservation of biological variability, we employed the following metrics: ARI for cell type labels, ASW for cell type labels, integrated cell type Local Inverse Simpson’s Index (cLISI), NMI for cell type labels, HVG conservation and trajectory conservation. These metrics allowed us to evaluate the methods’ capacity to preserve the biological information and capture the underlying heterogeneity across cell types.
ARI
ARI (42) measures the similarity between the assigned labels (e.g. cell types or batches) and the true labels, providing an indication of the accuracy of the integration. Similar to the pipeline described by Tran et al. (14), we first randomly subsampled 80% cells from the joint embeddings or the top 30 PCs outputs to perform k-means clustering using the stats package in R language environment where k is the number of unique cell types/batches. For clustering graph-based outputs, we utilized the Leiden algorithm implemented in the scanpy.tl.leiden function (40) with resolution parameters ranging from 0.2 to 2.0 in increments of 0.1. We selected the clustering result with the highest ARI with respect to known cell type labels for further analysis. ARI calculations were performed using the adjusted_rand_score function from the scikit-learn package. All the computing was repeated 20 times to ensure robustness. For the cell type evaluation, we calculated the average ARI with respect to cell types (denoted as ARIc). A high ARIc score indicates precise matching among cell types and strong conservation of cell type information, while a low score suggests random labeling.
For the batch effect removal evaluation, we modified the ARI computation to minimize the influence of cell type differences. Specifically, we calculated the ARI between the batch labels and cluster labels within each shared cell type separately. For each shared cell type, we first selected all cells belonging to that cell type across different batches. Then, we performed k-means clustering with k equal to the number of batches (species), which was followed by computing the ARI between the resulting cluster labels and the true batch labels. Finally, we averaged the ARI scores across all cell types to obtain the final batch ARI score (denoted as ARIb). To ensure that higher scores indicate better batch effect removal, we adjusted the ARIb using the equation:
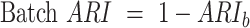 |
ASW
The Average Silhouette Width (ASW) (43) metric measures the quality of clustering by assessing the cohesion within clusters and the separation between clusters for individual cells. For a cell i, silhouette width method first calculates the minimum average distance to all other cells that do not belong to the same cluster with cell i. Next it is subtracted by the average distance of i from the other cells in the same clusters. Then it is further divided by the larger of these two values. We calculated the ASW by averaging the value of silhouette width for all the cells after clustering which is described as previous. ASW ranges from –1 to 1. The higher value of ASW represents the higher quality of clustering. It provides insights into the preservation of cell type information and the effectiveness of batch mixing. We also proposed a variation of ASW, denoted as ASW (species-specific cell type) by only focusing on the species-specific cell types. The workflow is the same as the original ones, except that this variation only considers this metric and performs the averaging solely on species-specific cell types.
In line with Tran et al. (14), we applied a consistent pipeline to compute ASW scores from both the batch and cell type perspectives. To ensure computational efficiency, we randomly subsampled the datasets to 80% of the original number of cells. The top 30 PCs or the entire joint embeddings of the down sampled datasets were used to calculate the distances between data points, enabling the computation of ASW scores. This process was repeated 20 times to obtain 20 ASW scores for both batch mixing and cell type conservation separately. All ASW scores were subsequently normalized to a range from 0 to 1, using below equation:
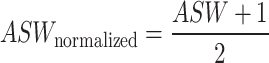 |
For batch mixing, we scaled the normalized batch ASW scores using below equation, as a higher score indicates better mixing among species:
 |
Finally, we used the average normalized ASWbatch and ASWcelltype for final evaluation.
LISI
Local Inverse Simpson’s Index (LISI) (36) scores provide insights into the randomness of cell pairs within the same group when the group is observed only once. LISI scores can be computed for both cell types (cLISI) and batches (iLISI) measuring how well the cell type is preserved and the batch mixing separately. The range of LISI scores is from 1 to the number of unique cell types or batches present in the datasets. A higher cLISI score means the group containing more cell types, representing a poor cell type information conservation. A higher iLISI score means the group containing more batches representing a good batch mixing. To calculate LISI scores, we followed the same pipeline as Tran et al. (14). For methods that output a pseudo-count matrix, we employed the first 30 PCs to compute the kNNs. Similarly, for methods that generate embeddings directly, we used the embedding to calculate the kNN. In the case of SAMap and BBKNN, we utilized its output, which is a kNN graph, to determine the LISI scores.
To normalize the LISI scores, we scaled them to a range of 0 to 1 using the equation below for cLISI and iLISI separately. For the LISI scores, higher value suggests better species mixing/celltype conservation in the integration.
 |
 |
NMI
NMI is a metric used to quantify the similarity between two sets of clusters. NMI score ranges from 0 to 1. If NMI equals to 1, it means the two clustering results are perfect agreement. If NMI close to 0, it means the clustering results are not more informative about each other than clustering by chance. In our evaluation, we employed NMI to compare the cell type labels as well as the batch (specie) labels with the Louvain clusters computed on the integrated datasets. By utilizing the scIB pipeline (13), we computed NMI scores for both cell types and batches.
For NMI cell type, the higher scores indicate a more accurate match between the clustering cells and specific cell types, implying a better preservation of cell type information across the integrated datasets. For NMI batch, we reversed it by subtracting it from 1. Thus, a higher NMI batch score indicates a higher degree of batch mixing by the integration methods.
kBET
The kNN batch effect test (kBET) (44) is a metric used to quantify the degree of batch mixing in integrated datasets. It assesses how well the samples from different batches intermingle with each other from repeated subsampling cells randomly. Higher kBET scores indicate a greater degree of batch mixing, suggesting a successful removal of batch effects and enhanced integration of the data from different batches. Conversely, lower kBET scores indicate a reduced level of batch mixing, signifying a better preservation of batch-specific signals and decreased unwanted batch effects in the integrated datasets.
Since kBET works on the kNN graphs, for the non-graph outputs (PCs based on the pseudo-count matrix, the joint embedding output), we utilized the top 30 dimensions of the PCs or the entire joint embeddings to calculate kNN graphs. We set the value of k (the number of nearest neighbors) to 50, which allows us to capture the local neighborhood relationships between cells. Further, since the k-nearest-neighborhood size could be different from the graph-based method outputs (for example, BBKNN and SAMap), we adopted the extension kBET pipeline outlined in the scIB (13) to compute the kBET scores. By applying kBET, we obtained a numerical score that reflects the degree of batch mixing in the integrated datasets.
Principal component regression
PC regression (44) is a metric used to quantify the level of batch effect removal achieved by integration methods. It leverages the hypothesis that batch variables are correlated with the PCs obtained from the integrated dataset. This metric only works for the non-graph-based output methods. To calculate the score, we followed the pipeline provided by scIB (13). First, we performed principal component analysis with default 50 PCs on the pseudo-count outputs and embedding outputs to obtain a set of PCs that capture the major sources of variation in the data. Next, we performed linear regression to model the relationship between the batch variables and each PC. Specifically, we calculated the contribution of the batch effect to each PC by multiplying the variance explained by the ith PC and the corresponding regression coefficient obtained from the linear regression model between the batch effect and the ith PC. The score is then computed by summarizing the variance contributions of the batch variables across all PCs. By assessing the extent to which batch variables are associated with the PCs, principal component regression score provides insights into the effectiveness of integration methods in mitigating batch effects. A higher score signifies a greater reduction in batch-related variation, indicating improved batch effect removal in the integrated dataset.
Graph connectivity
Graph connectivity (GC) (13) is a metric used to assess the degree of connectivity between cells of the same label in a kNN graph. In our analysis, we employed the pipeline provided by scIB (13) to calculate the graph connectivity score. First, the kNN graph was constructed based on the pseudo-count metric or the embedding outputs, where the number of nearest neighbors (k) was determined as 50. Then we subsampled the largest graph component from the kNN graph which only contains the nodes of a given cell label. Further, the number of the nodes in the subsampled graph component is divided by the total number of the cell label. For each cell label, we repeated this procedure and sum all the values followed by divided by the number of unique cell labels. Thus, the graph connectivity score ranges from 0 to 1, with higher values indicating the nodes with the same cell labels are well connected. We also proposed a variation of GC, denoted as GC (species-specific cell type) by only focusing on the species-specific cell types. The workflow is the same as the original ones, except that this variation only considers this metric and performs the averaging solely on species-specific cell types.
Highly variable gene conservation
HVG conservation is a metric used to evaluate how well the biological signal, represented by genes with high variability across cells, is preserved after integration of datasets. This metric only works on the pseudo-count metric outputs.
To calculate the HVG conservation score, we utilized the pipeline provided by scIB (13). First, we identified 500 HVGs within each individual dataset after the one-to-one orthologous gene filtering. For the datasets that contain <500 genes, we tried to identify half number of their total genes as HVGs.
Next, we calculated the HVGs from the integrated expression profiles. For each dataset, we then computed the number of overlapping genes between the HVGs identified before and after integration, which is further divided by the minimax number of the HVGs before and after integration. Finally, we average the values for each dataset as the HVG conservation score, which ranging from 0 to 1.
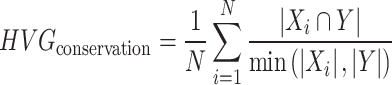 |
N is the total number of datasets. Xi is the number of HVGs in the dataset i before integration. Y is the number of the HVGs in the integrated dataset.
A higher HVG conservation score indicates a better preservation of the biological signal, as a larger proportion of HVGs are maintained across the integrated dataset.
Trajectory conservation
To evaluate the preservation of trajectory information after integration, we employed the concept of trajectory conservation. This metric quantifies the extent to which the integrated datasets maintain the original trajectory relationships observed in the individual species datasets.
In our analysis, we utilized the pipeline provided by scIB (13) to calculate the trajectory conservation score. We employed the diffusion pseudotime (DPT) function in Scanpy (sc.tl.dpt) to infer trajectories before and after integration. We set the most extremal cells in the cell type cluster as the starting cell of the trajectory after integration. To quantify the conservation of trajectories, the Spearman’s rank correlation coefficient between the trajectories calculated before and after integration was computed. This coefficient measures the monotonic relationship between the two sets of trajectory values, indicating the degree of conservation. The resulting trajectory conservation score was further scaled to a range from 0 to 1, with higher values indicating better preservation of trajectory information.
Ranking and metric aggregation
To comprehensively evaluate the performance of the integrated methods, we computed an overall score that considers both batch effect removal and biological variance conservation. Specifically, the batch effect removal metrics contain ARIbatch, ASWbatch, graph iLISI, NMIbatch, kBET, principal component regression score and graph connectivity. The biological variance conservation metrics contain ARIcelltype, ASWcelltype, graph cLISI, HVG conservation and trajectory conservation. For each sub-metric in batch effect removal metrics and bioconservation metrics, it was scaled using the min-max number for each experiment. Then, all the sub-metric scores are averaged separately for batch effect removal and bioconservation score. Further, we assigned a weight of 0.6 to biological variance conservation, indicating its greater importance compared to batch effect correction. By assigning this weight, we emphasized the significance of preserving the biological heterogeneity and capturing the underlying biological signals in the integrated datasets. A higher overall score indicates a method’s ability to effectively mitigate batch effects while preserving the biological variability in the integrated datasets, which is to prioritize the accurate representation of cell-type-specific gene expression patterns and functional differences across species.
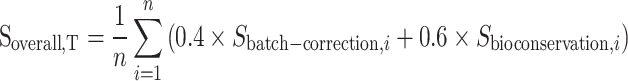 |
Soverall,T donates the overall score for a specific cross-species integration task T, which contains n different experiments/replicates. Sbatch-correction,i donates the average scaled score of the valid sub-metric from batch correction for a specific run i. Sbioconservation,i donates the average scaled score of the valid sub-metric from the bioconservation for a specific run i. For the final overall score for all the tasks, we averaged the overall scores.
Cell type tree phylogenetic inference
We followed the method described by Jasmine et al. (45) to construct the cell type tree for cat and dog lung tissues and seven species (Schmidtea mediterranea, Danio rerio, Ciona intestinalis, Mus musculus, Homo sapiens, Drosophila melanagaster and Caenorhabditid elegans) separately. Briefly, we filtered out the cell types in each species which has <100 cells based on the embeddings derived from SATURN. After that we performed the principal component analysis to the selected embeddings. The top 20 PCs were extracted for downstream analysis. For each cell type in each species, we randomly selected one cell to reduce the computational burden. We used contml from PHYLIP (version 3.698) to construct the cell type phylogenetic tree based on the Brownian motion model. Finally, in order to test the technical repeatability and biological repeatability of the tree, we calculated the jumble scores and scjackknife scores respectively for each tip in the tree following the previous description (45).
Results
Cross-species scRNA-seq integration benchmarking
We benchmarked nine widely used cross-species integration methods across 36 distinct tasks (Table 1) (1–3,5,30,46–57), spanning the entire taxonomic hierarchy. Each task embodied distinct challenges that are commonly faced in cross-species integration. One of the main challenges of benchmarking cross-species data integration is identifying the homolog-based gene space shared between species. We addressed the challenge through a specifically designed pipeline: all the datasets were processed with the same preprocessing protocol (13). We reduced the gene sets from both species to one-to-one orthologous with OrthoFinder (41) (see ‘Materials and methods’ section) for the methods requiring the same genes as input. For methods based on homologous genes between species, we identified homologous genes from different species based on protein sequence similarity.
Table 1.
Integration tasks for benchmarking
| Task | Species | Technology | Number of cells | Target |
|---|---|---|---|---|
| Cross genus | Felis catus (Cat) (46) and Panthera tigris altaica (Tiger) (46) | 10x Genomics | 27 200 | Different genus, same tissue |
| Felis catus (Cat) (47) and Panthera tigris altaica (Tiger) (46) | 10x Genomics | 55 469 | Different genus, different tissue | |
| Cross family | Felis catus (Cat) (46) and Canis lupus familiaris (Dog) (46) | 10x Genomics | 18 490 | Different family, same tissue |
| Felis catus (Cat) (47) and Canis lupus familiaris (Dog) (46) | 10x Genomics | 50 014 | Different family, different tissue | |
| Homo sapiens (Human) (48) and Macaque fascicularis (Monkey) (48) | 10x Genomics | 25 083 | Different family, same tissue | |
| Homo sapiens (Human) (48) and Macaque mulatta (Monkey) (48) | 10x Genomics | 18 159 | Different family, same tissue | |
| Cross order | Homo sapiens (Human) (49) and Mus musculus (Mouse) (50) | Microwell-seq | 84 638 | Different order, multiple tissue, contain nested batch |
| Nematostella vectensis (Sea anemone) (30) and Stylophora pistillata (Hard coral) (51) | MARS-seq | 26 661 | Different order, atlas level integration | |
| Homo sapiens (Human) (48) and Mus musculus (Mouse) (48) | 10x Genomics | 22 630 | Different order, same tissue | |
| Mus musculus (Mouse) (48) and Macaque fascicularis (Monkey) (48) | 10x Genomics | 10 295 | Different order, same tissue | |
| Mus musculus (Mouse) (48) and Macaque mulatta (Monkey) (48) | 10x Genomics | 4485 | Different order, same tissue | |
| Homo sapiens (Human) (48) and Sus scrofa (Pig) (48) | 10x Genomics | 19 273 | Different order, same tissue | |
| Mus musculus (Mouse) (48) and Sus scrofa (Pig) (48) | 10x Genomics | 5926 | Different order, same tissue, diverse cell type integration, both species have their unique cell types | |
| Macaque fascicularis (Monkey) (48) and Sus scrofa (Pig) (48) | 10x Genomics | 14 035 | Different order, same tissue, diverse cell type integration, both species have their unique cell types | |
| Macaque mulatta (Monkey) (48) and Sus scrofa (Pig) (48) | 10x Genomics | 4430 | Different order, same tissue, diverse cell type integration, both species have their unique cell types | |
| Cross class | Xenopus tropicalis (Frog) (1) and Danio rerio (Zebrafish) (5) | Indrop RNA-seq | 160 306 | Different class, embryo developmental time series data integration |
| Danio rerio (Zebrafish) (50) and Homo sapiens (Human) (48) | Microwell-seq 10x Genomics | 5586 | Different class, same cell types integration | |
| Danio rerio (Zebrafish) (50) and Macaque mulatta (Monkey) (48) | Microwell seq 10x Genomics | 6511 | Different class, same cell types integration | |
| Danio rerio (Zebrafish) (50) and Macaque fascicularis (Monkey) (48) | Microwell seq 10x Genomics | 4894 | Different class, same cell types integration | |
| Danio rerio (Zebrafish) (50) and Sus scrofa (Pig) (48) | Microwell seq 10x Genomics | 5007 | Different class, same cell types integration | |
| Danio rerio (Zebrafish) (50) and Felis catus (Cat) (46) | Microwell seq 10x Genomics | 10 031 | Different class, same cell types integration | |
| Cross phyla | Strongylocentrotus purpuratus (Sea urchin) (52) and Danio rerio (Zebrafish) (50) | 10x Genomics Microwell-seq | 71 225 | Across phyla, different sequencing technology, imbalanced dataset integration |
| Strongylocentrotus purpuratus (Sea urchin) (52) and Danio rerio (Zebrafish) (50) | 10x Genomics Microwell-seq | 125 359 | Across phyla, different sequencing technology, imbalanced dataset integration | |
| Strongylocentrotus purpuratus (Sea urchin) (52) and Danio rerio (Zebrafish) (50) | 10x Genomics Microwell-seq | 133 131 | Across phyla, different sequencing technology, imbalanced dataset integration | |
| Strongylocentrotus purpuratus (Sea urchin) (52) and Danio rerio (Zebrafish) (50) | 10x Genomics Microwell-seq | 276 935 | Across phyla, different sequencing technology, imbalanced dataset integration | |
| Strongylocentrotus purpuratus (Sea urchin) (52) and Danio rerio (Zebrafish) (50) | 10x Genomics Microwell-seq | 493 471 | Across phyla, different sequencing technology, imbalanced dataset integration | |
| Strongylocentrotus purpuratus (Sea urchin) (52) and Danio rerio (Zebrafish) (50) | 10x Genomics Microwell-seq | 710 007 | Across phyla, different sequencing technology, imbalanced dataset integration | |
| Strongylocentrotus purpuratus (Sea urchin) (52) and Danio rerio (Zebrafish) (50) | 10x Genomics Microwell-seq | 926 543 | Across phyla, different sequencing technology, imbalanced dataset integration | |
| Mus musculus (Mouse) (53) and Monomorium pharaonis (Ant) (54) | Microwell-seq snRNA-seq | 11 301 | Across phyla, different technology, common cell type, data quality influence | |
| Mus musculus (Mouse) (49) and Monomorium pharaonis (Ant) (54) | Microwell-seq snRNA-seq | 9487 | Across phyla, different technology, common cell type, data quality influence | |
| Drosophila melanogaster (Fly) (50) and Danio rerio (Zebrafish) (50) | Microwell-seq | 229 256 | Across phyla, time series data | |
| Caenorhabditis elegans (Roundworm) (55) and Homo sapiens (Human) (49) | sci-RNA-seq Microwell-seq | 374 515 | Across phyla, atlas level integration | |
| Octopus vulgaris (Octopus) (56) and Homo sapiens (Human) (49) | 10x Genomics Microwell-seq | 30 706 | Across phyla, different technology, same tissue (brain) integration | |
| Schmidtea mediterranea (Flatworm) (3) and Homo sapiens (Human) (49) | Drop-seq Microwell-seq | 394 562 | Across phyla, atlas level integration | |
| Ciona intestinalis (Sea vase) (2) and Nematostella vectensis (Sea anemone) (30) | 10x Genomics MARS-seq | 16 430 | Across phyla, similar stage, atlas level integration | |
| Trichoplax adhaerens (57) and Homo sapiens (Human) (49) | MARS-seq Microwell-seq | 348 608 | Across phyla, atlas level integration |
The performance of each integration method was assessed using 13 different metrics across two primary categories: batch effect removal and biological variance conservation (bioconservation). Batch effect removal for each cell identity label was quantified via metrics such as batch adjusted rand index (batch ARI) (58), k-nearest-neighbor batch effect test (kBET) (44) and average silhouette width across batches (batch ASW) (44). Beyond species/batch labels, we further evaluated batch effect removal using integration local inverse Simpson’s Index (iLISI) (36), batch normalized mutual information (batch NMI) (59), principal component regression (44) and graph connectivity. For assessing bioconservation, metrics such as cell type ARI (58), cell type integration local inverse Simpson’s Index (cLISI) (36), cell type normalized mutual information (cell type NMI) (59), average silhouette width across cell types (cell type ASW) (44), overlaps of HVGs per batch before and after integration, and conservation of trajectories were utilized.
We calculated an overall score to provide a comparative analysis of each method’s performance, considering batch effect removal and biological variance conservation elements. This score is devised by attributing a 40% weight to the average batch effect removal scores and a 60% weight to the average biological variance conservation scores. This weighting strategy mirrors the significance of these two aspects in appraising the efficacy of integration methods, providing a balanced and comprehensive overview of each method’s performance (13).
Integrating data across genera
To assess the efficacy of various methods in integrating data across genera, we started our study with a straightforward setting: benchmarking data integration from the same tissue but different genera within the same family. Specifically, we used lung samples from cat and tiger, each encompassing data from 12 distinct cell types generated using the same sequencing protocol. In this task, SATURN emerged as the top performer (Figure 2B and C; Supplementary Figure S1).
Figure 2.

Benchmarking results for the cross-genus integration task. (A) Overview of the methods ranked by average overall scores, with detailed average batch correction and average bioconservation scores for two cross-genus integration tasks. Overall scores are computed based on batch correction scores and bioconservation scores (‘Materials and methods’ section). (B and C) UMAP layouts visualizing unintegrated and integrated Felis catus (cat) and Panthera tigris altaica (tiger) lung datasets colored by species labels (B) and cell type labels (C).
The congruency of the overall cross genus species integration metric results (Figure 2A) and the integrated data plots (Figure 2B and C; Supplementary Figure S1) highlights the effectiveness of certain methods: high-performing methods adeptly integrated the different species while maintaining the biological variance at the cell type level.
Remarkably, the top five methods excelled in either batch correction or bioconservation but struggled to perform well in both simultaneously. This phenomenon may be attributed to the difficulty of differentiating species-specific variance from batch effects. Most methods available to date aim to construct a unified embedding across species by reducing variances, which comprise both species-specific biological variances and batch effects. Striking a balance between removing batch effects and preserving biological information, thus, becomes crucial. Methods like SATURN excelled in bioconservation but not in batch correction, while other methods like scVI and scGen showed the opposite pattern. Notably, batch effect removal metrics, like batch ARI and graph connectivity, played a crucial role in distinguishing the overall top five performers. SATURN, scVI, scGen and Seurat v4 CCA achieved higher scores on these metrics. Conversely, BBKNN exhibited residual species-specific clustering in all cell types, such as fibroblasts (Figure 2C and Supplementary Figure S2), suggesting its relatively lower effectiveness in fully mitigating batch effects.
Furthermore, in the context of conserving biological variance in cross-genus integration, SATURN outperformed other methods. It attained the highest scores in cell type ARI, cell type ASW, cLISI and cell type NMI, indicating a successful capture of underlying cell type heterogeneity across datasets.
In a more comprehensive task, we extended our analysis to integrate data from cat and tiger across three distinct cell types sourced from different tissues. Scanorama emerged as one of the top performers, followed by scGen (Supplementary Figures S1 and S2). Notably, as the integration task became more challenging, two methods from the previous top five performers (SAMap and Seurat v4 CCA) fell behind in the overall score even though they still performed well in bioconservation and batch correction separately.
As both datasets were based on the same 10x sequencing technology, this initial analysis underscored the complexity of integrating single-cell data across genera, revealing both the strengths and limitations of current methods in tackling such a task.
Integrating species beyond the genus
In the context of integrating species data beyond the genus, we designed intricate tasks to assess the performance of methods at various taxonomic levels, including cross-family, cross-order, cross-class and cross-phylum integration (Table 1). Similar to the cross-genus integration task, cross-family (e.g. cat and dog within the same order) and cross-order (e.g. sea anemone and hard coral within the same class) tasks encountered fewer technical batch effects, as those datasets originated from the same sequencing protocol. However, with the increased phylogenetic distance, the sequencing technologies varies due to natural differences across species, especially in cross-class (e.g. frog and zebrafish within the same phylum) and cross-phylum integration tasks (e.g. zebrafish and sea urchin).
In cross-family species integration scenarios, SATURN emerged as the best performer in most cases (Figure 3A and Supplementary Figures S3–S7). Notably, SATURN, scGen, Seurat v4 CCA and scVI consistently secured top four positions, mirroring their performance in the cross-genus integration task (Figure 2A and Supplementary Figure S3E). From cross-family to cross-order integration tasks, fastMNN, Harmony and SAMap stood out with superior performance and elevated their rankings. fastMNN and scGen consistently outperformed with higher scores in both bioconservation and batch correction, surpassing the average scores across all methods (Figure 3B). SATURN distinguished itself by excelling in bioconservation but showed relatively lower performance in batch correction. This observation suggests that SATURN prioritizes the preservation of meaningful biological signals over stringent batch correction. On the other hand, Scanorama, while achieving consistently high scores in batch correction, ranked lower. This is attributed to its subpar performance in bioconservation, indicating a potential trade-off where Scanorama excels in mitigating batch effects but struggles to effectively preserve crucial biological information, influencing its overall ranking (Supplementary Figures S3E and S8–S17).
Figure 3.

Benchmarking results for the integration tasks across family, order and class species pairs. (A) Performance of all methods in cross-family integration, ranked by average overall scores with detailed average scores for batch correction and bioconservation. (B) Scatter plot of the average overall batch correction score against the average overall bioconservation score for all cross-order species integration tasks. The error bars represent the standard errors across the tasks. The vertical dashed line represents the average batch correction score for all the methods in cross-order species integration tasks. The horizontal dashed line represents the average bioconservation score for all methods in cross-order species integration tasks. (C) Box plot of batch correction score and bioconservation score in cross-class species integration tasks. The purple dashed line represents average batch correction scores for all methods in cross-class species integration tasks. The pink dashed line represents average bioconservation scores for all methods in cross-class species integration tasks.
As we transitioned from cross-order to cross-class integration tasks, the disparity between batch correction and bioconservation increased. No method achieved the average scores in both aspects simultaneously. All methods, except BBKNN, SAMap and SATURN, exhibiting a higher score in bioconservation, performed better in batch correction than in bioconservation. This observation suggests that methods have their distinct preferences in batch correction and bioconservation as the taxonomic distances between two species increase (Figure 3C; Supplementary Figures S18–S24). Interestingly, SATURN and SAMap outperformed other methods when integrating the cell atlas of Xenopus tropicalis (frog) and Danio rerio (zebrafish) (Supplementary Figures S18 and S19). However, SAMap encountered challenges in achieving robust performance when integrating non-atlas level data, which consist of a limited number of cell types (Supplementary Figures S18 and S20–S24). This highlights SAMap’s effectiveness in handling well-defined atlases data but underscores limitations in scenarios where data are less comprehensive or lacks a predefined reference.
The average bioconservation scores across all methods declined as the integration moving from cross-class to cross-phylum. Notably, no method surpassed the average batch-correction and average bioconservation scores at the same time. SATURN, SAMap and BBKNN excelled in bioconservation while the other methods performed well in batch correction (Figure 4A; Supplementary Figures S25–S41).
Figure 4.

Benchmarking results for integration cross-phylum tasks. (A) Lollipop plot showing average performance in batch correction and bioconservation after scaling. Vertical dashed lines represent the average batch correction scores and bioconservation scores across all methods. (B) Bar plot of the overall batch correction scores and bioconservation scores across seven cross-phylum tasks. (C and D) Line plot of the overall scores (C) and bioconservation scores (D) for the integration of Homo sapiens and Macaque fascicularis, Homo sapiens and Mus musculus, Homo sapiens and Sus scrofa, Homo sapiens and Danio rerio, Homo sapiens and Octopus vulgaris, Homo sapiens and Schmidtea mediterranea from left to right, respectively.
Influence of phylogenetic distance on integration performance
We further investigated how increasing phylogenetic distance affects method performance by integrating Homo sapiens data with species at varied phylogenetic distances: Macaca fascicularis, Mus musculus, Sus scrofa, Danio rerio, Octopus vulgaris and Schmidtea mediterranea (Figure 4B–D and Supplementary Figure S25B).
As the phylogenetic distance between species increases, all methods show a decline in performance for both biological conservation and batch correction, indicating that integrating phylogenetically distant species is more challenging than integrating closely related ones. This trend is particularly evident in the biological conservation scores. For methods that utilize one-to-one orthologous genes as input, this decline can be explained by the decreasing number of one-to-one orthologous genes identified at greater phylogenetic distances (Figure 4C and D; Supplementary Figure S25B). Furthermore, we found that the decreasing trend also holds true for SAMap and SATURN. However, they generally exhibited greater robustness and outperformed the other methods. This result is supported by their superior performance in cross-phylum species integration tasks (Supplementary Figure S26). We hypothesize that their ability to handle homologous genes as input makes them more effective in integration tasks of phylogenetically distant species.
To test our hypothesis and to further assess the impact of input gene sets on the performance of SATURN and SAMap, we selected five integration tasks across five cross-species categories (see Supplementary Table S5 for task details) and used either one-to-one orthologs or homologous genes as input for these two methods. By comparing the model performance using the two different inputs, we aimed to determine whether their superior performance arises from the use of homologous genes or from their inherent algorithmic design.
Our analysis revealed that switching from homologous genes to one-to-one orthologs resulted in a performance decrease for both methods across all phylogenetic levels. Specifically, SATURN and SAMap exhibited average performance drops of 11.04% and 12.05%, respectively. For SATURN, there was a clear trend: the greater the phylogenetic distance between species, the more substantial the performance decline when using one-to-one orthologs (Supplementary Figure S42A). This suggests that homologous genes offer more comprehensive information for cross-species integration tasks than one-to-one orthologs as they capture a broader range of evolutionary relationships.
Additionally, we compared SATURN and SAMap using one-to-one orthologs as input (denoted as SATURN(one2one) and SAMap(one2one) separately) against other methods (Supplementary Figures S42B and Supplementary Figure S43–S47). Despite the reduced input gene set, SATURN(one2one) still ranked as one of the best performers among all methods benchmarked. This result underscores the robustness of SATURN’s algorithmic design, particularly its use of pretrained protein language models, in capturing biologically meaningful signals even with a limited gene set.
Time trajectory conservation, species-specific cell types, nested batch and imbalanced datasets influence
To investigate in detail into other factors that may influence cross-species integration, we first focused on one application of general interest: time trajectory preservation. Across development, time occurs as an important perturbational factor and thus causes subtle, nested variations among the population. A competitive integration method should be carefully designed to remove undesirable technical variation while preserving the time-related information inherent to different samples. Besides, cross-species integration tasks often involve datasets with nested batches within species, including variation arising from both biological and experimental differences. Moreover, datasets usually contain species-specific cell types, and these unique cell types should not be integrated but preserved for meaningful biological interpretations. Additionally, the integrated datasets may not have the same dataset size and sequencing depth. In this section, we evaluated the performance of the nine methods in handling the real time trajectory information from embryonic developmental datasets.
For the time series scRNA-seq data, cells from each time point bear both temporal information and batch effects. The main challenge of integrating such kind of data is to seamlessly integrate cells of identical types while retaining essential developmental time details. For the real-time trajectory conservation of frog and zebrafish embryonic developmental datasets (cross-class task) integration, scGen and Seurat v4 CCA performed better than SAMap and SATURN (Figure 5A and Supplementary Figure S48). This observation suggests that SAMap and SATURN may fail to learn effective representations that reflect the meaningful temporal progression when integrating the datasets, while scGen and Seurat v4 CCA achieve better trajectory conservation at the cost of reduced integration performance.
Figure 5.

Benchmarking results for integration tasks involving time series, nested batches and imbalanced datasets. (A) UMAP plot of the time trajectory of the embryo development data of zebrafish and frog (cross-class integration task). (B) Bar plot of the overall batch correction scores and the overall bioconservation score in Homo sapiens and Mus musculus integration with nested batches task. (C) Line plot for the overall bioconservation score in integrating 60 399 cells from sea urchin (Strongylocentrotus purpuratus) and different subsample percentage of the zebrafish (Danio rerio, total 1 082 680 cells) dataset. Subsample 6% from the zebrafish dataset is the balanced data size with the sea urchin dataset.
The presence of species-specific cell types poses a significant challenge in cross-species integration, as mixing those cells with the other species will lead to misinterpretations in downstream biological analyses. To evaluate how different integration methods handle species-specific cell types, we selected one representative integration task from each phylogenetic category (Supplementary Table S6).
scGen showed superior performance in isolating species-specific cell types in the cross-genus, cross-family and cross-order tasks, as indicated by higher ASW and GC scores for species-specific cell types (Supplementary Figure S49). This suggests that scGen's variational autoencoder framework effectively captures data structure and preserves unique cell types when sufficient orthologous genes are present. In contrast, SATURN outperformed other methods in the cross-class and cross-phylum tasks where phylogenetic distance was greater and the number of orthologous genes limited. Its use of contrastive learning and pretrained protein language models allows it to capture evolutionary distant gene relationships, enabling better isolation of species-specific cell types under these challenging conditions.
In some instances, the GC (species-specific cell type) and ASW (species-specific cell type) metrics provided contradictory results for the same method. For example, in certain tasks, SATURN exhibited a high GC (species-specific cell type) score but a low ASW (species-specific cell type) score. This discrepancy may result from the GC metric’s dependence on the predefined parameter k in the kNN graph, which might not optimally capture the desired structure for all datasets. Additionally, the GC metric can be affected by the global graph topology, making it less reliable for assessing the isolation of species-specific cell types in some cases. Therefore, we recommend prioritizing the ASW metric when evaluating the separation of species-specific cell types.
To assess the performance of nested batch correction, we used a dataset consisting of human and mouse samples from five common tissues. However, the mouse dataset included brain tissue, which was absent in the human dataset. A good integration method should successfully integrate the cells from corresponding tissues between mouse and human while isolating the cells from mouse brain tissue.
In this analysis, SATURN, scGen, and BBKNN emerged as the top three performers overall, showcasing exceptional proficiency in species matching (Figure 5B and Supplementary Figure S48). Notably, these three methods demonstrated impressive capabilities in preserving the distinct characteristics of mouse brain cells. However, BBKNN achieved successful integration only for a subset of cells from the two species. scGen encountered challenges in separately integrating bladder and kidney data. Additionally, SATURN integrated cell types successfully but failed to integrate muscle tissues between mouse and human (Supplementary Figure S48). This observation suggests that no method can effectively preserve spatial variance among cell types and manage complex integration scenarios encompassing variations in tissue composition and species-specific attributes.
In addition, we evaluated various methods on imbalanced datasets, utilizing zebrafish and sea urchin datasets (Figure 5C; Supplementary Figures S26, S48C and D, S34–S39). The zebrafish dataset, comprising 1 082 680 cells, contrasted starkly with the sea urchin dataset, which contained only 60 399 cells. In this task, we investigated whether the disparities in dataset size would impact the performance in bioconservation and batch correction. We achieved this by subsampling all cell types in the zebrafish dataset at intervals of 1%, 6%, 20%, 40%, 60% and 80%.
Further, we ran the integration pipeline for each method at these different scales three times to record the running time (Supplementary Figure S50) and computational resources (Supplementary Table S7) utilized and evaluated the algorithms’ robustness (Supplementary Table S8 and Supplementary Figures S51–S53). We found that methods like Harmony, scVI and Scanorama are highly efficient, scaling up to millions of cells with a running time of <1 h. In terms of robustness, graph-based methods like BBKNN and SAMap demonstrated consistently reliable performance, even when applied to datasets with millions of cells. In contrast, deep-learning algorithms, which involve mini-batch sampling and stochastic gradient optimization, exhibited more variability across runs, resulting in reduced robustness compared to graph-based methods.
From the bioconservation perspective, SATURN and BBKNN consistently preserved biological information well, particularly under extreme conditions while other methods did not generalize well. On the other hand, from the batch correction perspective, Scanorama displayed minimal changes, maintaining stability across all subsampling levels. Conversely, SAMap exhibited consistently improved performance in batch correction as subsampling increased. This nuanced exploration of method performance under varying subsampling conditions provides valuable insights into their robustness and adaptability in the face of imbalanced dataset sizes.
Further, we investigated the influence of data quality on the integration performance of the methods by integrating two distinct mouse datasets with an ant dataset of two kinds of neuronal cell types. The first mouse dataset (mouse1) had a median UMI/cell of 488, while the second mouse dataset (mouse2) was three times deeper sequenced than mouse1 with a median UMI/cell of 1499. Among the evaluated methods, SATURN and BBKNN exhibited a notable increase in overall score in both batch correction and bioconservation when transitioning from lower quality data (mouse1) to higher quality data (mouse2) (Supplementary Figures S48E–G, S40 and S41). Notably, SATURN excelled in bioconservation even with low sequencing depth, suggesting inherent robustness to data quality variations. In contrast, SAMap and Seurat v4 CCA exhibited increased batch correction scores and decreased bioconservation scores. scVI performed best with low sequencing depth data for batch correction. These findings imply that specific methods have varying sensitivity to data quality, with inconsistent effects. Some methods are affected in batch correction, while others are impacted in bioconservation or both, potentially due to distinct assumptions and models used by these methods. Researchers should consider the quality of their data when deciding on an appropriate integration method to ensure reliable and accurate integration of datasets.
Overall, this analysis underscored the pivotal role played by data characteristics, encompassing factors such as time trajectory variations, species-specific cell types, nested batch effects, imbalanced dataset size and data quality in shaping the performance of integration methods. It emphasized the necessity for meticulous evaluation of these factors when selecting an integration approach for cross species integration.
The cell type tree of life
Understanding the evolutionary relationships between cell types across different species is a fundamental question in biology. Cells are the basic units of life, and tracing homologous cell types across organisms can provide deep insights into how cell types have evolved, diversified or been conserved throughout evolution. Leveraging cross-species integration of single-cell RNA-seq data allow us to reconstruct phylogenetic relationships at the cellular level, offering a unique perspective on cell type evolution beyond traditional morphological or functional classifications.
We inferred two unrooted phylogenetic cell type trees. The first tree is a ‘fine-grained’ cell type tree of cat (Felis catus) and dog (Canis lupus familiaris) lung tissue datasets (Figure 6A). The second tree is a more ‘coarse-grained’ cell type tree from seven species spanning a larger phylogenetic distance (Schmidtea mediterranea, Danio rerio, Ciona intestinalis, Mus musculus, Homo sapiens, Drosophila melanogaster and Caenorhabditis elegans; Figure 6B). Based on our benchmarking results we used the integrated embeddings derived from SATURN for the generation of both cell type trees.
Figure 6.

Unrooted cell type trees for cat and dog lung tissue and seven phylogenetically distant species separately. (A) Seven cell types from cat and dog cluster in the cell phylogeny based on the integrated embedding derived from SATURN. (B) Forty-five cell types from seven model species (Schmidtea mediterranea, Danio rerio, Ciona intestinalis, Mus musculus, Homo sapiens, Drosophila melanogaster and Caenorhabditis elegans) cluster in the cell phylogeny based on the integrated embedding derived from SATURN. Species and cell types are labeled at the tips. Node support values are printed as ‘jumble score/scjackknife score’. MYA: million years ago.
The lung cell type tree from cat and dog encompasses seven cell type groups and was inferred from the top 20 PCs calculated from SATURN embeddings as phylogenetic characters (see ‘Materials and methods’ section). Robustness of the cell type tree was assessed by calculating ‘jumble scores’ and ‘scjackknife scores’ for technical and biological repeatability, respectively. The jumble score measures the robustness of a given tree topology when constructing the tree with the same set of cells but different algorithm initializations, while the scjackknife score describes the robustness of a given tree topology with different set of cells (45). Higher scores in both settings indicate more reliable tree topologies.
As anticipated, the ‘fine-grained’ cell type tree reveals that similar cell types cluster together, irrespective of branch length. This observation underscores the significance of cell type signals, as illustrated in Figure 6A. Moreover, the clustering of cell types is intricately linked to tissue-specific functions. Notably, T cells, endothelial cells, fibroblasts and ATII, all contributing to the internal defenses of the respiratory system, form a cohesive cluster. Similarly, secretory and ciliated cells, instrumental in external respiratory defenses, share a cluster, while ATI, focused on the gas exchange process, occupies a distinct cluster. This clustering strongly suggests that our tree construction method as well as SATURN integration adeptly captures the underlying functional characteristics of various cell types across species.
In contrast, the ‘coarse-grained’ multi species cell type tree exhibits conserved clustering only for certain cell types, as exemplified by neurons (Figure 6B). This clustering implies a common evolutionary origin and fundamental conservation among species. Additionally, the tree unveils species-specific cell type clustering, exemplified by the cell types from Schmidtea mediterranea. This intricate pattern hints at a complex interplay between conserved and species-specific cellular identities, likely reflecting adaptations to the distinct biological demands of specific species. Furthermore, the observation that certain cell types exhibited both conserved and species-specific clustering adds another layer of complexity. For example, two distinct muscle cell clades were identified. Notably, human endothelial cells and muscle cells from Ciona intestinalis and Caenorhabditis elegans expressed some shared marker genes, including ATP6V0D1, ERH, MGAT2, NUS1, PSMC3, PSMD8, RER1, RSL24D1, TCP1 and WDR43. These genes are relevant to proteasome function, various types of N-glycan biosynthesis, terpenoid backbone biosynthesis, and collecting duct acid secretion, indicating potential functional connections among these cell types. The dual nature of the cell types grouped in the muscle cells cluster suggests a modular functionality, where some aspects of cellular identity are evolutionarily stable, while others dynamically adapt to the specific needs of individual species. These findings highlight the nuanced and dynamic nature of cell type evolution, showcasing a balance between conservation and adaptation in the intricate tapestry of multicellular organisms.
Similar to the species taxonomy, the cell type phylogenetic tree enables a phylogenetic exploration of cell type relationships across different species, which will enable us to better understand the evolution at the cell type resolution. However, it is important to note that the current ‘coarse-grained’ cell type tree represents an initial attempt at construction. With a fine-grained view and evolutionary related species, we can get a better and accurate tree (60).
Discussion
Integrating single-cell RNA-seq data across species poses a formidable challenge, necessitating careful consideration of factors such as phylogenetic distance, batch effects, and preservation of biological signals. In this study, we systematically assessed and compared the performance of nine integration methods across a myriad of cross-species integration tasks, shedding light on their respective strengths and limitations.
Upon analyzing the results from 36 cross-species integration tasks, a discernible influence of phylogenetic distance on the ranking of integration methods emerged (Figure 7A and B; Supplementary Figure S54). Overall, methods leveraging biological sequence information exhibited superior performance in bioconservation, with notable examples being SATURN and SAMap. SATURN, a deep learning-based approach, employs a pre-trained language model to transform the homologous genes into universal embeddings, i.e. a low-dimensional representation of the data. Through weighing the connection between genes based on universal embeddings, SATURN can generate powerful cell embeddings crucial for meaningful cross-species comparisons. However, SATURN’s suboptimal performance in batch effect removal may be attributed to its reliance on contrastive learning which focuses on aligning similar cells by maximizing similarity between certain cell pairs. This approach may not effectively model and correct complex technical batch effects. In contrast, generative models like scGen and scVI explicitly separate biological signals from technical noise, leading to better batch correction performance.
Figure 7.

Overall performance of all methods and a guideline in cross-species scRNA-seq data integration tasks. (A) Scatter plot of the average overall batch correction score against average overall bioconservation score for the selected methods based on 36 integration tasks. Dashed lines indicate the average scores across all the methods. (B) The average overall scores and ranking of all methods in different cross-species integration tasks. (C) Scenario-specific decision-tree-style guidelines for cross-species scRNA-seq data integration.
Similarly, SAMap uses BLAST to extract protein sequence similarities between species to construct a gene homology graph. Further, through the gene homology graph, SAMap projects the cross-species scRNA-seq datasets to a joint space, which is connected by the mutual cross-species nearest neighborhoods. While robust in certain integration tasks, SAMap encounters challenges in simpler cross-genus, cross-family species integration and non-atlas level integration, attributed to the insensitivity of BLAST-derived features to taxonomic close species. Even when using one-to-one ortholog gene sets as input, SAMap and SATURN outperformed the other methods in most tasks, suggesting that the use of BLAST similarity scores and protein language models helps these methods to better capture biological signals. Conversely, Harmony and BBKNN demonstrate suboptimal performance across most tasks, likely due to their linear assumptions regarding batch effects, proving too simplistic for cross-species integration tasks. As the phylogenetic distance between two species increases, these linear assumptions fail to capture the heterogeneity across species effectively.
Our study underscores the importance of meticulous consideration of data characteristics, including sequencing depth, species-specific cell types, nested batch effects, imbalanced datasets and time-trajectory variations, for method selection. SATURN showcased robustness and flexibility in handling such challenges, whereas other methods displayed sensitivity. We also observed that different methods exhibited varied preferences in batch correction and bioconservation. SATURN, BBKNN and SAMap consistently outperformed others in bioconservation, prioritizing biological information preservation. In contrast, scGen, scVI, Seurat v4 CCA and Scanorama exceled in batch correction, emphasizing the removal of batch effects. A trade-off between batch effects removal and biological conservation, especially in overlapping scenarios, becomes apparent (13).
In order to provide a reference for researchers to select appropriate methods for cross-species integration across various scenarios, we introduced a decision tree based on our benchmark results considering both batch correction scores and bioconservation scores since these two aspects measure how well the two species’ cell is mix and how well the biological information is preserved after integration (Figure 7C). We first provided recommendations based on the phylogenetic distance of the given species pairs. For example, when integrating species from different families but the same order (cross-family), we recommend using SATURN, scGen, scVI or Seurat v4 CCA. When integrating cross-phylum species, we recommend using SATURN, SAMap or BBKNN. Additionally, we provide guidelines for common scenarios encountered in cross-species integration studies. For example, given two datasets with imbalanced scales, where one has millions of cells and another contains only several thousand cells, we recommend employing SATURN, SAMap or fastMNN for the integration task. Researchers could further use a combination of top performing methods to reduce potential biases associated with any specific method. We further provide a guideline only considering the methods’ performance in bioconservation for researchers who are more interested in the biological information conservation metrics (Supplementary Figure S55).
Integrating multiple species presents challenges due to the decreasing number of one-to-one orthologous genes as more species are included, potentially compromising integration quality. Alternatively, methods that utilize homologous genes beyond one-to-one orthologs, such as SATURN and SAMap, are more robust when integrating multiple batches and handling large phylogenetic distances. These approaches help mitigate the loss of informative genes and maintain integration quality across multiple datasets.
Cross-species integration in scRNA-seq data analysis is a powerful tool for unraveling biological complexities. Particularly, using cross-species data allows for tracing the evolution of cell types across species and time. By comparing gene expression patterns across species, insights into the evolutionary conservation of cellular processes emerge, facilitating the identification of conserved pathways and shared regulatory networks. This approach transcends the limitations of studying individual species, offering a broader perspective on cellular diversity and function. Moreover, it enables knowledge transfer from well-studied to less-explored species, deepening our understanding of their biology and uncovering species-specific innovations and adaptations.
We would like to note that our benchmark study has focused only on cross-species integration of scRNA-seq data. Future work should extend the assessment to other data types like scATAC-seq. Additionally, addressing challenges related to the identification of one-to-one orthologous gene relationships is crucial, as the current integration methods may overlook valuable information in one-to-many and many-to-many homologous relationships. Based on the fact that no method performs well in both bioconservation and batch correction, a natural extension could be constructing a self-supervised learning-based method, for example, generative pretraining (61) as well as contrastive learning-based models (62,63), and further refining these algorithms for integrated embeddings. Furthermore, the cell type tree we crafted does not capture the full spectrum, given its exclusive reliance on scRNA-seq data in the current methodology. Future iterations could enhance comprehensiveness by incorporating additional data, such as cell ontology information, for a more holistic tree construction. Moreover, the intrinsic heterogeneity in single-cell transcriptomics poses challenges to the reproducibility of tree construction. Similar to genetic variation, cell types may undergo drift, split or loss over evolutionary time, resulting in a paraphyletic relationship among labeled cells of the same type. This dynamic nature can lead to the absence of one-to-one corresponding cell types across species (60). This inherent variability might contribute to the presence of some counter-factual branches observed in our ‘coarse-grained’ tree.
In conclusion, our comprehensive evaluation of integration methods in cross-species integration tasks provides valuable insights into their performance across different phylogenetic distances and data qualities. This study thus contributes to the understanding of the strengths and limitations of various integration methods, aiding researchers in selecting the most appropriate approach for their specific cross-species integration studies.
Supplementary Material
Acknowledgements
This research used the computational resources from the Supercomputing Laboratory at King Abdullah University of Science & Technology (KAUST) in Thuwal, Saudi Arabia.
Contributor Information
Huawen Zhong, BioEngineering Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Wenkai Han, Computer Science Program, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Klarman Cell Observatory, Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA.
David Gomez-Cabrero, BioEngineering Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Unit of Translational Bioinformatics, Navarrabiomed—Fundación Miguel Servet, Universidad Pública de Navarra (UPNA), IdiSNA, Pamplona, Spain.
Jesper Tegner, BioEngineering Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Computer Science Program, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Unit of Computational Medicine, Department of Medicine, Center for Molecular Medicine, Karolinska Institutet, Karolinska University Hospital, L8:05, SE-171 76 Stockholm, Sweden; Science for Life Laboratory, Tomtebodavagen 23A, SE-17165 Solna, Sweden.
Xin Gao, Computer Science Program, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Center of Excellence on Smart Health, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Center of Excellence for Generative AI, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Guoxin Cui, BioEngineering Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Marine Science Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Manuel Aranda, BioEngineering Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia; Marine Science Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia.
Data availability
Datasets are collected from published studies, as described in Supplementary Table S2 and Supplementary Table S3. The preprocessed datasets for each task are available at https://figshare.com/s/6187811b6c3fae02a4d3. The output data from all runs are available in Supplementary Table S9-Supplementary Table S11. The source code and the corresponding step by step manual is available at https://figshare.com/s/2f65bfa7032ffbd199c9.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
This study was funded by King Abdullah University of Science and Technology (KAUST). Funding for open access charge: King Abdullah University of Science and Technology (KAUST).
Conflict of interest statement. The authors declare no conflicts of interest.
References
- 1. Briggs J.A., Weinreb C., Wagner D.E., Megason S., Peshkin L., Kirschner M.W., Klein A.M. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 2018; 360:eaar5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao C., Lemaire L.A., Wang W., Yoon P.H., Choi Y.A., Parsons L.R., Matese J.C., Levine M., Chen K. Comprehensive single-cell transcriptome lineages of a proto-vertebrate. Nature. 2019; 571:349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fincher C.T., Wurtzel O., de Hoog T., Kravarik K.M., Reddien P.W. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science. 2018; 360:eaaq1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu M., Zheng X., Fan C.-M., Zheng Y. Lineage dynamics of the endosymbiotic cell type in the soft coral Xenia. Nature. 2020; 582:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wagner D.E., Weinreb C., Collins Z.M., Briggs J.A., Megason S.G., Klein A.M. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science. 2018; 360:981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Plass M., Solana J., Wolf F.A., Ayoub S., Misios A., Glažar P., Obermayer B., Theis F.J., Kocks C., Rajewsky N. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science. 2018; 360:eaaq1723. [DOI] [PubMed] [Google Scholar]
- 7. Siebert S., Farrell J.A., Cazet J.F., Abeykoon Y., Primack A.S., Schnitzler C.E., Juliano C.E. Stem cell differentiation trajectories in Hydra resolved at single-cell resolution. Science. 2019; 365:eaav9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Musser J.M., Schippers K.J., Nickel M., Mizzon G., Kohn A.B., Pape C., Ronchi P., Papadopoulos N., Tarashansky A.J., Hammel J.U. Profiling cellular diversity in sponges informs animal cell type and nervous system evolution. Science. 2021; 374:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arendt D., Bertucci P.Y., Achim K., Musser J.M. Evolution of neuronal types and families. Curr. Opin. Neurobiol. 2019; 56:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shafer M.E. Cross-species analysis of single-cell transcriptomic data. Front. Cell Dev. Biol. 2019; 7:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J., Sun H., Jiang M., Li J., Zhang P., Chen H., Mei Y., Fei L., Lai S., Han X. Tracing cell-type evolution by cross-species comparison of cell atlases. Cell Rep. 2021; 34:108803. [DOI] [PubMed] [Google Scholar]
- 12. Tarashansky A.J., Musser J.M., Khariton M., Li P., Arendt D., Quake S.R., Wang B. Mapping single-cell atlases throughout Metazoa unravels cell type evolution. eLife. 2021; 10:e66747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luecken M.D., Büttner M., Chaichoompu K., Danese A., Interlandi M., Müller M.F., Strobl D.C., Zappia L., Dugas M., Colomé-Tatché M. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods. 2022; 19:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tran H.T.N., Ang K.S., Chevrier M., Zhang X., Lee N.Y.S., Goh M., Chen J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020; 21:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeng H. What is a cell type and how to define it?. Cell. 2022; 185:2739–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Woych J., Ortega Gurrola A., Deryckere A., Jaeger E.C.B., Gumnit E., Merello G., Gu J., Joven Araus A., Leigh N.D., Yun M. et al. Cell-type profiling in salamanders identifies innovations in vertebrate forebrain evolution. Science. 2022; 377:eabp9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lotfollahi M., Naghipourfar M., Luecken M.D., Khajavi M., Büttner M., Wagenstetter M., Avsec Ž., Gayoso A., Yosef N., Interlandi M. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 2022; 40:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Michielsen L., Lotfollahi M., Strobl D., Sikkema L., Reinders M., Theis F.J., Mahfouz A. Single-cell reference mapping to construct and extend cell type hierarchies. NAR Genom. Bioinform. 2023; 5:lqad070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018; 15:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang F., Wang W., Wang F., Fang Y., Tang D., Huang J., Lu H., Yao J. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intellig. 2022; 4:852–866. [Google Scholar]
- 21. Pliner H.A., Shendure J., Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods. 2019; 16:983–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pasquini G., Arias J.E.R., Schäfer P., Busskamp V. Automated methods for cell type annotation on scRNA-seq data. Comput. Struct. Biotechnol. J. 2021; 19:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018; 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryu Y., Han G.H., Jung E., Hwang D. Integration of single-cell RNA-seq datasets: a review of computational methods. Mol. Cells. 2023; 46:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng C., Chen W., Jin H., Chen X. A review of single-cell rna-seq annotation, integration, and cell–cell communication. Cells. 2023; 12:1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Forcato M., Romano O., Bicciato S. Computational methods for the integrative analysis of single-cell data. Brief. Bioinf. 2021; 22:bbaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stuart T., Satija R. Integrative single-cell analysis. Nat. Rev. Genet. 2019; 20:257–272. [DOI] [PubMed] [Google Scholar]
- 28. Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M. The human cell atlas. eLife. 2017; 6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindeboom R.G., Regev A., Teichmann S.A. Towards a human cell atlas: taking notes from the past. Trends Genet. 2021; 37:625–630. [DOI] [PubMed] [Google Scholar]
- 30. Sebé-Pedrós A., Saudemont B., Chomsky E., Plessier F., Mailhé M.-P., Renno J., Loe-Mie Y., Lifshitz A., Mukamel Z., Schmutz S. Cnidarian cell type diversity and regulation revealed by whole-organism single-cell RNA-seq. Cell. 2018; 173:1520–1534. [DOI] [PubMed] [Google Scholar]
- 31. Song Y., Miao Z., Brazma A., Papatheodorou I. Benchmarking strategies for cross-species integration of single-cell RNA sequencing data. Nat. Commun. 2023; 14:6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haghverdi L., Lun A.T., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018; 36:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hie B., Bryson B., Berger B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 2019; 37:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Polański K., Young M.D., Miao Z., Meyer K.B., Teichmann S.A., Park J.-E. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics. 2020; 36:964–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M. Integrated analysis of multimodal single-cell data. Cell. 2021; 184:3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-r., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019; 16:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lotfollahi M., Wolf F.A., Theis F.J. scGen predicts single-cell perturbation responses. Nat. Methods. 2019; 16:715–721. [DOI] [PubMed] [Google Scholar]
- 38. Rosen Y., Brbić M., Roohani Y., Swanson K., Li Z., Leskovec J. Toward universal cell embeddings: integrating single-cell RNA-seq datasets across species with SATURN. Nat. Methods. 2024; 21:1492–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lotfollahi M., Hao Y., Theis F.J., Satija R. The future of rapid and automated single-cell data analysis using reference mapping. Cell. 2024; 187:2343–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018; 19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emms D.M., Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019; 20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rand W.M. Objective criteria for the evaluation of clustering methods. J. Am. Statist. Assoc. 1971; 66:846–850. [Google Scholar]
- 43. Rousseeuw P.J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987; 20:53–65. [Google Scholar]
- 44. Büttner M., Miao Z., Wolf F.A., Teichmann S.A., Theis F.J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods. 2019; 16:43–49. [DOI] [PubMed] [Google Scholar]
- 45. Mah J.L., Dunn C.W. Cell type evolution reconstruction across species through cell phylogenies of single-cell RNA sequencing data. Nat. Ecol. Evol. 2024; 8:325–338. [DOI] [PubMed] [Google Scholar]
- 46. Chen D., Sun J., Zhu J., Ding X., Lan T., Wang X., Wu W., Ou Z., Zhu L., Ding P. Single cell atlas for 11 non-model mammals, reptiles and birds. Nat. Commun. 2021; 12:7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eaton D.M., Berretta R.M., Lynch J.E., Travers J.G., Pfeiffer R.D., Hulke M.L., Zhao H., Hobby A.R., Schena G., Johnson J.P. Sex-specific responses to slow progressive pressure overload in a large animal model of HFpEF. Am. J. Physiol. Heart Circul. Physiol. 2022; 323:H797–H817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Zyl T., Yan W., McAdams A., Peng Y.-R., Shekhar K., Regev A., Juric D., Sanes J.R. Cell atlas of aqueous humor outflow pathways in eyes of humans and four model species provides insight into glaucoma pathogenesis. 2020; 117:10339–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Han X., Zhou Z., Fei L., Sun H., Wang R., Chen Y., Chen H., Wang J., Tang H., Ge W. Construction of a human cell landscape at single-cell level. Nature. 2020; 581:303–309. [DOI] [PubMed] [Google Scholar]
- 50. Wang R., Zhang P., Wang J., Ma L., E W., Suo S., Jiang M., Li J., Chen H., Sun H. Construction of a cross-species cell landscape at single-cell level. Nucleic Acids Res. 2023; 51:501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Levy S., Elek A., Grau-Bové X., Menéndez-Bravo S., Iglesias M., Tanay A., Mass T., Sebé-Pedrós A. A stony coral cell atlas illuminates the molecular and cellular basis of coral symbiosis, calcification, and immunity. Cell. 2021; 184:2973–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Foster S., Oulhen N., Wessel G. A single cell RNA sequencing resource for early sea urchin development. Development. 2020; 147:dev191528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fei L., Chen H., Ma L., E W., Wang R., Fang X., Zhou Z., Sun H., Wang J., Jiang M. Systematic identification of cell-fate regulatory programs using a single-cell atlas of mouse development. Nat. Genet. 2022; 54:1051–1061. [DOI] [PubMed] [Google Scholar]
- 54. Li Q., Wang M., Zhang P., Liu Y., Guo Q., Zhu Y., Wen T., Dai X., Zhang X., Nagel M. A single-cell transcriptomic atlas tracking the neural basis of division of labour in an ant superorganism. Nat. Ecol. Evol. 2022; 6:1191–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cao J., Packer J.S., Ramani V., Cusanovich D.A., Huynh C., Daza R., Qiu X., Lee C., Furlan S.N., Steemers F.J. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017; 357:661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Styfhals R., Zolotarov G., Hulselmans G., Spanier K.I., Poovathingal S., Elagoz A.M., De Winter S., Deryckere A., Rajewsky N., Ponte G. Cell type diversity in a developing octopus brain. Nat. Commun. 2022; 13:7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sebé-Pedrós A., Chomsky E., Pang K., Lara-Astiaso D., Gaiti F., Mukamel Z., Amit I., Hejnol A., Degnan B.M., Tanay A. Early metazoan cell type diversity and the evolution of multicellular gene regulation. Nat. Ecol. Evol. 2018; 2:1176–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hubert L., Arabie P. Comparing partitions. J. Classification. 1985; 2:193–218. [Google Scholar]
- 59. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 60. Church S.H., Mah J.L., Dunn C.W. Integrating phylogenies into single-cell RNA sequencing analysis allows comparisons across species, genes, and cells. Plos Biology. 2024; 22:e3002633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cui H., Wang C., Maan H., Pang K., Luo F., Wang B. scGPT: towards building a foundation model for single-cell multi-omics using generative AI. Nat. Methods. 2024; 21:1470–1480. [DOI] [PubMed] [Google Scholar]
- 62. Han W., Cheng Y., Chen J., Zhong H., Hu Z., Chen S., Zong L., Hong L., Chan T.-F., King I. Self-supervised contrastive learning for integrative single cell RNA-seq data analysis. Brief. Bioinf. 2022; 23:bbac377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rosen Y., Roohani Y., Agrawal A., Samotorcan L., Consortium T.S., Quake S.R., Leskovec J. Universal cell embeddings: a foundation model for cell biology. 2023; bioRxiv doi:29 November 2023, preprint: not peer reviewed 10.1101/2023.11.28.568918. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets are collected from published studies, as described in Supplementary Table S2 and Supplementary Table S3. The preprocessed datasets for each task are available at https://figshare.com/s/6187811b6c3fae02a4d3. The output data from all runs are available in Supplementary Table S9-Supplementary Table S11. The source code and the corresponding step by step manual is available at https://figshare.com/s/2f65bfa7032ffbd199c9.