

Abstract
Phosphorescent gold(iii) complexes possess long-lived emissive excited states, making them ideal for use as molecular sensors and photosensitizers for organic transformations. Literature reports indicate that gold(iii) emitters exhibit good catalytic activity in homogeneous photochemical reactions. Heterogeneous metal–organic framework (MOF)-supported gold(iii) photocatalysts are considered to show high recyclability in photochemical reactions and potentially provide new selectivities. Here, we report the design and development of visible-light-absorbing MOF-covalently linked gold(iii) photocatalysts. A MOF-supported gold(iii) complex exhibits a longer phosphorescence lifetime than its homogeneous counterpart, reaching approximately 110 μs under argon and 12 μs under air when suspended in acetonitrile. This is attributed to the localization of the gold(iii) complex within the MOF nano-cages. The MOF-derived gold(iii) photosensitizer exhibits good catalytic performance in intramolecular and intermolecular [2 + 2] cycloaddition reactions to construct functionalized cyclobutanes and azetidines, both of which are important building blocks for pharmaceuticals. These photochemical [2 + 2] cycloaddition reactions catalyzed by the gold(iii)-MOF are governed by an energy transfer mechanism and do not require redox modulators. Under similar reaction conditions, the crossed [2 + 2] photocycloaddition reaction of two activated alkenes proceeds smoothly, and the cyclobutane product is obtained within 3 hours of irradiation under dilute conditions (0.2 mol dm−3 of alkene).
Phosphorescent gold(iii) complexes possess long-lived emissive excited states, making them ideal for use as photocatalysts for organic transformations.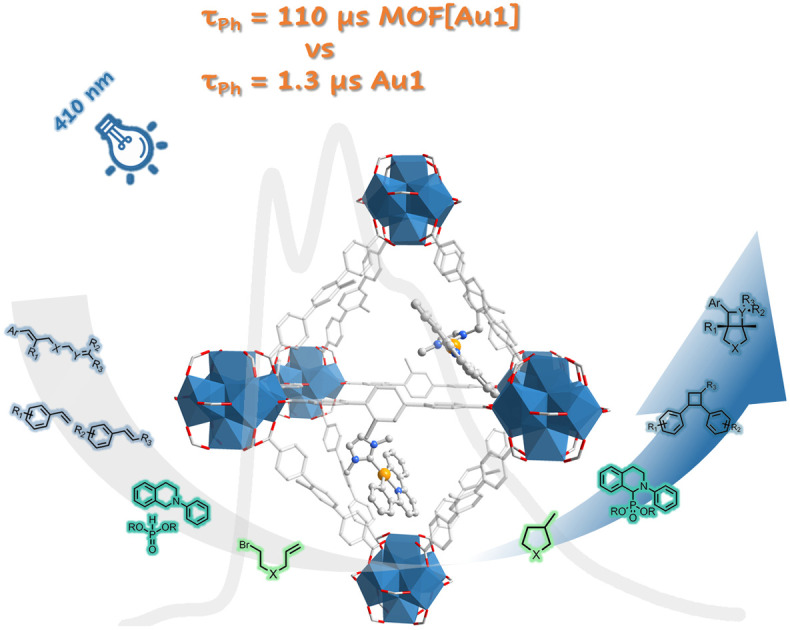
Introduction
Triplet emitters with long phosphorescence lifetimes (τph) are attractive for their potential use as photocatalysts in photo-redox catalysis and photosensitizers in energy transfer catalysis. Given the electrophilic nature of the gold(iii) ion, the frontier orbitals of luminescent gold(iii) complexes have a minute metal parentage, so they usually exhibit long-lived ligand-centered phosphorescence (3IL) with lifetimes (at room temperature) of up to hundreds of microseconds;1 this is significantly longer than the sub-microsecond to several microsecond lifetimes of Ir(iii) triplet emitters.2 In addition, luminescent gold(iii) complexes exhibit intersystem crossing with close to 100% efficiency, which is brought about by the large spin–orbit coupling constant of gold (around 5100 cm−1). This allows the conversion of all singlet-excited state molecules into their triplet manifolds. These attractive photophysical properties make luminescent gold(iii) complexes good photosensitizers for triplet-energy-transfer-mediated photochemical reactions. It is worth noting that the absorption of gold(iii) complexes can be easily tuned to the visible light region through ligand modification.1b,c Therefore, gold(iii) complexes can be developed as photocatalysts for visible-light-induced organic transformation reactions. It has been reported in the literature that gold(iii) emitters exhibit high catalytic activity for homogeneous photochemical reactions.1
Exploring the catalytic capabilities of metal–organic frameworks (MOFs) has become a rapidly growing field over the past decade.3 This is due to the inherently superior properties of MOFs, including hyperreactivity, high porosity, huge internal surface area, and customizable pore sizes.3,4 The pore-confined nanocages of MOFs provide a valuable platform for selective reaction processes and catalytic innovation.3,5 MOF-supported catalysts are of particular interest due to their ease of recovery and recycling and their potential for efficient large-scale production in organic synthesis.6 Furthermore, heterogeneous photocatalysts have advantages over homogeneous catalytic systems because their catalytic performance is less susceptible to the solubility and concentration of the photo-catalytically active species. Given the advantages offered by MOF-based heterogeneous catalytic systems, we are interested in exploring the potential of integrating photo-functional gold(iii) complexes into MOFs to study photo-redox catalysis and photochemical energy transfer reactions.
Several strategies for implanting catalytically active species into MOFs have been reported. One such approach is the “ion exchange” method, in which a charged catalyst is encapsulated in the pores of a MOF through an ion exchange reaction with the existing counterions present in the parent framework. In 2015, Che and co-workers reported the use of this simple method to construct a series of gold(iii)-MOF composites that exhibited high photocatalytic activity for the aerobic oxidation of secondary amines to imines and oxidative C–H functionalization.7 However, gold(iii)-MOFs developed using this method may encounter stability issues because the cationic gold(iii) complexes inside the MOF nano-cages may exchange with other cations generated during the reaction. This ion exchange process may lead to the release of the gold(iii) complex from the MOF, negatively affecting the reusability of the gold(iii) catalyst. Alternatively, following the isoreticular principle, MOFs can be functionalized prior to framework construction; additional linkers covalently bound to gold photosensitizers/photocatalysts are employed in MOF synthesis. This “pre-synthetic functionalization” method allows for the functionalization of targeted MOFs while retaining their basic structure, crystallinity, and porosity to a certain extent. For instance, Toste and co-workers introduced bidentate gold(iii) complexes into MOFs using a mixed-linker synthetic strategy.8 They found that the judicious design of gold(iii)-containing biphenyl linkers improved the stability of C^C-based gold(iii) complexes relative to their homogeneous gold(iii) counterparts. The enhanced stability directly translates into their superior catalytic performance in intramolecular cycloaddition reactions compared to homogeneous gold(iii) catalysts. Another approach is to modify the structure of a given MOF by introducing catalysts at the reactive sites mounted on the organic linkers.9 However, this method poses challenges in preparing a variety of MOF-derived metal complexes because the post-synthetic reaction is performed in the solid state, which brings difficulties to the metalation reaction. In some cases, if additional metal catalysts are used in post-synthetic reactions (such as Pd catalysts used in Suzuki coupling), their complete removal may be difficult. This raises concerns regarding the interpretation of experimental results, as it becomes unclear whether the observed catalytic activity is attributed to residual metal catalysts or to the MOF itself.
We here disclose the solvothermal synthesis of two gold(iii)-functionalized MOFs, namely UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6%, following a mixed-linker synthetic strategy. Pincer-type gold(iii) complexes were incorporated into terphenyl-based linkers through facile reactions between pendent imidazolium groups and gold(iii) chloride complexes. The incorporation of gold(iii) complexes in the MOF creates a framework with the same microstructure as that of the parent MOF UiO-68-Me, thereby retaining its high porosity and crystallinity but with changes in pore size and surface area. It was found that the phosphorescence lifetime of Au1 attached to the MOF was significantly increased compared to that of the homogeneous counterpart Au1. The reusable heterogeneous gold(iii) catalyst exhibits excellent performance for intramolecular and intermolecular [2 + 2] cycloaddition reactions, singlet oxygen-sensitized oxidation reactions, and alkyl halide reductive cyclization reactions under visible light irradiation.
Results and discussion
Synthesis of linkers (H2TPDC-[Au1], H2TPDC-[Au2]) and gold(iii)-functionalized MOFs
Pincer gold(iii) complexes containing N-heterocyclic carbene (NHC) linked to terphenyl dicarboxylic acid ligands, including H2TPDC-[Au1] and H2TPDC-[Au2], were prepared following a four-step procedure with dimethyl 2′-methyl-[1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylate (H2TPDC) as the starting material (Scheme S1†). Pincer gold(iii) complexes, Au1 or Au2, are covalently attached to the terphenyl building blocks via pendent imidazolium groups. This is easily achieved by refluxing a methanol solution of [Au(C^N^C)Cl], potassium tert-butoxide, and terphenyl ligand. The crystal structure of Me-TPDC-[Au1] is shown in Scheme 1. Due to steric hindrance, the [Au(C^N^C)] plane adopts a twisted orientation relative to the carbene ligand. The [Au(C^N^C)] fragment is aligned parallel to one of the phenyl rings of the terphenyl unit, with an interplanar contact of about 4.1 Å.
Scheme 1. Synthetic pathways for linkers H2TPDC-[Au1] and H2TPDC-[Au2]. (Inset) The crystal structure of Me-TPDC-[Au1].

UiO-68-Me is a face-centered cubic topology MOF, which is constructed from Zr6O4(OH)4 clusters and terphenyl building units. It was selected for the development of gold(iii)-functionalized MOFs for photocatalytic investigations because of its ultrahigh surface area, exceptional thermal stability, high resistance to water and organic solvents, and high stability (the ability to maintain its crystalline structure) even under high-pressure conditions.10a,b Furthermore, UiO-68-Me has a three-dimensional porous structure with highly ordered distribution of tetrahedral and octahedral cages with calculated pore sizes of approximately 12 and 14 Å, respectively.10c The large cages in UiO-68-Me not only provide ample space to accommodate gold(iii) complexes but also facilitate the easy diffusion of substrates. Using a mixed-linker synthetic strategy, UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% were successfully synthesized (Scheme 2, see the ESI† for more details). For comparison, UiO-68-Me was also prepared according to the reported method.10d The current design involves integrating the organogold(iii) complex into the terphenyl ligand via a pendent imidazolium unit, largely retaining the key terphenyl building blocks in the established UiO-68 MOFs, which enables the synthesis of gold(iii)-functionalized MOFs in a more accessible manner and preserves the high porosity and crystallinity of the parent framework.
Scheme 2. Synthesis of gold(iii)-MOFs UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% using a mixed-linker strategy.

Characterization, stability, and porosity
Fig. 1a depicts the powder X-ray diffraction (PXRD) data of the as-synthesized UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6%, whose diffraction patterns are consistent with the simulated data for UiO-68-Me. The comparison suggests that UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6%, developed using the mixed-linker synthetic protocol, largely maintain the highly ordered structure of the parent framework UiO-68-Me. The morphologies and particle sizes of UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% were characterized using field-emission scanning electron microscopy (FE-SEM).
Fig. 1. (a) Experimentally obtained PXRD patterns of UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% and the simulated pattern of UiO-68-Me; (b) N2 sorption isotherms of UiO-68-Me, UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% at 77 K; (c) pore size distribution of UiO-68-Me, UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% analyzed using the NLDFT method based on nitrogen adsorption measurements; (d) illustration of the effect of increasing the loadings of gold(iii) complexes on pore size distributions.

Fig. S1† shows the octagon-like structures of these gold(iii)-MOFs. Notably, the average particle sizes of UiO-68[Au1] decrease from 20 to 9 μm upon changing the feed ratio of linkers H2TPDC-[Au1] and H2TPDC used in the MOF synthesis from 1 : 20 to 1 : 5. It can be speculated that the growth of UiO-68[Au1] crystal sizes may be impeded by the high concentration of the linker H2TPDC-[Au1] during the crystallization process.
The chemical composition and metal distribution of these MOFs were characterized using energy-dispersive X-ray spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), and inductively coupled plasma-atomic emission spectroscopy (ICP-MS). The measurement of SEM coupled with EDS elemental mapping shows UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% with an even distribution of Au, Zr, C, and O (Fig. S2–5†). Even when using low feed ratios of gold(iii)-modified linkers H2TPDC-[Au1] and H2TPDC-[Au2], the observed uniform distribution of Au in UiO-68[Au1]1.2% and UiO-68[Au2]2.6% suggests the feasibility of the mixed-linker synthetic method employed for gold(iii)-functionalized UiO-68 MOFs. XPS analysis was conducted to determine the oxidation state of Au species in UiO-68[Au1]4.5% and UiO-68[Au2]2.6% as representatives. As shown in Fig. S6,† their Au 4f7/2 photoelectron peaks are located at binding energy values of 84.2–84.6 eV, which are comparable to the Au 4f7/2 peaks of the respective gold(iii) chloride complexes (85.2 eV), revealing that gold(iii) complexes are present in UiO-68[Au1]4.5% and UiO-68[Au2]2.6%. ICP-MS analysis shows the Au : Zr ratios of 1 : 25.6, 1 : 13.1, 1 : 6.3, and 1 : 15.1 for UiO-68[Au1]1.2%, UiO-68[Au1]2.4%, UiO-68[Au1]4.5%, and UiO-68[Au2]2.6%, respectively (Table S2†). These characterization efforts collectively demonstrate the successful construction of gold(iii)-MOFs using the mixed-linker synthetic protocol.
The digestion experiment, accompanied by NMR analysis, offers an effective way to verify the constituent building blocks in mixed-linker MOFs. In this study, UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% were subjected to digestion under K3PO4/D2O/DMSO-d6, and subsequently, clear solutions were obtained. The 1H NMR spectra of digested UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% solutions indicate the presence of intact gold(iii)-substituted terphenyl ligands (Fig. S10 and 11†), revealing that the gold(iii)-functionalized linkers are released from the MOFs due to the decomposition of Zr-based clusters by tripotassium phosphate. The digestion experiment showed a lower percentage of gold(iii)-functionalized linkers in the MOF compared to the percentage used in the feed ratio for MOF synthesis. UiO-68[Au1]4.5% exhibited good stability, maintaining its high crystallinity and structural integrity when exposed to various organic solvents as well as acidic (pH 3–5) and basic (pH 9–11) solutions. Its robustness was even evident under high-temperature conditions (100 °C for 6 h). These results are illustrated in Fig. S12 and S16.†UiO-68[Au1]4.5% was treated with NaBH4, and the resulting MOF sample was subjected to TEM and XPS analysis. TEM characterization shows particle aggregation within the MOFs, and XPS analysis suggests the presence of Au(0) in the MOFs (Fig. S8 and S9†).
In order to study the porosity of gold(iii)-MOFs, the single-component equilibrium adsorption behaviors of activated MOFs were studied in detail. The MOF was activated through solvent exchange using MeCN and then thermally treated at 100 °C under vacuum. PXRD studies show that the crystallinity of MOFs can be maintained after thermal treatment (Fig. S13–S17†). The permanent porosity of UiO-68-Me and gold(iii)-functionalized MOFs was evaluated through nitrogen gas sorption measurements at 77 K and 1 bar (Fig. 1b and Table S3†). UiO-68-Me, UiO-68[Au1]1.2–4.5%, and UiO-68[Au2]2.6% show typical reversible type I adsorption–desorption isotherms of nitrogen at 77 K (Fig. 1b), indicating their microporous structure. Activated UiO-68-Me shows a N2 volumetric uptake of 1081 cm3 g−1 (at P/P0 = 0.9), corresponding to a pore volume of 1.67 cm3 g−1 and a Brunauer–Emmett–Teller (BET) surface area of 4323 cm2 g−1. These values are comparable to those reported in the literature.10d Compared with the parent framework UiO-68-Me, lower N2 sorption was observed for UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6%, showing reduced pore volume (1.36–1.58 cm3 g−1) and BET surface area (3471–3947 cm2 g−1). Interestingly, increasing the gold content in UiO-68[Au1] from 1.2 to 4.5 wt% results in a further decrease in the BET surface area and pore volume of the MOFs. Based on these experimental findings, it can be inferred that pendent gold(iii) complexes tend to be located in the micropores of MOFs. The pore size distributions of UiO-68-Me, UiO-68[Au1]1.2–4.5%, and UiO-68[Au2]2.6% were derived from experimental sorption data by fitting the isotherms using the NLDFT model. UiO-68-Me has two different sizes of cages: a tetrahedral cage (12 Å) and an octahedral cage (14 Å). When the pincer gold(iii) complex Au1 was grafted onto the MOF linker, the distribution of pores with a size of 14 Å gradually decreased as the content of Au1 increased (Fig. 1c). The trend is evident in the results for UiO-68[Au1]1.2–4.5%. These data reveal the preferential location of Au1 within the octahedral cage, thereby reducing the respective pore size, as shown in Fig. 1d.
The porous structure of MOFs features a high surface area, allowing them to efficiently adsorb small molecules. The kinetic adsorption capacity of UiO-68-Me and UiO-68[Au1]4.5% towards other small molecules was further examined for comparison under pressures up to 0.1 or 1.1 bar at 298.15 K (Fig. 2). The results show that UiO-68[Au1]4.5%, which has narrower pore sizes compared to UiO-68-Me, is ineffective for selective molecule sieving of C2H6/C2H4, C3H8/C3H6, and C6H6/C6H12. However, when comparing the adsorption capacities of UiO-68-Me and UiO-68[Au1]4.5% for CH4, C2H6, and C3H8 (Fig. 2d–f), there is an interesting observation: compared with UiO-68-Me, UiO-68[Au1]4.5% exhibits a lower CH4 uptake, while showing a comparable uptake capacity for C2H6 and a higher adsorption capacity for C3H8. These behaviors exhibited by UiO-68[Au1]4.5% may be attributed to the increased C–H/π interactions between gas molecules within the UiO-68[Au1]4.5% pores and the aromatic rings of [Au(C^N^C)(NHC)]+; the order is C3H8 > C2H6 > CH4.11 Hydrogen is a green energy carrier, and the H2 storage capability of MOFs has been actively studied.12 In this work, the H2 uptake of UiO-68[Au1]4.5% was slightly higher (0.081 mg g−1 at 1 bar) compared to that of UiO-68-Me (0.066 mg g−1 at 1 bar), as shown in Fig. 2a.
Fig. 2. Single-component sorption isotherms for (a) H2, (b) CO2 and N2, (c) C6H6 and C6H12, (d) CH4, (e) C2H4 and C2H6, and (f) C3H6 and C3H8 of UiO-68-Me and UiO-68[Au1]4.5% at 298.15 K. “Ads” and “des” stand for adsorption and desorption, respectively.

Photophysical properties
As shown in Fig. S19,† the UV-visible diffuse reflectance spectrum of UiO-68[Au1]4.5% shows additional absorption bands in the lower-energy region of 380–430 nm. This is somewhat different from UiO-68-Me, whose absorption band extends to around 400 nm. Notably, the low-energy structured absorption bands observed in UiO-68[Au1]4.5% are similar in profile and energy to those found in the corresponding homogeneous gold(iii) complex counterpart Au1. This finding also provides evidence for the successful introduction of gold(iii) complexes into MOF matrices.
Upon photoexcitation, UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% exhibit dual emissions at room temperature when suspended in deoxygenated MeCN, with the higher-energy emission band peaking at around 370 nm and the other located in the green (or yellow) energy region (Fig. 3a–c). Table 1 summarizes the phosphorescence lifetime (τph), emission quantum yield (Φph, ΦF), and emission energy (τem, ph) of the gold(iii)-MOFs investigated in this work. Since the emission maximum, spectral profile, and nanosecond emission lifetime (<10 ns) of the blue emission band are very similar to those of the terphenyl linker Me-TPDC, the emission band centered at around 370 nm can be reasonably attributed to fluorescence derived from the terphenyl skeleton of gold(iii)-MOFs. The lower-energy vibronic emission bands (E0–0 of 477–532 nm) of these gold(iii)-MOFs resemble the emission energy of their respective homogeneous gold(iii) carbene complexes (i.e.Au1 and Au2).13 Together with small radiative decay rate constants on the order of 102 s−1 and long emission lifetimes of up to 110 μs, which are typical traits of 3IL-emitting gold(iii) complexes,1 the lower-energy structured emission bands correspond to phosphorescence originating from the 3IL excited state of the MOF pendent gold(iii) complex.
Fig. 3. (a) Fluorescence spectra of gold(iii)-MOFs and the unsubstituted terphenyl building unit recorded with an excitation wavelength at 290 nm; (b and c) emission spectra of Me-TPDC-[Au1] and Me-TPDC-[Au2] dissolved in degassed MeCN and emission spectra of UiO-68[Au1]4.5% and UiO-68[Au2]2.6% suspended in MeCN under argon, with an excitation wavelength of 380 nm for (b) and 390 nm for (c); (d) absorption spectrum of Me-TPDC-[Au1] in MeCN and emission spectra of Me-TPDC in MeCN and UiO-68-Me suspended in MeCN; (e) singlet oxygen emission spectra generated by 5,10,15,20-tetraphenylporphyrin (H2TPP) used as a reference and by UiO-68[Au1]4.5% and UiO-68[Au2]2.6% in oxygen-saturated MeCN; (f) emission spectra of UiO-68[Au1]2.4% (suspended in MeCN) in the presence of different concentrations of nitrobenzene (0.0245–0.145 mol dm−3), with an excitation wavelength of 355 nm; inset: plot of emission quenching of UiO-68[Au1]2.4% by nitrobenzene at different concentrations.

Table 1. Photophysical properties of linkers (Me-TPDC-[Au1] and Me-TPDC-[Au2]) and MOFs (UiO-68-Me, UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6%) studied in this worka.
| MOFs/compound | Conditions | λ em, ph (nm) | Φ ph (%) | τ ph (μs) | λ em, F (nm) | Φ F b (%) |
|---|---|---|---|---|---|---|
| UiO-68[Au1]4.5% | Air | 479, 511, 545(sh) | 0.3 | 12.1 | 372 | 4.1 |
| Degassed | 1.3 | 56.9 | ||||
| UiO-68[Au1]2.4% | Air | 479, 511, 545(sh) | <0.1 | 1.14 | 375 | 12.3 |
| Degassed | 1.2 | 41.5 | ||||
| UiO-68[Au1]1.2% | Air | 477, 511, 549(sh) | <0.1 | 1.35 | 374 | 16.2 |
| Degassed | 2.2 | 113.3 | ||||
| UiO-68[Au2]2.6% | Air | 532, 569, 613(sh) | <0.1 | 0.02 | 373 | 15.3 |
| Degassed | 2.2 | 103.0 | ||||
| UiO-68-Me | Air | 369 | 95.6 | |||
| Me-TPDC-[Au1] | Air | 478, 510, 543(sh) | <0.1 | 0.4 | ||
| Degassed | 0.3 | 1.3 | ||||
| Me-TPDC-[Au2] | Air | 520, 560, 602 | <0.1 | 0.9 | ||
| Degassed | 3.6 | 178.5 | ||||
| Me-TPDC | Air | 358 | 58.5 |
For the measurements, the MOFs were suspended in MeCN, and the gold(iii) complexes were dissolved in MeCN at a concentration of 2 × 10−5 mol dm−3; λem, ph and λem, F refer to phosphorescence and fluorescence emission maxima, respectively; Φph and ΦF denote phosphorescence and fluorescence quantum yields, respectively; τph represents phosphorescence lifetime; “sh” stands for shoulder; values of Φ below 0.001 fall outside instrumental accuracy.
Fluorescence quantum yields (ΦF) were recorded with the samples exposed to air.
It was found that the functionalization of UiO-68-Me with gold(iii) complexes (Au1 or Au2) profoundly suppressed the fluorescence originating from the terphenyl skeleton, and ΦF decreased from 95.6% in UiO-68-Me to less than 20% in UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6%. As shown in Fig. 3d, the emission spectrum of the terphenyl linker Me-TPDC has significant spectral overlap with the absorption spectrum of Me-TPDC-[Au1], showing that the energy transfer process from the terphenyl fragment to Au1 is feasible. This may be the reason for the reduced fluorescence intensity observed in UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% compared to UiO-68-Me. Upon increasing the Au1 content in UiO-68[Au1] from 1.2 wt% to 4.5 wt%, the blue fluorescence intensity further decreases, with ΦF reducing from 16% to 4%. This may be due to the reduced molecular distance between the terphenyl unit (as an energy donor) and Au1 (as an energy acceptor), leading to more efficient energy transfer. Compared with the terphenyl ligand Me-TPDC, two gold(iii)-substituted linkers, Me-TPDC-[Au1] and Me-TPDC-[Au2], also show reduced fluorescence intensity.
Incorporating the gold(iii) complex Au1 into MOFs has been shown to significantly increase the τph of Au1, as shown in Table 1. Under degassed conditions, the τph of UiO-68[Au1]1.2–4.5% (42–110 μs) is more than 30 times longer than the τph of Au1 in MeCN (1.3 μs). It has been reported that the emission lifetime increases when gold(iii) complexes are encapsulated in MOFs using a cation exchange method.7 Notably, the phosphorescence emission of UiO-68[Au1]4.5% exhibits less susceptibility to oxygen quenching compared to UiO-68[Au1]1.2–2.4%. When exposed to air, the τph of UiO-68[Au1]4.5% decreased by five times, whereas the τph of UiO-68[Au1]1.2% and UiO-68[Au1]2.4% decreased by approximately 80 and 40 times, respectively. The different responses to oxygen quenching observed in UiO-68[Au1]1.2–4.5% may be due to differences in their nano-space dimensions. As mentioned above, UiO-68[Au1]4.5% has the smallest pore volume and BET surface area among UiO-68[Au1]. The reduction of nano-cages may help prevent oxygen diffusion into the MOF cage, thereby effectively suppressing the phosphorescence quenching of UiO-68[Au1]4.5% by oxygen.
Considering that UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6% have long triplet excited state lifetimes and relatively high triplet energy with E0–0 at 477–532 nm (2.33–2.60 eV) compared to the 1O2 phosphorescence emission energy (λem at around 1 eV), these MOF-supported gold(iii) complexes are of appeal for serving as 1O2-generating photosensitizers upon visible light irradiation. In this regard, the generation of 1O2 under light irradiation of UiO-68[Au1]4.5% and UiO-68[Au2]2.6% was examined. Fig. 3e shows the 1O2 emission spectrum centered at around 1270 nm, which was recorded with an oxygen-saturated MeCN suspension of gold(iii)-MOFs under 380 nm light irradiation. The recorded 1O2 emission demonstrates that these gold(iii)-MOFs can serve as heterogeneous photosensitizers for singlet oxygen evolution in photocatalytic reactions. The subsequent sections will delve into the photochemical oxidation reaction mediated by gold(iii)-MOFs.
As UiO-68[Au1] has a relatively long triplet lifetime compared to its homogeneous counterpart Au1, we studied the emission quenching of UiO-68[Au1]2.4% suspended in MeCN in the presence of nitrobenzene under air. The addition of nitrobenzene resulted in emission quenching of UiO-68[Au1]2.4% without any change in the emission maximum (Fig. 3f). Significant emission quenching occurs at nitrobenzene concentrations in the range of 0.0245–0.075 mol dm−3, and then, as the analyte concentration further increases, the quenching shows a trend toward a maximum. This observation may be a result of diffusion control within the MOF pores.
Photo-catalytic organic transformation reactions
To evaluate the photoreactivity and recyclability of the as-prepared gold(iii)-MOFs, their utility as photocatalysts in various reactions, including [2 + 2] cycloaddition, singlet oxygen-sensitized oxidation reaction, and alkyl bromide reductive cyclization, was investigated. Our research begins with the light-driven alkene [2 + 2] cycloaddition reaction. This reaction is the premier method for constructing cyclobutanes, which are prevalent among successful drug candidates.14 The formation of cyclobutane from alkenes can be achieved through photosensitized energy transfer15 or electron transfer16 mechanisms. Our developed UiO-68[Au1]1.2–4.5% shows a triplet energy of 62 kcal mol−1, which is determined from the onset of the emission spectrum, as indicated in Fig. 3b. This value indicates that photoexcited UiO-68[Au1]1.2–4.5% is capable of energy transfer to those alkenes with relatively low triplet energies of around 50–60 kcal mol−1.17 According to the excited state reduction potential of Au1 (referred to as [Au(C^N^C)(NHC)]+), where E(Au*/Au−) = +1.29 V (versus SCE),13a another possible mechanistic scenario involving electron transfer can be ruled out. From a thermodynamic perspective, the alkene substrate examined here, 1e, which has a first oxidation peak potential of +1.42 V (versus SCE),15a is hardly oxidized by the excited state of Au1. For these reasons, the gold(iii)-MOF-mediated light-driven [2 + 2] cycloaddition reaction studied here preferentially proceeds through an energy transfer mechanism rather than an electron transfer pathway.
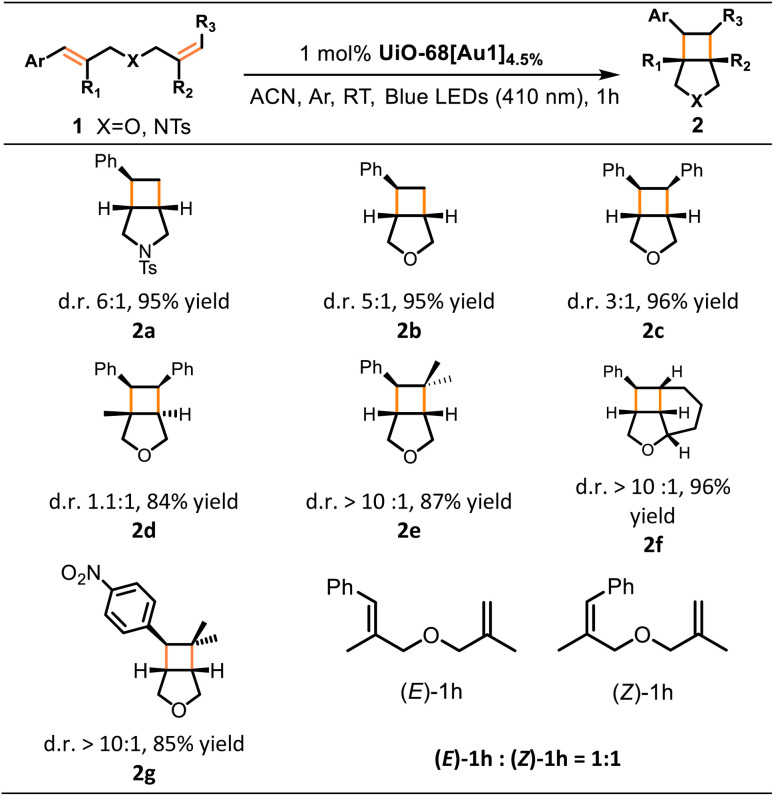
Initial experiments were performed with 1a in the presence of catalytic amounts (1 mol%) of UiO-68[Au1]4.5% in MeCN for 1 h under 410 nm irradiation. Considering that the long-lived triplet excited state of the gold(iii) photosensitizer would be quenched by oxygen, the reaction was carried out under argon to obtain bicyclic compound 2a in a high yield of 95% and a diastereomeric ratio (d.r.) of 6 : 1, as shown in Table 2. Notably, the yield of 2a decreased only slightly (by 7%, Table S4†) when the reaction was conducted under air. This marginal decrease can be attributed to the extended emission lifetime of UiO-68[Au1]4.5% in the presence of oxygen, approximately 12 μs. Omission of either the photosensitizer UiO-68[Au1]4.5% or light irradiation produced only trace amounts of product 2a (Table S4†), confirming the necessity of both components for the reaction to occur. Using UiO-68-Me instead of UiO-68[Au1]4.5% resulted in only trace amounts of 2a. We also compared the catalytic efficiency of UiO-68[Au1]4.5% with other widely studied photocatalysts, such as Ir[(dFppy)2dtbbpy]PF6 and thioxanthone. The yields of product 2a were comparable (Table S4†). As a control, Au1-mediated photocycloaddition of 1a produced 2a in a comparable yield (96%) under otherwise identical conditions, indicating that the incorporation of Au1 into the MOF did not alter its catalytic activity (Table S4†). Notably, the incorporation of gold(iii) complexes into UiO-68 MOFs was found to confer a higher degree of stability to the gold(iii) catalyst. The supportive findings include: (i) in a 150 min reaction with 1a (Fig. 5a), the use of UiO-68[Au1]4.5% as the catalyst led to the formation of 2a with a turnover number of 484. This turnover number is at least as good as (if not better than) the 274 turnover number obtained using Au1. Notably, the photo-activity of homogeneous Au1 decreased significantly after 30 minutes of reaction; and (ii) after photolysis, the color of the reaction solution mixture involving UiO-68[Au1]4.5% remained unchanged, while the reaction carried out with the homogeneous counterpart Au1 turned deep purple, indicating that the homogeneous gold(iii) catalyst decomposed during the photolysis (Fig. 5c and d).
Table 2. Photo-induced intramolecular [2 + 2] cycloadditions of alkenes enabled by UiO-68[Au1]4.5%a.
|
Conditions: reactions were performed with 0.1 mmol of 1a–1h and 1 mol% UiO-68[Au1]4.5% in MeCN (2 mL) at room temperature under argon and 410 nm irradiation for 1 h; product yields and diastereomeric ratios (d.r.) were determined by 1H NMR analysis from the crude product using chlorodiphenylmethane as an internal standard.
Fig. 5. (a) Product turnovers in the [2 + 2] cycloaddition of 1a to 2a by UiO-68[Au1]4.5% or Au1 (referring to [Au(C^N^C)(NHC)]PF6); (b) substrate conversion and product yield in recycling experiments with ten reaction cycles using UiO-68[Au1]4.5% as the photocatalyst in the given reaction; (c and d) photographs of the photocycloaddition of 1a involving UiO-68[Au1]4.5% or Au1, taken after the reaction; (e) PXRD patterns of UiO-68-Me (simulated) and UiO-68[Au1]4.5% before and after catalysis (measured).

In order to verify the reaction mechanism, an emission quenching experiment was performed on Au1 with increasing concentration of 1a (Fig. 4a). A linear correlation with the concentration of 1a was observed with a quenching rate constant (kq) of 4.1 × 109 dm3 mol−1 s−1, suggesting that the quenching of the triplet excited state of Au1 by 1a could occur upon light irradiation. The nanosecond transient absorption difference (ns-TA) spectrum of Au1 in the presence of 1a was recorded and is shown in Fig. 4b. This spectrum shares a common profile with the TA spectrum of Au1 only (Fig. 4c). The main distinguishing feature between these two TA spectra (Fig. 4b and c) is the decay lifetime of the absorption band. Specifically, after adding 1a, the decay lifetime of the absorption band at 440 nm is reduced from 0.76 μs to 0.4 μs, which is consistent with the large kq obtained in the quenching experiment. The reaction mechanism is shown in Fig. S21.† Upon visible light irradiation, the gold(iii) catalyst is excited to the singlet excited state and then undergoes rapid intersystem crossing to reach its triplet manifold. The reactive triplet state of alkene 1a is accessed through a Dexter-type energy transfer process from the triplet excited state of the gold(iii) catalyst to the alkene substrate. The resulting triplet-excited-state alkene reacts with the ground-state alkene to form a biradical intermediate, which undergoes ring closure to yield bicyclic cycloaddition product 2a.
Fig. 4. (a) Stern–Volmer plots of emission quenching of Au1 (2 × 10−5 mol dm−3 in MeCN) by different substrates. The fitted kq values for Au1 upon addition of 1a, 3d, 2-vinylnaphthalene (6b), or ethyl cinnamate (6a) are 4.1 × 109, 3.0 × 109, 1.1 × 1010, or 8.7 × 109 dm3 mol−1 s−1, respectively; (b) ns-TA spectrum of Au1 (2 × 10−5 mol dm−3) in the presence of 1a (3 × 10−4 mol dm−3); (c) ns-TA spectrum of Au1 only (2 × 10−5 mol dm−3).

The cycling stability of the heterogeneous photocatalyst UiO-68[Au1]4.5% was evaluated. UiO-68[Au1]4.5% can be readily recycled through washing with MeCN for subsequent reactions. After ten consecutive reaction cycles, both the substrate (1a) conversion and product yield retained high values of 96% and 89%, respectively (Fig. 5b). The remarkable recyclability and catalytic activity of gold(iii)-MOF may be attributed to its high stability under the reaction conditions used. A comprehensive stability assessment of the recycled UiO-68[Au1]4.5% was performed using PXRD, XPS, ICP-MS, SEM, and TEM measurements. PXRD analysis demonstrated that the recycled UiO-68[Au1]4.5% maintained high crystallinity and structural integrity (Fig. 5e). Comparison of the XPS spectra of UiO-68[Au1]4.5% before and after ten catalytic runs showed no significant differences in the peak profiles and Au 4f binding energy values associated with the gold(iii) oxidation state (Fig. S23†). ICP-MS analysis indicated minimal leaching (only 4%) of Au1 from the MOFs (Table S5†). SEM and TEM images of UiO-68[Au1]4.5% showed that gold nanoparticles were not deposited onto the MOF surface. The gold(iii) complex appears to remain well-contained within the MOF structure. As shown in Fig. S24 and S25,† a decrease in the particle size of the MOF was observed after ten reaction runs, and the decrease in particle size did not seem to compromise the catalytic performance of the MOF to some extent. To highlight the effect of the Au1 catalyst covalently linked to the MOF, UiO-68-Me and Au1 were physically combined and the resulting mixtures were subjected to catalytic studies for comparison. When this physically mixed system was used to replace UiO-68[Au1]4.5% as the photosensitizer, the substrate conversion and product yields of 2a dropped significantly to around 30% after two reaction runs (Fig. S26†).
The substrate scope of the UiO-68[Au1]-catalyzed intramolecular cycloadditions of alkenes was examined. As shown in Table 2, styrene derivatives (1b–1g) containing different functional groups underwent [2 + 2] cycloaddition smoothly, and the bicyclic products 2b–2g were obtained in high yields (84–96%). It is worth noting that the sterically demanding substituents do not impede the reaction (products 2d and 2e). Notably, when using the substrate (E)-1h in which R1 and R2 are both methyl groups, 1H NMR analysis after irradiation showed no evidence of the desired cyclobutane product, instead a mixture of (E)-1h and (Z)-1h was observed.
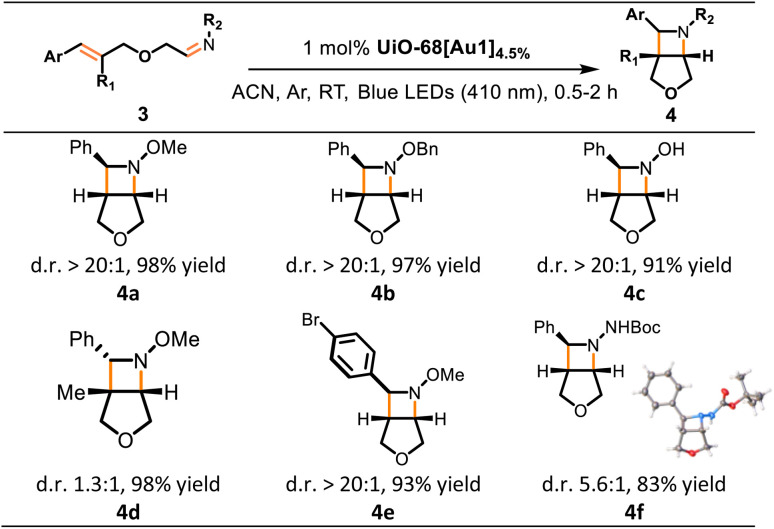
Azetidine compounds are widely found in natural products and biologically active compounds.18 Then, we expanded our studies to the synthesis of these compounds through intramolecular [2 + 2] cycloaddition with oximes or hydrazones,18 catalyzed by gold(iii)-MOF photosensitizers. Following the modified reaction conditions reported by Schindler and co-workers,19UiO-68[Au1]4.5% was found to smoothly convert substrates 3a–3f into azetidine-containing bicyclic products 4a–4f in a short irradiation time of up to 2 h. The desired azetidines 4a–4f were formed in high-to-excellent yields (83–98%). Cyclization of oximes 3a–3c and 3e yielded azetidine products (4a–4c and 4e) with high diastereoselectivity (>20 : 1 d.r). The structure of one isomer of 4f was verified by X-ray crystallographic analysis (Table 3). In addition, Stern–Volmer studies were performed on Au1 by increasing 3d (as a representative example) concentration, and the large kq value of 3.0 × 109 dm3 mol−1 s−1 confirmed the efficient quenching of the Au1 excited state by 3d (Fig. 4a).
Table 3. Light-driven azetidine formation mediated by UiO-68[Au1]4.5%.a.
|
Conditions: reactions were performed with 0.1 mmol of 3a–3f (used as mixtures of E/Z oxime or hydrazone isomers) and 5 mg of UiO-68[Au1]4.5% in MeCN (2 mL) at room temperature under argon and 410 nm irradiation for 0.5–2 h; product yields and diastereomeric ratios (d.r.) were determined by 1H NMR analysis from the crude reaction mixture using chlorodiphenylmethane as an internal standard. The single-crystal structure of product 4f is presented.
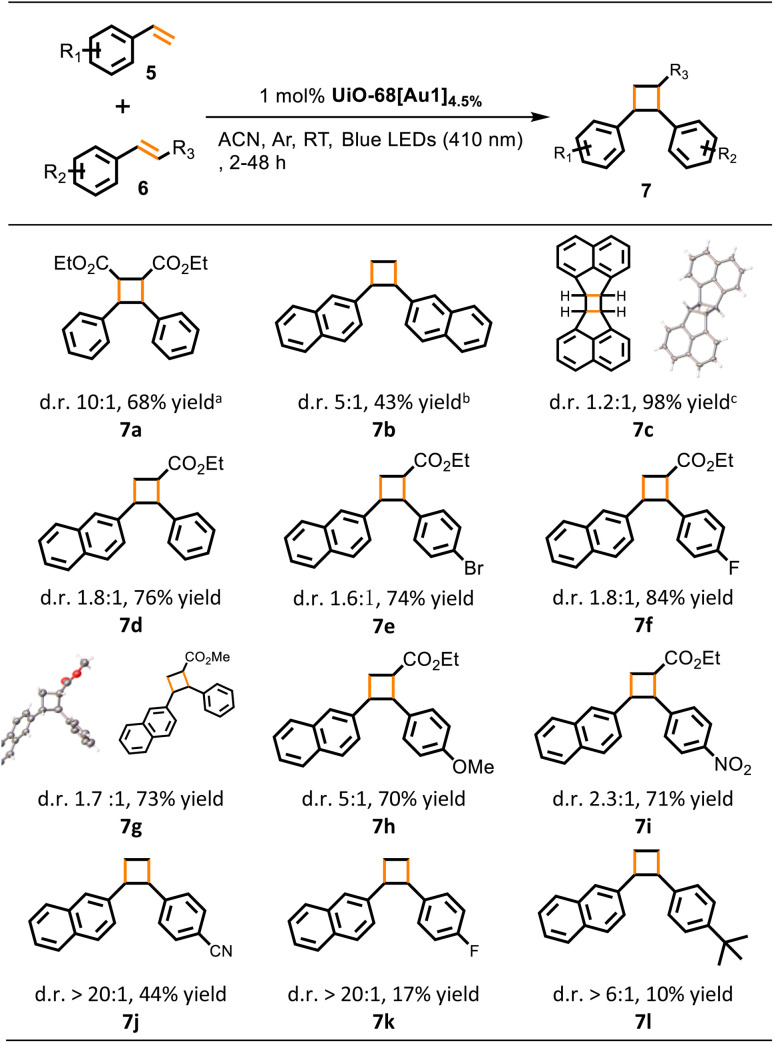
The intermolecular [2 + 2] photocycloaddition reaction provides a direct and straightforward method for the synthesis of four-membered cyclic lead compounds using readily available starting materials. However, cycloadditions performed in an intermolecular manner are more challenging than intramolecular reactions. After confirming the applicability of the gold(iii)-MOF in the [2 + 2] photocycloaddition of alkenes to synthesize cyclobutane, we studied the homo-coupling [2 + 2] cycloaddition reactions of ethyl cinnamate 6a, 2-vinylnaphthalene, and cyclic alkene 6c (Table 4). Significant phosphorescence quenching was observed for Au1 when substrate 6a or 2-vinylnaphthalene was added, with a large kq of (8.7–11) × 109 dm3 mol−1 s−1 (Fig. 4a). Under optimal reaction conditions, that is, utilizing 1 mol% UiO-68[Au1]4.5%, 410 nm irradiation, and MeCN as solvent, homodimerization of 6a and 6c led to the formation of cyclobutane in a yield of 68% (7a) and as high as 98% (7c). However, complete conversion of 2-vinylnaphthalene was observed with an overall product yield of 43% only (7b).
Table 4. UiO-68[Au1]4.5%-catalyzed homo and cross [2 + 2] dimerization of alkenesa.
|
Conditions: reactions (homodimerization for 7a–7c) were carried out with 0.2 mmol of 2-vinylnaphthalene, 6a or 6c and 5 mg of UiO-68[Au1]4.5% in MeCN (2 mL) at room temperature under argon and 410 nm irradiation for 30 ha/16 hb/2 hc; reactions (cross dimerization for 7d–7l) were performed with 0.1 mmol of 2-vinylnaphthalene, 0.3 mmol of aryl terminal alkene or methyl/ethyl cinnamate, and 1 mol% UiO-68[Au1]4.5% in MeCN (2 mL) at room temperature under argon and 410 nm irradiation for 3 h; product yields and diastereomeric ratios (d.r.) were determined by 1H NMR analysis from the crude reaction mixture using chlorodiphenylmethane as an internal standard. The crystal structures of anti-7c15e and 7g are presented.
Encouraged by the promising results of the homo-coupling cycloaddition, we proceeded to study the cross-coupling [2 + 2] cycloaddition reactions catalyzed by UiO-68[Au1]4.5% (Table 4). We selected the heterocycloaddition of 2-vinylnaphthalene with ethyl cinnamate as the benchmark reaction. 2-Vinylnaphthalene was used as a limited substrate, and the ratio of 2-vinylnaphthalene to ethyl cinnamate was optimized to 1 : 3. After 3 h of irradiation under dilute conditions (0.05 mol dm−3 of 2-vinylnaphthalene), the yield of product 7d was 76%. For methyl cinnamate, the yield of product 7g was 73%, which was comparable to the yield of 7d (76%) when using ethyl cinnamate. Para-functionalized ethyl cinnamate with halogens (bromo and fluoro), electron-donating groups (i.e. OMe), or electron-withdrawing groups (i.e., NO2) is well-tolerated and the corresponding cross-coupled products (7e, 7f, 7h, and 7i) were obtained in high yields (71–84%). Compared with previous reports using copper(i)-based MOFs15c or iridium(iii) photocatalysts,15d gold(iii)-MOF catalyzed coupling reactions between two different aryl alkenes occurred in significantly shorter irradiation times. In these cross [2 + 2] dimerization reactions of alkenes, small amounts of homo-coupling side products containing ester groups were found in 5–18% yields (see the ESI†), and trace amounts of 7b were detected. The UiO-68[Au1]4.5%-catalyzed homocycloaddition of vinylbenzene did not occur, and only trace amounts of vinylbenzene conversion were observed. This result may be due to the relatively high triplet energy of vinylbenzene. In addition, the cross photo-cycloaddition reaction of 2-vinylnaphthalene with other substituted styrenes was also studied. After irradiation at 410 nm for 3 h, the corresponding cross-cycloaddition products 7j, 7k, and 7l were obtained in yields of 10–44% (Table 4).
Given the ability of these gold(iii)-MOFs to generate 1O2 (Fig. 3e), which is known for its utility in oxidation reactions, our focus shifted to exploring the catalytic activity of UiO-68[Au1]4.5% in photochemical oxidation reactions under oxygen-saturated conditions. The use of 1O2 has been reported to promote the oxidation of secondary amines to imines and the selective partial oxidation of sulfides to sulfoxides.1a,b,20UiO-68[Au1]4.5% delivered decent photocatalytic performance for these two oxidation reactions. With 1 mol% UiO-68[Au1]4.5%, irradiation of oxygen-saturated solutions containing dibenzylamine or methyl(p-tolyl)sulfane can form the desired product in an excellent yield (90–95%; 8a, 8b) (Scheme 3).
Scheme 3. Visible-light-driven oxidative organic transformations using UiO-68[Au1]4.5% performed in MeCN.

We further extended the study of gold(iii)-MOFs to light-driven aerobic C–H phosphorization of tertiary amines with phosphite esters. For 2-phenyl-1,2,3,4-tetrahydroisoquinoline, its α-C–H bond is oxidized by 1O2 to form iminium ions,21 which are easily captured by nucleophiles such as dialkyl phosphates. This results in a new bond being installed directly in the α-position of the amine. In this work, 1 mol% UiO-68[Au1]4.5% catalyzed the aerobic C–H phosphorization reaction of 2-phenyl-1,2,3,4-tetrahydroisoquinoline in O2-saturated methanol under light irradiation; phosphonated products containing various dialkyl phosphonates were formed, with a yield of 76–97% (8c–8h, Scheme 3). When using air-filled methanol, the product yield of 8c (93%) was slightly lower than that (97%) achieved under oxygen-saturated conditions (Table S7†). Control experiments confirmed that light, photocatalysts, and oxygen are all necessary for this reaction to proceed. UiO-68[Au1]4.5% showed good recyclability and maintained a high conversion of 88% after three consecutive catalytic cycles. The excellent recyclable photocatalytic performance of gold(iii)-MOF may be attributed to its robust framework under the photocatalytic reaction conditions, as evidenced by PXRD (Fig. S27†).
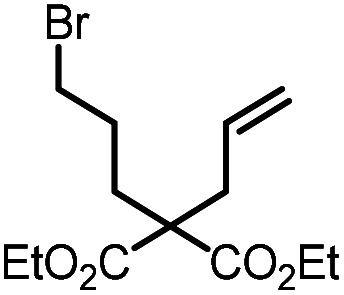
To evaluate the potential of gold(iii)-MOFs to catalyze photo-redox transformations, we selected the reductive cyclization of 9a as the benchmark reaction. In the presence of UiO-68[Au1]4.5% (1 mol%) and stoichiometric diisopropylamine (DIPEA, 2 equiv.), which serves as both a sacrificial electron donor and a hydrogen atom source, the cyclic compound 10a was obtained in 71% yield in the MeCN/MeOH mixture (1 : 1 v/v) under an argon atmosphere and visible light irradiation. By adding an additional hydrogen donor, 1,4-cyclohexadiene (1,4-CHD, 2 equiv.), the yield of 10a was increased to 97%. Control experiments showed that when the reaction was carried out without light, DIPEA, or UiO-68[Au1]4.5%, no cyclization product 10a was produced, as shown in Table S8.† Stern–Volmer quenching experiments were performed on Au1 with increasing concentration of DIPEA or 9a. Significant emission quenching of Au1 by DIPEA was observed (kq = 7.7 × 109 dm3 mol−1 s−1, Fig. S28†), whereas no detectable emission quenching of Au1 was found in the presence of 9a. Electrochemical studies indicate that the oxidation of DIPEA (Epa = +0.8 V versus SCE at 100 mV s−1, Fig. S31†) by the excited state of Au1 (E(Au*/Au−) = +1.29 V versus SCE) is thermodynamically favourable. No oxidation waves for Au1 were recorded within the potential window of DMF, which makes it difficult to predict the excited state oxidation potential of Au1 (E(Au+/Au*)). However, the absence of emission quenching of Au1 by alkyl bromide 9a excludes the possibility of reduction of 9a by Au1*.
Combined analysis of emission quenching experiments and electrochemical studies suggests that reductive quenching of Au1* by DIPEA initiates the reaction, rather than oxidative quenching of Au1* by alkyl bromides, as illustrated in Fig. S29.† Bromoalkanes have reduction potentials of around −1.9 to −2.5 V versus SCE.22 In path A (Fig. S29†), the reduced gold(iii) intermediate, [AuIII]−, is a strong reducing agent that can promote the reductive cleavage of carbon–bromide bonds to generate carbon-centered radical intermediates. This activation also facilitates the regeneration of the gold(iii) photo-catalyst. The resulting alkyl radical then undergoes intramolecular carbon–carbon bond formation reactions.
Alternatively, the α-aminoalkyl radical (intermediate II) derived from DIPEA, generated by reductive quenching of Au1*, can also serve as a halogen-atom transfer (XAT) reagent.23 The XAT process between the α-aminoalkyl radical and the alkyl bromide would produce a carbon radical and an iminium salt, as shown in Fig. S30† (path B). The alkyl radical formed by subsequent intramolecular cyclization can be reduced by [AuIII]− to produce a carbanion intermediate, which is then protonated to give the desired cyclic product. To obtain more information on this possibility, we performed cyclic voltammetry experiments on 9a and DIPEA. At a scan rate of 20–100 mV s−1, DIPEA displays an irreversible oxidation wave located at around 0.73–0.80 V versus SCE; this anodic peak current does not increase with the addition of 9a (approximately 10 equivalents relative to DIPEA; 50 mM of 9a is the same concentration as the reaction conditions used) (Fig. S31†). If this XAT process occurs, the oxidized DIPEA species will be consumed through the reaction with the substrate 9a, which will result in an increase in the anodic peak current after the addition of 9a. Although this alternative pathway is unlikely to be supported based on the current data (Fig. S31†), the reaction between the oxidized DIPEA species and substrate 9a may be slow compared to the CV scanning times, resulting in no detectable increase in the anodic peak current upon addition of 9a. From a mechanistic perspective, this alternative reaction pathway (Fig. S30†) cannot be ruled out.
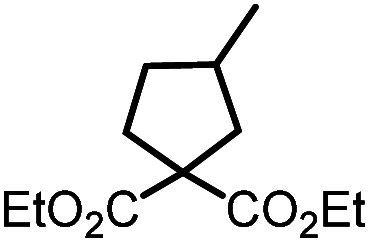
Having established the optimized reaction conditions, we proceeded with the investigation of the substrate scope (Table 5, 9b–9f). Respective cyclic compounds (10c–10f) were obtained in high yields (90–97%) within short irradiation times of up to 2 h. The reaction catalyzed by UiO-68[Au1]4.5% exhibits good functional group tolerance, allowing the formation of five-membered and six-membered rings (10d, 10e), radical cyclization of bromoalkynes (i.e., 10c), and cyclization of sterically demanding bromoalkenes (i.e., 10f). It is worth noting that the conversion of 9b into the corresponding cyclic product 10b required a prolonged irradiation time of 6 h, and the yield was 85%. PXRD analysis confirmed the structural integrity of the MOFs after photolysis under amine-based catalytic conditions (Fig. S32†).
Table 5. Visible-light-promoted reductive cyclization of alkyl halides facilitated by UiO-68[Au1]4.5%a.
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Conversion; yield | ||
| 1 |
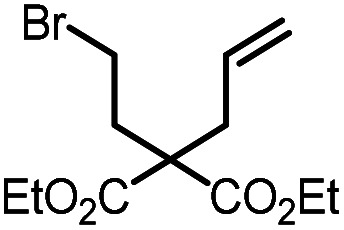
|
9a |
|
10a | 99%; 97% |
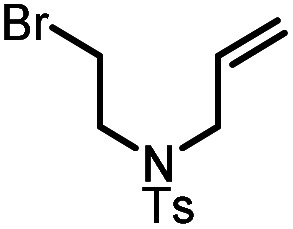
| 2 |
|
9b |
|
10b | 95%; 85% |
| 3 |
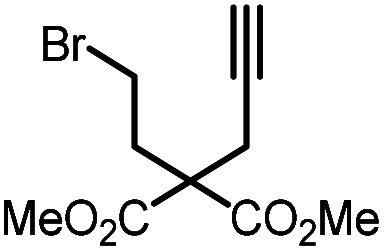
|
9c |
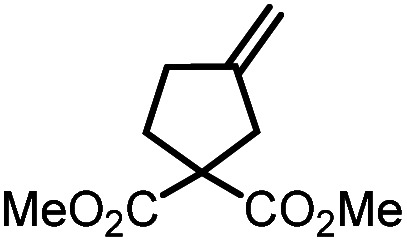
|
10c | 99%; 96% |
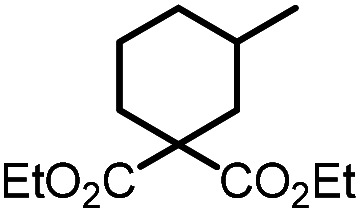
| 4 |
|
9d |
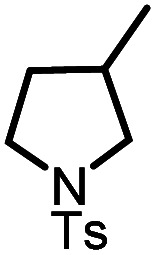
|
10d | 99%; 97% |
| 5 |
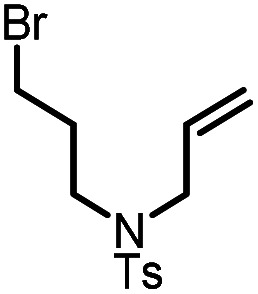
|
9e |
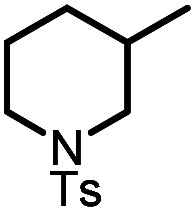
|
10e | 95%; 90% |
| 6 |
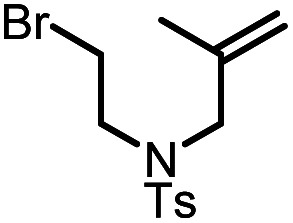
|
9f |
|
10f | 99%; 95% |
Conditions: 9a–9f (0.1 mmol), diisopropylamine (DIPEA) (0.2 mmol), 1,4-cyclohexadiene (1,4-CHD) (0.2 mmol), and UiO-68[Au1]4.5% (5 mg) in a MeCN/MeOH mixture (1 : 1, 1 mL each) at room temperature under argon and 410 nm irradiation for 0.5–6 h; conversions and product yields were determined by 1H NMR analysis from the crude product using chlorodiphenylmethane as an internal standard.
Conclusions
Two novel gold(iii)-functionalized MOFs, including UiO-68[Au1]1.2–4.5% and UiO-68[Au2]2.6%, were developed following a mixed-linker synthetic strategy. The gold(iii) emitter Au1 grafted onto the MOF demonstrates a longer phosphorescence lifetime than its homogeneous counterpart, reaching approximately 110 μs under argon and 12 μs under air at room temperature when suspended in MeCN. The extended lifetime is attributed to the localization of gold(iii) complexes within the MOF nanocages, which is supported by gas sorption measurements. The extended emission lifetime provides benefits for light-driven organic transformations that rely on energy transfer or electron transfer mechanisms mediated by photo-excited catalysts. The heterogeneous photocatalyst UiO-68[Au1]4.5% shows good performance in intramolecular and intermolecular [2 + 2] cycloaddition reactions, singlet oxygen-sensitized oxidation reactions, and reductive cyclization of alkyl bromides. Furthermore, the gold(iii)-MOF catalyst exhibits remarkable recyclability, as it can be easily separated and reused for ten consecutive reaction cycles without significant reduction in catalytic efficiency. The yields of the cyclobutane product remain high, maintaining around 90% after ten cycles of reuse. This excellent recyclability demonstrates the stability and robustness of the gold(iii)-MOF photocatalyst reported in this work, which are key features for practical applications requiring long-term operation and reuse of the catalyst. This work highlights the great potential of heterogeneous gold(iii)-MOFs as versatile catalytic platforms for photo-redox or energy transfer organic transformations under visible light irradiation, helping to develop milder and more sustainable chemical production.
Data availability
The data that support the findings of this study are available in the ESI† of this article.
Author contributions
C.-M. Che conceived and supervised the project. J.-R. Chen designed and synthesized gold(iii)-MOFs, and conducted the characterization and catalytic studies of the developed MOFs. D. Zhou performed the photophysical and mechanistic studies. J.-R. Chen and D. Zhou wrote the original draft of the manuscript. D. Zhou and C.-M. Che revised and refined the final manuscript. J.-R. Chen, D. Zhou, Y. Liu, X.-C. Huang, and C.-M. Che provided lead contributions to data analysis and project discussion. X.-C. Huang, M. Li and Y. Xiao offered suggestions on MOF synthesis and characterization. Y. Xiao provided assistance in solving the X-ray crystal structure of Me-TPDC-[Au1]. All authors have read and approved the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by a research grant from the Innovation and Technology Commission (HKSAR, China) to the State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Hong Kong Research Grants Council (General Research Fund, HKU 17305521 and 17309823), “Laboratory for Synthetic Chemistry and Chemical Biology” funded by the Health@InnoHK of Innovation and Technology Commission (HKSAR, China), the Guangdong Major Project of Basic and Applied Basic Research (2019B030302009), the Key (Guangdong-Hong Kong Joint) Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province (2023B1212120011), the National Natural Science Foundation of China (22071142), and the Special Fund Project for Science and Technology Innovation Strategy of Guangdong Province (STKJ2023077). The photophysical measurements were conducted in part using the instruments from the Hong Kong Quantum AI Lab Limited funded by the AIR@InnoHK Program of Hong Kong SAR Government. The authors also thank the Southern University of Science and Technology for financial support.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06058j
Notes and references
- (a) To W.-P. Tong G. S.-M. Lu W. Ma C. Liu J. Chow A. L.-F. Che C.-M. Angew. Chem., Int. Ed. 2012;51(11):2654–2657. doi: 10.1002/anie.201108080. [DOI] [PubMed] [Google Scholar]; (b) To W.-P. Chan K. T. Tong G. S. M. Ma C. Kwok W.-M. Guan X. Low K.-H. Che C.-M. Angew. Chem., Int. Ed. 2013;52(26):6648–6652. doi: 10.1002/anie.201301149. [DOI] [PubMed] [Google Scholar]; (c) To W.-P. Tong G. S. M. Cheung C.-W. Yang C. Zhou D. Che C.-M. Inorg. Chem. 2017;56(9):5046–5059. doi: 10.1021/acs.inorgchem.7b00180. [DOI] [PubMed] [Google Scholar]
- (a) Idris M. Kapper S. C. Tadle A. C. Batagoda T. Ravinson D. S. M. Abimbola O. Djurovich P. I. Kim J. Coburn C. Forrest S. R. Thompson M. E. Adv. Opt. Mater. 2021;9(8):2001994. doi: 10.1002/adom.202001994. [DOI] [Google Scholar]; (b) Yang X. Guo H. Liu B. Zhao J. Zhou G. Wu Z. Wong W.-Y. Adv. Sci. 2018;5(5):1701067. doi: 10.1002/advs.201701067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu Y. Yang C. Liu J. Zhang M. Liu W. Li W. Wu C. Cheng G. Yang Q. Wei G. Che C.-M. Chem. Sci. 2021;12(30):10165–10178. doi: 10.1039/D1SC01426A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Whelan É. Steuber F. W. Gunnlaugsson T. Schmitt W. Coord. Chem. Rev. 2021;437:213757. doi: 10.1016/j.ccr.2020.213757. [DOI] [Google Scholar]; (b) Bavykina A. Kolobov N. Khan I. S. Bau J. A. Ramirez A. Gascon J. Chem. Rev. 2020;120(16):8468–8535. doi: 10.1021/acs.chemrev.9b00685. [DOI] [PubMed] [Google Scholar]; (c) Luo Y.-H. Dong L.-Z. Liu J. Li S.-L. Lan Y.-Q. Coord. Chem. Rev. 2019;390:86–126. doi: 10.1016/j.ccr.2019.03.019. [DOI] [Google Scholar]; (d) Lee J. Farha O. K. Roberts J. Scheidt K. A. Nguyen S. T. Hupp J. T. Chem. Soc. Rev. 2009;38(5):1450–1459. doi: 10.1039/B807080F. [DOI] [PubMed] [Google Scholar]; (e) Manna K. Zhang T. Greene F. X. Lin W. J. Am. Chem. Soc. 2015;137(7):2665–2673. doi: 10.1021/ja512478y. [DOI] [PubMed] [Google Scholar]
- (a) Furukawa H. Cordova K. E. O'Keeffe M. Yaghi O. M. Science. 2013;341(6149):1230444. doi: 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]; (b) Deng H. Grunder S. Cordova K. E. Valente C. Furukawa H. Hmadeh M. Gándara F. Whalley A. C. Liu Z. Asahina S. Kazumori H. O'Keeffe M. Terasaki O. Stoddart J. F. Yaghi O. M. Science. 2012;336(6084):1018–1023. doi: 10.1126/science.1220131. [DOI] [PubMed] [Google Scholar]; (c) Deng H. Doonan C. J. Furukawa H. Ferreira R. B. Towne J. Knobler C. B. Wang B. Yaghi O. M. Science. 2010;327(5967):846–850. doi: 10.1126/science.1181761. [DOI] [PubMed] [Google Scholar]; (d) Eddaoudi M. Kim J. Rosi N. Vodak D. Wachter J. O'Keeffe M. Yaghi O. M. Science. 2002;295(5554):469–472. doi: 10.1126/science.1067208. [DOI] [PubMed] [Google Scholar]
- (a) Dang D. Wu P. He C. Xie Z. Duan C. J. Am. Chem. Soc. 2010;132(41):14321–14323. doi: 10.1021/ja101208s. [DOI] [PubMed] [Google Scholar]; (b) Metzger E. D. Brozek C. K. Comito R. J. Dincă M. ACS Cent. Sci. 2016;2(3):148–153. doi: 10.1021/acscentsci.6b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu C.-D. Hu A. Zhang L. Lin W. J. Am. Chem. Soc. 2005;127(25):8940–8941. doi: 10.1021/ja052431t. [DOI] [PubMed] [Google Scholar]
- (a) Xiao J.-D. Li R. Jiang H.-L. Small Methods. 2023;7(1):2201258. doi: 10.1002/smtd.202201258. [DOI] [PubMed] [Google Scholar]; (b) Corma A. García H. Llabrés i Xamena F. X. Chem. Rev. 2010;110(8):4606–4655. doi: 10.1021/cr9003924. [DOI] [PubMed] [Google Scholar]
- Sun C.-Y. To W.-P. Wang X.-L. Chan K.-T. Su Z.-M. Che C.-M. Chem. Sci. 2015;6(12):7105–7111. doi: 10.1039/C5SC02216A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. S. Kapustin E. A. Pei X. LIopis S. Yaghi O. M. Toste F. D. Chem. 2020;6(1):142–152. doi: 10.1016/j.chempr.2019.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ma B. Xia Q. Wang D. Jin J.-K. Li Z. Liang Q.-J. Sun M.-Y. Liu D. Liu L.-J. Shu H.-X. Yang J. Li D. He J. Angew. Chem., Int. Ed. 2023;62(21):e202300233. doi: 10.1002/anie.202300233. [DOI] [PubMed] [Google Scholar]; (b) Fei H. Cohen S. M. J. Am. Chem. Soc. 2015;137(6):2191–2194. doi: 10.1021/ja5126885. [DOI] [PubMed] [Google Scholar]; (c) Yu X. Cohen S. M. J. Am. Chem. Soc. 2016;138(38):12320–12323. doi: 10.1021/jacs.6b06859. [DOI] [PubMed] [Google Scholar]
- (a) Ye X. Liu D. Cryst. Growth Des. 2021;21(8):4780–4804. doi: 10.1021/acs.cgd.1c00460. [DOI] [Google Scholar]; (b) Cavka J. H. Jakobsen S. Olsbye U. Guillou N. Lamberti C. Bordiga S. Lillerud K. P. J. Am. Chem. Soc. 2008;130(42):13850–13851. doi: 10.1021/ja8057953. [DOI] [PubMed] [Google Scholar]; (c) Schaate A. Roy P. Godt A. Lippke J. Waltz F. Wiebcke M. Behrens P. Chem.–Eur. J. 2011;17(24):6643–6651. doi: 10.1002/chem.201003211. [DOI] [PubMed] [Google Scholar]; (d) Tan C. Han X. Li Z. Liu Y. Cui Y. J. Am. Chem. Soc. 2018;140(47):16229–16236. doi: 10.1021/jacs.8b09606. [DOI] [PubMed] [Google Scholar]
- Nishio M. Phys. Chem. Chem. Phys. 2011;13(31):13873–13900. doi: 10.1039/C1CP20404A. [DOI] [PubMed] [Google Scholar]
- (a) Suh M. P. Park H. J. Prasad T. K. Lim D.-W. Chem. Rev. 2012;112(2):782–835. doi: 10.1021/cr200274s. [DOI] [PubMed] [Google Scholar]; (b) Rowsell J. L. C. Eckert J. Yaghi O. M. J. Am. Chem. Soc. 2005;127(42):14904–14910. doi: 10.1021/ja0542690. [DOI] [PubMed] [Google Scholar]; (c) Hu Y. H. Zhang L. Adv. Mater. 2010;22(20):E117–E130. doi: 10.1002/adma.200902096. [DOI] [PubMed] [Google Scholar]
- (a) Au V. K.-M. Wong K. M.-C. Zhu N. Yam V. W.-W. J. Am. Chem. Soc. 2009;131(25):9076–9085. doi: 10.1021/ja9027692. [DOI] [PubMed] [Google Scholar]; (b) Yan J. J. Chow A. L.-F. Leung C.-H. Sun R. W.-Y. Ma D.-L. Che C.-M. Chem. Commun. 2010;46(22):3893–3895. doi: 10.1039/C001216E. [DOI] [PubMed] [Google Scholar]
- (a) van der Kolk M. R. Janssen M. A. C. H. Rutjes F. P. J. T. Blanco-Ania D. ChemMedChem. 2022;17(9):e202200020. doi: 10.1002/cmdc.202200020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hui C. Liu Y. Jiang M. Wu P. Trends Chem. 2022;4(8):677–681. doi: 10.1016/j.trechm.2022.04.006. [DOI] [Google Scholar]; (c) Dembitsky V. M. J. Nat. Med. 2008;62:1–33. doi: 10.1007/s11418-007-0166-3. [DOI] [PubMed] [Google Scholar]
- (a) Lu Z. Yoon T. P. Angew. Chem., Int. Ed. 2012;51(41):10329–10478. doi: 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martynova E. A. Voloshkin V. A. Guillet S. G. Bru F. Beliš M. Hecke K. V. Cazin C. S. J. Nolan S. P. Chem. Sci. 2022;13(23):6852–6857. doi: 10.1039/D2SC00864E. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guo J. Tang W. Y. Li Z. Wu X. Liu L.-J. To W.-P. Shu H.-X. Low K.-H. Chow P. C. Y. Lo T. W. B. He J. Nat. Catal. 2024;7(3):307–320. doi: 10.1038/s41929-024-01112-9. [DOI] [Google Scholar]; (d) Liu Z. Zhou C. Lei T. Nan X.-L. Chen B. Tung C.-H. Wu L.-Z. CCS Chem. 2019;2(1):582–588. doi: 10.31635/ccschem.019.201900049. [DOI] [Google Scholar]; (e) Guo J. Fan Y.-Z. Lu Y.-L. Zheng S.-P. Su C.-Y. Angew. Chem., Int. Ed. 2020;59(22):8661–8669. doi: 10.1002/anie.201916722. [DOI] [PubMed] [Google Scholar]
- (a) Riener M. Nicewicz D. A. Chem. Sci. 2013;4(6):2625–2629. doi: 10.1039/C3SC50643F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ischay M. A. Ament M. S. Yoon T. P. Chem. Sci. 2012;3(9):2807–2811. doi: 10.1039/C2SC20658G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni T. Caldwell R. A. Melton L. A. J. Am. Chem. Soc. 1989;111(2):457–464. doi: 10.1021/ja00184a008. [DOI] [Google Scholar]
- (a) Brown A. Brown T. B. Calabrese A. Ellis D. Puhalo N. Ralph M. Watson L. Bioorg. Med. Chem. Lett. 2010;20(2):516–520. doi: 10.1016/j.bmcl.2009.11.097. [DOI] [PubMed] [Google Scholar]; (b) Jean Jr D. J. St. Fotsch C. J. Med. Chem. 2012;55(13):6002–6020. doi: 10.1021/jm300343m. [DOI] [PubMed] [Google Scholar]; (c) Zhu Y.-Z. Johnson B. M. Yang T. J. AAPS J. 2008;10:178–192. doi: 10.1208/s12248-008-9016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker M. R. Richardson A. D. Schindler C. S. Nat. Commun. 2019;10(1):5095. doi: 10.1038/s41467-019-13072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Zehnder T. N. Blacque O. Venkatesan K. Dalton Trans. 2014;43(31):11959–11972. doi: 10.1039/C4DT01187B. [DOI] [PubMed] [Google Scholar]; (b) Skolia E. Gkizis P. L. Kokotos C. G. Org. Biomol. Chem. 2022;20(29):5836–5844. doi: 10.1039/D2OB01066F. [DOI] [PubMed] [Google Scholar]
- Rueping M. Zhu S. Koenigs R. M. Chem. Commun. 2011;47(30):8679–8681. doi: 10.1039/C1CC12907D. [DOI] [PubMed] [Google Scholar]
- (a) Revol G. McCallum T. Morin M. Gagosz F. Barriault L. Angew. Chem., Int. Ed. 2013;52(50):13342–13345. doi: 10.1002/anie.201306727. [DOI] [PubMed] [Google Scholar]; (b) Zidan M. McCallum T. Swann R. Barriault L. Org. Lett. 2020;22(21):8401–8406. doi: 10.1021/acs.orglett.0c03030. [DOI] [PubMed] [Google Scholar]; (c) Tran H. McCallum T. Morin M. Barriault L. Org. Lett. 2016;18(17):4308–4311. doi: 10.1021/acs.orglett.6b02021. [DOI] [PubMed] [Google Scholar]
- (a) Huang L.-Q. Yang D.-Y. Dong C.-L. He Y.-H. Guan Z. Adv. Synth. Catal. 2023;365(15):2553–2559. doi: 10.1002/adsc.202300519. [DOI] [Google Scholar]; (b) Su Y.-L. Tram L. Wherritt D. Arman H. Griffith W. P. Doyle M. P. ACS Catal. 2020;10(22):13682–13687. doi: 10.1021/acscatal.0c04243. [DOI] [Google Scholar]; (c) Neff R. K. Su Y.-L. Liu S. Rosado M. Zhang X. Doyle M. P. J. Am. Chem. Soc. 2019;141(42):16643–16650. doi: 10.1021/jacs.9b05921. [DOI] [PubMed] [Google Scholar]; (d) Wang X.-Y. He Y.-Q. Zhou Y. Lu L. Song X.-R. Zhou Z.-Z. Tian W.-F. Xiao Q. Org. Lett. 2023;25(21):3847–3852. doi: 10.1021/acs.orglett.3c01191. [DOI] [PubMed] [Google Scholar]; (e) Guo Y. Zhu J. Wang Y. Li Y. Hu H. Zhang P. Xu J. Li W. ACS Catal. 2024;14(2):619–627. doi: 10.1021/acscatal.3c05003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the ESI† of this article.