

Abstract
Alzheimer's disease (AD) is a complex, incurable neurological condition characterized by cognitive decline, cholinergic neuron reduction, and neuronal loss. Its exact pathology remains uncertain, but multiple treatment hypotheses have emerged. The current treatments, single or combined, alleviate only symptoms and struggle to manage AD due to its multifaceted pathology. The developmental drugs target pivotal disease factors involved in the envisaged hypotheses and include targets such as amyloid aggregation, hyperphosphorylated tau proteins, and receptors like cholinergic, adrenergic, etc. Present-day research focuses on multi-target directed ligands (MTDLs), which inhibit multiple factors simultaneously, helping slow the disease's progression. This review attempts to collate the recent information related to proposed hypotheses for AD etiology. It systematically organizes the advances in various therapeutic options for AD, with a particular emphasis on clinical candidates. Also, it is expected to help medicinal chemists design novel AD treatments based on available information, which could be helpful to AD patients.
This review collates the recent information related to proposed hypotheses for AD etiology and advances in various therapeutic options, with a particular emphasis on clinical candidates.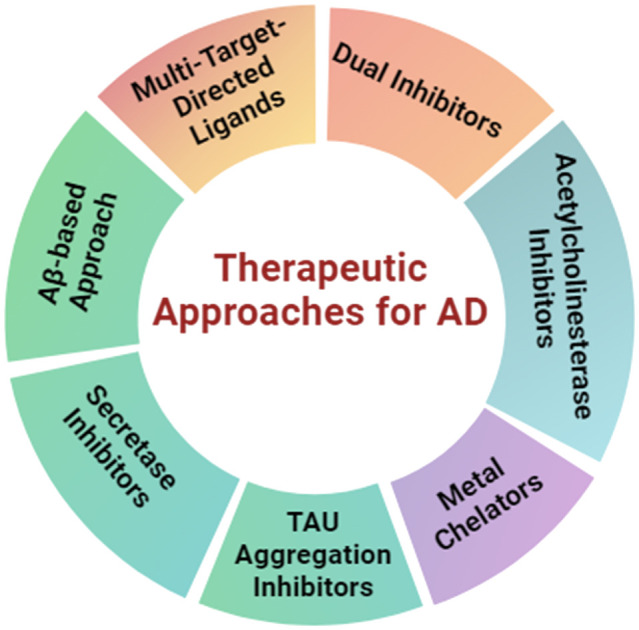
1. Introduction
AD is a progressive neurodegenerative condition, particularly in those 65 years or older, and is a leading cause of dementia.1 It gradually erodes cognitive abilities and interpersonal engagement due to brain cell degeneration. As of 2023, about 55 million people worldwide have dementia, with AD accounting for 60–70% of these cases. By 2050, this number is expected to reach 139 million due to the aging global population. Females are at a higher risk of AD. In India, over 4 million individuals have various forms of dementia, with Alzheimer's as the most widespread type.2
A century since AD's discovery, the disease's precise pathogenesis remains elusive and not fully comprehended. A combination of factors, encompassing the abnormal folding and aggregation of proteins, frequently associated with oxidative stress and the generation of free radicals, result in AD.3 Further, bioenergetics, irregularities in mitochondrial function, and neuroinflammation processes are involved in its complications. Understanding these factors, aided by the latest research on AD pathogenesis, has laid the foundation for research into potential treatments. However, the absence of animal models accurately mirroring the human AD pathogenesis remains a significant hurdle.4 Given the multifaceted nature of the disease, concentrating solely on one causative factor has proven ineffective or comparatively less impactful. Also, designing selective drugs that target causative factors is a significant challenge. Developing multi-target directed ligands (MTDLs) is another challenge, as it can lead to various side effects.5 Biologicals have also been explored, such as aducanumab, lecanemab and donanemab, which are the sole approved disease-modifying drugs for Alzheimer's. These human monoclonal antibodies are specifically designed to target aggregated beta-amyloid proteins found in the brain lesions associated with AD. Other available treatments, mainly cholinergic drugs such as galantamine, rivastigmine, and donepezil, primarily focus on managing symptoms, as the degeneration of brain cells in Alzheimer's is irreversible. The inability to reverse brain cell damage remains a significant task in Alzheimer's therapy.6
The present review concisely discusses the latest findings and hypotheses concerning the underlying causes of AD to offer valuable insights and information to medicinal chemists. It discusses advancements in a broad spectrum of therapeutic strategies for AD, emphasizing promising candidates that have progressed to clinical trials, shedding light on their potential to offer effective treatment options.
2. Pathogenesis of AD
The precise mechanisms underlying the development of Alzheimer's disease (AD) are still not entirely elucidated. However, this complex condition has been linked to various symptoms and pathological features. These include the formation of neurofibrillary tangles (NFTs) within brain cells, the accumulation of amyloid-β (Aβ) in the form of senile plaques, increased oxidative stress, inflammation within neurons, and a decline in the functioning of cholinergic pathways, etc. While these factors are recognized in AD, the interplay and precise sequence of events leading to the disease are still under active investigation. A comprehensive understanding of AD's pathogenesis is crucial for the development of effective treatments and interventions for this devastating condition.7 The following are proposed hypotheses with the current understanding of AD and the present understanding of the interactions (Fig. 1).
Fig. 1. Pathogenesis of AD summarizing the central role of amyloid beta plaques.

A. The amyloid hypothesis
The widely accepted hypothesis for AD is the amyloid-cascade pathway, centered around the amyloid precursor protein (APP). Under normal conditions, amyloid precursor protein (APP) is cleaved by α-secretase, followed by γ-secretase, yielding harmless fragments.8 In AD, cleavage by β-secretase (BACE 1) followed by γ-secretase leads to the formation of amyloid β (Aβ) peptides, which aggregate and accumulate as extracellular plaques. Aβ is a hallmark of AD and comprises 37 to 43 amino acids, with the isoform Aβ42 being the most problematic. β-Amyloid peptides aggregate into plaques, triggering inflammation, brain damage, synaptic dysfunction, and tau protein hyperphosphorylation, leading to neurofibrillary tangle formation (Fig. 1).9 BACE 1 is a key target for therapies aiming to reduce amyloid plaque production to slow AD progression.10 The important milestones in the development of the amyloid cascade hypothesis and its practical utilization are presented in Table 1.
Table 1. Important events in developing the amyloid cascade hypothesis and practical implementations11.
| Year | Key research findings |
|---|---|
| 1906 | Senile plaques were first established |
| 1984 | Amyloid β protein (Aβ) was isolated as the principal constituent found within the plaques in the brains of individuals with AD |
| 1987 | The observation that the formation of Aβ resulted from the processing of amyloid precursor proteins |
| 1990 | Evidence of the neurotoxic properties of Aβ aggregates was presented |
| 1992 | The hypothesis of the amyloid cascade was put forward |
| 1995 | AD patients exhibit a significant decrease in CSF Aβ42 levels, revealing an established connection between Aβ and inflammation |
| 1997 | The discovery of the ability of Aβ42 to inhibit long-term potentiation |
| 1998 | The primary instigator of neuronal damage was identified as Aβ oligomers |
| 2001 | Evidence was provided to establish a connection between Aβ and the formation of neurofibrillary tangles |
| 2004 | The inaugural amyloid PET tracer, Pittsburgh compound-B (PIB), was formulated |
| 2016 | Light therapy led to a reduction in Aβ accumulation in both animal models of AD and patients |
| 2017 | The transfer of Aβ from the periphery through the blood–brain barrier into the brain is reported |
| 2018 | The ultrasensitive technology, Simoa, was devised for quantifying Aβ at sub-femtomolar concentrations |
| 2021 | Aducanumab became the inaugural FDA-approved medication for diminishing Aβ plaques |
| 2023 | FDA approval has been granted to lecanemab for the treatment of AD |
B. The tau (τ) protein hypothesis
The τ-protein is essential for microtubule stabilization, provides structural support, aids axonal transport, and promotes neuronal growth. Senile plaques trigger τ-protein hyperphosphorylation, leading to its aggregation with cytoskeletal proteins and reduced microtubule interaction. It elevates free τ-protein levels, promoting self-aggregation and fibril formation. Consequently, axonal transport is impaired, resulting in axonal degeneration due to disruption of nutrient transport. The compromised neurons eventually form neurofibrillary tangles (NTs), contributing to neurodegeneration (Fig. 1).12
C. The cholinergic deficit hypothesis
The cholinergic hypothesis posits that AD progression is primarily linked to the loss of cholinergic neurons and reduced acetyltransferase activity, resulting in decreased acetylcholine (ACh) (Fig. 2). As per the hypothesis, loss of limbic and neocortical cholinergic innervations due to neurofibrillary degeneration in the basal forebrain and associated loss of cholinergic neurotransmission results in decreased cognitive function. This degeneration primarily affects memory and cognition-related regions, like the hippocampus and frontal cortex, leading to compromised choline uptake, impaired ACh release, receptor imbalances, and disrupted neurotrophin support.13
Fig. 2. The cholinergic hypothesis of AD with their reported inhibitors.

D. Adrenergic hypothesis
In advanced AD, significant degeneration of noradrenergic neurons occurs in the locus coeruleus (LC), with a 30% loss during the transition to amnestic mild cognitive impairment (MIC) and an additional 25% reduction in AD progression. AD patients display variable adrenergic receptor expression, including decreased α1 adrenergic receptors (α1ARs) in the prefrontal cortex, hippocampus, and cerebellar hemisphere. Also, increased α1AR binding sites in specific layers, decreased α2 adrenergic receptors (α2ARs) in the nucleus basalis of Meynert, reduced β1 adrenergic receptors (β1ARs) in the cortex, and increased β2 adrenergic receptors (β2ARs) in the cortex and hippocampus. The role of beta-adrenergic receptor alterations in AD is still controversial. However, clinical investigations indicate that Amyloid beta peptide (Aβ) initiates subtle modifications in synaptic function during AD. Specifically, Aβ interacts with β2 adrenergic receptors within the central noradrenergic system, influencing synaptic functions in prefrontal cortical neurons. This interaction leads to the internalization and degradation of β2-adrenergic receptors, subsequently impairing adrenergic and glutamatergic activities and impacting cognitive function in AD.14
E. Glutamatergic hypothesis
Glutamatergic networks in the hippocampal regions are essential for cognitive function, with N-methyl d-aspartate receptors (NMDARs) playing a pivotal role in synaptic strength and long-term potentiation (LTP) crucial for memory. AD patients exhibit lower levels of vesicular glutamate transporters (VGLUT-1 and VGLUT-2) in the prefrontal cortex, indicating disrupted glutamatergic synapses. Soluble Aβ oligomers further disrupt glutamatergic networks, inhibiting LTP and inducing NMDAR hyperactivation. It also causes activation of ligand-gated or ionotropic glutamate receptors (iGluRs), predominantly the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subtype. The excess NMDAR activity raises intracellular Ca++ levels, triggering nitric oxide synthesis, free radical generation, oxidative stress (OS), apoptosis, and excitotoxic neuronal death. These changes further the AD pathogenesis caused by Aβ plaques by disrupting synaptic plasticity and neuronal function.15Fig. 3 illustrates the effect of normal synaptic transmission and long-term potentiation of the glutamatergic network in the brain.
Fig. 3. Effect of normal synaptic transmission and long-term potentiation of the glutamatergic network in the brain.

F. Calcium homeostasis hypothesis
Calcium serves as a crucial intracellular messenger and is important in vital physiological processes. Its precise control relies on complex mechanisms, with mitochondria and the endoplasmic reticulum (ER) playing central roles. ATPase Ca2+ pumps and the Na+–Ca2+ exchanger aid calcium efflux, while the ER membrane facilitates calcium exchange. Disruptions in these processes can lead to harmful intracellular calcium buildup, triggering protein cleavage, oxidative stress, energy disruption, and activation of proteins like β-amyloid and τ-protein. Aβ exacerbates calcium overload in AD by inducing oxidative stress and membrane pore formation, linking calcium dysregulation to AD pathology (Fig. 1).16
G. Oxidative stress hypothesis
Neurodegenerative disorders often involve an imbalance between reactive oxygen species (ROS) generation and antioxidant availability, resulting in cellular damage. AD exhibits disturbances in antioxidant enzyme activity, driven by Aβ's activation of NMDA receptors, which fosters ROS production. OS also contributes, in turn, to increased Aβ production and aggregation, as well as τ-protein hyperphosphorylation and polymerization (Fig. 1).17
H. Microgliosis
Microglia are the immune cells residing within the brain and are activated by β-amyloid plaques (Fig. 4). Upon activation, microglial cells release pro-inflammatory mediators, ROS, proteases, and complements, and secondly, as a defense mechanism, break down amyloid plaques. Though clusters of microglial cells gather around β-amyloid plaques, these are unable to break down the plaques effectively. This phenomenon, referred to as “frustrated phagocytosis,” contributes to neurodegenerative alterations.18
Fig. 4. Effects of activation of microglia.

I. Metal chelation hypothesis
β-Amyloid precipitates at low Zn2+ concentrations, Cu2+ and Fe3+ enhance aggregation, especially at slightly acidic pH (6.8). Elevated Cu2+ and Zn2+ levels in Alzheimer's patients are linked to apoE4 allele induction. β-Amyloid also possesses redox activity, reducing Cu2+ and Fe3+, generating H2O2, leading to ROS formation, exacerbating the pathology (Fig. 1).19
J. Prion-like behaviour of plaques
Prions are self-propagating proteins linked to neurodegenerative disorders like Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), and fatal familial insomnia (FFI).20 Aβ takes on a pathogenic conformation akin to prions, spreading in a cross-synaptic manner from specific brain regions. Tau protein also follows the same pattern with initially formed neurofibrillary tangles (NFTs), which are linked to cognitive decline.21
K. Ghrelin/GHSR1α signaling
GHSR1α, the ghrelin receptor in the hippocampus, regulates learning and memory through unique signaling involving Ca2+/calmodulin-dependent protein kinase II (CaMKII) and dopamine receptor D1 (DRD1). AD involves early hippocampal damage, possibly related to GHSR1α loss, leading to synaptic stress and memory problems. AD patients have been reported to have increased hippocampal GHSR1α, possibly as an Aβ defense mechanism (Fig. 5).22
Fig. 5. Amyloid plaques interacting with ghrelin receptors.

L. Cerebral capillaries constriction
Cerebrovascular disorders can lead to brain function alterations, and early signs of AD include angiogenesis damage and reduced cerebral blood flow. AD patients exhibit abnormal capillary contraction. Animal studies showed that introducing exogenous Aβ can lower cerebral blood flow in rats, triggering Aβ production. Aβ presence generates ROS via NOX, releasing endothelin (ET), which contracts pericytes via ETA receptors. This contraction causes pericyte necrosis, sustaining capillary constriction and resulting in ischemia. Crucially, Aβ oligomers are central to this complex process, linking vascular dysfunction to AD pathology (Fig. 1).23
M. Histamine and its receptors role in neuroinflammation in AD
In Alzheimer's disease patients, neurofibrillary tangles, the buildup of neuroinflammatory mediators in microglia, and extracellular amyloid plaques are commonly observed. Neuroinflammation is a persistent feature and plays a vital role in the disease's progression. While histamine is generally thought to trigger inflammatory responses in the peripheral system, increasing evidence suggests that it has a dual role in modulating microglial inflammatory responses in the central nervous system. H1R activation stimulates microglial activation and pro-inflammatory effects, whereas H2R activation produces inhibitory and anti-inflammatory outcomes. H3R is prominently expressed in both microglia and astrocytes. Acute to moderate microglial activation enhances the anti-inflammatory responses of M2 microglia, while chronic microglial activation contributes to neuroinflammation in Alzheimer's disease. The activation of microglia and astrocytes is considered a central driver of neuroinflammatory processes. When microglia are excessively activated, they produce high levels of cytotoxic factors, such as nitrogen oxides and prostaglandins, which damage neurons, leading to degeneration and cell death.24
N. Glycogen synthase kinase-3β and tau: an essential pair in AD
GSK-3β is a ubiquitous serine/threonine kinase initially recognized for its role in phosphorylating and inhibiting glycogen synthase. As a key regulator of numerous cellular processes, GSK-3β is central to cell metabolism and signaling, playing significant roles in both healthy and disease states. It has been implicated in various human disorders, including neurodegenerative diseases like AD. GSK-3β is thought to serve as a molecular link between Aβ and tau in the development of AD. Aβ activates GSK-3β, which then phosphorylates tau. Additionally, GSK-3β promotes the production of β-amyloid by upregulating β-amyloid cleaving enzyme-1 (BACE1) and presenilin-1 (PS1) and contributes to Aβ toxicity. The upregulation of GSK-3β also induces tau hyperphosphorylation, disrupts neuronal synaptic plasticity, and plays a role in the early onset of AD symptoms. Recent studies have shown that tau has acetyltransferase activity, enabling it to self-acetylate. This process also acetylates β-catenin, stabilizing it and allowing tau to exert its anti-apoptotic effects. These findings suggest that tau may directly acetylate GSK-3β, enhancing its activity and triggering a vicious cycle that contributes to chronic neurodegeneration, as seen in the progression of AD.25
O. Role of serotonin in AD
Serotonin (5-HT), also known as 5-hydroxytryptamine, is a biogenic amine that acts as a neurotransmitter. It functions at neuronal synapses, influencing cognition, mood, and sleep by binding to receptors on both neuronal and non-neuronal cell membranes. The 5-HT receptors involved in regulating physiological signaling pathways are classified into two main types: G-protein coupled receptors and ligand-gated ion channels. These receptors are further divided into seven groups: 5-HT1 (including 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, and 5-HT1F), 5-HT2 (including 5-HT2A, 5-HT2B, and 5-HT2C), 5-HT3, 5-HT4, 5-HT5 (including 5-HT5A and 5-HT5B), 5-HT6, and 5-HT7. Since the 5-HT7 receptor subtype is associated with neurogenesis and hippocampal neuronal function, its role in apoptosis and long-term potentiation in Alzheimer's disease has been investigated. Studies have shown that activating the 5-HT7 receptor can help alleviate synaptic dysfunction in Alzheimer's by reducing apoptosis in the hippocampus. However, this effect appears to be more linked to the treatment of psychotic symptoms in AD rather than cognitive enhancement. Hyperactivity of pyramidal neurons in the CA1 region of hippocampal circuit is an early sign of Alzheimer's disease. Abnormal serotonin signaling may contribute to this increased neural activity in the CA1 region of mice. Disruptions in 5-HT/5-HT3aR and/or 5-HT/5-HT1aR signaling have been found to cause heightened excitability of pyramidal neurons and a reduction in serotonin signaling in the hippocampus. This disrupted signaling could lead to cognitive impairment by promoting increased neural activity in the CA1 pyramidal neurons.26
P. Role of gamma-aminobutyric acid (GABA) in AD
GABA has been identified in 1950 and regarded as the main inhibitory neurotransmitter in the mammalian brain, essential for coordinating the activity of human cortical networks. Consequently, the GABAergic system has been linked to various behavioral and cognitive functions, including the regulation of alertness, anxiety, learned fear, and memory. The GABAergic system in the hippocampus is essential for preserving the excitatory/inhibitory (E/I) balance and synchronizing the activity of multiple populations of pyramidal neurons both within the hippocampus and across other regions of the brain. The alterations in the GABAergic system in Alzheimer's disease and normal aging have been extensively studied through animal models. Current literature indicates that GABA levels decline as part of the normal aging process. This decline is particularly evident in patients with AD and mild cognitive impairment (MCI), especially in the cingulate cortex and the medial parietal lobe. However, it remains unclear whether these changes can predict the onset of dementia in healthy older adults. Disruptions in neural networks have also been reported to emerge during the preclinical and mild stages of Alzheimer's disease. Although animal models offer compelling evidence that dysregulated GABAergic signaling, potentially influenced by APOEε4, contributes to hippocampal hyperactivity and subsequent memory impairments in aging animals, this mechanism has not been directly tested in humans, either cross-sectionally or longitudinally.27
3. Therapeutic approaches for the treatment of AD
3.1. Current therapeutic strategies
The current treatment options for AD are focused on preventing disease progression via monoclonal antibodies and on providing symptomatic relief to the patients.
3.1.1. Drugs that may change disease progression
Aducanumab, lecanemab and donanemab are anti-amyloid antibodies that remove amyloid plaques from the brain. Aducanumab was approved by the US-Food and Drug Administration (USFDA) in 2021 for mild cognitive impairment or mild dementia of Alzheimer's. However, it has not been approved in Europe due to a lack of strong evidence of its efficacy. It is an IgG1 antibody binding to the Aβ at amino acids 3–7. Lecanemab and donanemab also acts on amyloid plaques, albeit differently, by inhibiting aggregated soluble and insoluble forms of Aβ peptide with high selectivity to Aβ protofibrils. US-FDA approved it in July 2023 for patients with early Alzheimer's disease. The common side effects of these antibodies include amyloid-related imaging abnormalities (ARIA), which can lead to swelling and bleeding in the brain, as well as swelling of the face, headaches, infusion-related reactions, and vision changes.28–31
3.1.2. Drugs that may mitigate some of the symptoms of the disease
3.1.2.1. Cognitive symptoms
These alleviate memory and thinking symptoms without affecting the progression of the disease. They offer temporary relief by targeting neurotransmitters or receptors, enhancing patient comfort, dignity, and independence. These include cholinesterase inhibitors (donepezil, galantamine, and rivastigmine) and the NMDA receptor antagonist memantine (Fig. 6). Cholinesterase inhibitors preserve acetylcholine levels, which decline in AD and contribute to cognitive loss.32 Memantine blocks excessive glutamate receptor activation, preventing neuronal death caused by glutamate excess. These medications address cognitive symptoms and temporarily improve AD patients' quality of life. The cholinesterase inhibitors have been approved for use in mild to severe dementia due to Alzheimer's, whereas memantine is approved for moderate to severe dementia. A combination of donepezil with memantine is also approved for moderate to severe dementia due to Alzheimer's.33,34
Fig. 6. USFDA-approved therapy for improving cognitive symptoms.35.

3.1.2.2. Non-cognitive (behavioral and psychological) symptoms
In AD patients, sleep changes such as difficulty in sleeping, taking longer daytime naps, changes in sleep cycle, etc., may aggravate mood swings. Suvorexant, a potent dual orexin receptor (OXR1 and OXR2) antagonist, is recommended for treating sleep changes in AD patients. It promotes sleep by binding to the receptors and blocking the binding of orexin A and B, neuropeptides that promote wakefulness, to the receptors. Recently, suvorexant has been found to decrease tau phosphorylation and Aβ in humans.36
3.2. Therapeutic strategies based on hypotheses proposed
AD drug development aims at disease modification or symptom management by targeting Aβ and tau protein.37 Developing new central nervous system (CNS) drugs is challenging but vital to finding more effective AD treatments.38 Most of the drugs that reached phase II trials failed to show promising results in phase III, like BIIB092, crenezumab, TRx0237, azeliragon, verubecestat, atabecestat, BI 409306, etc.39 The following section discusses various therapeutic options based on the understanding of proposed hypotheses and also lists developmental candidates, particularly in clinical trials.
3.2.1. Aβ-Based approach
Targeting Aβ with vaccines and antibodies is a promising approach for halting AD progression, exemplified by the recent entry of monoclonal antibodies into the market and ongoing clinical trials.40Fig. 7 illustrates the downstream events of APP processing and the possible therapeutic interventions being considered for drug development. Some small molecules targeting Aβ peptide for AD treatment are in clinical trials (Table 2).
Fig. 7. Downstream events of APP cleavage and therapeutic options being explored.

Table 2. Aβ-Targeting small molecules in clinical trials (https://clinicaltrials.gov/).
| Drug name | Structure | Mode of action | Route | Status | Ref. |
|---|---|---|---|---|---|
| Acitretin |
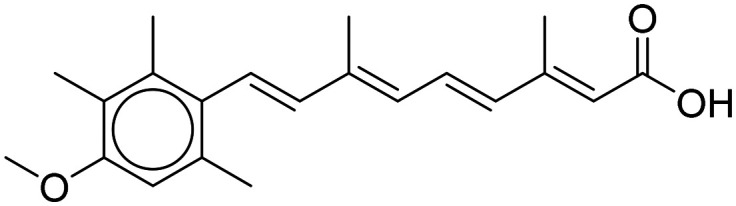
|
Reduces Aβ generation | Oral | Phase II | 41 |
| Lenalidomide |
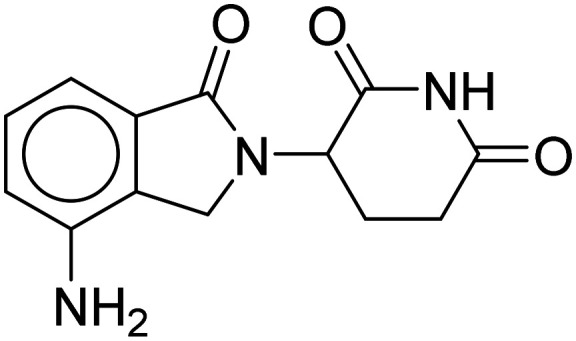
|
BACE1 inhibitor | Oral | Phase II | 42 |
| Levetiracetam |
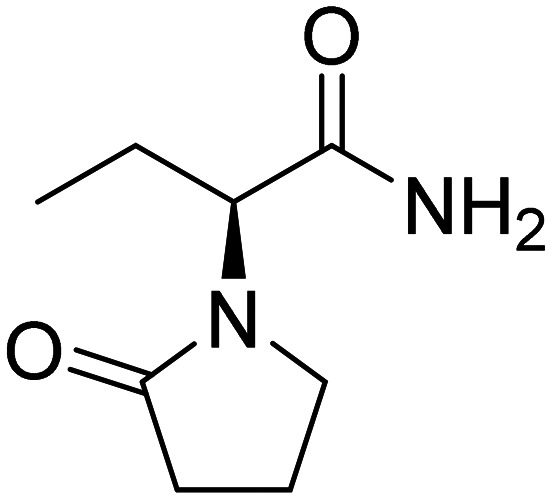
|
Reduce Aβ generation | Oral | Phase II | 43 |
| NIC5-15 (1R,2S,3R,4S,5S,6S)-6-methoxy cyclohexane-1,2,3,4,5-pentaol |
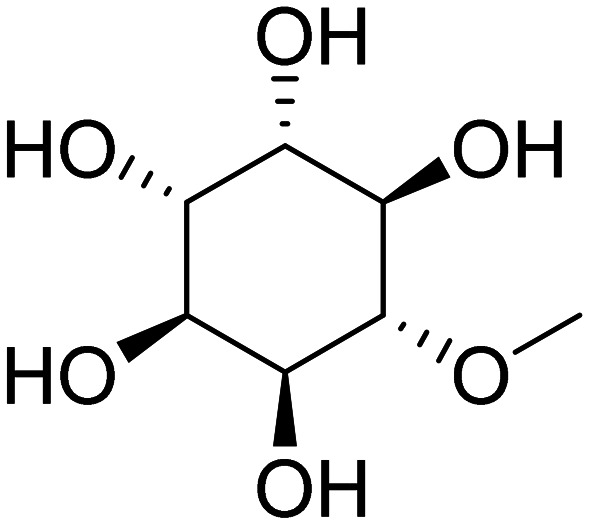
|
Modulator of γ-secretase | Oral | Phase II | 44 |
| Posiphen |
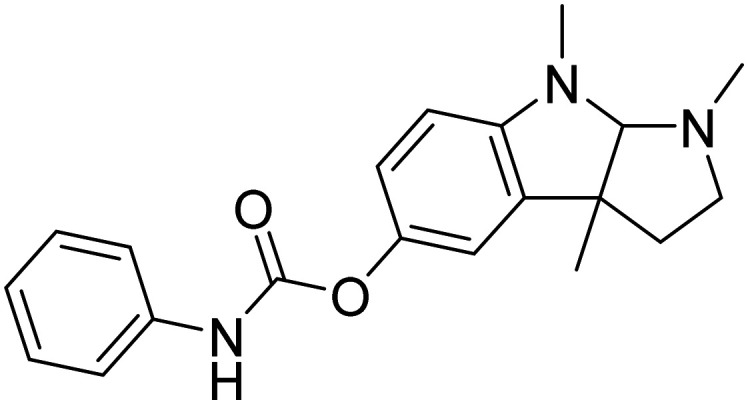
|
Hinders the translation of the APP | Oral | Phase II | 20 |
| ALZT-OP1 [cromolyn + (R)-ibuprofen] |
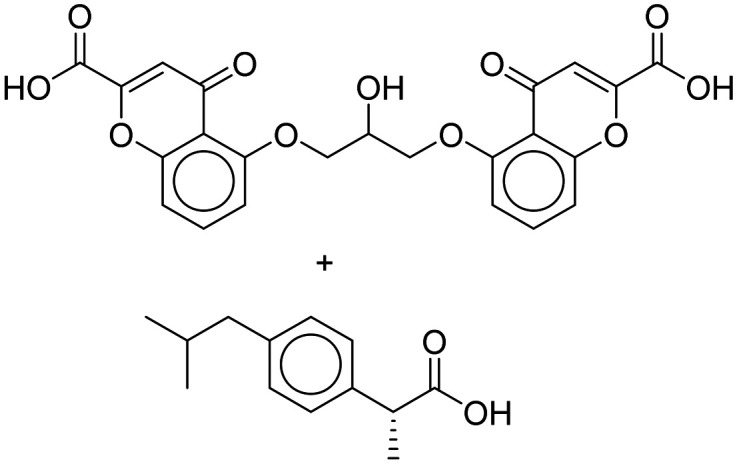
|
Facilitate the elimination of Aβ or its aggregates | Oral | Phase III | 45 |
| Bexarotene |
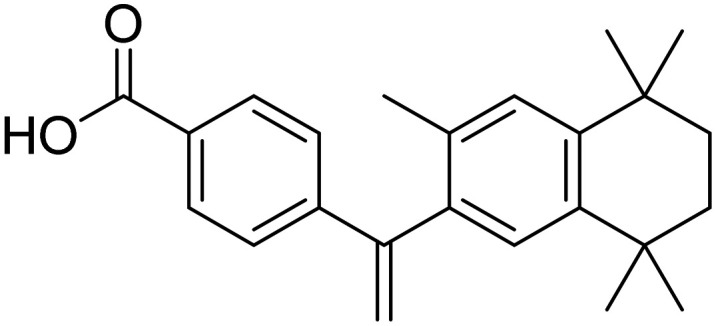
|
Facilitate the elimination of Aβ or its aggregates | Oral | Phase II | 46 |
| ALZ-801 3-aminopropane-1-sulfonic acid |
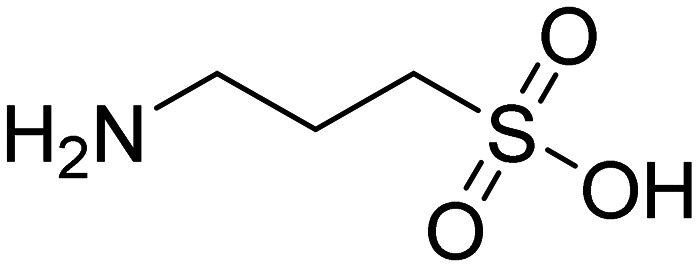
|
Disrupt or hinder the formation of Aβ aggregates | Oral | Phase II | 47 |
| Contraloid | Peptide (all d enantiomeric) sequence: PTLHTHNRRRRR | Terminates toxic and replicating amyloid beta (Aβ) oligomer prions by disassembling aggregates into non-toxic Aβ monomers | Oral | Phase I | 48 |
| PBT2 5,7-dichloro-2-((dimethylamino)methyl)quinolin-8-ol |
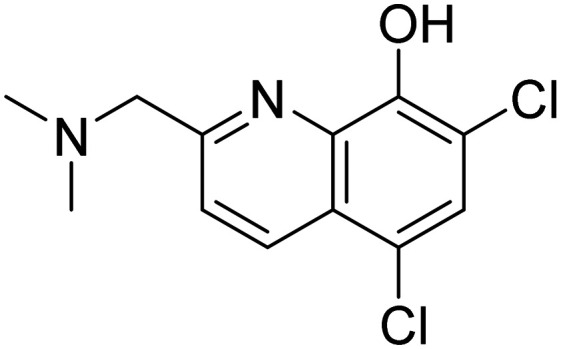
|
Decreasing metal-facilitated Aβ aggregation | Oral | Phase II | 49 |
| Varoglutamstat |
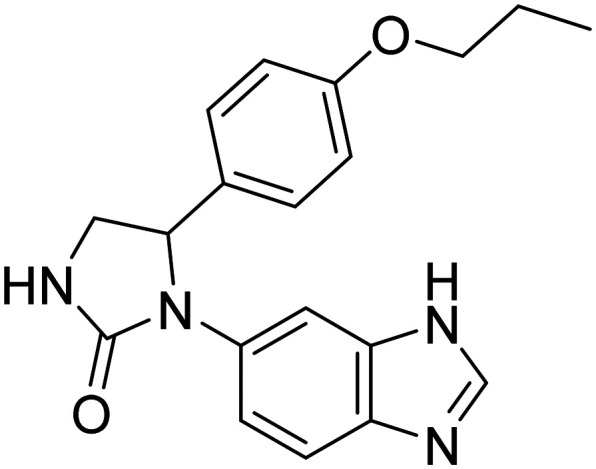
|
Prevents the formation of a particularly toxic and aggregation-prone variant of Aβ known as pGlu-Aβ | Oral | Phase II | 50 |
| ALX-001 (4R,5R)-5-(2-chlorophenyl)-4-(5-(phenylethynyl)pyridin-3-yl)oxazolidin-2-one |
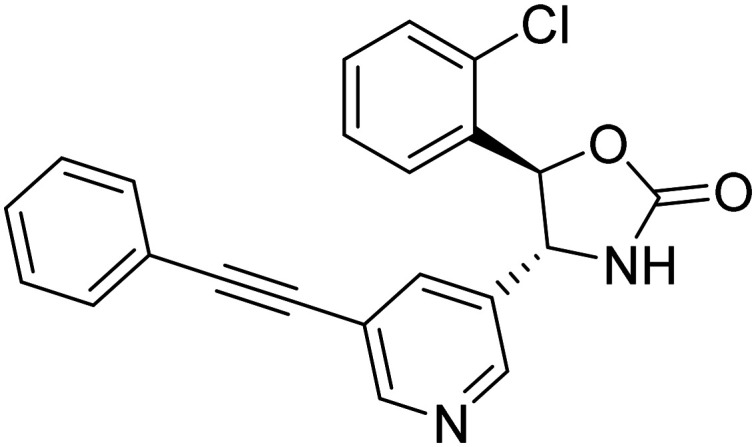
|
Designed to block the pathogenic binding of toxic amyloid-β oligomers and cellular prion protein to glutamate receptor | Oral | Phase II | 51 |
| CT1812 2-(tert-butoxy)-4-(3-methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol fumarate |
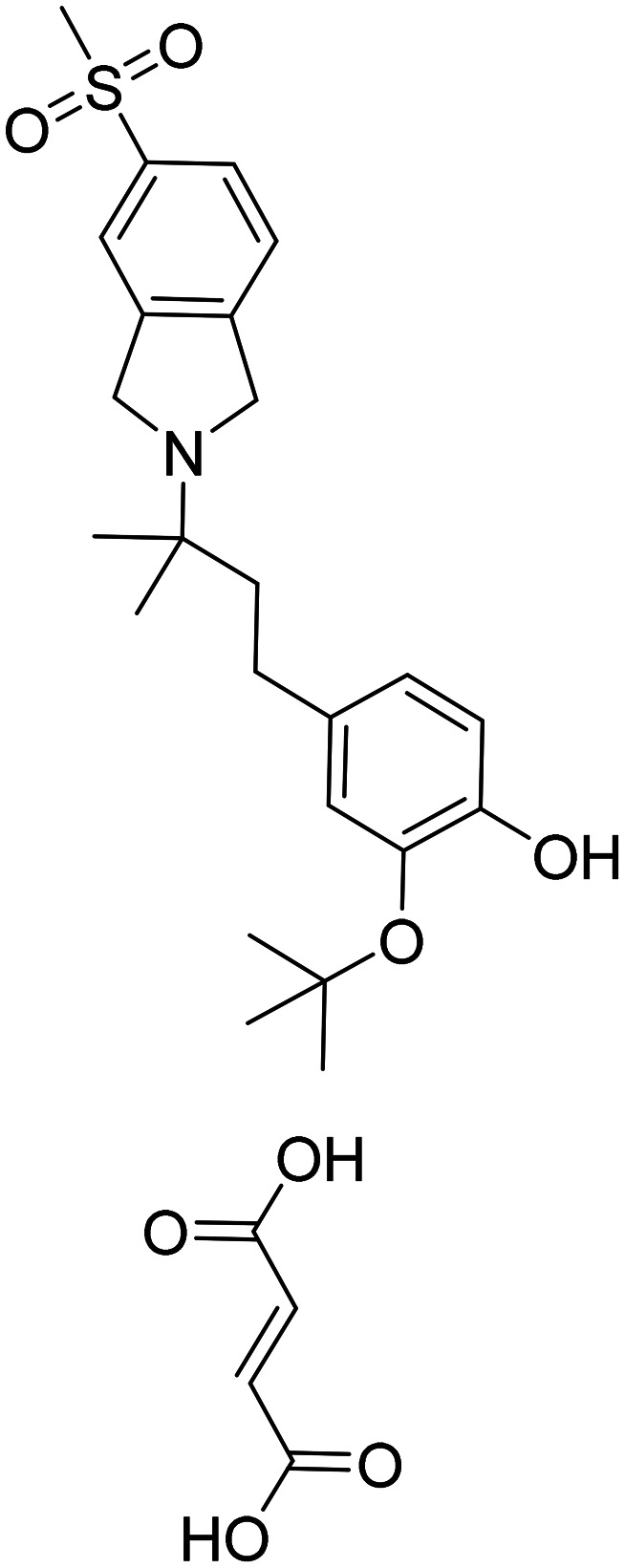
|
Prevents the interaction between oligomeric Aβ and its receptors, leading to a reduction in synaptic toxicity caused by Aβ | Oral | Phase I | 52 |
| Nasal insulin | Restructuring of synapses and utilization of glucose | Intranasal | Phase III | 53 | |
| Simufilam |
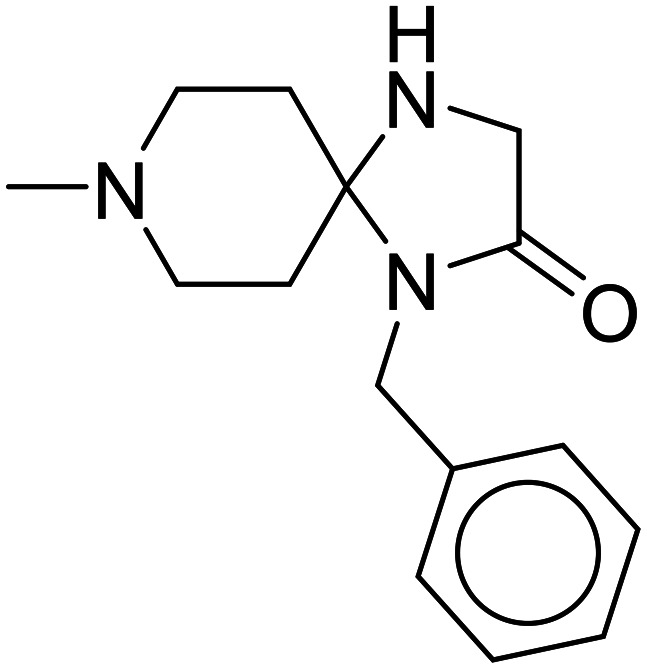
|
Diminishes tau buildup, decreases neuroinflammation | Oral | Phase III | 54 |
3.2.2. Secretase inhibitors
Secretases are a group of proteolytic enzymes responsible for processing APP. Three key secretases, namely α-secretase, γ-secretase, and β-secretase, are involved in this process. In healthy conditions, APP undergoes cleavage by α- and γ-secretases. However, the cleavage pattern in AD involves β- and γ-secretases [Fig. 8].55
Fig. 8. APP processing by α-, β- and γ-secretases.

α-Secretase
Activation of α-secretase is represented by the metalloprotease ADAM10, which cleaves APP within the Aβ domain, inhibiting Aβ generation and yielding neuroprotective APP fragments. Therefore, α-secretase cleavage of APP is beneficial by impeding the formation of Aβ peptides and protecting against neurotoxic agents. It has led to the proposal of enhancing α-secretase activity as a treatment strategy to shift the balance towards the nonamyloidogenic pathway, potentially influencing the progression of AD (Table 3).56
Table 3. List of α-secretase activators/enhancers in clinical trials57.
| Compound name | Structure | Status |
|---|---|---|
| LY2811376 4-(2,4-difluoro-5-(pyrimidin-5-yl)phenyl)-4-methyl-5,6-dihydro-4H-1,3-thiazin-2-amine |
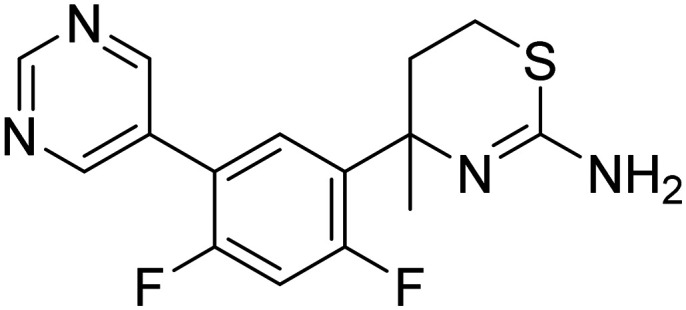
|
Phase I |
| MK-8931 N-(3-(3-amino-2,5-dimethyl-1,1-dioxido-5,6-dihydro-2H-1,2,4-thiadiazin-5-yl)-4-fluorophenyl)-5-fluoropicolinamide |
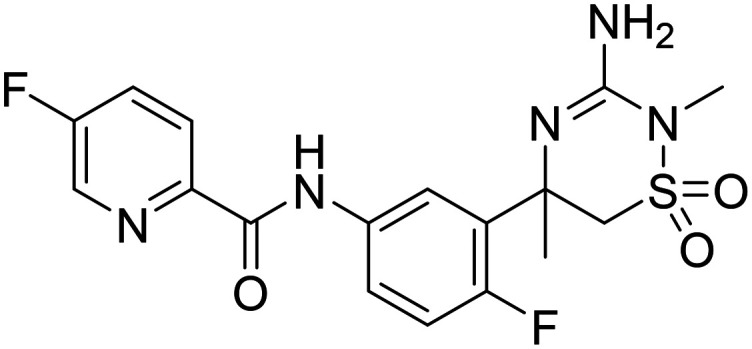
|
Phase I |
| LY2886721 N-(3-(2-amino-4a,5-dihydro-4H-furo[3,4-d][1,3]thiazin-7a(7H)-yl)-4-fluorophenyl)-5-fluoropicolinamide |
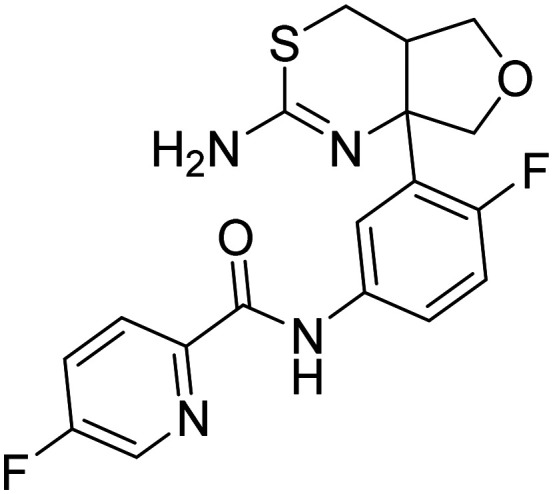
|
Phase I |
| Etazolate |
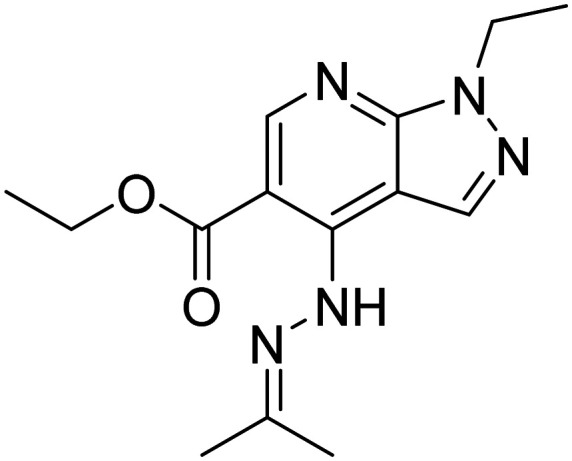
|
Phase II |
| AZ-4217 N-(3-(2-amino-5,5-difluoro-4-methyl-5,6-dihydro-4H-1,3-oxazin-4-yl)-4-fluorophenyl)-5-cyanopicolinamide |
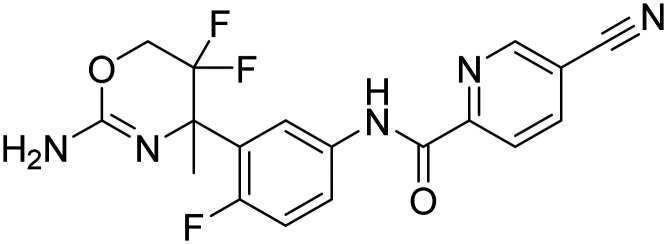
|
Phase II |
| APH1105 | A bryostatin analog | Phase II |
β-Secretase
The 1999 discovery of β-secretase (BACE) was pivotal in AD research, identified independently by five research groups using various names like BACE, β-secretase, Asp2, or memapsin 2.58 BACE, a type 1 transmembrane aspartic protease with 501 amino acids, functions optimally under low pH conditions in intracellular compartments. It is predominantly expressed in brain neurons, and modulating its expression directly impacts Aβ production.59 BACE's discovery led to the identification of its homolog, BACE2. Still, due to its low expression in brain neurons and distinct APP cleavage activity, BACE1 remains the main β-secretase, a potential therapeutic target to reduce cerebral Aβ levels in AD.60
Despite several drugs developed to inhibit BACE activity, such as elenbecestat, atabecestat, lanabecestat, and verubecestat, failing to reach the market, researchers remain committed to the pursuit of BACE-inhibitory drugs due to the critical role it has in the disease development.61 The structures of some of the representative β-secretase inhibitors are given in Fig. 9, indicating the use of various scaffolds.
Fig. 9. A few representative β-secretase inhibitors for AD.62,63.

γ-Secretase
γ-Secretase, composed of presenilin-I (PS-I), nicastrin, anterior pharynx-I, and presenilin enhancer-2, plays a pivotal role in AD as it executes the final intramembrane cleavage of APP.64 γ-Secretase cleavage, which follows cleavage by α-secretase, yields the C99 fragment, leading to a minor fraction of Aβ-42. Mutations in presenilins, the catalytic core of γ-secretase, contribute to elevated Aβ-42 levels in familial AD (FAD). However, γ-secretase influences other substrates like notch, delta, and tumor necrosis factor-alpha converting enzyme (TACE).65 Targeting γ-secretase to reduce Aβ formation is challenging due to the potential disruption of vital functions of these substrates.66 As a result, drugs designed as γ-secretase modulators (such as avagacestat, semagacestat, and flurizan) have faced setbacks during clinical trials.67 The status of different γ-secretase inhibitors is shown in Table 4.
Table 4. γ-Secretase inhibitors for AD in clinical trials.
| Drug name | Structure | Mode of action | Remarks | Status | Ref. |
|---|---|---|---|---|---|
| Semagacestat |
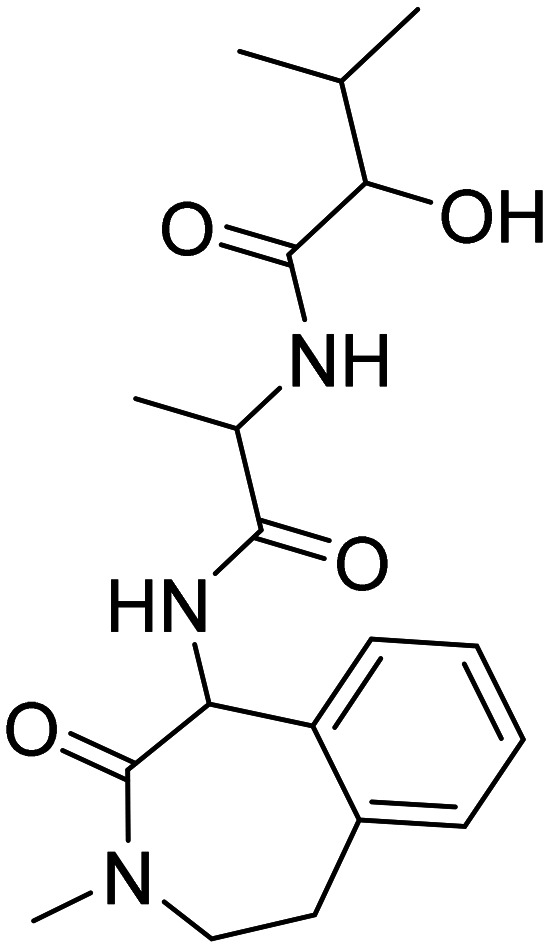
|
Reduces Aβ levels in AD and induces changes in the peptidome of human cerebrospinal fluid | In phase III trials, the outcome was inferior to placebo | Terminated in phase III | 67 |
| MK-0752 3-((1s,4r)-4-((4-chlorophenyl)sulfonyl)-4-(2,5-difluorophenyl)cyclohexyl)propanoic acid |
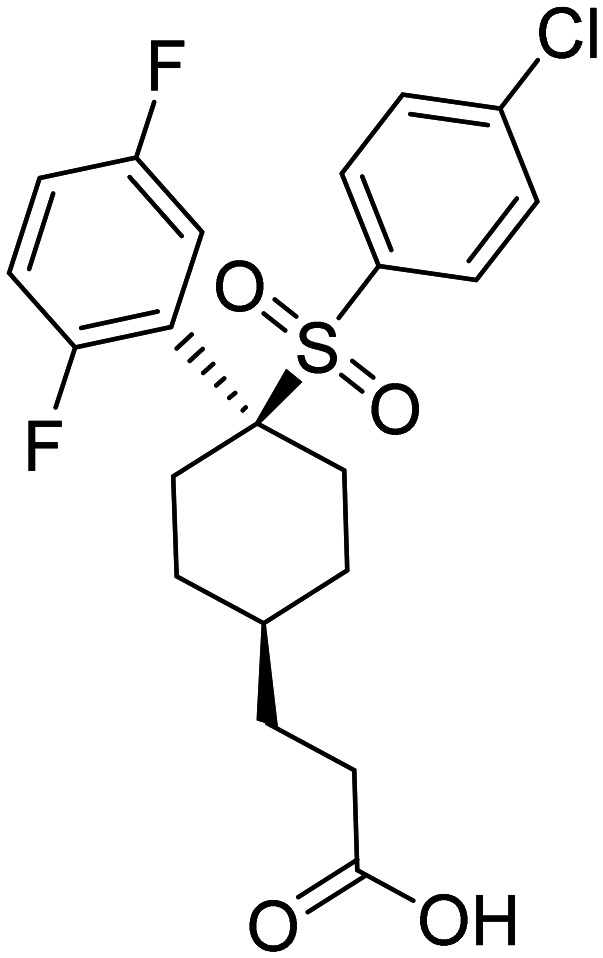
|
Lowers Aβ1-40 levels in healthy participants, and currently undergoing testing for its potential in cancer treatment | The medication was linked to gastrointestinal discomfort and feelings of fatigue | Terminated in phase I | 68 |
| E 2012 (S,E)-1-(1-(4-fluorophenyl)ethyl)-3-(3-methoxy-4-(4-methyl-1H-imidazol-1-yl)benzylidene)piperidin-2-one |
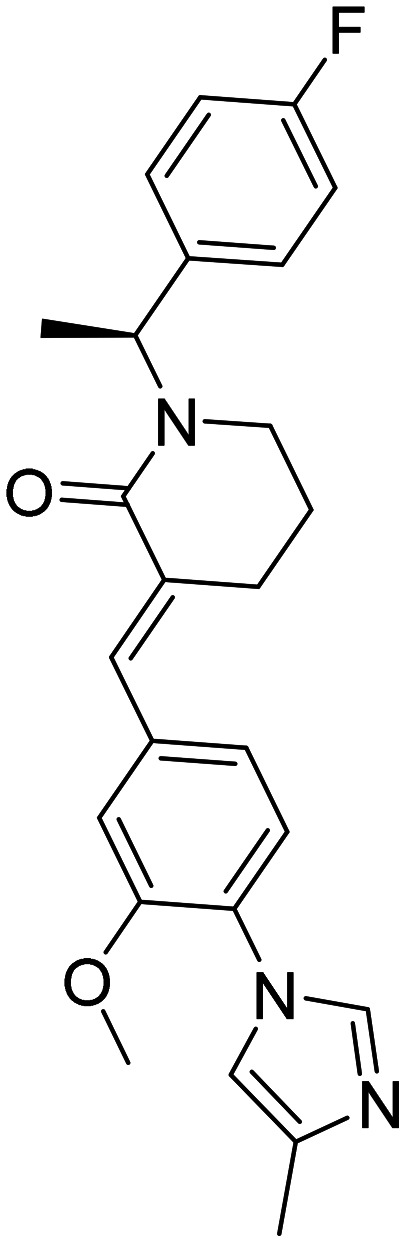
|
A gamma-secretase inhibitor/modulator that preserves notch function without impacting notch processing | Lenticular opacity | Terminated in phase I | 69 |
| Avagacestat, BMS-708163, 2-((4-chloro-N-(2-fluoro-4-(1,2,4-oxadiazol-3-yl)benzyl)phenyl)sulfonamido)-5,5,5-trifluoropentanamide |
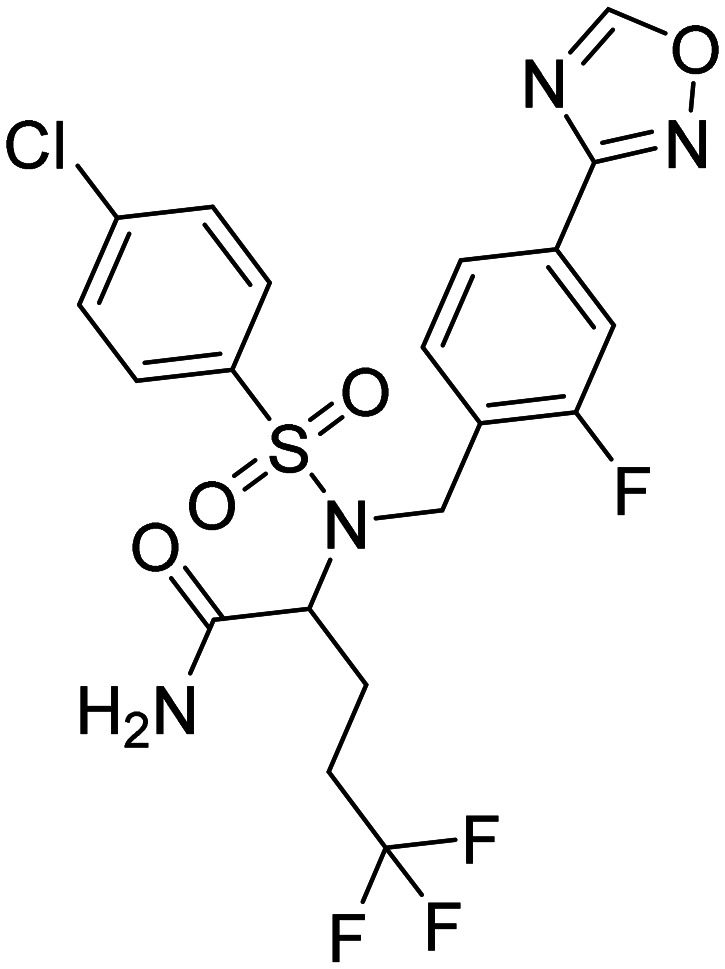
|
Preserving notch function leads to a reduction in Aβ levels in the cerebrospinal fluid of healthy participants | Amyloid-related imaging abnormalities (ARIA) | Terminated in phase II | 69 |
| Nirogacestat, PF-3084014 2-((6,8-difluoro-1,2,3,4-tetrahydronaphthalen-2-yl)amino)-N-(1-(2-methyl-1-(neopentylamino)propan-2-yl)-1H-imidazol-4-yl) pentanamide |
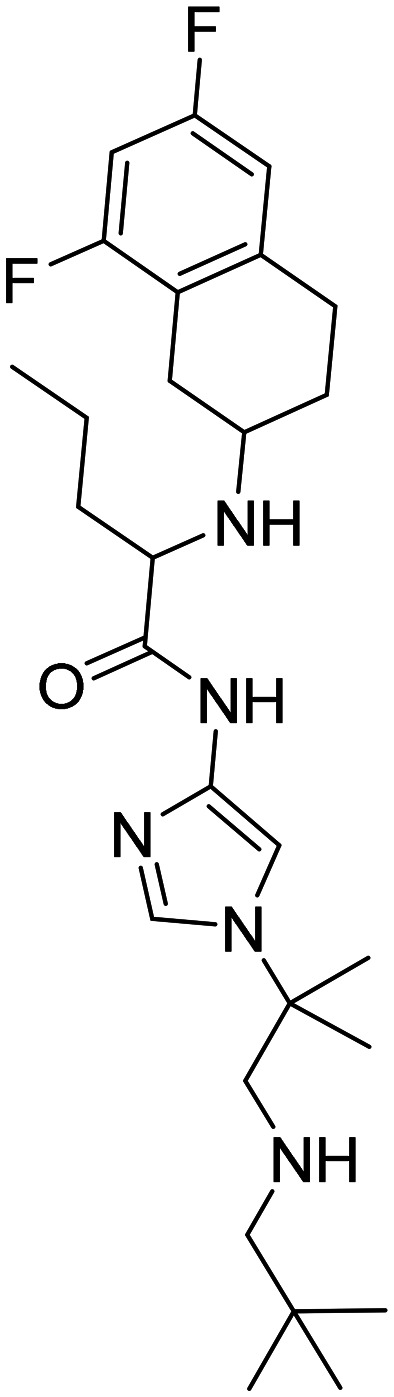
|
Reversible, orally bioavailable, noncompetitive, and selective γ-secretase inhibitor | Aβ1-40 levels show no decline in the cerebrospinal fluid of patients with AD | US FDA has approved its use for desmoid tumors. Terminated in phase II for AD patients | 69 |
3.2.3. Metal chelators
Metal ions, such as Zn2+, Cu2+, and Fe3+, promote the aggregation of β-amyloid. Zn2+ can lead to aggregation even at low physiological concentrations, while Cu2+ and Fe3+ enhance aggregation under mildly acidic conditions. Elevated Cu2+ and Zn2+ levels have been linked to developing the apoE4 allele associated with AD. β-Amyloid also participates in redox reactions mediated by metal ions, generating ROS.70 To counteract metal interactions with Aβ, strategies like using iron-chelating agents are under investigation (Table 5).71
Table 5. Clinically approved small molecule-based chelating agents in metal chelation therapy.
| Name | Structure |
|---|---|
| Deferoxamine |
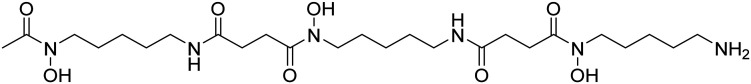
|
| Deferiprone |
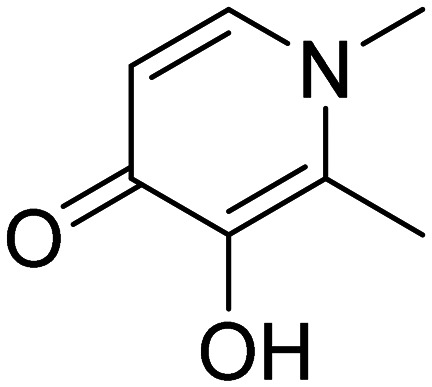
|
| d-Penicillamine |
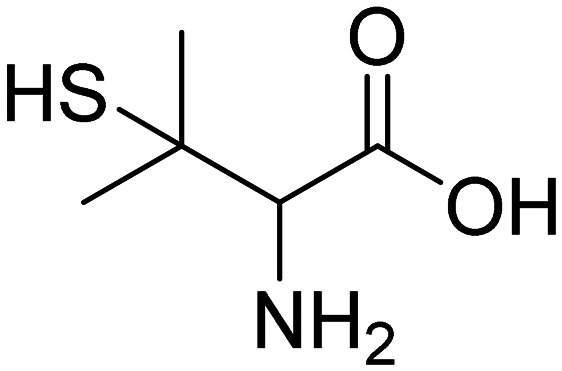
|
| Deferasirox |
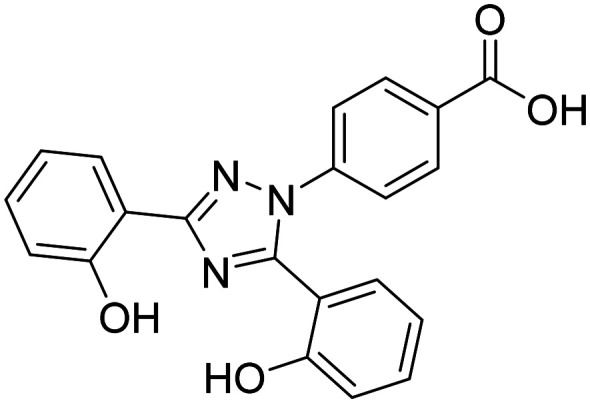
|
| Ethylenediaminetetraacetic acid |
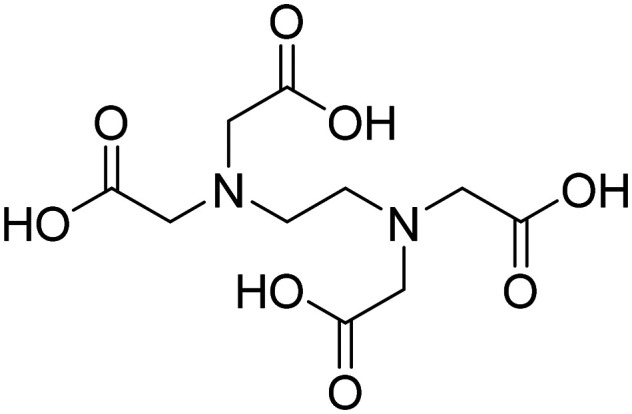
|
| Triethylenetetramine (TETA) |
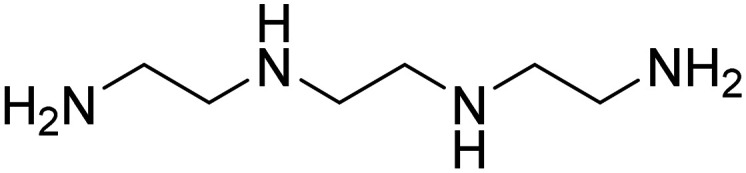
|
3.2.4. Acetylcholinesterase inhibitors
The cholinergic hypothesis in AD focused on the loss of cholinergic neurons. It reduced acetyltransferase (ChAT) activity, primarily in the nucleus basalis of Meynert, a vital source of cholinergic input to the neocortex.72 This degeneration is associated with cognitive decline, impacting memory, attention, and learning-related brain regions such as the hippocampus and frontal cortex. Issues encompass impaired choline uptake, compromised acetylcholine release, receptor imbalances, disrupted neurotrophin support, and axonal transport deficits. Reduced ChAT and increased AChE activity result in acetylcholine depletion, impairing cognitive function.73 Treatment primarily relies on acetylcholinesterase inhibitors (AChEIs) to restore acetylcholine levels. Some approved AChEIs include tacrine, donepezil, rivastigmine, galantamine, etc., of which tacrine has been withdrawn.74
AD's complexity involves various pathological factors, including Aβ plaques, neurofibrillary tangles, elevated AChE and MAO activity, and increased reactive oxygen species and metal ions. Single-target drugs often prove insufficient due to AD's multifaceted pathology. Many drugs failed in phase three clinical trials due to inadequate target interaction, toxic side effects, or a lack of differentiation from placebos.75 FDA-approved treatments offer symptomatic relief but do not halt disease progression. Addressing AD's multifaceted nature demands a comprehensive approach. Combination therapy has limitations, including drug interactions and pharmacokinetic challenges.76 Multi-target directed ligands (MTDLs) and dual inhibitors are emerging as a promising strategy, targeting multiple disease-related pathways with a single molecule. MTDLs often focus on AChE, MAO enzymes, and the Aβ cascade pathway, offering potential for more effective and streamlined AD treatment by addressing the intricate network of pathological factors involved.77
3.3. Dual inhibitors
Given the intricate nature of AD and the multitude of factors contributing to its progression, addressing a single causative aspect is unlikely to yield a complete cure or effectively slow down the disease's advancement.78 Therefore, recent reports involve the development of molecules capable of targeting two distinct factors concurrently. This innovative strategy offers the potential to diminish required dosage concentrations, enhancing the likelihood of effective treatment and disease progression attenuation. Moreover, it could result in cost savings and alleviate the financial burden on patients.79–81 Examples of such dual inhibitors are given in Fig. 10.
Fig. 10. Structures of some of the representative dual inhibitors.82–92.

3.4. Multi-target-directed ligands
Similar to dual inhibitors, multi-target-directed ligands (MTDLs), often termed “dirty drugs”, offer promise for AD treatment.93 These compounds combine pharmacophore moieties from different bioactive compounds, enhancing therapeutic safety and efficacy compared to single-target drugs.94 MTDLs offer a comprehensive approach to addressing the intricate network of factors contributing to AD pathogenesis (Fig. 11).95
Fig. 11. Structures of some of the representative multi-target-directed ligands.86,96,97.

4. Candidates under clinical investigation for AD
Despite recent clinical trial failures, several promising candidates are in the AD drug development pipeline. Table 6 collates the available information (updated as of Nov 2024) on various developmental candidates in clinical trials, phase-wise and by category. The list is in addition to the candidates mentioned in the above sections (https://clinicaltrials.gov/).
Table 6. Drugs under clinical trial for AD treatment.
| Drug name | Structure | Mode of action | Class | National Clinical Trial number |
|---|---|---|---|---|
| Phase I | ||||
| Disease-modifying biologicals | ||||
| Mode of action | ||||
| MK-2214 | Anti-tau monoclonal antibody | Tau | NCT05466422 | |
| SHR-1707 | Prevent Aβ plaque and activate microglia to phagocytize various forms of Aβ | Amyloid | NCT06199037 | |
| NIO752 | Antisense oligonucleotide to tau, decreasing the abundance of tau aggregates at neuromuscular junction | Tau | NCT06372821 | |
| IBC-Ab002 | Humanized IgG1 antibody inhibits an immune checkpoint protein (PD-L1), stimulating the immune system | Inflammation/immunity | NCT05551741 | |
| ALN-APP | Small interfering RNA targeting the amyloid precursor protein mRNA | Amyloid | NCT05231785 | |
| CpG1018 | Immune adjuvant made up of short, unmethylated cytosine–phosphate–guanine oligodeoxynucleotides, stimulate clearance of amyloid pathology | Inflammation/immunity | NCT05606341 | |
| AL003 | Monoclonal antibody (MAb) targeting SIGLEC-3 (CD33), reactivates microglia and immune cells in the brain, improves microglial clearance of toxic proteins | Inflammation | NCT03822208 | |
| AAV-hTERT | Extending telomeres may benefit AD, reduce Aβ-induced neurotoxicity, and effects on multiple cellular pathways | Epigenetic | NCT04133454 | |
| Lu AF87908 | MAb to reduce tau | Tau | NCT04149860 | |
| LY3372993 | MAb to reduce amyloid (N3pG-AB) | Amyloid | NCT06653153 | |
| AAVrh.10hAPOE2 | MAb targeting SIGLEC-3 (CD33), reactivates microglia and immune cells in the brain, improves microglial clearance of toxic proteins | Epigenetic | NCT05400330 | |
| XPro1595 | TNF inhibitor, reduces neuroinflammation | Inflammation | NCT05318976 | |
| GSK933776 | MAb against N-terminus of Aβ | Amyloid | NCT00459550 | |
| ALZ-101 | Vaccine against soluble Aβ oligomers | Amyloid | NCT05328115 | |
| VT301 | Regulatory T-cells (Tregs) | Inflammation | NCT05016427 | |
| CAD106 | CAD106 combines multiple copies of Aβ1-6 peptide derived from the N-terminal B cell epitope of Aβ, coupled to a Qβ virus-like particle. In animals, CAD106 induced Aβ-antibody titers without activating Aβ-reactive T cells | Amyloid | NCT01097096 | |
| KHK6640 | MAb that consists of anti-Aβ-peptide | Amyloid | NCT02377713 | |
| NPT088 | An Ig-fusion general amyloid interaction motif (GAIM) based dimer | Amyloid | NCT03008161 | |
| Disease-modifying drugs | ||||
| BDPP (2S,4S)-(−)-2,4-bis(diphenylphosphino)pentane |
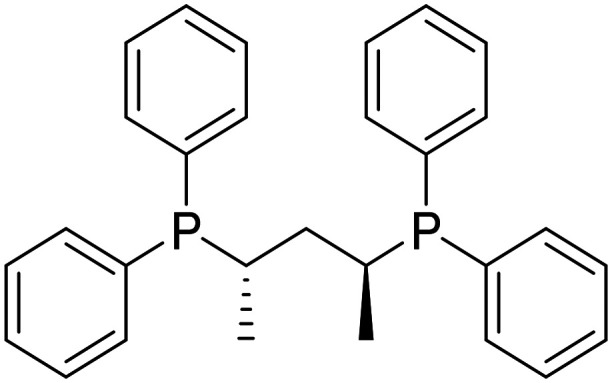
|
Prevents Aβ and tau aggregation | Proteostasis/proteinopathies | NCT02502253 |
| BEY2153 | Aβ and tau aggregation inhibitor, inhibits neuronal death | Proteostasis/proteinopathies | NCT04476303 | |
| Dabigatran |
|
Direct thrombin inhibitor, reduce neurovascular damage | Vasculature | NCT03752294 |
| Dexmedetomidine |
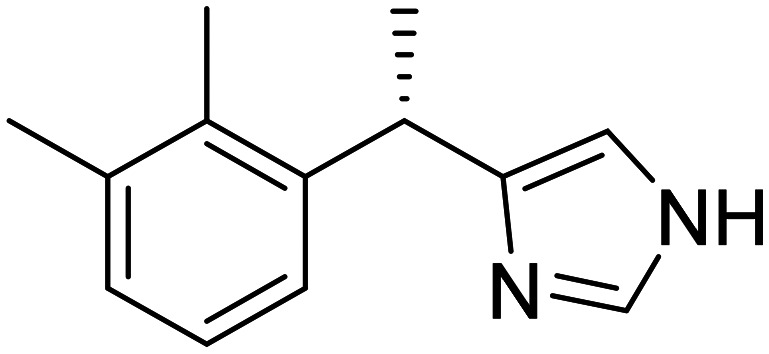
|
Selective α2-adrenergic receptor agonist, neuroprotection | Circadian rhythm | NCT06052254 |
| Edicotinib |
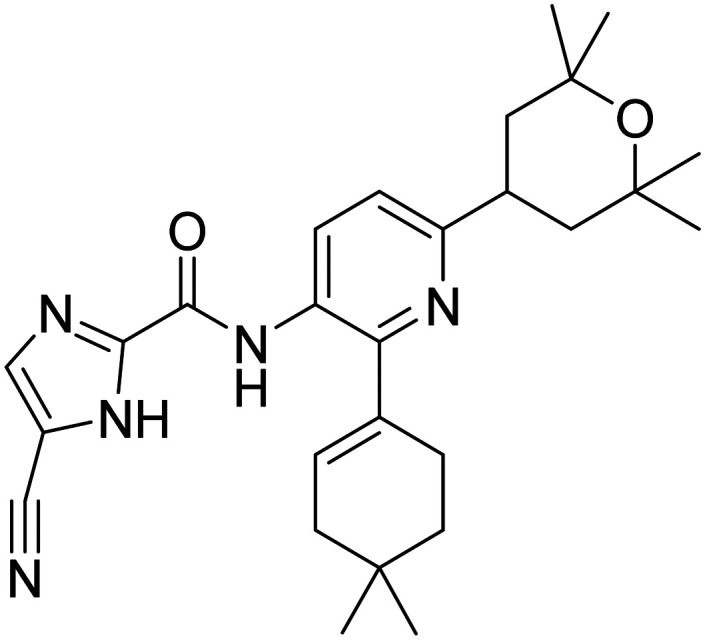
|
CSF-1R antagonist, attenuates microglial proliferation and neurodegeneration | Inflammation | NCT04121208 |
| Efavirenz |
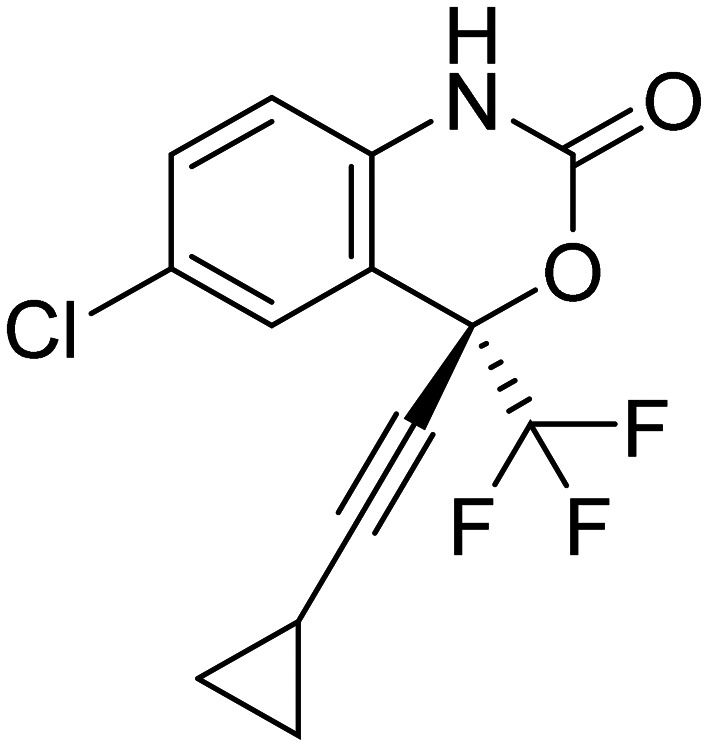
|
NNRTI, promotes cholesterol removal, enhances amyloid reduction | Epigenetics | NCT03706885 |
| Emtricitabine |
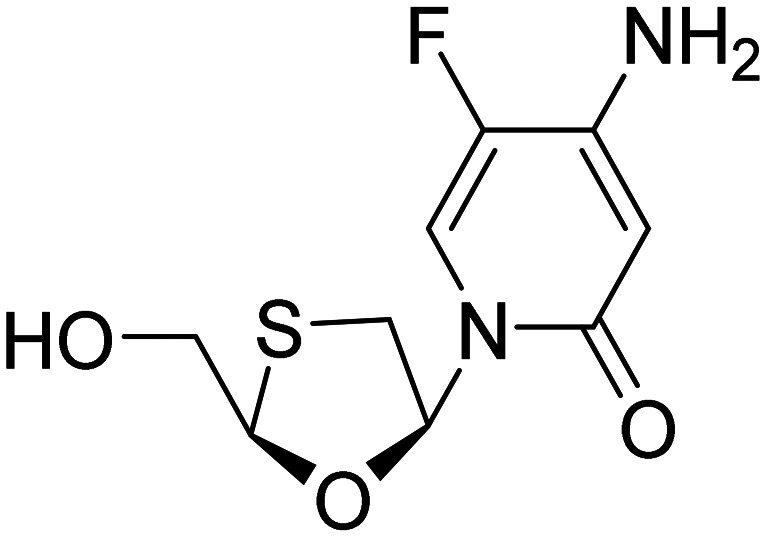
|
NRTI, reduces neuroinflammation | Inflammation | NCT04500847 |
| MK-4334 |
|
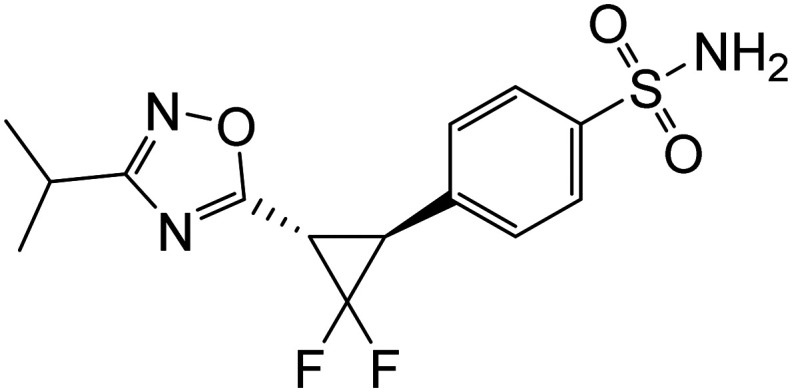
Positive allosteric modulators of α7 nAChR | Growth factors and hormones | NCT03740178 |
| NNI-3624-(3-cyano-6,7-dimethylquinolin-2-yl)-N-(2-ethoxyphenyl)-1,4-diazepane-1-carbothioamide |
|
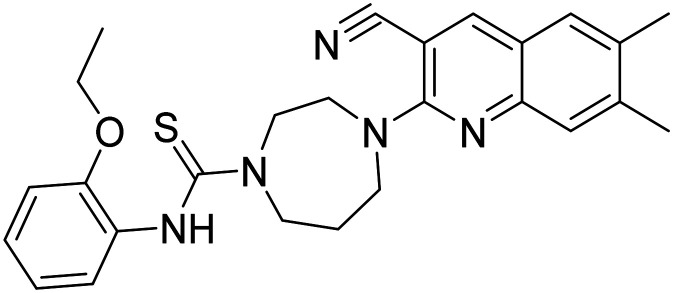
Enhance neurogenesis, activates progenitor cells | Neurogenesis | NCT04074837 |
| REM0046127 | Regulates calcium dyshomeostasis, tau and Aβ reduction | Synaptic plasticity/neuroprotection | NCT04672135 | |
| Salsalate |
|
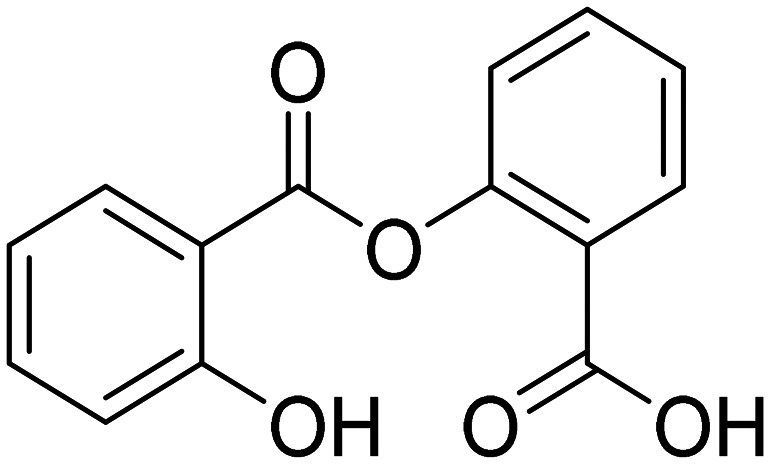
NSAID to reduce inflammation | Inflammation | NCT03277573 |
| TC-5619 N-((2S,3R)-2-(pyridin-3-ylmethyl)quinuclidin-3-yl)benzofuran-2-carboxamide |
|
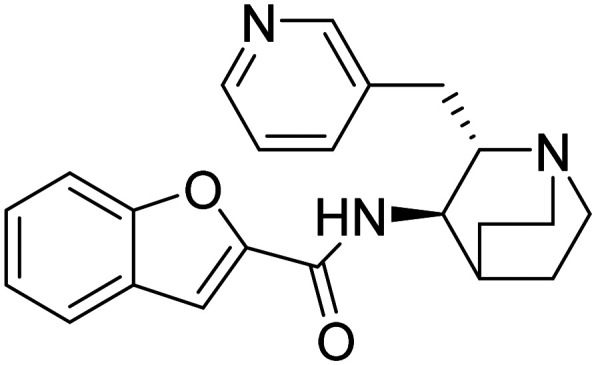
Partial agonist at the α7 subtype of the neural nicotinic acetylcholine receptors | Cognition enhancement | NCT01254448 |
| TPI-287 (1S,4S,7S,7aR,7a1S,10aS,11aR,13aS,13bR)-1-(benzoyloxy)-4-(((2R,3S)-3-((tert-butoxycarbonyl)amino)-2-hydroxy-5-methylhexanoyl)oxy)-2-hydroxy-5,7a1,14,14-tetramethyl-9-vinyl-1,3,4,7,7a,7a1,10a,11,11a,13b-decahydro-2H-8,10,12-trioxa-2,6-methanocyclobuta[b]cyclodeca[de]naphthalene-7,13a(13H)-diyl diacetate |
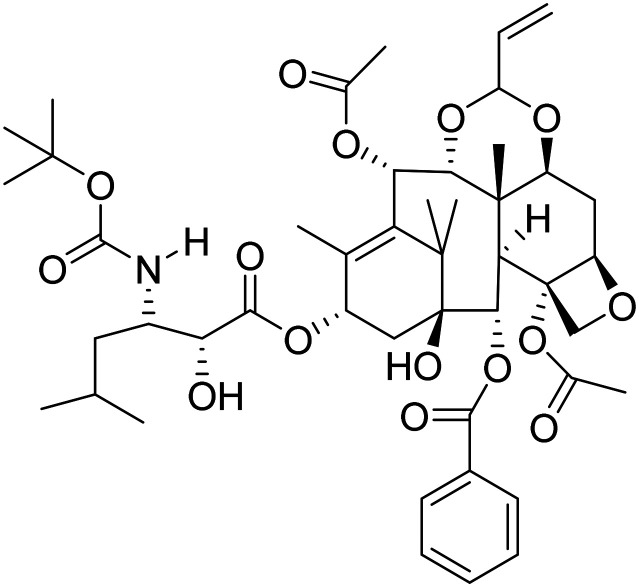
|
Stabilization of microtubule | Tau | NCT01966666 |
| CMS121 4-(4-(cyclopentyloxy)quinolin-2-yl)benzene-1,2-diol |
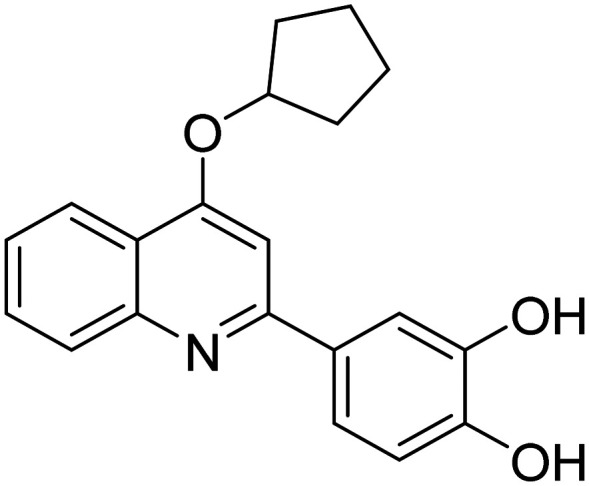
|
Fatty acid synthase inhibitor it protects against excess lipid peroxidation and inflammation and alleviates cognition | Inflammation | NCT05318040 |
| Oxaloacetate |
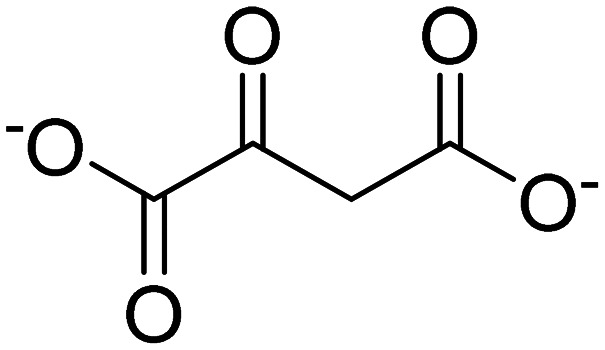
|
Improves mitochondrial function | Bioenergetics | NCT02593318 |
| Telmisartan |
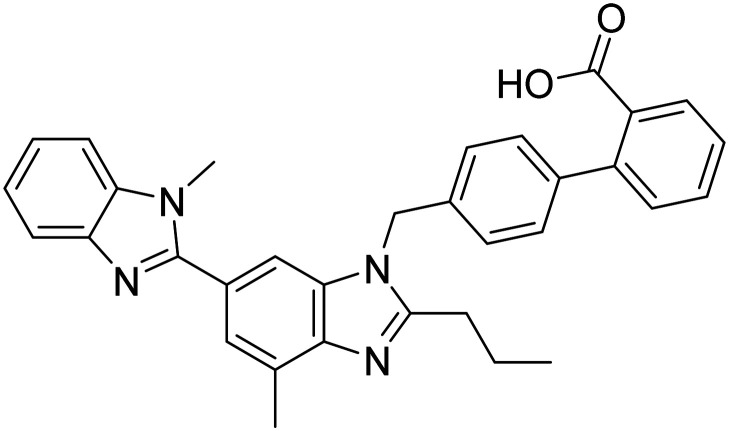
|
Angiotensin II receptor blocker | Vasculature | NCT02471833 |
| Trehalose |
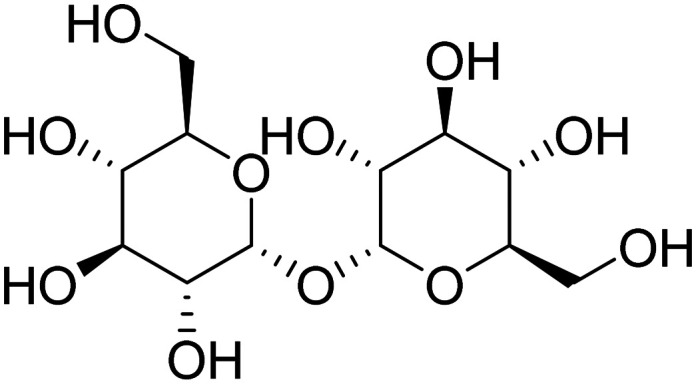
|
Induces autophagy and promotes clearance of aggregated proteins | Cell death | NCT05332678 |
| Vorinostat |
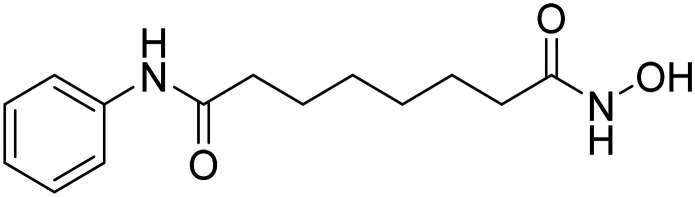
|
Histone deacetylase (HDAC) inhibitor, enhanced synaptic plasticity | Epigenetics | NCT03056495 |
| Symptom-reducing small molecules | ||||
| MK-1942 | Improves mitochondrial structure and function | Neurotransmitter receptors | NCT04308304 | |
| MK-8189 | Phosphodiesterase 10A inhibitor | Behavioural changes | NCT05227118 | |
| Memogain |
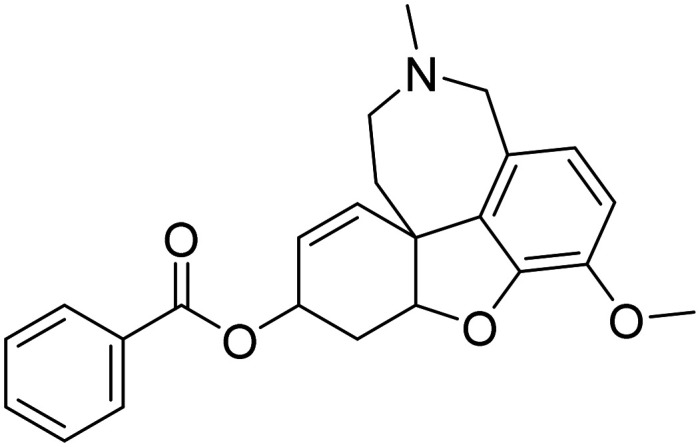
|
AChE/BuChE MTDL | Neurotransmitter receptors | — |
| Phase II | ||||
| Disease-modifying biologicals | ||||
| AL002 | MAb targeting TREM2 receptors to promote microglial clearance of Aβ | Inflammation | NCT05744401 | |
| ACI-35 | Active immunotherapy targeting tau | Tau | NCT04445831 | |
| ABvac40 | Active immunotherapy to remove Aβ | Amyloid | NCT03461276 | |
| AADvac1 | This is an active vaccine designed to elicit an immune response against pathologically modified forms of tau protein | Tau | NCT01850238 | |
| BCG vaccine | Vaccination against tuberculosis infection, immunomodulator | Inflammation | NCT05004688 | |
| Bryostatin 1 | Protein kinase C inhibitor, facilitates synaptogenesis | Synaptic plasticity/neuroprotection | NCT04538066 | |
| Daratumumab | MAb targeting CD38, regulates microglial activity | Inflammation/immunity | NCT04070378 | |
| Etanercept | Inhibits the function of a pro-inflammatory cytokine called tumor necrosis factor alpha (TNF-α) | Inflammation | NCT01068353 | |
| Crenezumab | MAb targeting soluble Aβ oligomers | Amyloid | NCT01723826 | |
| Gosuranemab | MAb targeting truncated form of tau | Tau | NCT03352557 | |
| Gantenerumab | Human IgG1 Ab against Aβ fibrils | Amyloid | NCT01760005 | |
| Pepinemab | MAb directed at semaphorin 4D to reduce inflammation | Inflammation | NCT04381468 | |
| GV1001 | hTERT peptide vaccine, mimics extra-telomeric functions to inhibit neurotoxicity, apoptosis, and reactive oxygen species | Epigenetic | NCT05303701 | |
| GB301 | Regulatory T cells, reduce neuroinflammation | Inflammation/immunity | NCT04468659 | |
| IVIG | Polyclonal antibody, remove amyloid | Amyloid | NCT01300728 | |
| JNJ-63733657 | MAb targeting soluble tau | Tau | NCT04619420 | |
| IONIS MAPTRx | Antisense oligonucleotide targeting tau expression, MAPT RNA inhibitor | Tau | NCT03186989 | |
| R07126209 | Anti-Aβ MAb with enhanced BBB penetration | Amyloid | NCT04639050 | |
| Semorinemab | MAb to remove extracellular tau | Tau | NCT03828747 | |
| Tilavonemab | MAb to remove tau and prevent propagation | Tau | NCT02880956 | |
| Zagotenemab | MAb to remove tau and reduce tau propagation | Tau | NCT03518073 | |
| Disease-modifying drugs | ||||
| Allopregnanolone |
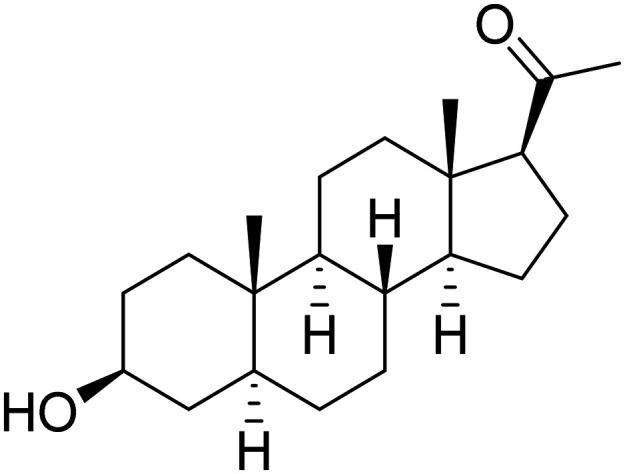
|
GABA-A receptor modulator, promote neurogenesis and reduce inflammation | Growth factors/hormones | NCT04838301 |
| Telmisartan + perindopril |
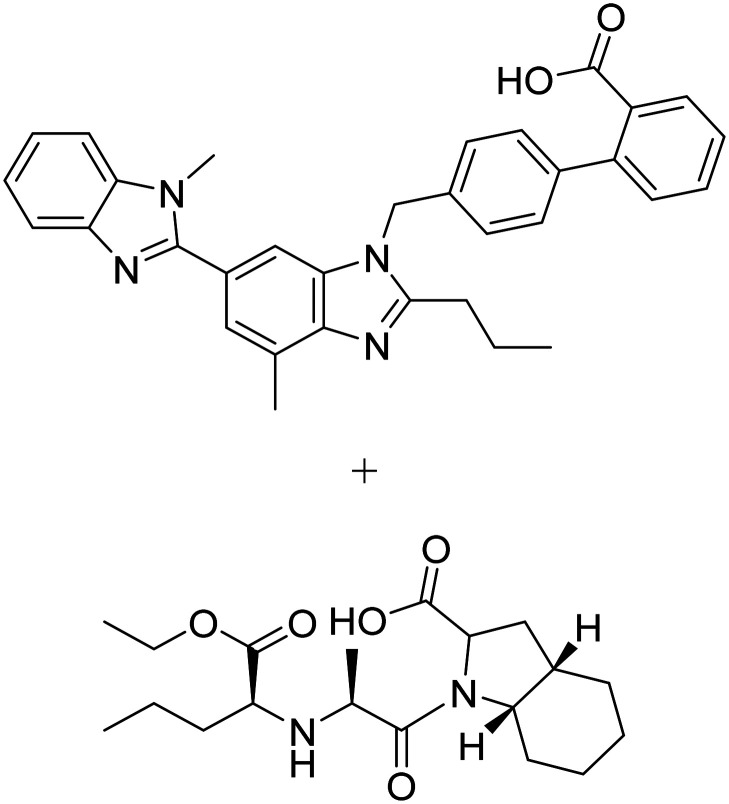
|
Telmisartan: angiotensin II receptor blocker | Vasculature | NCT02085265 |
| Perindopril: angiotensin converting enzyme inhibitor | ||||
| Empagliflozin |
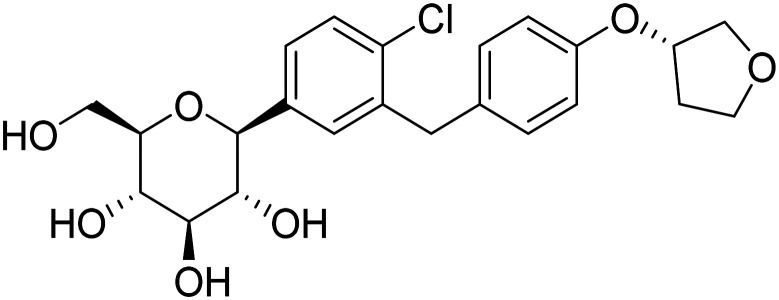
|
SGLT2 inhibitor, improve glycemic control, enhance neuronal function | Metabolism and bioenergetics | NCT05081219 |
| Sodium phenylbutyrate + tauroursodeoxycholic acid |
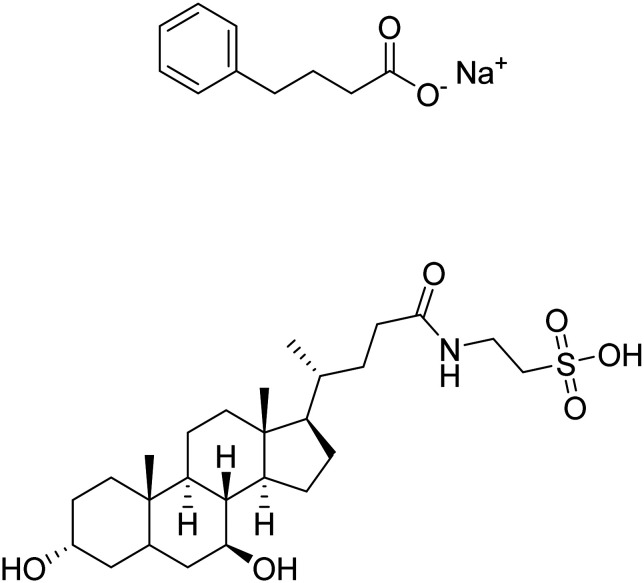
|
Reduce cell death associated with mitochondrial dysfunction, modulate neuroinflammation | Cell death | NCT03533257 |
| Fosgonimeton |
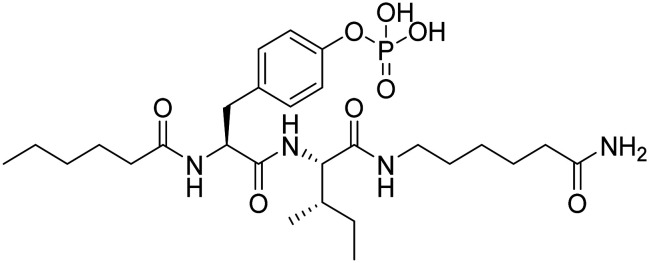
|
Activates signalling via the hepatocyte growth factor system to regenerate neurons and enhance synaptic plasticity | Synaptic plasticity/neuroprotection | NCT04488419 |
| AR1001 | PDE-5 inhibitor it improves synaptic plasticity | Synaptic plasticity/neuroprotection | NCT05531526 | |
| Blarcamesine |
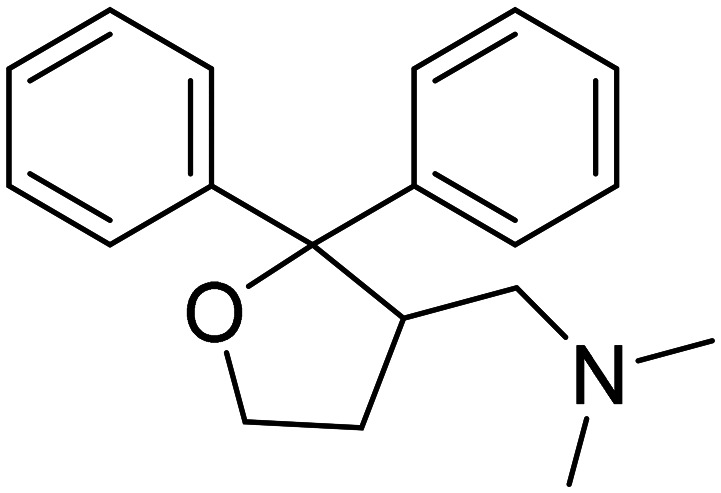
|
Sigma-1 receptor agonist, M2 antagonist, ameliorate oxidative stress, protein misfolding, mitochondrial dysfunction and inflammation | Synaptic plasticity/neuroprotection | NCT04314934 |
| Zatolmilast |
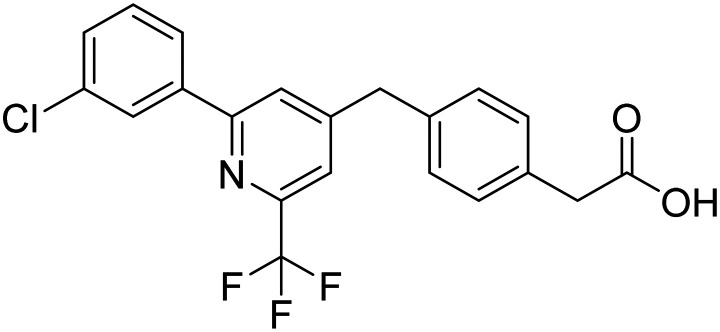
|
PDE-4 inhibitor, prolongs cAMP activity and improves neuronal plasticity | Synaptic plasticity/neuroprotection | NCT03817684 |
| Benfotiamine |
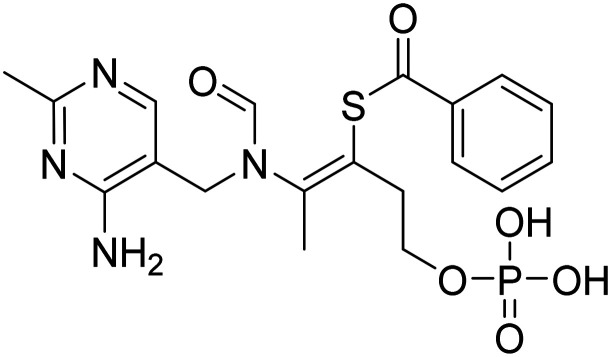
|
Synthetic thiamine to improve neuronal function | Metabolism and bioenergetics | NCT06223360 |
| Bromocriptine |
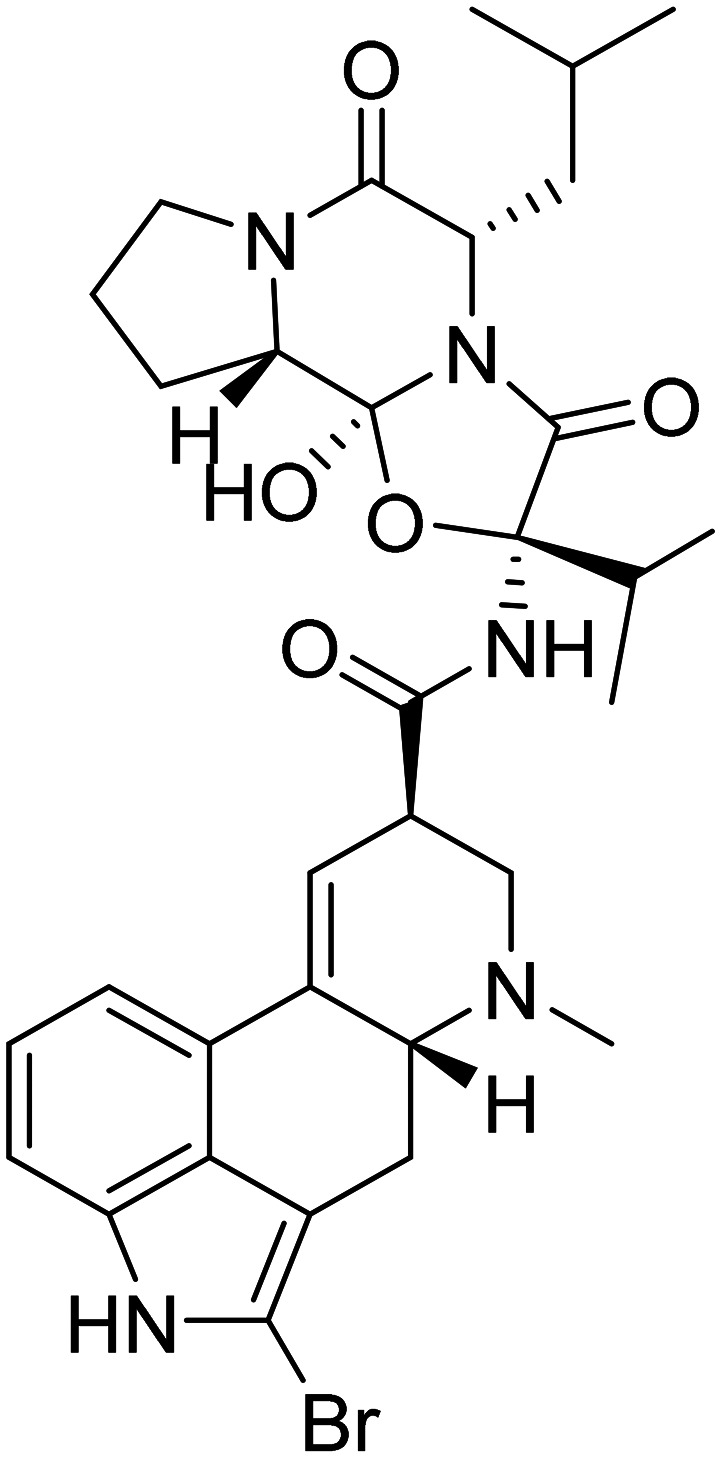
|
Dopamine agonist with anti-Aβ effects | Neurotransmitter receptors | NCT04413344 |
| DHA (docosahexanoic acid) |

|
Omega 3 fatty acid, improve synaptic function, antioxidant | Oxidative stress | NCT00440050 |
| Dapagliflozin |
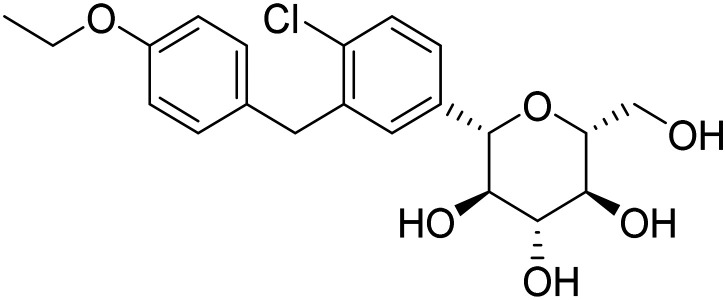
|
SGLT2 inhibitor, to improve insulin sensitivity and CNS glucose metabolism | Metabolism and bioenergetics | NCT03801642 |
| Elayta |
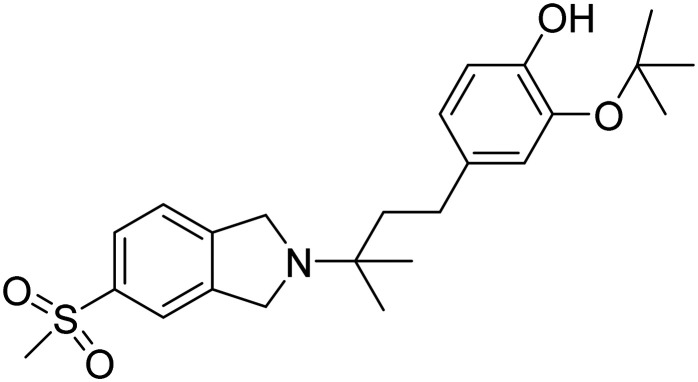
|
Sigma-2 receptor antagonist, competes with oligomeric Aβ binding, protect against Aβ-induced synaptic toxicity | Synaptic plasticity/neuroprotection | NCT05531656 |
| Edonerpic acid |
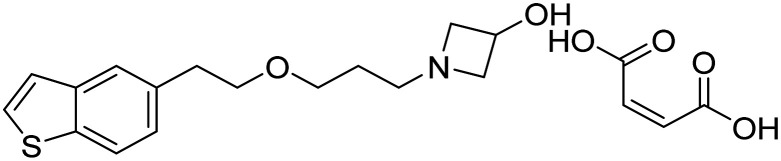
|
Neurotrophic agent, activates sigma receptors to preserve synaptic plasticity, protect against Aβ toxicity | Synaptic plasticity/neuroprotection | NCT00663936 |
| Dasatinib + quercetin |
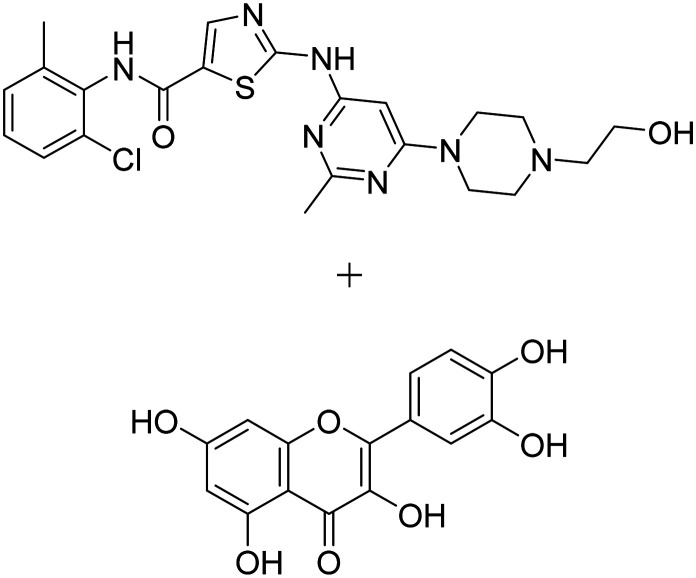
|
Dasatinib: tyrosine kinase inhibitor | Inflammation/immunity | NCT05422885 |
| Quercetin: flavonoid, senolytic therapy approach to reduce senescent cells and tau aggregation | ||||
| Lamivudine |
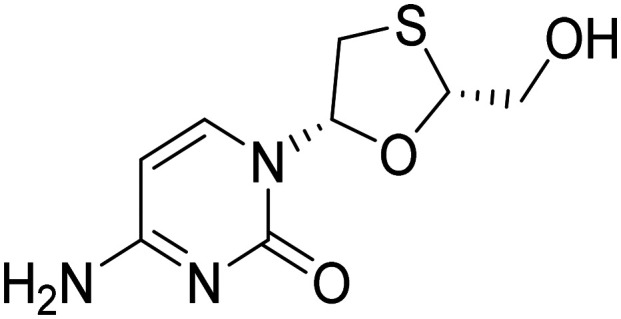
|
Nucleoside reverse transcriptase inhibitor, reduces genetic rearrangements | Epigenetic | NCT04552795 |
| l-Serine |
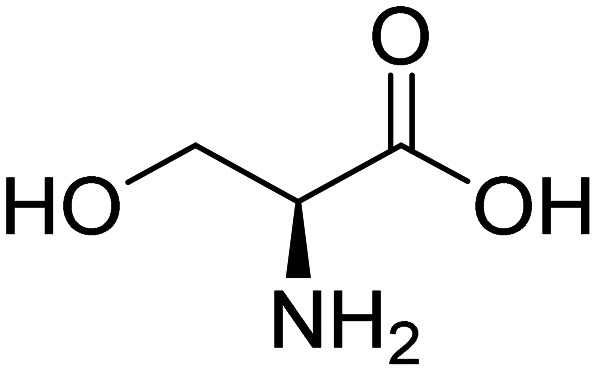
|
Dietary amino acid, reduce brain inflammation and preserve nerve cells | Inflammation | NCT05331144 |
| Liraglutide |
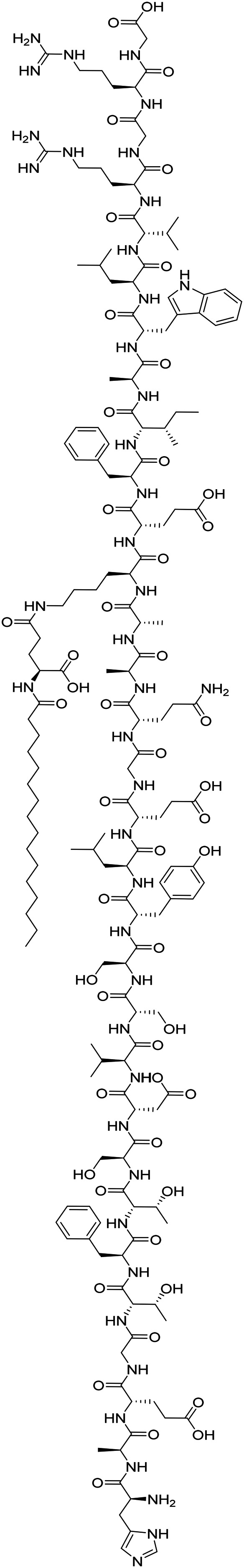
|
Glucagon-like peptide 1 receptor agonist, improve CNS glucose metabolism | Metabolism and bioenergetics | NCT01469351 |
| Lupron |
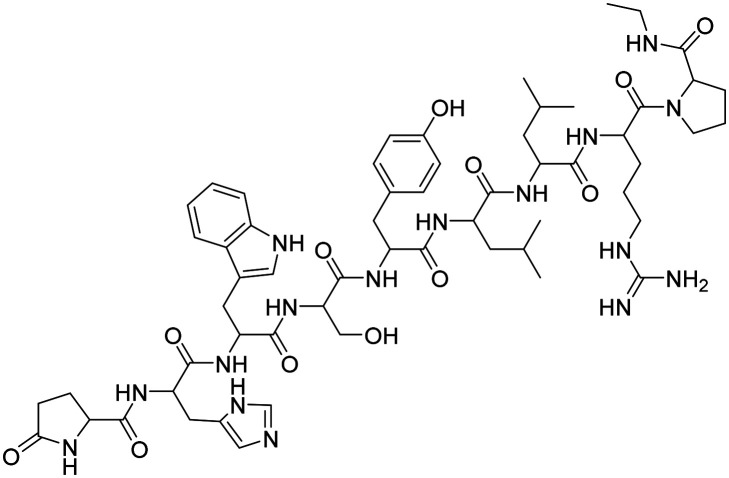
|
GnRH receptor agonist, reduce effects of elevated GnRH and gonadotropins on the brain | Growth factors and hormones | NCT00063310 |
| Montelukast |
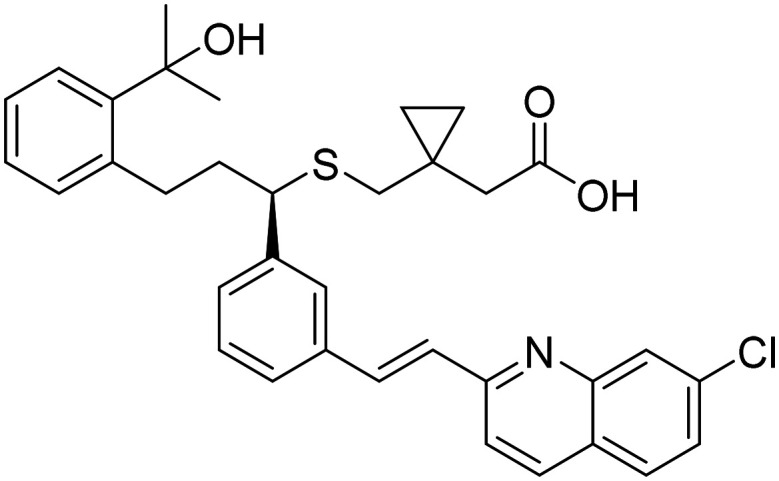
|
Cysteinyl leukotriene type 1 (cysLT-1) receptor antagonist, effects on inflammatory processes, neuronal injury, blood–brain-barrier integrity, and Aβ protein accumulation | Inflammation | NCT03991988 |
| Nilotinib |
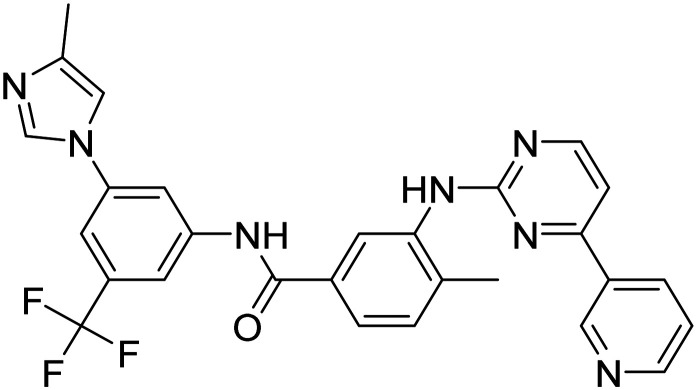
|
Tyrosine kinase inhibitor, autophagy enhancer, promotes clearance of Aβ and tau | Proteostasis/proteinopathies | NCT05143528 |
| Neflamapimod |
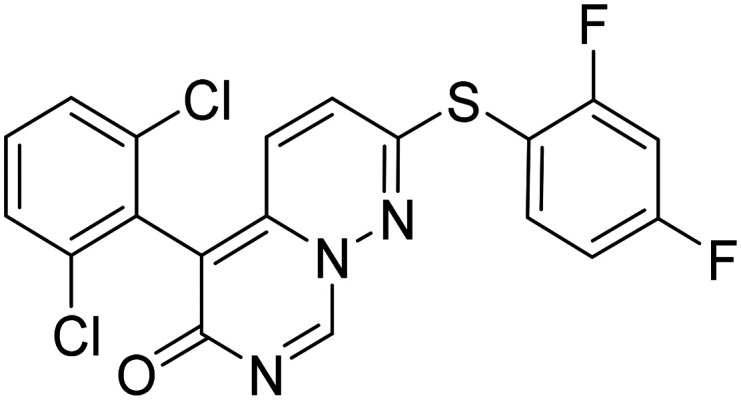
|
p38 MAPK-α inhibitor, enhance endolysosomal function to reduce synaptic dysfunction | Synaptic plasticity/neuroprotection | NCT03402659 |
| Nicotinamide |
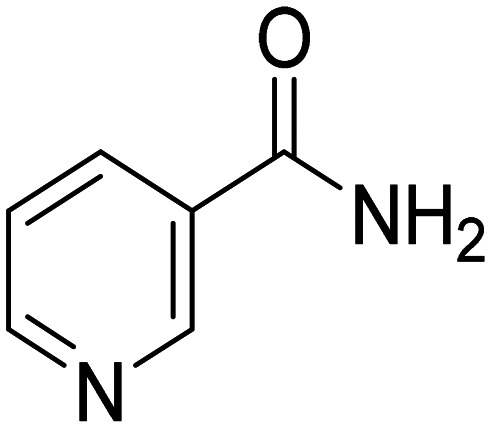
|
HDAC inhibitor, reduce tau-induced microtubule depolymerization and tau phosphorylation | Tau | NCT00580931 |
| Tacrolimus |
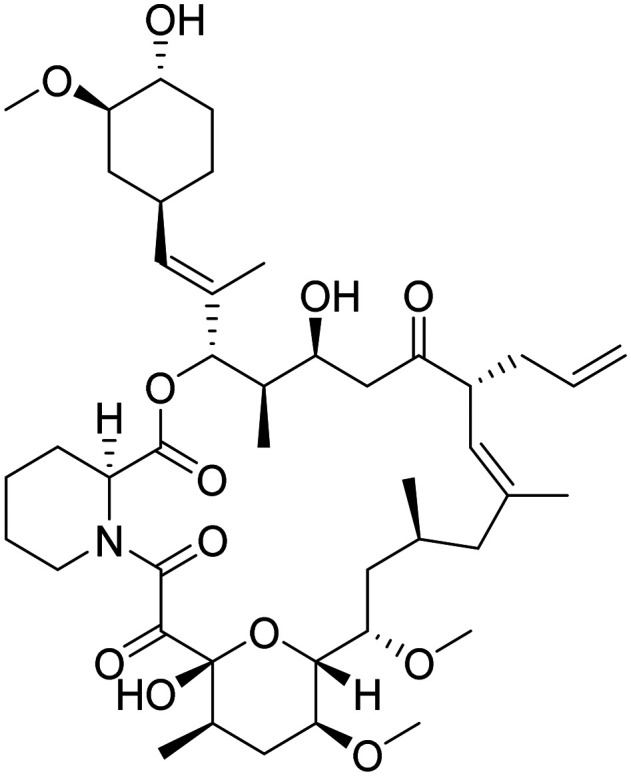
|
Calcineurin inhibitor, prevent Aβ-induced dendritic spine loss and synaptic dysfunction | Synaptic plasticity/neuroprotection | NCT04263519 |
| PU-AD8-((6-iodobenzo[d][1,3]dioxol-5-yl)thio)-9-(3-(isopropylamino)propyl)-9H-purin-6-amine |
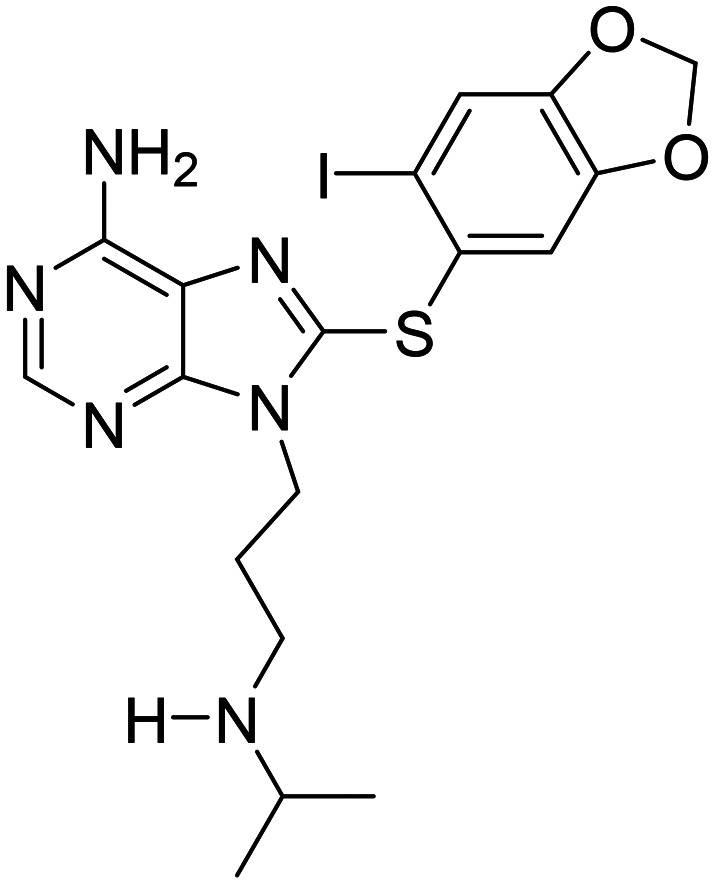
|
Heat shock protein 90 inhibitor, prevent aggregation and hyperphosphorylation of tau | Tau | NCT03935568 |
| PF-044479436-((3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one |
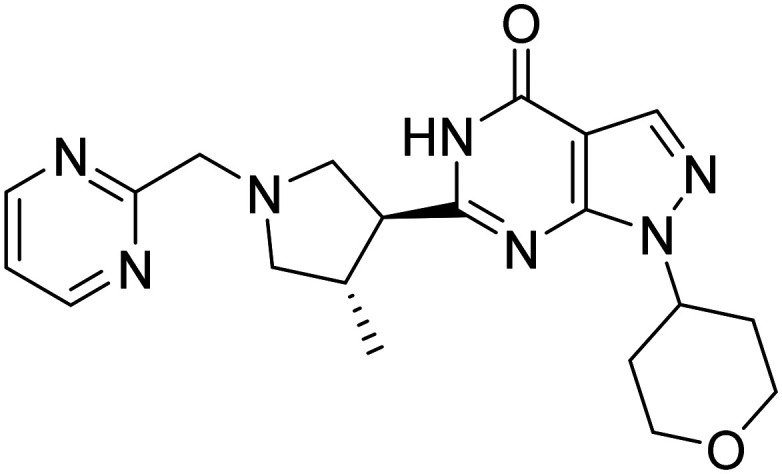
|
Selective phosphodiesterase 9 (PDE9) inhibitor | Cognition enhancement | NCT00988598 |
| Perindopril |
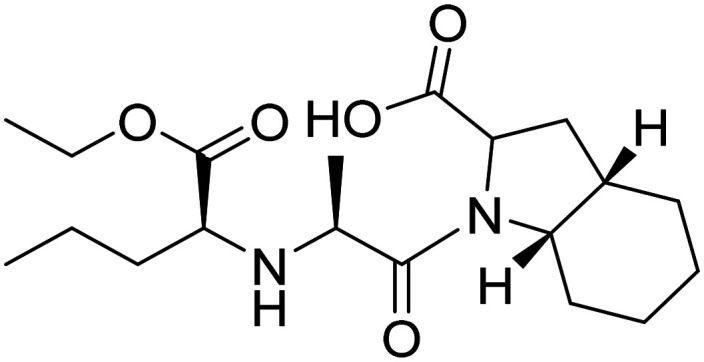
|
Angiotensin converting enzyme inhibitor | Vasculature | NCT02085265 |
| PQ912(S)-1-(1H-benzo[d]imidazol-6-yl)-5-(4-propoxyphenyl)imidazolidin-2-one |
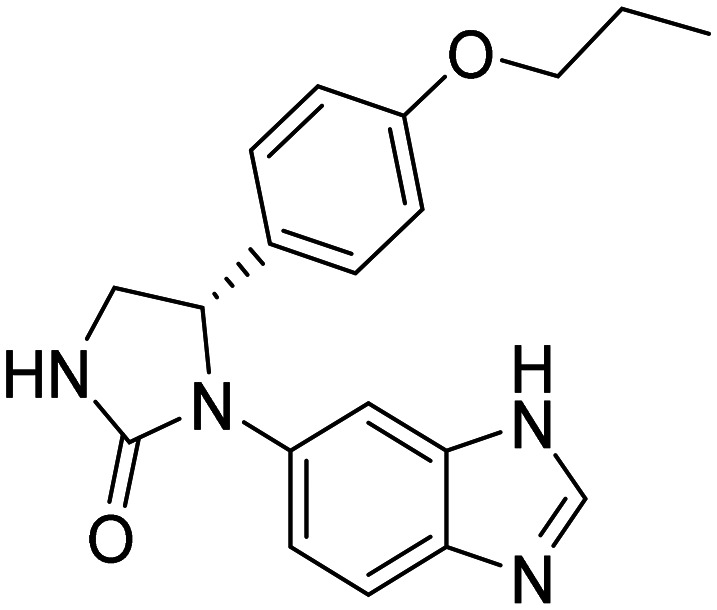
|
Glutaminyl cyclase (QC) enzyme inhibitor to reduce pyroglutamate Aβ (pGlu-Aβ) production | Amyloid | NCT02389413 |
| Rapamycin |
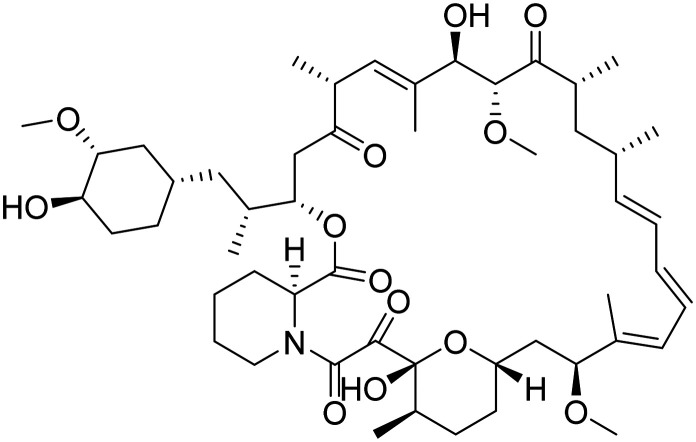
|
mTOR inhibitor, ameliorate metabolic and vascular effects of aging | Proteostasis/proteinopathies | NCT04629495 |
| S-Equol |
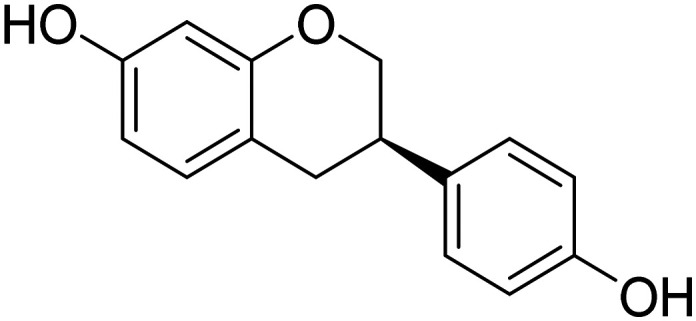
|
Agonist of non-hormonal estrogen receptor B located on mitochondria to potentiate mitochondrial function | Metabolism and bioenergetics | NCT03101085 |
| Sovateltide |
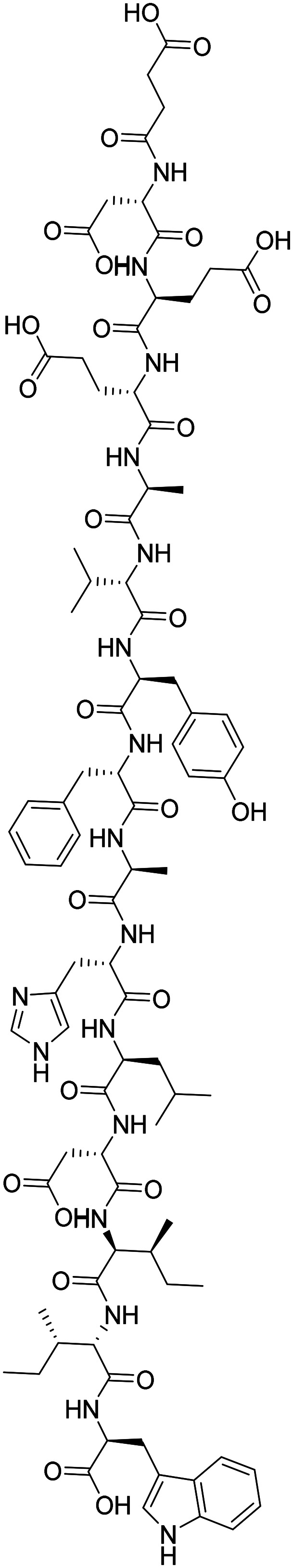
|
Endothelin B receptor agonist, augments activity of neuronal progenitor cells | Neurogenesis | NCT04052737 |
| Vafidemstat |
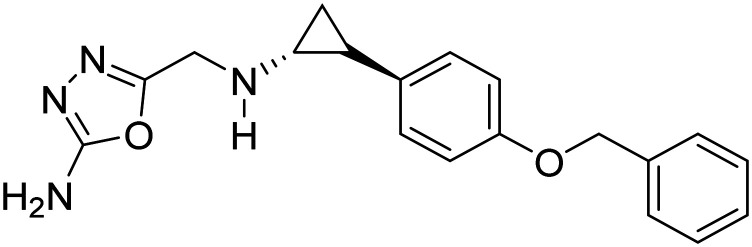
|
HDAC demethylase inhibitor and MAO-B inhibitor, neuroprotective | Synaptic plasticity/neuroprotection | NCT03867253 |
| Valacyclovir |
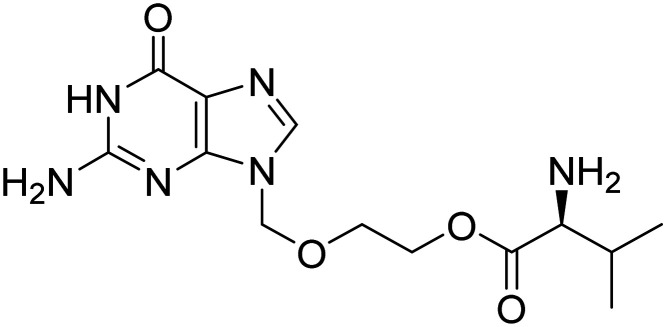
|
Antiviral against HSV-1 and -2 infection, prevent Aβ aggregation and plaque deposition | Infection/immunity | NCT03282916 |
| Symptom-reducing small molecules | ||||
| AD-35phosphenoperoxoic acid compound with 6′-(2-(1-(pyridin-2-ylmethyl)piperidin-4-yl)ethyl)spiro[cyclopropane-1,5′-[1,3]dioxolo[4,5-f]isoindol]-7′(6′H)-one |
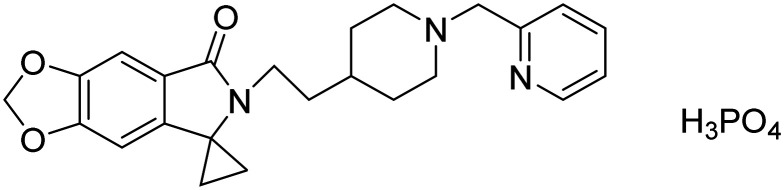
|
Acetylcholinesterase inhibitor | Neurotransmitter receptors | NCT03625401 |
| Huperzine A |
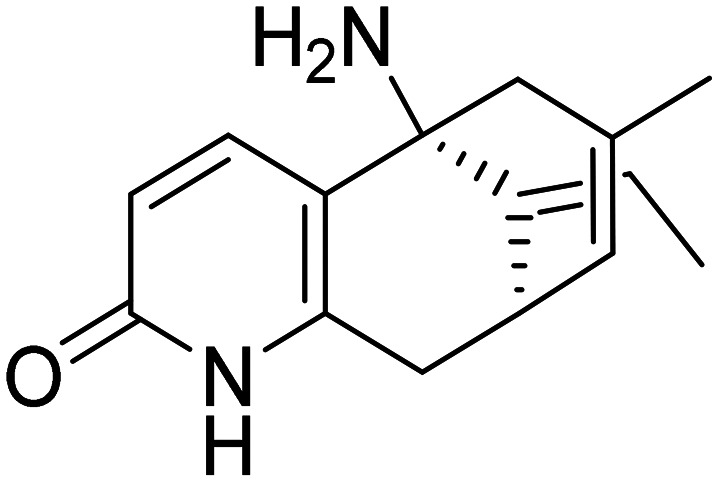
|
NMDA receptor antagonist and AChE inhibitor (MTDL) | Neurotransmitter receptors | NCT00083590 |
| Dexmedetomidine |
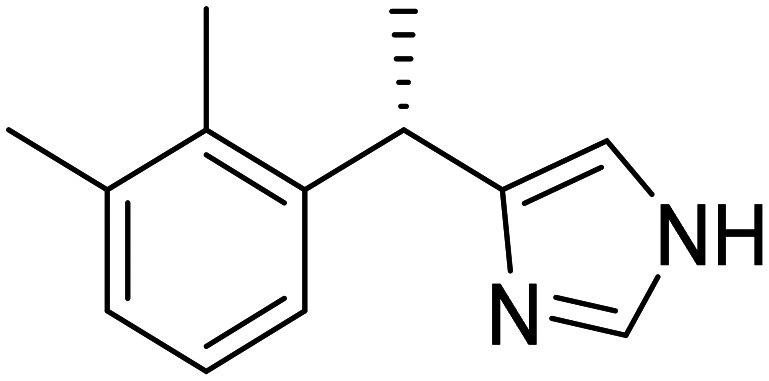
|
Sublingual dexmedetomidine, selective α2-adrenergic receptor agonist | Neurotransmitter receptors | NCT06052254 |
| CORT108297(R)-4a-(ethoxymethyl)-1-(4-fluorophenyl)-6-((4-(trifluoromethyl)phenyl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinoline |
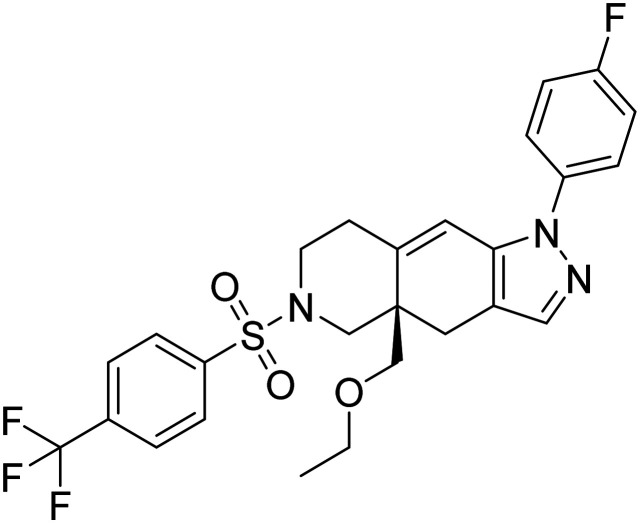
|
Selective glucocorticoid receptor antagonist, reduce neuroendocrine stress responses | Hormone (cognition enhancement) | NCT04601038 |
| Dronabinol |
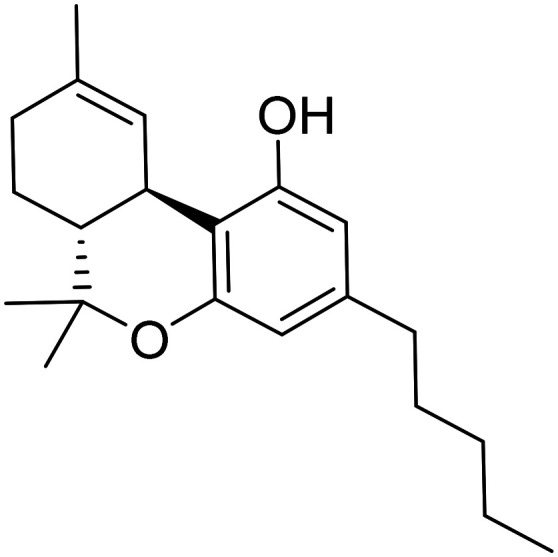
|
CB1 and CB2 endocannabinoid receptor partial agonist | Neurotransmitter receptors | NCT02792257 |
| Nicotine |
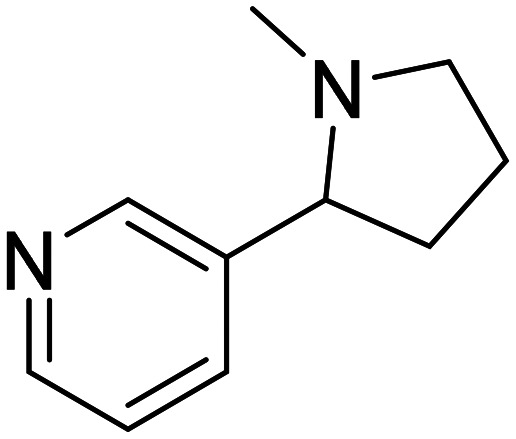
|
Nicotinic acetylcholine receptor agonist enhances cognition | Neurotransmitter receptors | NCT02720445 |
| Prazosin |
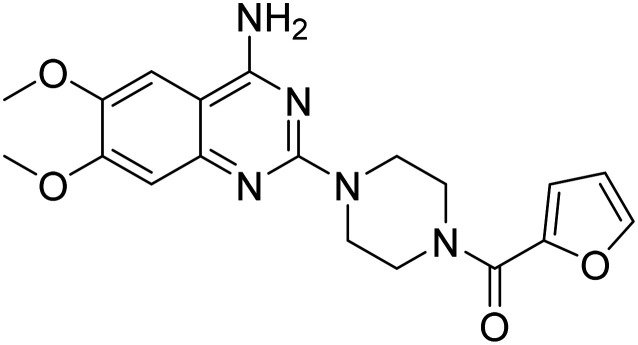
|
α-1 adrenoreceptor antagonist | Neurotransmitter receptors | NCT01126099 |
| SAGE-718 | First-in-class, oxysterol-based positive allosteric modulator (PAM) of N-methyl-d-aspartate (NMDA) receptors | Neurotransmitter receptors | NCT05619692 | |
| Phase III | ||||
| Disease-modifying biologicals | ||||
| Donanemab | MAb specific for pyroglutamate form of Aβ | Amyloid | NCT06566170 | |
| Gantenerumab | MAb directed at Aβ plaques and oligomers | Aβ | NCT01760005 | |
| Lecanemab | MAb directed at Aβ protofibrils | Aβ | NCT05269394 | |
| UB311 | Vaccine that stimulates a T-helper type 2 regulatory immune response over a T-helper type 1 pro-inflammatory response, and to avoid cross-reactivity with similar endogenous antigens, i.e., autoimmune responses | Amyloid | NCT02551809 | |
| Disease-modifying drugs | ||||
| Atuzaginstat |
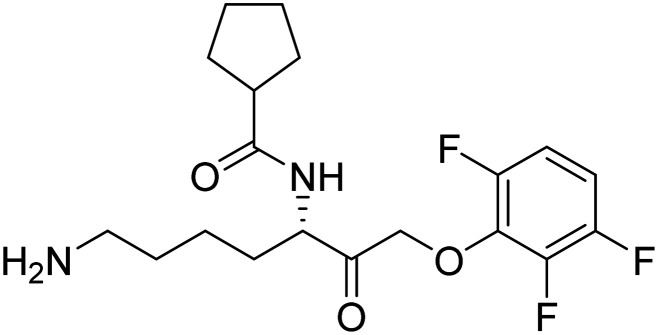
|
It reduces neuroinflammation and hippocampal degeneration | Inflammation/infection due to P. gingivalis | NCT03823404 |
| Blarcamesine |
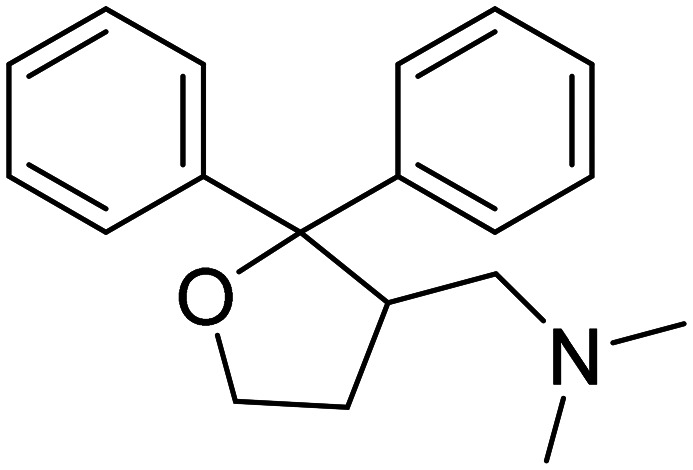
|
Sigma-1 receptor agonist, M2 autoreceptor antagonist. It ameliorates oxidative stress, protein misfolding, mitochondrial dysfunction, and inflammation | Synaptic plasticity | NCT04314934 |
| GV-971sodium (2S,3S,4S,5S,6R)-6-((1-carboxylato-2,4-dihydroxyhexan-3-yl)oxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylate oxo-l3-methanolate |
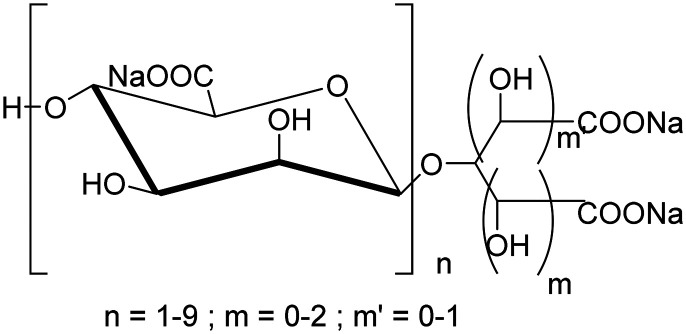
|
Algae-derived acidic oligosaccharides, changes microbiome to reduce peripheral and central inflammation | Inflammation | NCT04520412 |
| Icosapent ethyl |
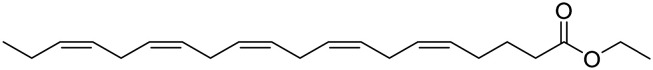
|
Purified form of the omega-3 fatty acid EPA. It improves synaptic function and reduces inflammation | Oxidative stress | NCT02719327 |
| Losartan + amlodipine + atorvastatin |
|
Losartan: angiotensin II receptor blocker | Vasculature | NCT02913664 |
| Amlodipine: calcium channel blocker | ||||
| Atorvastatin: anti-hyperlipidemic (HMG-CoA reductase inhibitor) | ||||
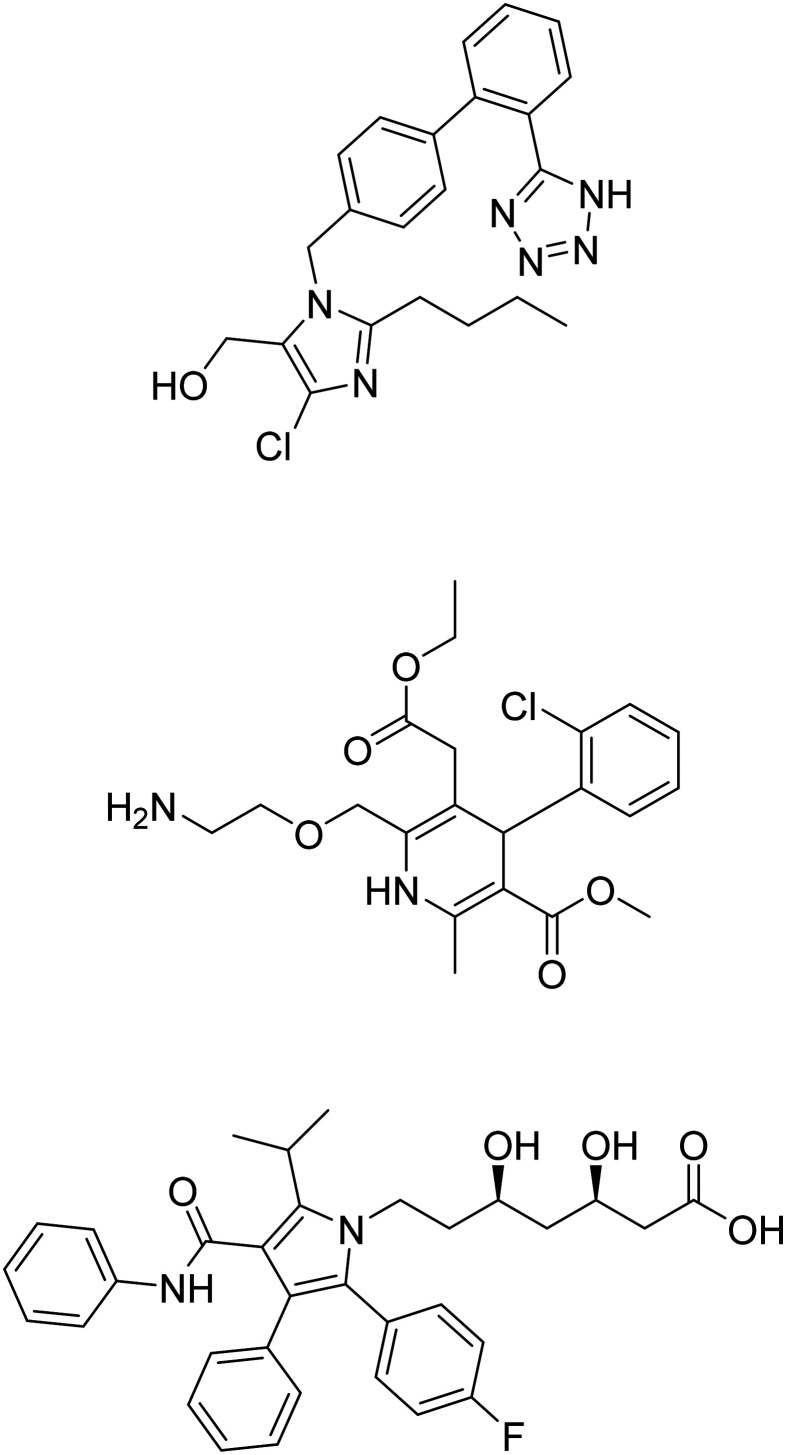
| Metformin |
|
Insulin sensitizer to improve CNS glucose metabolism | Metabolism and bioenergetics | NCT04098666 |
| Omega-3 fatty acids |
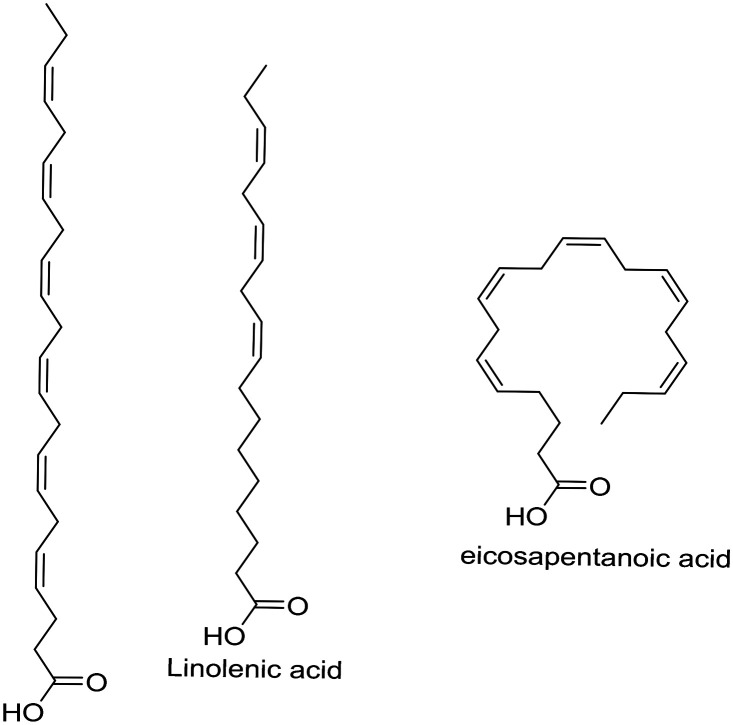
|
Antioxidant | Oxidative stress | NCT01058941 |
| NE3107 |
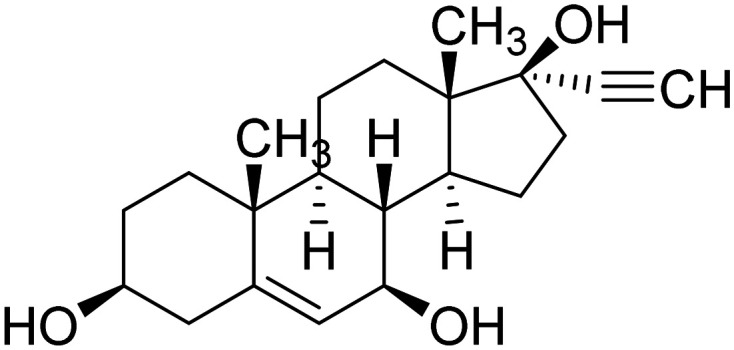
|
MAPK-1/3 inhibitor. It reduces pro-inflammatory NFκB activation | Inflammation | NCT04669028 |
| Tricaprilin |
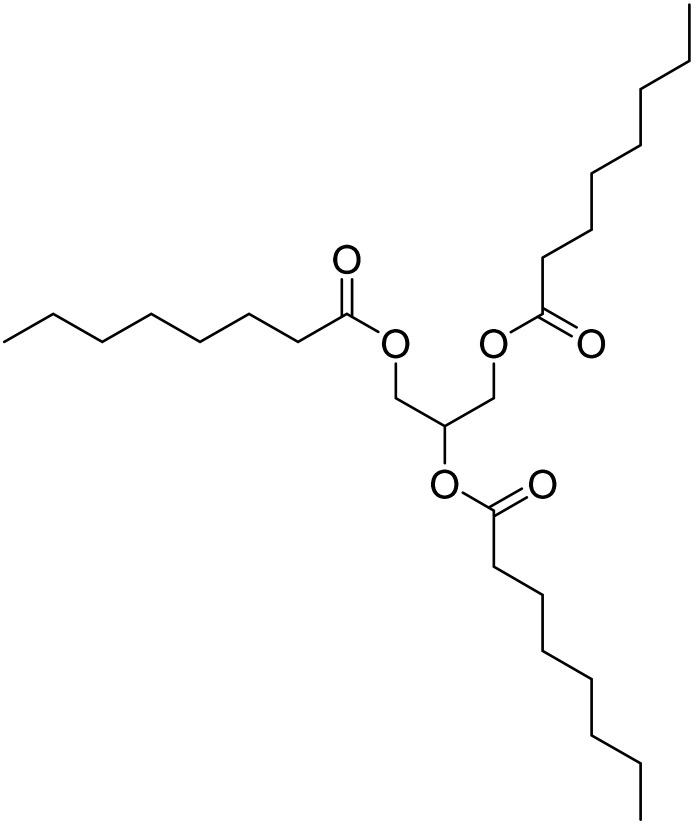
|
Caprylic triglyceride. It induces ketosis and improves mitochondrial and neuronal function | Metabolism and bioenergetics | NCT05809908 |
| Troriluzole |
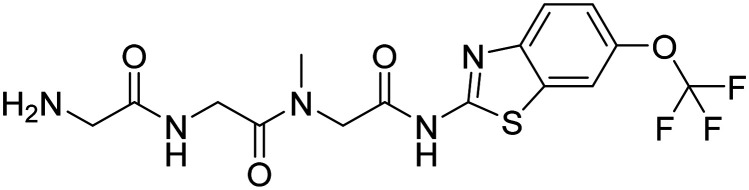
|
Glutamate modulator; prodrug of riluzole. It improves synaptic function | Synaptic plasticity/neuroprotection | NCT03605667 |
| Leuco-methylthioninium (LMTX) mesylate |
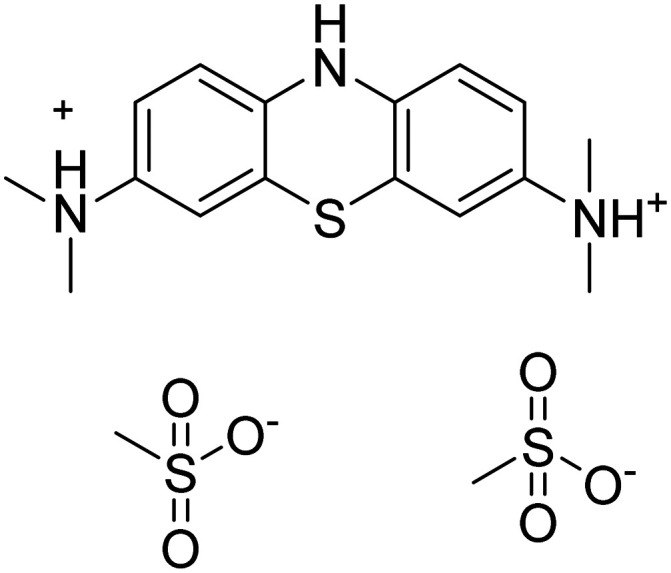
|
Tau protein aggregation inhibitor | Tau | NCT03539380 |
| Symptom-reducing small molecules | ||||
| Brexpiprazole |
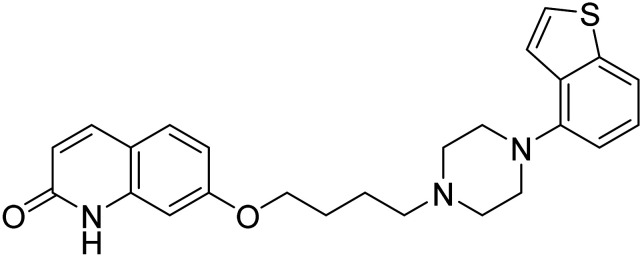
|
Atypical antipsychotic, D2 receptor partial agonist | Neurotransmitter receptors | NCT03548584 |
| Dextromethorphan + quinidine |
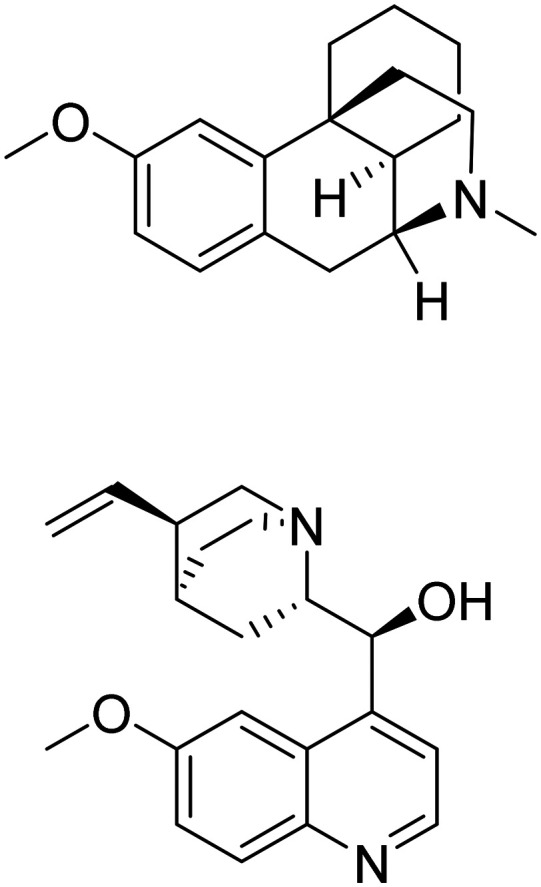
|
Sigma1 receptor agonist, NMDA receptor antagonist | Neurotransmitter receptors | NCT01584440 |
| Donepezil + memantine |
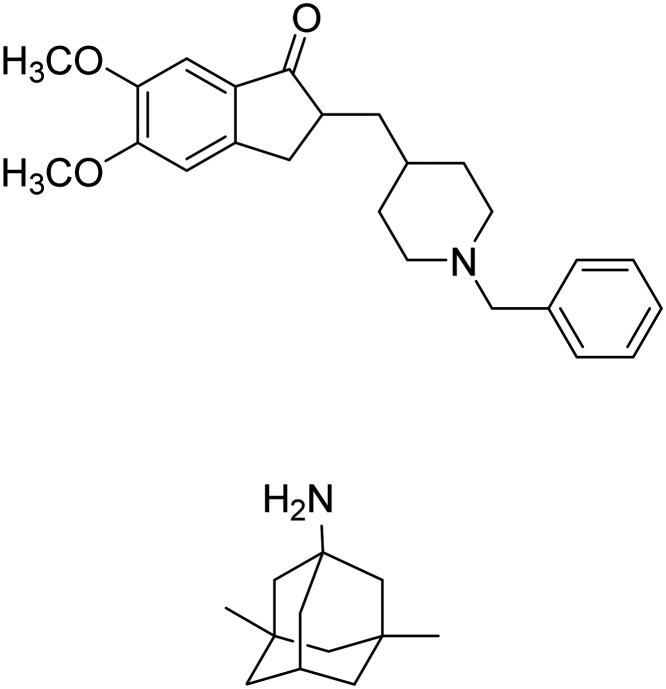
|
Donepezil: binds to AChE and reversibly inactivates it | Cognition enhancement | NCT02580305 |
| Memantine: blocks current flow through NMDA receptor channels | ||||
| Caffeine |
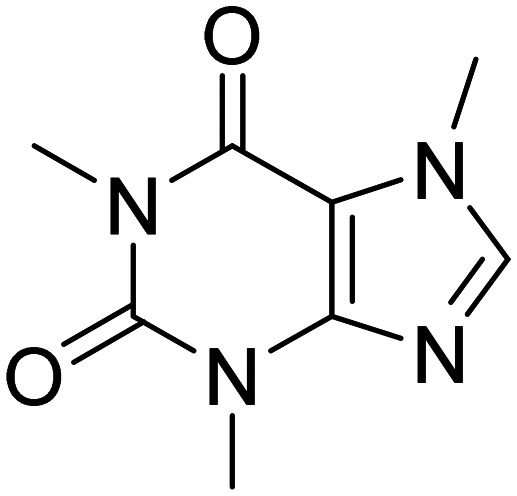
|
Pleiotropic effect on CNS | Metabolism and bioenergetic, cognition enhancement | NCT00692510 |
| Escitalopram |
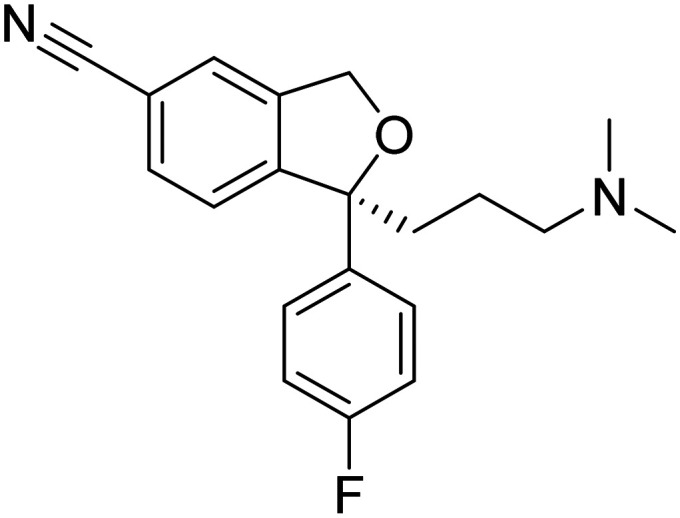
|
Selective serotonin reuptake inhibitor | Neurotransmitter receptors | NCT05004987 |
| Guanfacine |
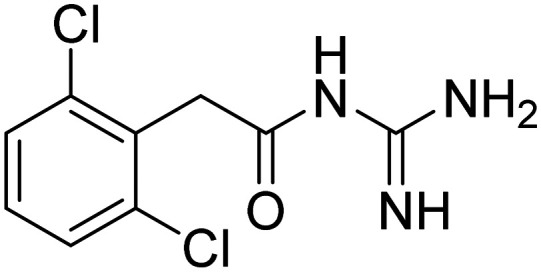
|
α-2 adrenergic agonist | Cognition enhancement | NCT03116126 |
| Mirtazapine |
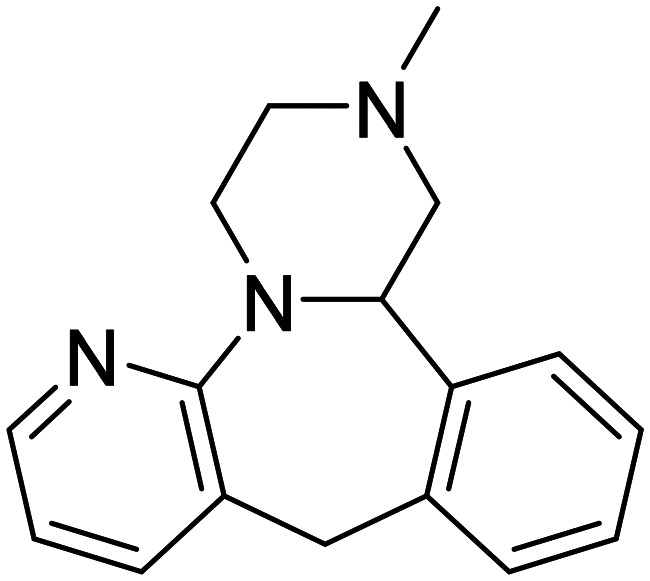
|
α-1 antagonist | Neurotransmitter receptors | NCT01505504 |
| Nabilone |
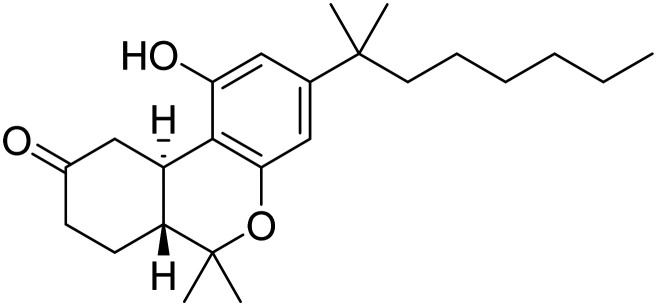
|
Synthetic cannabinoid, CB1 and CB2 receptor agonist. It suppresses neuronal excitability and neurotransmitter release | Neurotransmitter receptors | NCT04516057 |
| Octohydroamino-acridine succinate |
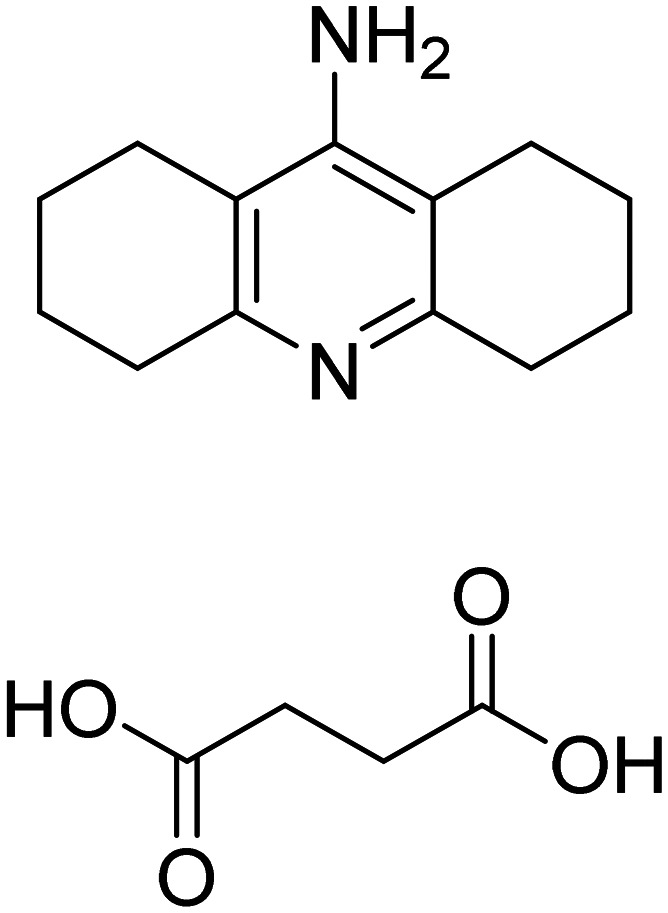
|
AChE inhibitor | Cognition enhancement | NCT01569516 |
Future perspective
Given the complexity of the condition and the numerous potential risk factors involved, current research is concentrated on various causative factors of AD. Many molecules in the developmental stage are designed to disrupt AD progression by targeting one or multiple agents linked to the condition. These potential targets encompass the accumulation of beta-amyloid and tau proteins (distinctive signs of Alzheimer's), neuroinflammation, immune reactions, metabolic alterations, and various other elements. Recent basic and clinical research has generated extensive insights into the biological mechanisms underlying Alzheimer's disease. Despite advances in understanding the molecular basis of Alzheimer's disease, efforts to develop effective drugs or non-pharmacological interventions to prevent, halt, or slow its progression have remained largely unsuccessful. Extensive pharmaceutical studies and clinical trials, whether successful or not, are invaluable as they help identify promising drugs or eliminate ineffective ones, guiding the way forward in the fight against Alzheimer's disease. The outcomes of “failed” clinical trials should be leveraged to refine and adjust future approaches. Priority should be given to preventing or slowing neurodegeneration, especially in individuals at risk, as treating advanced Alzheimer's such as restoring damaged neurons and synapses will likely remain a challenging task, at least in the near term. Non-pharmacological approaches that utilize modern technologies, such as non-invasive or minimally invasive surgical procedures, should be actively integrated into the strategies for combating Alzheimer's disease. Maintaining a healthy lifestyle through diet, sleep, and exercise can be beneficial and may help delay the onset of Alzheimer's symptoms, potentially through mechanisms like neurogenesis. If a significant delay of one to two decades or more can be achieved, it could effectively equate to the eradication of Alzheimer's disease for many elderly individuals. It is anticipated that the forthcoming treatments will entail a combination of drugs or devices targeting multiple objectives, accompanied by risk reduction approaches akin to those employed in current therapies for numerous cancer and AIDS cases. In clinical studies on donanemab, a trend toward slower Tau accumulation was noted. Therefore, future research should focus on exploring the relationship between reduced Aβ plaques and Tau levels to achieve meaningful benefits for Alzheimer's patients. The accelerated FDA approval of aducanumab, donanemab and lecanemab has brought new hope for Alzheimer's drug development, and we anticipate more effective and affordable treatments for patients in the future.
Conclusion
Single-target drugs often prove insufficient due to AD's multifaceted pathology. Many drugs have failed in clinical trials due to inadequate target interaction, toxic side effects, or a lack of differentiation from placebos. FDA-approved treatments offer symptomatic relief but do not halt disease progression. Addressing AD's multifaceted nature demands a comprehensive approach. Therefore, this review aims to emphasize recent progress in exploring innovative AD treatments from the perspective of medicinal chemistry. Various aspects of addressing AD have been consolidated, such as multiple hypotheses, single targets, and MTDLs. Aβ aggregation inhibitors, metal chelators, and neuroprotective mechanisms appear to hold promise, and MTDLs are expected to play a crucial role in the management of AD. In conclusion, although a cure for Alzheimer's disease through drug therapy has not yet been found, significant progress is being made. It is anticipated that new treatments with high efficacy, low adverse effects, and economic viability will be developed soon. This review may serve as a valuable resource for the medicinal chemistry community for exploring strategies to manage AD.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†
Author contributions
Amit Sharma: data curation, formal analysis, investigation, writing original draft. Santosh Rudrawar: conceptualization, validation, formal analysis, supervision, writing – review & editing. Hemant R. Jadhav: conceptualization, validation, formal analysis, supervision, writing – review & editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00630e
References
- Shanmugam J. V. Duraisamy B. Simon B. C. Bhaskaran P. Alzheimer's disease classification using pre-trained deep networks. Biomed. Signal Process. Control. 2022;71:103217. doi: 10.1016/j.bspc.2021.103217. [DOI] [Google Scholar]
- https://www.alz.org/alzheimers-dementia/facts-figures, accessed (07 May 2024)
- Domico M. and Hill V., What You Need to Know about Alzheimer's Disease, Bloomsbury Publishing USA, 2022, 10.1016/S0002-9343(97)00262-3 [DOI] [Google Scholar]
- Galle S. A. Geraedts I. K. Deijen J. B. Milders M. V. Drent M. L. The interrelationship between insulin-like growth factor 1, apolipoprotein E ε4, lifestyle factors, and the aging body and brain. J. Prev. Alzheimers Dis. 2020;7:265–273. doi: 10.14283/jpad.2020.11. [DOI] [PubMed] [Google Scholar]
- Olczak K. J. Taylor-Bateman V. Nicholls H. L. Traylor M. Cabrera C. P. Munroe P. B. Hypertension genetics past, present and future applications. J. Intern. Med. 2021;290:1130–1152. doi: 10.1111/joim.13352. [DOI] [PubMed] [Google Scholar]
- Sadiq I. Z. Free radicals and oxidative stress: Signaling mechanisms, redox basis for human diseases, and cell cycle regulation. Curr. Mol. Med. 2023;23:13–35. doi: 10.2174/1566524022666211222161637. [DOI] [PubMed] [Google Scholar]
- Thoe E. S. Fauzi A. Tang Y. Q. Chamyuang S. Chia A. Y. Y. A review on advances of treatment modalities for Alzheimer's disease. Life Sci. 2021;276:119129. doi: 10.1016/j.lfs.2021.119129. [DOI] [PubMed] [Google Scholar]
- Golde T. E. Disease-modifying therapies for Alzheimer's disease: More questions than answers. Neurotherapeutics. 2022;19:209–227. doi: 10.1007/s13311-022-01201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier N. G. Hodgson N. A. Gitlin L. N. Characteristics of activities for persons with dementia at the mild, moderate, and severe stages. Gerontologist. 2017;57:987–997. doi: 10.1093/geront/gnw133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek G. E. Decourt B. Sabbagh M. N. Lecanemab (BAN2401): an anti–beta-amyloid monoclonal antibody for the treatment of Alzheimer disease. Expert Opin. Invest. Drugs. 2023;32:89–94. doi: 10.1080/13543784.2023.2178414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasb M. Tao W. Chen N. Alzheimer's Disease Puzzle: Delving into Pathogenesis Hypotheses. Aging Dis. 2024;15(1):43–73. doi: 10.14336/AD.2023.0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G. Leugers C. J. Tau and tauopathies. Prog. Mol. Biol. Transl. Sci. 2012;107:263–293. doi: 10.1016/B978-0-12-385883-2.00004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig L. A. Hong N. S. McDonald R. J. Revisiting the cholinergic hypothesis in the development of Alzheimer's disease. Neurosci. Biobehav. Rev. 2011;35:1397–1409. doi: 10.1016/j.neubiorev.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Li S. The β-adrenergic hypothesis of synaptic and microglial impairment in Alzheimer's disease. J. Neurochem. 2023;165:289–302. doi: 10.1111/jnc.15782. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-J. Lin C.-H. Lane H.-Y. Involvement of cholinergic, adrenergic, and glutamatergic network modulation with cognitive dysfunction in Alzheimer's disease. Int. J. Mol. Sci. 2021;22:2283. doi: 10.3390/ijms22052283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honarnejad K. Herms J. Presenilins: role in calcium homeostasis. Int. J. Biochem. Cell Biol. 2012;44:1983–1986. doi: 10.1016/j.biocel.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Khan M. F. Wang G. Environmental agents, oxidative stress and autoimmunity. Curr. Opin. Toxicol. 2018;7:22–27. doi: 10.1016/j.cotox.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. V. Hanson J. E. Sheng M. Microglia in Alzheimer's disease. J. Cell Biol. 2018;217:459–472. doi: 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.-L. Fan Y.-G. Zhao L.-X. Zhang Q. Wang Z.-Y. The metal ion hypothesis of Alzheimer's disease and the anti-neuroinflammatory effect of metal chelators. Bioorg. Chem. 2023;131:106301. doi: 10.1016/j.bioorg.2022.106301. [DOI] [PubMed] [Google Scholar]
- Maccecchini M. L. Chang M. Y. Pan C. John V. Zetterberg H. Greig N. H. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-β peptide and τ levels: target engagement, tolerability and pharmacokinetics in humans. J. Neurol., Neurosurg. Psychiatry. 2012;83:894–902. doi: 10.1136/jnnp-2012-302589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traikapi A. Konstantinou N. Gamma oscillations in Alzheimer's disease and their potential therapeutic role. Front. Syst. Neurosci. 2021;15:782399. doi: 10.3389/fnsys.2021.782399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C., Merz G. E., Aoyagi A., DeGrado W. F. and Prusiner S. B., Aβ and Tau Prions Causing Alzheimer's Disease, in Alzheimer's Disease: Methods and Protocols, Springer US, New York, NY, 2022, pp. 293–337 [DOI] [PubMed] [Google Scholar]
- Fan L. Mao C. Xinchao H. Zhang S. Yang Z. Zhengwei H. Sun H. New insights into the pathogenesis of Alzheimer's disease. Front. Neurol. 2020;10:1312. doi: 10.3389/fneur.2019.01312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z. An Q. Zhang W. Li Y. Zhang Q. Yan H. Histamine and Receptors in Neuroinflammation: Their Roles on Neurodegenerative Diseases. Behav. Brain Res. 2024;465:114964. doi: 10.1016/j.bbr.2024.114964. [DOI] [PubMed] [Google Scholar]
- Zhou Q. Li S. Li M. Ke D. Wang Q. Yang Y. Liu G.-P. Wang X.-C. Liu E. Wang J.-Z. Human tau accumulation promotes glycogen synthase kinase-3β acetylation and thus upregulates the kinase: a vicious cycle in Alzheimer neurodegeneration. EBioMedicine. 2022;78:103970. doi: 10.1016/j.ebiom.2022.103970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaldijk E. Vermeiren Y. The Role of Serotonin within the Microbiota-gut-brain Axis in the Development of Alzheimer'S Disease: A Narrative Review. Ageing Res. Rev. 2022;75:101556. doi: 10.1016/j.arr.2021.101556. [DOI] [PubMed] [Google Scholar]
- Jiménez-Balado J. Eich T. S. GABAergic Dysfunction, Neural Network Hyperactivity and Memory Impairments in Human Aging and Alzheimer's Disease. Semin. Cell Dev. Biol. 2021;116:146–159. doi: 10.1016/j.semcdb.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte N. Nortley R. Attwell D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer's disease. Acta Neuropathol. 2020;140:793–810. doi: 10.1007/s00401-020-02215-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo-Moreno T. González-Acedo A. Rivas-Domínguez A. García-Morales V. García-Cozar F. J. Ramos-Rodríguez J. J. Melguizo-Rodríguez L. Therapeutic approach to Alzheimer's disease: Current treatments and new perspectives. Pharmaceutics. 2022;14:1117. doi: 10.3390/pharmaceutics14061117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perneczky R. Jessen F. Grimmer T. Levin J. Flöel A. Peters O. Froelich L. Anti-amyloid antibody therapies in Alzheimer's disease. Brain. 2023;146:842–849. doi: 10.1093/brain/awad005. [DOI] [PubMed] [Google Scholar]
- Alexander G. C. Emerson S. Kesselheim A. S. Evaluation of aducanumab for Alzheimer disease: scientific evidence and regulatory review involving efficacy, safety, and futility. JAMA, J. Am. Med. Assoc. 2021;325:1717–1718. doi: 10.1001/jama.2021.3854. [DOI] [PubMed] [Google Scholar]
- Breijyeh Z. Karaman R. Comprehensive review on Alzheimer's disease: Causes and treatment. Molecules. 2020;25:5789. doi: 10.3390/molecules25245789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H. Yamanish Y. Iimura Y. Kawakami Y. Donepezil hydrochloride (E2020) and other acetylcholinesterase inhibitors. Curr. Med. Chem. 2000;7:303–339. doi: 10.2174/0929867003375191. [DOI] [PubMed] [Google Scholar]
- Marco-Contelles J. do Carmo Carreiras M. Rodríguez C. Villarroya M. García A. G. Synthesis and pharmacology of galantamine. Chem. Rev. 2006;106:116–133. doi: 10.1021/cr040415t. [DOI] [PubMed] [Google Scholar]
- Anand P. Singh B. A review on cholinesterase inhibitors for Alzheimer's disease. Arch. Pharmacal Res. 2013;36:375–399. doi: 10.1007/s12272-013-0036-3. [DOI] [PubMed] [Google Scholar]
- Lucey B. P. Liu H. Toedebusch C. D. Freund D. Redrick T. Chahin S. L. Mawuenyega K. G. Suvorexant acutely decreases tau phosphorylation and Aβ in the human CNS. Ann. Neurol. 2023;94:27–40. doi: 10.1002/ana.26641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L. S. Mangialasche F. Andreasen N. Feldman H. Giacobini E. Jones R. Mantua V. Clinical trials and late-stage drug development for A lzheimer's disease: an appraisal from 1984 to 2014. J. Intern. Med. 2014;275:251–283. doi: 10.1111/joim.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremic D. Jiménez-Díaz L. Navarro-López J. D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer's disease: A systematic review. Ageing Res. Rev. 2021;72:101496. doi: 10.1016/j.arr.2021.101496. [DOI] [PubMed] [Google Scholar]
- Kim J. M. Jeon H. Kim H. Y. Kim Y. S. Failure, Success, and Future Direction of Alzheimer Drugs Targeting Amyloid-β Cascade: Pros and Cons of Chemical and Biological Modalities. ChemBioChem. 2023;24:e202300328. doi: 10.1002/cbic.202300328. [DOI] [PubMed] [Google Scholar]
- Ghosh S. Ali R. Verma S. Aβ-oligomers: A potential therapeutic target for Alzheimer's disease. Int. J. Biol. Macromol. 2023:124231. doi: 10.1016/j.ijbiomac.2023.124231. [DOI] [PubMed] [Google Scholar]
- Athar T. Al Balushi K. Khan S. A. Recent advances on drug development and emerging therapeutic agents for Alzheimer's disease. Mol. Biol. Rep. 2021;48(7):5629–5645. doi: 10.1007/s11033-021-06512-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decourt B. Wilson J. Ritter A. Dardis C. DiFilippo F. P. Zhuang X. Cordes D. Lee G. Fulkerson N. D. St Rose T. Hartley K. Sabbagh M. N. MCLENA-1: A Phase II Clinical Trial for the Assessment of Safety, Tolerability, and Efficacy of Lenalidomide in Patients with Mild Cognitive Impairment Due to Alzheimer's Disease. Open Access J. Clin. Trials. 2020;12:1–13. doi: 10.2147/oajct.s221914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel K. Ranasinghe K. G. Beagle A. J. La A. Ah Pook K. Castro M. Mizuiri D. Honma S. M. Venkateswaran N. Koestler M. Zhang W. Mucke L. Howell M. J. Possin K. L. Kramer J. H. Boxer A. L. Miller B. L. Nagarajan S. S. Kirsch H. E. Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity: A Randomized Clinical Trial. JAMA Neurol. 2021;78(11):1345–1354. doi: 10.1001/jamaneurol.2021.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W. Yang H. Yang J. Small-molecule drugs development for Alzheimer's disease. Front. Aging Neurosci. 2022;14:1019412. doi: 10.3389/fnagi.2022.1019412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.alz.org/, (accessed 30 May 2024)
- Cummings J. L. Zhong K. Kinney J. W. Heaney C. Moll-Tudla J. Joshi A. Pontecorvo M. Devous M. Tang A. Bena J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer's disease. Alzheimers Res. Ther. 2016;8:1–9. doi: 10.1186/s13195-016-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. A. Yu J. Y. Versavel M. Abushakra S. Kocis P. Power A. Kaplan P. L. Amedio J. Tolar M. Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer's disease. Clin. Pharmacokinet. 2018;57:315–333. doi: 10.1007/s40262-017-0608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzsche J. Jürgens D. Willuweit A. Adermann K. Fuchs C. Simons S. Windisch M. Hümpel M. Rossberg W. Wolzt M. Willbold D. Safety and pharmacokinetics of the orally available antiprionic compound PRI-002: A single and multiple ascending dose phase I study. Alzheimers Dement. 2020;6:e12001. doi: 10.1002/trc2.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannfelt L. Blennow K. Zetterberg H. Batsman S. Ames D. Harrison J. Masters C. L. Targum S. Bush A. I. Murdoch R. Wilson J. Ritchie CW; PBT2-201-EURO study group. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7(9):779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- Scheltens P. Hallikainen M. Grimmer T. Duning T. Gouw A. A. Teunissen C. E. Wink A. M. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer's disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res. Ther. 2018;10:1–14. doi: 10.1186/s13195-018-0431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurrier J. Nicholson L. S. Fang X. T. Stoner A. J. Toyonaga T. Holden D. Siegert T. R. Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci. Transl. Med. 2022;14:eabi8593. doi: 10.1126/scitranslmed.abi8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo N. J. Yuede C. M. LaBarbera K. M. Limegrover C. S. Rehak C. Yurko R. Waybright L. Preclinical and clinical biomarker studies of CT1812: A novel approach to Alzheimer's disease modification. Alzheimer's Dementia. 2021;17:1365–1382. doi: 10.1002/alz.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellar D. Register T. Lockhart S. N. Aisen P. Raman R. Rissman R. A. Brewer J. Craft S. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer's disease: A randomized trial. Sci. Rep. 2022;12:1346. doi: 10.1038/s41598-022-05165-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-Y. Pei Z. Lee K.-C. Lopez-Brignoni E. Nikolov B. Crowley C. A. Marsman M. R. Barbier R. Friedmann N. Burns L. H. PTI-125 reduces biomarkers of Alzheimer's disease in patients. J. Prev. Alzheimers Dis. 2020;7:256–264. doi: 10.14283/jpad.2020.6. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T. Molecular pathogenesis and disease-modifying therapies of Alzheimer's disease and related disorders. JMA J. 2022;5:307–313. doi: 10.31662/jmaj.2022-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khezri M. R. Mohebalizadeh M. Ghasemnejad-Berenji M. Therapeutic potential of ADAM10 modulation in Alzheimer's disease: a review of the current evidence. Cell Commun. Signaling. 2023;21:60. doi: 10.1186/s12964-023-01072-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda A. Montiel E. Ulrich H. Paz C. Selective Secretase Targeting for Alzheimer's Disease Therapy. J. Alzheimer's Dis. 2021;81(1):1–17. doi: 10.3233/JAD-201027. [DOI] [PubMed] [Google Scholar]; , PMID: 33749645
- Zhang Y. Chen H. Li R. Sterling K. Song W. Amyloid β-based therapy for Alzheimer's disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023;8:248. doi: 10.1038/s41392-023-01484-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli S. Satir T. M. Camacho R. Fruhwürth S. Bergström P. Zetterberg H. Agholme L. APP-BACE1 Interaction and Intracellular Localization Regulate Aβ Production in iPSC-Derived Cortical Neurons. Cell. Mol. Neurobiol. 2023:1–16. doi: 10.1007/s10571-023-01374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen Y. C. Kammeyer A. M. Tirlangi J. Ghosh A. K. Mesecar A. D. A Structure-Based Discovery Platform for BACE2 and the Development of Selective BACE Inhibitors. ACS Chem. Neurosci. 2021;12(4):581–588. doi: 10.1021/acschemneuro.0c00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evin G. Future Therapeutics in Alzheimer's Disease: Development Status of BACE Inhibitors. BioDrugs. 2016;30(3):173–194. doi: 10.1007/s40259-016-0168-3. [DOI] [PubMed] [Google Scholar]; , PMID: 27023706
- Iraji A. Khoshneviszadeh M. Firuzi O. Khoshneviszadeh M. Edraki N. Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 2020;97:103649. doi: 10.1016/j.bioorg.2020.103649. [DOI] [PubMed] [Google Scholar]
- Gehlot P. Kumar S. Vyas V. K. Choudhary B. S. Sharma M. Malik R. Guanidine-based β amyloid precursor protein cleavage enzyme 1 (BACE 1) inhibitors for the Alzheimer's disease (AD): A Review. Bioorg. Med. Chem. 2022:117047. doi: 10.1016/j.bmc.2022.117047. [DOI] [PubMed] [Google Scholar]
- Haass C. Kaether C. Thinakaran G. Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harbor Perspect. Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie P. Vartak A. Li Y. M. γ-Secretase inhibitors and modulators: Mechanistic insights into the function and regulation of γ-Secretase. Semin. Cell Dev. Biol. 2020;105:43–53. doi: 10.1016/j.semcdb.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J. Y. γ-Secretase in Alzheimer's disease. Exp. Mol. Med. 2022;54(4):433–446. doi: 10.1038/s12276-022-00754-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley D. B. Sundell K. L. Sethuraman G. Dowsett S. A. May P. C. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr. Med. Res. Opin. 2014:2021–2032. doi: 10.1185/03007995.2014.939167. [DOI] [PubMed] [Google Scholar]
- Wang D. Xu J. Liu B. He X. Zhou L. Hu X. Qiao F. Zhang A. Xu X. Zhang H. Wicha M. S. Zhang L. Shao Z. M. Liu S. IL6 blockade potentiates the anti-tumor effects of γ-secretase inhibitors in Notch3-expressing breast cancer. Cell Death Differ. 2018;25(2):330–339. doi: 10.1038/cdd.2017.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordvall G. Lundkvist J. Sandin J. Gamma-secretase modulators: a promising route for the treatment of Alzheimer's disease. Front. Mol. Neurosci. 2023;16:1279740. doi: 10.3389/fnmol.2023.1279740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush A. I. Tanzi R. E. Therapeutics for Alzheimer's disease based on the metal hypothesis. Neurotherapeutics. 2008;5:421–432. doi: 10.1016/j.nurt.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gucký A. Hamuľaková S. Targeting Biometals in Alzheimer's Disease with Metal Chelating Agents Including Coumarin Derivatives. CNS Drugs. 2024;38:507–532. doi: 10.1007/s40263-024-01093-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemers E. Drug Development in AD: Point of View from the Industry. J. Prev. Alzheimers Dis. 2015;2:216–218. doi: 10.14283/jpad.2015.80. [DOI] [PubMed] [Google Scholar]
- Henley D. B. Sundell K. L. Sethuraman G. Dowsett S. A. May P. C. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr. Med. Res. Opin. 2014;30:2021–2032. doi: 10.1185/03007995.2014.939167. [DOI] [PubMed] [Google Scholar]
- Becker R. E. Greig N. H. Increasing the success rate for Alzheimer's disease drug discovery and development. Expert Opin. Drug Discovery. 2012;7:367–370. doi: 10.1517/17460441.2012.672409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atri A. Molinuevo J. L. Lemming O. Wirth Y. Pulte I. Wilkinson D. Memantine in patients with Alzheimer's disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimers Res. Ther. 2013;5:1–11. doi: 10.1186/alzrt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A.-L. Gulbahce N. Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertini C. Salerno A. de Sena P. Pinheiro M. Bolognesi M. L. From combinations to multitarget-directed ligands: A continuum in Alzheimer's disease polypharmacology. Med. Res. Rev. 2021;41:2606–2633. doi: 10.1002/med.21699. [DOI] [PubMed] [Google Scholar]
- Li F. Zhao C. Wang L. Molecular-targeted agents combination therapy for cancer: developments and potentials. Int. J. Cancer. 2014;134:1257–1269. doi: 10.1002/ijc.28261. [DOI] [PubMed] [Google Scholar]
- Maramai S. Benchekroun M. Gabr M. T. Yahiaoui S. Multitarget therapeutic strategies for Alzheimer's disease: Review on emerging target combinations. Biomed. Res. Int. 2020:5120230. doi: 10.1155/2020/5120230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay R. R. Popovic-Nikolic M. R. Nikolic K. Uliassi E. Bolognesi M. L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018;7:1–14. doi: 10.1186/s40169-017-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. Jiang X. He S. Jiang H. Feng F. Liu W. Wei Q. Sun H. Rational design of multitarget-directed ligands: strategies and emerging paradigms. J. Med. Chem. 2019;62:8881–8914. doi: 10.1021/acs.jmedchem.9b00017. [DOI] [PubMed] [Google Scholar]
- Malafaia D. Albuquerque H. M. T. Silva A. M. S. Amyloid-β and tau aggregation dual-inhibitors: A synthetic and structure-activity relationship focused review. Eur. J. Med. Chem. 2021;214:113209. doi: 10.1016/j.ejmech.2021.113209. [DOI] [PubMed] [Google Scholar]
- Mishra P. Kumar A. Panda G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer's disease (1998–2018) Bioorg. Med. Chem. 2019;27:895–930. doi: 10.1016/j.bmc.2019.01.025. [DOI] [PubMed] [Google Scholar]
- Najafi Z. Saeedi M. Mahdavi M. Sabourian R. Khanavi M. Tehrani M. B. Moghadam F. H. Design and synthesis of novel anti-Alzheimer's agents: Acridine-chromenone and quinoline-chromenone hybrids. Bioorg. Chem. 2016;67:84–94. doi: 10.1016/j.bioorg.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Deng Y. Jiang Y. Zhao X. Wang J. Design, Synthesize and Bio-Evaluate 1, 2-Dihydroisoquinolin-3 (4H)-One Derivates as Acetylcholinesterase and β-Secretase Dual Inhibitors in Treatment with Alzheimer's Disease. J. Biosci. Med. 2016;4:112. doi: 10.4236/jbm.2016.41014. [DOI] [Google Scholar]
- Kumar N. Kumar V. Anand P. Kumar V. Dwivedi A. R. Kumar V. Advancements in the development of multi-target directed ligands for the treatment of Alzheimer's disease. Bioorg. Med. Chem. 2022;61:116742. doi: 10.1016/j.bmc.2022.116742. [DOI] [PubMed] [Google Scholar]
- Sang Z. Wang K. Dong J. Tang L. Alzheimer's disease: updated multi-targets therapeutics are in clinical and in progress. Eur. J. Med. Chem. 2022;238:114464. doi: 10.1016/j.ejmech.2022.114464. [DOI] [PubMed] [Google Scholar]
- Kogen H. Toda N. Tago K. Marumoto S. Takami K. Ori M. Yamada N. Design and synthesis of dual inhibitors of acetylcholinesterase and serotonin transporter targeting potential agents for Alzheimer's disease. Org. Lett. 2002;20:3359–3362. doi: 10.1021/ol026418e. [DOI] [PubMed] [Google Scholar]
- He F. Ran Y. Li X. Wang D. Zhang Q. Lv J. Chenggong Y. Design, synthesis and biological evaluation of dual-function inhibitors targeting NMDAR and HDAC for Alzheimer's disease. Bioorg. Chem. 2020;103:104109. doi: 10.1016/j.bioorg.2020.104109. [DOI] [PubMed] [Google Scholar]
- Kaur S. Bansal Y. Design, molecular Docking, synthesis and evaluation of xanthoxylin hybrids as dual inhibitors of IL-6 and acetylcholinesterase for Alzheimer's disease. Bioorg. Chem. 2022;121:105670. doi: 10.1016/j.bioorg.2022.105670. [DOI] [PubMed] [Google Scholar]
- Lv P. Xia C.-L. Wang N. Liu Z.-Q. Huang Z.-S. Huang S.-L. Synthesis and evaluation of 1, 2, 3, 4-tetrahydro-1-acridone analogues as potential dual inhibitors for amyloid-beta and tau aggregation. Bioorg. Med. Chem. 2018;26:4693–4705. doi: 10.1016/j.bmc.2018.08.007. [DOI] [PubMed] [Google Scholar]
- Iraji A. Khoshneviszadeh M. Firuzi O. Khoshneviszadeh M. Edraki N. Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 2020;97:103649. doi: 10.1016/j.bioorg.2020.103649. [DOI] [PubMed] [Google Scholar]
- Luo Z. Sheng J. Sun Y. Chuanjun L. Yan J. Liu A. Luo H.-b. Huang L. Li X. Synthesis and evaluation of multi-target-directed ligands against Alzheimer's disease based on the fusion of donepezil and ebselen. J. Med. Chem. 2013;56:9089–9099. doi: 10.1021/jm401047q. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Soacha D. A. Scheiner M. Decker M. Multi-target-directed-ligands acting as enzyme inhibitors and receptor ligands. Eur. J. Med. Chem. 2019;180:690–706. doi: 10.1016/j.ejmech.2019.07.040. [DOI] [PubMed] [Google Scholar]
- Leon R. Garcia A. G. Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer's disease. Med. Res. Rev. 2013:139–189. doi: 10.1002/med.20248. [DOI] [PubMed] [Google Scholar]
- Dorababu A. Promising heterocycle-based scaffolds in recent (2019–2021) anti-Alzheimer's drug design and discovery. Eur. J. Pharmacol. 2022;920:174847. doi: 10.1016/j.ejphar.2022.174847. [DOI] [PubMed] [Google Scholar]
- Pathak C. Kabra U. D. A comprehensive review of multi-target directed ligands in the treatment of Alzheimer's disease. Bioorg. Chem. 2024:107152. doi: 10.1016/j.bioorg.2024.107152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†