

Abstract
Background
HIV-1 RT is a heterodimeric enzyme, comprising of the p66 and p51 subunits. Earlier, we have shown that the β7-β8 loop of p51 is a key structural element for RT dimerization (Pandey et al., Biochemistry 40: 9505, 2001). Deletion or alanine substitution of four amino acid residues of this loop in the p51 subunit severely impaired DNA binding and catalytic activities of the enzyme. To further examine the role of this loop in HIV-1 RT, we have increased its size such that the six amino acids loop sequences are repeated in tandem and examined its impact on the dimerization process and catalytic function of the enzyme.
Results
The polymerase and the RNase H activities of HIV-1 RT carrying insertion in the β7-β8 loop of both the subunits (p66INS/p51INS) were severely impaired with substantial loss of DNA binding ability. These enzymatic activities were restored when the mutant p66INS subunit was dimerized with the wild type p51. Glycerol gradient sedimentation analysis revealed that the mutant p51INS subunit was unable to form stable dimer either with the wild type p66 or mutant p66INS. Furthermore, the p66INS/p66INS mutant sedimented as a monomeric species, suggesting its inability to form stable homodimer.
Conclusion
The data presented herein indicates that any perturbation in the β7-β8 loop of the p51 subunit of HIV-1 RT affects the dimerization process resulting in substantial loss of DNA binding ability and catalytic function of the enzyme.
Background
Human immunodeficiency virus type-1 reverse transcriptase (HIV-1 RT) is a product of the gag-pol polyprotein precursor, which is subsequently cleaved by the pol-encoded protease to yield the active form of the enzyme [1,2]. This multifunctional enzyme is responsible for copying the single stranded viral RNA genome into double stranded proviral DNA [3,4]. HIV-1 RT is a heterodimer consisting of a 66 and 51 kDa polypeptide chain designated as p66 and p51, respectively. The p51 subunit is generated via endoproteolytic cleavage of the p66 subunit between Phe 440 and Tyr 441 [5,6]. The larger subunit (p66) contains both polymerase and RNase H activities, while the smaller subunit (p51) lacks these functions, in context of the heterodimer [7,8]. However, both the p66 and p51 monomers are functionally inactive when dissociated from each other [9]. Several years have passed since it was first suggested that agents that could specifically disrupt the dimerization of HIV-1 RT might prove a worthwhile antiretroviral strategy [10], though such agents have yet to be developed.
Despite the fact that p51 shares an identical amino acid sequence with the N-terminal portion of p66, the two subunits assume different global folding patterns in the formation of the asymmetric heterodimer [9]. Structural determination through X-ray crystallography has revealed that the p66 subunit of HIV-1RT has its polymerase domain in an "open" conformation, with its subdomains forming a large cleft which accommodates DNA. In contrast, the p51 subunit assumes a compact folded conformation that causes the active site residues in this subunit to be buried and therefore, nonfunctional [11-13]. It has been proposed that the open conformation of p66 is supported by interactions with a closed and compact p51 molecule [12,14,15]. The two subunits interact mainly via their connection subdomains. Additional contacts, between the thumb subdomain of p51 and RNase H subdomain of p66 are also substantial [11,12].
Although there have been conflicting reports regarding the DNA polymerase activity of recombinant preparations of the p51 homodimer [16,17], it has become clear that p51 mainly plays a supportive role in context of the p66/p51 heterodimer. Assembly of chimeric heterodimers formed by mixing subunits of HIV-1 RT and FIV-1 RT, has demonstrated that the p51 subunit of HIV-1 RT helps to preserve the functional integrity of the HIV-1 RT heterodimer [18]. Despite the fact that several functions have been proposed for the p51 subunit, the mechanism whereby p51 performs these functions has remained largely undefined. Some of the proposed functions for p51 include: (i) stabilizing the t-RNA primer binding for the initiation of reverse transcription [11], (ii) enhancement of strand displacement DNA synthesis [19,20], and (iii) as a processivity factor in DNA synthesis [21].
Cys→Ser mutation at position 280 in the p51 subunit has been shown to alter the RNase H activity of the heterodimeric enzyme, indicating that this residue in the thumb subdomain of p51 plays an important role in support of the RNase H activity of p66 [22]. The emergence of a strain of HIV-1 resistant to the non-nucleoside RT inhibitor TSAO (Tertbutyldimethyl silyspiro amino oxathioledioside) displaying Glu→Lys mutation at position 138 in the p51 subunit of HIV-1 RT has also been reported [23], thus implicating p51 to play a more direct role in drug binding and/or the enzymatic activities of HIV-1RT. This report was initially surprising, since Glu138 of p51 was thought to be quite distant from the purported dNTP-binding pocket of HIV-1RT, as well as the NNRTI binding pocket. However, in light of our recent findings implicating this loop region of p51 as a critical structural element supporting the catalytic functions of p66, it seems feasible that mutation at position 138 in p51 effectively altered the binding of TSAO through its influence on the p66 catalytic subunit [14].
Examination of the crystal structure of HIV-1 RT reveals the presence of a small groove like region on the floor of the polymerase cleft of p66 [11]. The β7-β8 loop of p51, comprising of six amino acids denoted as SINNET appears to fit into this groove-like region and likely stabilizes the polymerase domain of p66. In an earlier communication, we have shown that the p51 subunit of HIV-1 RT is required to load the p66 subunit on to the template primer for DNA synthesis [14]. Our recent studies indicate that the β7-β8 loop of the p51 subunit is essential for the catalytic function of the p66 subunit. Deletion of this loop or substitution of four amino acid residues with alanine within the β7-β8 loop of p51 severely impaired the DNA polymerase activity of the enzyme as a consequence of the inability of the enzyme to form stable dimers [15]. These findings clearly establish the absolute requirement of the β7-β8 loop of p51 for RT dimerization.
Nonetheless, the question regarding the optimal size and composition requirement of this loop for efficient dimerization remains unanswered. In the present article, we have addressed the impact of increasing the size of the β7-β8 loop on the dimerization process. As a preamble to these studies, we have increased the size of this loop by repeating its six amino acid sequence in tandem. The rationale for duplicating the loop sequence was to increase the size of this loop without significantly disrupting the interactions seen with the wild type β7-β8 loop. The resulting mutant derivatives of HIV-1 RT containing insertion of six amino acids in the β7-β8 loop in either or both the subunits were analyzed for their ability to form stable dimers and other biochemical characteristics. In this article, we present evidence that HIV-1 RT mutants, carrying insertion of six amino acids in the β7-β8 loop specifically in the second subunit, do not form stable dimers. This inability to dimerize substantially decreases the enzymes affinity for DNA consequently impairing its polymerase and RNase H activities.
Results
Glycerol gradient ultra-centrifugation analysis
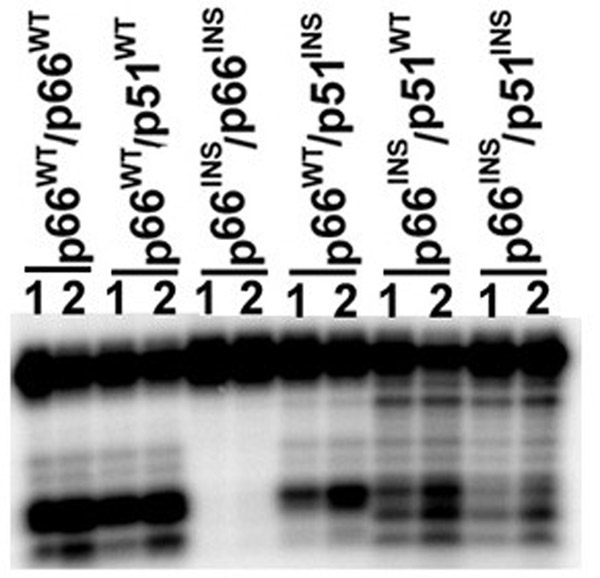
The ability of the HIV-1 RT mutants, carrying insertion of six residues in the β7-β8 loop in either one or both the subunits was analyzed by glycerol gradient sedimentation analysis. Fractions of 200 μL were collected from the bottom of the tube and aliquots of every third fraction were subjected to SDS PAGE. The results shown in Figure 1A, indicate that the sedimentation peak for the wild type p66/p66 homodimer was between fractions 16–19 of the gradient (Panel A), whereas the wild type p51 species sedimented between fractions 22–28, as a monomeric protein (Panel C). The sedimentation pattern for the p66WT/p51INS mutant indicated two distinct peaks. While the p66WT subunit predominantly sedimented in between fractions 16–19, at the predicted position of the homodimer, the p51INS subunit sedimented between fractions 22–28, indicating a monomeric conformation (Panel D). This sedimentation profile indicates that the p66WT/p51INS mutant carrying the 6 amino acid insertion in the β7-β8 loop of its p51 subunit is unable to form stable heterodimer. Under these conditions, the p66WT subunit of the p66WT/p51INS mutant tends to homodimerize. The mutants, p66INS/p66INS (Panel B) and p66INS/p51INS (Panel F), exhibited a distinct sedimentation peak between fractions 22–28 of the gradient, indicating that these enzymes are unable to form stable dimers. In contrast, the p66INS/p51WT mutant (Panel E), in which only the p66 subunit carried an insertion in the β7-β8 loop sedimented between fractions 16–19 of the gradient, indicating that these two subunits can form stable heterodimers. These analyses imply that the proper size of the β7-β8 loop in the second subunit of HIV-1 RT is important for the formation of a stable dimeric enzyme.
Figure 1.

(A) Sedimentation profile of the wild type and mutant HIV-1 RT. Mutant enzymes carrying 6 amino acids insertion in the β7-β8 loop of either or both the subunits were applied on 10–30% linear glycerol gradient (5 mL) and centrifuged at 48,000 rpm in SW 50.1 rotor for 22 hours. Gradients were fractionated from the bottom and subjected to SDS polyacrylamide gel electrophoresis and Coomassie Blue staining. (B) Polymerase Activity profile of the glycerol gradient fractions. Every third fraction between 7 and 33 of the glycerol gradient (Fig. 1A) was diluted 10-fold and analyzed for its polymerase activity on poly (rA)(dT)18 as described under Materials and Methods. The reaction products were resolved on an eight percent denaturing poly-acrylamide urea gel and subjected to PhosphorImaging.
In order to correlate the sedimentation profile of these insertion mutants with their functional activity, we analyzed the polymerase activity in the various gradient fractions. These results are presented in Fig. 1B. The polymerase activity profile of the gradient fractions of the wild-type p66/p66 and the p66INS/p51WT mutant revealed major polymerase activity peaks corresponding to fractions 16–19 (Fig. 1B). This activity peak correlates with the protein band intensity seen in Fig. 1A (panels A and E) and is also in agreement with the sedimentation pattern of these two enzymes. Interestingly, the activity profile of the p66WT/p51INS mutant also yielded a peak corresponding to gradient fractions 16–19 (Fig. 1B), thus substantiating our contention that the p66WT subunit of the p66WT/p51INS mutant tends to self-dimerize and form the catalytically active p66 homodimer. The wild type p51 and the two mutants, p66INS/p66INS and p66INS/p51INS, the sedimentation profile of which indicated a monomeric conformation (Fig. 1A) were conspicuously devoid of any polymerase activity (Fig. 1B). These results imply that the β7-β8 loop of the second subunit of HIV-1 RT is critical in forming functionally active dimeric enzyme.
DNA polymerase activities of wild type HIV-1 RT and its insertion mutants
DNA polymerase activity of the wild type HIV-1 RT and its mutant derivatives were quantitatively determined on both heteropolymeric RNA (U5-PBS RNA) and DNA (49-mer) templates, primed with 32P labeled 17-mer PBS primer. As documented in Table 1, insertion of the six amino acid segment in both the subunits (p66INS/p66INS or p66INS/p51INS), resulted in significant loss of polymerase activity corresponding to greater than 90% reduction. Interestingly, when the p66INS mutant was dimerized with the wild type p51 (p66INS/p51WT), the polymerase activity was restored to wild type levels. The same result was obtained when p66INS was dimerized with p51 having Asp→Ala mutation at amino acid position 186 (one of the catalytically crucial carboxylate triad) but having an intact β7-β8 loop. The rationale for using p51D186A mutant was to ascertain that the wild type polymerase activity observed with p66INS/p51WT was not due to residual polymerase activity of p51. These results suggest that insertion of six residues in the β7-β8 loop of p51 but not in p66 is detrimental to the function of the heterodimeric enzyme.
Table 1.
Effect of duplication of the β7–β8 loop in either or both the subunits of HIV-1 RT on the polymerase activity of the enzyme
| Percent of wild type polymerase Activity | |||
| Enzyme Species | Poly (rA).(dT)18 | U-5 PBSRNA/17-mer | U-5 PBS DNA/17-mer |
| p66WT/p51WT | 100 | 100 | 100 |
| p66WT/p66WT | 92.6 | 82.5 | 85.6 |
| p66INS/p66INS | 1.5 | 11 | 10 |
| p66INS/p51INS | 0.6 | 1.2 | 2 |
| p66INS/p51WT | 98 | 106 | 97 |
| p66INS/p51D186A | 103 | 116 | 96 |
| p66WT/p51INS | 60 | 57 | 65 |
The polymerase activities of the wild type HIV-1 RT and its mutant derivatives carrying insertion of 6 amino acids in the β7-β8 loop of either or both the subunits were determined with the indicated template primer at saturating dNTP concentrations. Reactions were carried out at 37°C for 3 min and the total picomoles of nucleotide incorporated were determined. The values are expressed as a percentage of the wild type polymerase activity and are an average of three independent experiments.
As shown in figure 2, evaluation of the polymerase activity of the wild type HIV-1RT and its insertion mutants by primer extension assay generally mirrored the results observed in the TCA precipitation assay, summarized in Table 1. Insertion of six amino acid residues in the β7-β8 loops of both the subunits (p66INS/p66INS or p66INS/p51INS) resulted in significant impairment of polymerase activity of the mutant enzymes. On an RNA template, both these enzymes exhibited total lack of polymerase activity (Fig. 2B), though the p66INS/p51INS mutant exhibited residual primer extension capability on a DNA template (Fig. 2A), while the p66INSp/66INS mutant was inactive on this template. Consistent with the results of the TCA precipitation assay, both the mutants, p66INS/p51WT and p66INS/p51D186A, exhibited near wild type (p66WT/p51WT) polymerase activity on both RNA and DNA templates. Thus, the impairment of the polymerase activity seen with these mutants carrying insertion in either both the subunits or specifically in the second subunit may be related to the inability of these mutants to form stable dimers resulting in a closed polymerase cleft.
Figure 2.

Analysis of polymerase products catalyzed by insertion mutants of HIV-1 RT. Primer extension reactions catalyzed by the wild type and mutant enzymes were on DNA (panel A) and RNA (panel B) templates, primed with 5'-32P labeled 17-mer primer. Each set of reactions was carried out for 30 seconds (lane 1) and 60 seconds (lane 2) at 37°C and quenched by the addition of equal volume of Sanger's gel loading dye. The reaction products were resolved on an 8% polyacrylamide-7M urea gel and subjected to PhosphorImager analysis.
Effect of insertion in either or both the subunits of HIV-1 RT on the DNA binding function of the enzyme
Earlier we have shown that substitution of four amino acids on the β7-β8 loop with alanine reduced the DNA binding ability of the enzyme. Similar results were obtained when four amino acids were deleted from the loop. We therefore, concluded that the effect was exerted via p51 since alanine substitution or deletion specifically in the p66 subunit had no effect on DNA binding and polymerase activity of the enzyme. We speculated that alanine substitution or deletion in the loop of the p51 subunit may shorten the interacting sphere of the loop which may not be able to induce opening of the polymerase cleft in p66, which is essential for DNA binding. Given the fact that shortening of this loop in p51 prevented DNA binding, we were interested in examining the effect of six amino acids insertion on DNA binding. Therefore, we determined the equilibrium dissociation constants (Kd) of E-TP binary complexes for the wild type enzyme and its mutant derivatives by gel mobility shift assay. For this purpose, we used a 33-mer heteropolymeric DNA template primed with 5' 32P-labeled 21-mer DNA. Results shown in figure 3 and table 2 indicate a 24-fold reduction in DNA binding affinity when both the subunits carried insertion (p66INS/p51INS) in their β7-β8 loops. Similar results were obtained with the p66INS/p66INS mutant. Interestingly, the DNA binding affinity was restored to the wild type levels, when the mutant p66 subunit was dimerized with the wild type p51 (p66INS/p51WT). We expected that the p66WT/p51INSmutant would also exhibit reduction in its affinity for DNA, since insertion of six residues in the p51 subunit was speculated to be detrimental for the dimerization process. However, the DNA binding affinity of the p66WT/p51INS mutant was similar to the wild type enzyme. Our glycerol gradient sedimentation analysis had revealed that the p66WT/p51INS mutant does not form stable dimers, rather the p66WT subunit tends to self dimerize. Thus, the wild type DNA binding affinity seen in case of the p66WT/p51INS mutant may be attributed to the presence of these p66WT/p66WT homodimeric species. Based on our observations from the sedimentation analysis, we propose that the loss of DNA binding function in case of the HIV-1RT insertion mutants results from a failure to form stable dimers. The wild type p51 alone exhibited very low DNA binding affinity with a Kd[DNA]1300-fold higher than the wild type heterodimeric enzyme. These results suggest that the optimal size of the β7-β8 loop in the second subunit of the HIV-1 RT dimer is crucial for opening the polymerase cleft of the p66 subunit.
Figure 3.

DNA binding affinity of mutant HIV-1 RT carrying insertion in the β7–β8 loop of either or both the subunits. The 49-mer DNA primed with 5'-32P labeled 21-mer DNA primer was incubated with varying concentrations of the individual enzyme at 4°C for 10 min. The mixture was electrophoresed under non-denaturing conditions on a 6% (w/v) polyacrylamide gel and analyzed on phosphorImager (Left panel). The positions of the free template primer (TP) and enzyme bound template-primer (E-TP binary complex) are indicated for the wild type enzyme. The percent of E-TP complex formed as a function of enzyme concentration was plotted for determining the Kd values (Right panel).
Table 2.
DNA and dNTP binding affinities of the mutant HIV-1 RT carrying insertion in the β7–β8 loop of either or both the subunits
| Enzyme Species | Kd [DNA] (nM) | App. Kd [dNTP] (μM) |
| p66WT/p51WT | 2.0 | 6.0 |
| p66WT/p66WT | 2.6 | 5.8 |
| p66INS/p51INS | 48.0 | 8.0 |
| p66INS/p66INS | 34.0 | ND |
| p66INS/p51WT | 4.0 | 9.0 |
| p66WT/p51INS | 3.0 | 5.0 |
The dissociation constant of E-TP binary complex (Kd [DNA]) and the apparent dNTP binding affinity (Kd [dNTP]) for the mutant HIV-1 RT was determined by gel mobility shift assay. For Kd [DNA] determination, 5' 32P-labeled dideoxy terminated 33/21-mer DNA template-primer was incubated with varying concentrations of the individual WT or mutant enzyme and resolved on a native polyacrylamide gel at 4°C. The slower migrating E-TP complexes were detected by phosphorImaging and quantified using ImageQuant software. For determining the apparent Kd [dNTP], the concentration of the individual enzyme used was such that retarded 100% of the labeled TP. The extent of stable E-DNA-dNTP ternary complex formed in the presence of varying concentrations of the next correct nucleotide (dGTP) was determined following the addition of the DNA trap. This was estimated by calculating the percent of the un-dissociated template primer in the ternary complex using ImageQuant software. The values shown are the average of two independent experiments. ND indicates that the apparent Kd [dNTP] in case of p66INS/p66INS could not be determined by this analysis.
Ternary complex formation by the wild type and mutant enzymes
In the crystal structures of the ternary complex of HIV-1 RT (E-DNA-dNTP) the finger subdomain moves by 20Å towards the palm subdomain [24]. In this finger closing conformation, the DNA is locked in a stable ternary complex poised for catalysis. An in vitro assay using dideoxy terminated primer annealed with the template which allows the next correct dNTP to bind in the ternary complex without actual DNA synthesis has recently been reported [25]. Using this assay system, we have evaluated the ability of the insertion mutants to form the ternary complexes and the effect of DNA trap on such complexes. Since binding of dNTP to the enzyme is an ordered mechanism which occurs only after DNA binding, the extent of labeled TP remaining bound to the enzyme in the presence of dNTP and DNA trap represents the extent of ternary complex formed. The E-TP binary complex was formed at enzyme concentrations which binds 100% of the labeled template primer. The preformed E-TP complex was then incubated in the presence of next correct dNTP followed by addition of 300-fold molar excess of unlabeled TP as the DNA trap. We found that E-TP binary complex was completely competed out by the DNA trap (data not shown) while a significant amount of the E-TP binary complex converted to E-TP-dNTP ternary complex was resistant to competition with DNA trap (Fig. 4) suggesting the stability of the ternary complex. Table 2 lists the apparent dNTP binding affinity for the WT enzyme and its insertion mutants determined from data shown in Fig. 4. It was observed that although the DNA binding affinity was severely affected in case of the p66INS/p51INS mutant, its apparent dNTP binding affinity in the ternary complex did not change with respect to the wild type enzyme. However, the p66INS/p66INS mutant was unable to form a ternary complex. These data suggest that these two mutants may have a different conformation and mode of interaction in the ternary complex. It is apparent that the p66INS/p66INS binds to TP in a nonproductive manner which may have a direct impact on dNTP binding in the ternary complex.
Figure 4.
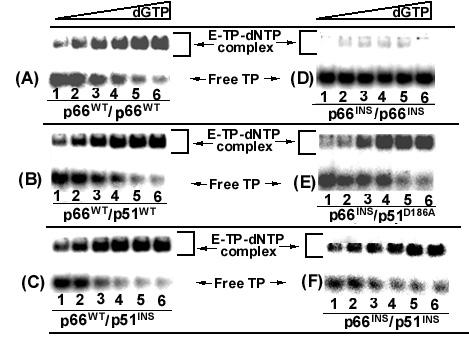
Apparent dNTP binding affinity of mutant HIV-1 RT carrying insertion in the β7–β8 loop of either or both the subunits. The 33-mer DNA primed with 5'-32P labeled dideoxy (ddC) terminated 21-mer DNA primer was incubated with the individual enzyme at 4°C for 10 min. The binding affinity [Kd (dNTP)] of the wild type enzyme and its mutant derivatives in the ternary complex was determined by incubating the preformed E-TP binary complex of the individual enzyme species at different concentrations (0.26–800 μM) of the next correct dNTP (dGTP). Following incubation with dGTP, 300 fold molar excess of the unlabeled TP was used as trap to remove the readily dissociable binary complexes from the ternary complex population. The extent of stable ternary complexes formed were resolved on 6% native polyacrylamide gel and analyzed by phosphorImaging. The [Kd [dNTP]], in the ternary complex for each enzyme was determined by quantifying the E-TP-dNTP ternary complexes as a function of increasing concentrations of dGTP and fitting the data to the equation for single-site ligand binding using Sigma-Plot. Lanes 1–6 represent the ternary complex formed in the presence of dGTP at 0.2, 1.28, 6.4, 32, 160 and 800 μM, concentrations.
Steady state kinetic analysis of HIV-1 RT and its insertion mutants
In order to determine whether alteration in DNA binding without any change in the apparent dNTP binding affinity of the insertion mutants is consistent with their kinetic parameters, we analyzed the steady-state kinetic parameters of these mutants. The results of this investigation are summarized in Table 3. On poly (rA).(dT)18, only the p66INS/p66INS mutant showed a significant increase in Km [dNTP]. This observation is in agreement with the apparent dNTP binding affinity data in the ternary complex, where p66INS/p66INS mutant was found to be defective in forming a productive ternary complex. This observation is also consistent with our suggestion that p66INS/p66INS binds nonproductively to TP that may influence the formation of ternary complexes. Interestingly, the p66INS/p51INSmutant carrying insertion in both the subunits did not display the same reduction in dTTP binding affinity. However, the p66INS/p66INS and p66INS/p51INS mutants displayed nearly 6,000-fold and 400-fold reduction in catalytic efficiency (kcat/Km) compared to their wild type counterparts, respectively, on this template primer. A 10-fold reduction in catalytic efficiency in case of the p66INS/p51WT was noted only on poly (rA). (dT)18 and may be template-primer specific. None of the enzymes displayed a significant reduction in Km [dNTP] when the heteropolymeric DNA\DNA template primer was used, although the p66INS/p66INS and p66INS/p51INS mutants exhibited drastic reduction in catalytic efficiency. The p66WT/p51INSdisplayed no change in either dNTP binding or catalytic efficiency. These results are in keeping with our analysis of the polymerase and DNA binding assay for this mutant. Once again, we believe that the wild type p66 subunit in this enzyme preparation tends to homodimerize since the p51INS fails to participate in stable dimer formation. This phenomenon masks the deleterious effect of the insertion mutation.
Table 3.
Steady-State kinetic parameters of mutant HIV-1 RT carrying insertion in the β7–β8 loop of either or both the subunits
| Poly (rA).(dT)18 | U-5 PBS 49-mer DNA/17 mer | |||||
| Enzyme | Km (dTTP) (μM) | kcat (S-1)102 | kcat/Km (S-1M-1) × 104 | Km (dNTP) (μM) | kcat (S-1)103 | kcat/Km (S-1M-1) × 104 |
| p66WT/p66WT | 5.01 | 135.7 | 27.08 | 2.10 | 15.8 | 0.754 |
| p66WT/p51WT | 5.48 | 103.7 | 18.93 | 3.91 | 25.0 | 0.641 |
| p66INS/p66INS | 46.39 | 0.198 | 0.0043 | 6.05 | 0.400 | 0.0063 |
| p66INS/p51WT | 2.57 | 43.7 | 1.702 | 5.47 | 15.00 | 0.270 |
| p66WT/p51INS | 4.86 | 89.8 | 18.490 | 2.35 | 15.40 | 0.655 |
| p66INS/p51INS | 3.35 | 0.15 | 0.0448 | 5.71 | 0.300 | 0.0046 |
Kinetic parameters of the wild type HIV-1RT and its mutant derivatives were determined on homopolymeric RNA (poly rA) and heteropolymeric DNA (U5-PBS 49-mer) templates with (dT)18 and 17 mer DNA as primers, respectively. The values obtained are the average of three independent experiments.
RNase H activity of the insertion mutants
Since the polymerase activity of the homo- and hetero-dimeric enzymes carrying insertion in the β7-β8 loop in both the subunits (p66INS/p66INS and p66INS/p51INS) was drastically impaired, it was of interest to examine how this insertion affects their RNase H activity. To evaluate this, we employed a 30-mer RNA-DNA hybrid, and examined the cleavage of the 5'-32P-RNA strand of the duplex by the wild type enzyme and its mutant derivatives. The result of this analysis is presented in Fig. 5. Similar to their polymerase activities, the RNase H activities of the p66INS/p66INS and p66INS/p51INS mutants were severely impaired. This is not surprising, since our analysis of the DNA binding function of these two mutants had indicated a substantial loss of DNA binding affinity, which in turn is expected to affect both the polymerase and RNase H functions. Dimerization of the p66INS subunit with the wild type p51 (p66INS/p51WT) carrying an intact β7-β8 loop resulted in substantial recovery of the RNase H activity. The RNase H activity seen in case of the p66WT/p51INS mutant was not surprising since the p66WT subunit of the p66WT/p51INSmutant tends to self-dimerize and form the catalytically active p66 homodimer.
Figure 5.
RNase H activity of the wild-type HIV-1 RT and its mutant derivatives. The individual enzymes were incubated with 5'-32P-labeled 30-mer RNA annealed with 30-mer complementary DNA strand, at 37°C for 30 sec and 1 min as described in Materials and Methods. The cleavage products were resolved on an 8% denaturing polyacrylamide-urea gel and analyzed on a phosphorImager. Lanes 1 and 2 represent reactions carried out for 30 sec and 1 min, respectively.
Discussion
In an earlier investigation on the role of the p51 subunit of HIV-1 RT, we demonstrated that decrease in size of its β7-β8 loop impairs the catalytic function of the heterodimer [15]. In the present studies, we demonstrate that maintaining the wild type size of this loop in the p51 subunit is critical for dimerization of the enzyme and its catalytic activity. Duplication of the β7-β8 loop sequence selectively in the p66 subunit did not affect the dimer formation, DNA binding or polymerase activity of the p66INS/p51WT mutant. However, insertion of the same amino acid residues in the β7-β8 loop of p51 prevented stable dimerization of the p51INS subunit with either p66INS or p66WT and adversely impacted the DNA binding, polymerase and RNase H activities. Earlier, we have shown that p51 facilitates the loading of the p66 subunit on to the template primer [14]. Therefore, the impaired polymerase activity and template-primer binding affinity of HIV-1 RT mutants carrying insertion in p51 may be due to their inability to load the catalytic p66 (p66INS) on the template primer. These altered biophysical/enzymatic properties of these insertion mutants may be attributed to the reduced dimer stability.
Crystal structures of HIV-1 RT show that p66 and p51 assume different folding patterns and tertiary structures [11,12]. It has been proposed that p66 in a monomeric form exists in a closed conformation similar to p51 [26]. Following dimerization with another molecule of p51 (or p66), it assumes an open conformation [11,14,15]. The polymerase domain of p51 is buried within its core. This difference in tertiary structures between the two subunits makes the dimer asymmetric. The amino acid residues at the contact interface differ with respect to their position and location in the 3-D structure. The counterpart of amino acid residues of p66 located at the contact interface are buried in the p51 folded conformation, whereas those of p51 are scattered in the p66 'open' conformation (Fig. 6).
Figure 6.
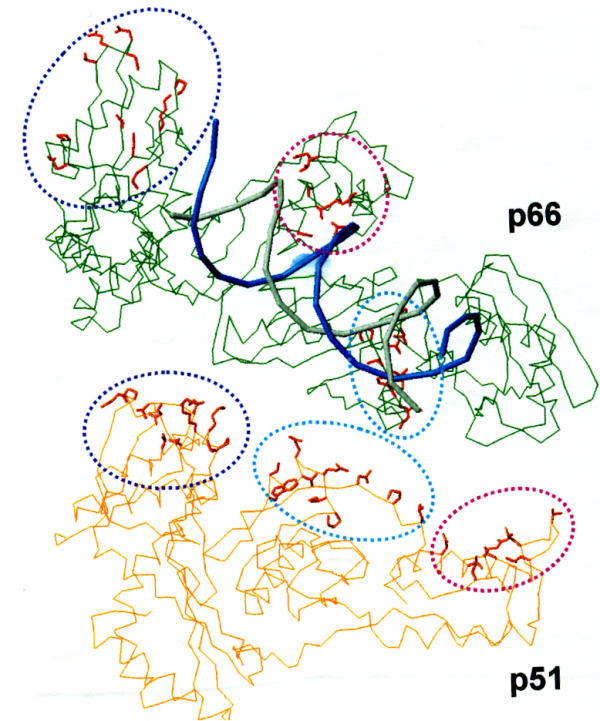
Amino acid residues in the p51 subunit in contact with the p66 subunit. The Cα backbone of p51 and p66 is shown in orange and green, respectively. Amino acids residues of p51 making contact with the p66 subunit are represented as 'sticks' and are shown encircled. The location of analogous residues in p66 are also encircled. It may be noted that these residues in p66 are scattered. The residues circled within 'blue' are from fingers; those in light blue' are from 'connection' and those in pink are from 'thumb' subdomains. The template (blue) and primer (gray) are also displayed in this figure. The residues in blue circle are V21, K22, P25, P52, E53, N57, T131, N136, N137, E138, T139, in light blue circle are P392, I393, Q394, E396, T397, T400, W401, N418, P420, L422, and those in pink circle are N255, Q258, V261, N265, V276, L283, T286, L289.
Of the several domain interactions between p66 and p51, the β7-β8 loop of p51 is strategically positioned to interact with the residues on the floor of the palm subdomain of p66. It has been suggested that the stability of the dimer is related to the buried surface area between the two subunits [11,12]. In the nevirapine-bound HIV-1 RT crystal structure, the total contact surface area between the subunits is approximately ~4600 Å2. The two major contact regions between the subunits which provide it stability are their connection subdomains and the thumb of p51 and RNase H domain of p66. These contacts account for approximately two third of the total buried surface area. Interestingly, the marginal decrease in the total surface area due to deletion of four residues in the β7-β8 loop does not account for the dimer instability, thus suggesting that polar interactions of residues in the β7-β8 loop of p51 with the palm subdomain of p66 may play a role in conferring stability to the heterodimer. The observation that a single point mutation at L289 of p66, a residue not in direct contact with p51, also destabilizes the dimer [27,28] indicates that other factors may also contribute towards dimer stability.
In order to analyze the impact of inserting the six amino acid peptide in the β7-β8 loop of p51, we used the molecular modeling approach. A search in the database of known protein structures employing the 'loop-search' algorithm of SYBYL yielded 100 loops, of which only five were sterically permissible. The loop exhibiting the best homology was incorporated in the modeled structure (Fig. 7). This loop of 9 amino acids (RFNAHGDVN) from the protein S. lectin formed a short anti-parallel two strand β-sheet. This inserted loop lies in the vicinity of the palm subdomain of p66 and exhibits additional hydrophobic and polar interactions with residues in the palm subdomain of p66, not seen in the wild type structure. These additional interactions are expected to enhance the stability of the dimer. However, sedimentation analysis indicates that the insertion mutants form unstable dimer. This implies that the insertion may have altered the relative position of the subdomains in the two subunits thereby perturbing the dimer stability. In summary, the β7-β8 loop of p51 is an important structural element involved in imparting stability to the heterodimer and in opening the polymerase cleft of p66 for catalysis.
Figure 7.

Molecular model of the inserted peptide in the β7–β8 loop of the p51 subunit. Molecular modeling of the extended loop was performed by the 'Loop Search' option of SYBYL version 6.5 (Tripos Associates, St. Louis, MO). A total of 100 loops, matching the Cα distances constrain between the end points of the loop residues, were obtained. The loops were examined for 'bumps' with the neighboring protein structure. Selection of the appropriate loop was based on the least root mean square deviation and maximum homology with the inserted loop sequence. A loop satisfying these criteria containing two small anti-parallel β-strands is shown in cyan. The side chains of amino acid residues of p66 are shown in green and those of p51 (wild type) are shown in orange. The side chains of interacting residues in the inserted peptide are colored violet and are indicated by their position number in parentheses. Interactions seen in the wild type loop are shown in blue dotted line while additional interactions are shown in black dotted lines.
Materials and methods
PfuTurbo DNA polymerase and PCR reagents were obtained from Stratagene, Inc. Restriction endonucleases, DNA modifying enzymes and HPLC-purified dNTPs were purchased from Roche Molecular Biochemicals. Fast flow chelating sepharose (iminodiacetic-Sepharose) for immobilized metal affinity chromatography (IMAC), Phosphocellulose and Q-sepharose was purchased from Amersham Pharmacia Biotech. The α-32P-dNTPs and γ-32P-ATP were purchased from Perkin Elmer life sciences. The DNA oligomers were synthesized at the Molecular Resource Facility at UMDNJ. All other reagents and chemicals were of the highest available purity grade and purchased from Fisher, Millipore Corp., Roche Molecular and Bio-Rad.
Plasmid and clones
The expression vector pET-28a and E. coli expression strain BL21 (DE3) were obtained from Novagen. The HIV-1RT expression clones (pKK223-3 RT66 and pET-28a-RT51) constructed in this laboratory [29-31] were used for PCR amplification and construction of the insertion mutants in the p66 and p51 subunits of HIV-1 RT. An HIV-RNA expression clone pHIV-PBS was a generous gift from Dr. M. A. Wainberg [32].
Insertion of 6 amino acid residues in the β7–β8 loop
The pKK-RT66 clone containing two unique restriction sites, Hpa1 and Stu1, at codons 136 and 140 in the RT coding region [15] was used for insertion of 6 amino-acid residues in the β7-β8 loop of the p66 and p51 subunit. The pKK-RT66 clone was digested with HpaI restriction enzyme to generate a blunt end at codon 136. For insertion, two complementary pre-kinased 18-mer synthetic DNA oligos having the following sequences: 5'-ATA AAC AAT GAG ACA ATA-3 (sense strand) and 3'-TAT TTG TTA CTC TGT TAT-5' (antisense strand) were hybridized. The 18-mer duplex DNA encoding the insertion peptide (Ile-Asn-Asn-Glu-Thr-Ile) was ligated with Hpa1 digested pKK-RT66 in between codon 135 and 136. The positive clones were screened in E. coli HB101 by the absence of an Hpa1 site and the correct orientation of the insertion was confirmed by DNA sequencing. This construct expresses the p66+6aa subunit without His tag sequences. A His-tag at the N-terminal of the p66+6aa subunit was introduced by sub cloning the Bal-I and Hind III fragment of pKKRT66+6aa into pET-28a-RT66 expression cassette. A unique Sac I site was also introduced in pKK-RT66 template at codon 440. The construction of P51+6aa was carried out by removal of the 360 bp fragment from pKK-RT66+6aa by restriction digestion with SacI followed by re-ligation of the vector ends. The insertion mutant in pET28a and pKK223-3 vectors were introduced into E. coli BL-21 (DE3) pLys S and E. coli JM109, respectively, for expression. Induction of the enzyme protein was carried out as described before for the wild type HIV-1RT [29]. The enzyme with the hexahistidine-tag was purified from bacterial lysates by immobilized metal affinity chromatography [33], while non-hexahistidine-tagged enzyme was purified using the phosphocellulose and Q-Sepharose columns as described previously [24].
Preparation of the heterodimeric enzyme with subunit specific insertion
The p51 subunit with a hexahistidine-tag and a non-tagged p66 were used to generate the heterodimers containing insertion in either or both of the subunits. For each set of heterodimers, 260 μg of p51 was mixed with 660 μg of p66 in the buffer containing 50 mM Tris HCl, pH 7.8, 60 mM KCl and 5 mM MgCl2. The rationale for using a 1:3 ratio of p51 to p66 was to saturate the His-tagged p51 with the non-tagged p66, ensuring heterodimer formation and eliminating excess p66 during IMAC purification. The mixture was incubated for 16 hours at 4°C and applied to (0.5 mL) Ni2+ iminodiacetic-Sepharose (IDA-Sepharose) column, which was pre-equilibrated with the binding buffer (20 mM Tris HCl pH 7.8, 500 mM NaCl and 5 mM Imidazole). The column was washed with 15 mL of the same buffer to remove the excess of p66 that was not dimerized with p51 bound to the IDA-sepharose column. The heterodimeric RT was then eluted from the column with elution buffer (20 mM Tris HCl pH 7.8, 500 mM NaCl and 250 mM imidazole). Fractions of 0.5 mL were collected and an aliquot of each fraction was analyzed by SDS-PAGE using Coomassie Blue stain. The fractions containing approximately equal band intensity of p66 and p51 were dialyzed against a storage buffer (50 mM Tris HCl pH 7.0, 200 mM NaCl and 50% Glycerol) and this enzyme preparation was used in all experiments.
Glycerol gradient ultra centrifugation
Fifty micrograms of the enzyme protein in 100 μL of buffer (50 mM Tris HCl, pH 7.8, 1 mM DTT and 400 mM NaCl) was carefully loaded onto 5 mL of 10–30% glycerol gradients prepared in the same buffer. The gradients were centrifuged at 48,000 rpm in an SW48 rotor for 22 h at 4°C. Fractions (200 μL) were collected from the bottom of the tube and aliquots of these fractions were electrophoresed using SDS PAGE and Coomassie Blue stain to identify the protein peak.
The polymerase activity in the gradient fractions were analyzed by extension of the labeled (dT)18 annealed to poly (rA) template. Every third fraction between 7 and 33 of the glycerol gradient was diluted 10-fold and analyzed for its polymerase activity. Reactions were carried out at 37°C for 2 min at 20 μM dTTP concentration and quenched with Sanger's gel loading dye [35]. The reaction products were resolved by denaturing polyacrylamide-urea gel electrophoresis and analyzed on a PhosphorImager (Molecular Dynamics, Inc.).
DNA polymerase assay
Polymerase activity of the HIV-1RT WT and insertion mutant enzymes was determined using two different template-primers: U-5PBS HIV-1 RNA and synthetic 49-mer U5-PBS DNA templates primed with the 17-mer PBS primer [36]. Assays were carried out in a 50 μL volume containing 50 mM Tris HCl, pH 7.8, 100 μg/mL bovine serum albumin, 5 mM MgCl2, 1 mM dithiothreitol, 60 mM KCl, 100 nM template-primer, 50 μM of each of the four dNTPs with one of them being 32P-labeled (0.1 μCi/nmol dNTP) and 21 nM enzyme. Reactions were incubated at 37°C for 3 min and terminated by the addition of ice-cold 5% trichloroacetic acid containing 5 mM inorganic pyrophosphate. Following termination, the reaction mixtures were filtered on Whatman GF/B filters. The filters were then dried, immersed in scintillation fluid and counted in a liquid scintillation counter.
Gel analysis of RNA and DNA dependent polymerase activities
The U5-PBS HIV-1 RNA and heteropolymeric synthetic U5-PBS HIV-1 DNA templates primed with the 17-mer PBS DNA primer were used to assess the polymerase activities of the wild type and mutant heterodimeric enzymes. The primers were 5'-labeled using γ-32P-ATP and T4 polynucleotide kinase according to the standard protocol [37]. Polymerase reactions were carried out by incubating 2.5 nM template primer with 50 nM of the wild type HIV-1RT or its mutant derivative in a total reaction volume of 6 μL containing 25 mM Tris-HCl, pH 7.5, 10 mM dithiothreitol, 100 μg/mL bovine serum albumin, 5 mM MgCl2 and 50 μM of each dNTP. Reactions were initiated by the addition of enzyme and terminated by the addition of an equal volume (6 μL) of Sanger's gel loading dye [35]. The reaction products were resolved by denaturing poly acrylamide-urea gel electrophoresis and analyzed on a PhosphorImager (Molecular Dynamics, Inc.).
Template-Primer (TP) binding affinity of the wild type enzyme and its mutant derivatives
The dissociation constants (Kd) of the E-TP binary complexes of the wild type HIV-1 RT and its mutant derivatives were determined as described by Tong et al. [25]. The heteropolymeric 33-mer DNA (0.4 nM) annealed to 5'-32P-labeled 21-mer primer (0.3 nM) was incubated with varying concentrations of the wild type enzyme and its mutant derivatives in a total volume of 10 μL containing 50 mM Tris-HCl, pH 7.8, 5 mM MgCl2 and 0.01 % BSA. Following incubation of the mixture for 10 min at 4°C, equal volume of 2× gel-loading dye containing 0.25% bromophenol blue and 20% glycerol was added. The E-TP binary complexes formed were resolved at 4°C on 6% native polyacrylamide gel using Tris-Borate buffer (85 mM Tris, 85 mM Boric acid, pH 8.0). The amounts of the TP in the binary complex (E-TP) and in free form with respect to the varying concentrations of the enzyme protein were determined by PhosphorImager (Molecular Dynamics, Pharmacia) analysis of the gel. The fraction of the bound DNA was plotted against enzyme concentration and the Kd [DNA] value was determined as the RT concentration at which 50% of DNA is bound.
Ternary complex formation assay
The ternary complex (E-DNA-dNTP) formation was assessed by incubating the binary complexes of enzyme and dideoxy terminated template primer in the presence of next correct dNTP [25]. The binary complexes were formed by incubating 10–50 nM of the wild type enzyme or its mutant derivatives with 0.3 nM of 5'-32P-labeled dideoxy terminated 33-mer/21-mer template-primer as described above. The chosen concentration of enzyme was such that resulted in almost complete shift during E-TP complex formation. The E-TP-dNTP ternary complex formation was assessed by the addition of dNTP complementary to the next template base (in this case dGTP, 200 μM). Following incubation with dNTP at 4°C for 10 min, 300 fold molar excess of a DNA trap was added to the incubation mixture to assess the stability of the binary and ternary complexes formed by the enzyme. The complexes were resolved on a 6% native polyacrylamide followed by phosphorImaging. The extent of E-TP-dNTP ternary complexes formed was quantified using ImageQuant software.
RNase H activity assay
We used a 5'-32P labeled 30-mer synthetic U5-PBS RNA template annealed with a complementary 30-mer DNA to determine the RNase H activity of the enzymes [31]. The reaction mixture contained labeled RNA-DNA hybrid (10 K Cerenkov cpm), 50 mM Tris-HCl pH 8.0, 60 mM KCl, 10 mM dithiothreitol, 0.1 mg/ml bovine serum albumin, 5 mM MgCl2, and 20 ng of enzyme in a final volume of 5 μl. Reactions were carried out at 37°C for 30 sec and 1 min and terminated by the addition of equal volume of Sanger's gel loading dye [35]. The cleavage products were analyzed on an 8% denaturing polyacrylamide-urea gel and scanned on a phosphorImager (Molecular Dynamics Inc.).
Acknowledgments
Acknowledgements
This research was supported by a grant from the National Cancer Institute, NIH (CA72821).
Contributor Information
Pradeep K Pandey, Email: pandeypk@umdnj.edu.
Neerja Kaushik, Email: kaushik@umdnj.edu.
Kamalendra Singh, Email: ksingh@umdnj.edu.
Bechan Sharma, Email: sharmabe@umdnj.edu.
Alok K Upadhyay, Email: upadhyaak@umdnj.edu.
Suriender Kumar, Email: kumarsu@umdnj.edu.
Dylan Harris, Email: Dylan_Harris@millipore.com.
Virendra N Pandey, Email: pandey@umdnj.edu.
References
- Chandra A, Gerber T, Chandra P. Biochemical heterogeneity of reverse transcriptase purified from the AIDS virus, HTLV-III, FEBS Lett. 1986;197:84–88. doi: 10.1016/0014-5793(86)80303-9. [DOI] [PubMed] [Google Scholar]
- Muller B, Restle T, Weiss S, Gautel M, Sczakiel G, Goody R. Co-expression of the subunits of the heterodimer of HIV-1 reverse transcriptase in Escherichia coli. J Biol Chem. 1989;264:13975–13978. [PubMed] [Google Scholar]
- Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumor Viruses. Nature. 1970;226:1209–1211. doi: 10.1038/2261209a0. [DOI] [PubMed] [Google Scholar]
- Temin HM, Mizytani S. RNA-directed DNA polymerase in virions of Rouse sarcoma virus. Nature. 1970;226:2111–1213. doi: 10.1038/2261211a0. [DOI] [PubMed] [Google Scholar]
- Farmerie WG, Leob D, Casavant NC, Hutchinson CAd, Egell MH, Swanstrom R. Expression and processing of AIDS virus reverse transcriptase in E. coli. Science. 1987;236:305–308. doi: 10.1126/science.2436298. [DOI] [PubMed] [Google Scholar]
- Mizrahi V, Lazarus GM, Miles LM, Meyers CA, Debouck C. Recombinant HIV-1 reverse transcriptase: purification, primary structure, and polymerase/ribonuclease H activities. Arch Biochem Biophys. 1989;273:347–358. doi: 10.1016/0003-9861(89)90493-1. [DOI] [PubMed] [Google Scholar]
- Di Marzo Veronese FD, Copeland TD, DeVico AL, Rahman R, Oroszlan S, Gallo RC, Sarngadharan MG. Characterization of Highly Immunogenic p66/p51 as the Reverse Transcriptase of HTLV-III/LAV. Science. 1986;231:1289–1291. doi: 10.1126/science.2418504. [DOI] [PubMed] [Google Scholar]
- Le Grice SFJ, Nass T, Wohlgensinger B, Schatz O. Subunit-selective mutagenesis indicates minimal polymerase activity in heterodimer-associated p51 HIV-1 reverse transcriptase. EMBO J. 1991;10:3905–3911. doi: 10.1002/j.1460-2075.1991.tb04960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restle T, Muller B, Goody RS. RNase H activity of HIV reverse transcriptases is confined exclusively to the dimeric forms. FEBS Lett. 1992;300:97–100. doi: 10.1016/0014-5793(92)80172-D. [DOI] [PubMed] [Google Scholar]
- Restle T, Muller B, Goody RS. Dimerization of human immunodeficiency virus type 1 reverse transcriptase: A target for chemotherapeutic intervention. J Biol Chem. 1990;265:8986–8988. [PubMed] [Google Scholar]
- Kohlstaedt LA, Wang J, Eriedman JM, Rice PA, Steitz TA. Crystal structural at 3.5 A resolution of HIV-1 RT complexed with an inhibitor, Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- Wang J, Smerdon SJ, Jager J, Kohlstaedt LA, Rice PA, Friedman JM. Structural Basis of Asymmetry in the Human Immunodeficiency Virus Type 1 Reverse Transcriptase Heterodimer, Proc Natl Acad Sci USA. 1994;91:7242–7246. doi: 10.1073/pnas.91.15.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni RG, Ding J, Jacobo-Molina A, Hughes SH, Arnold E. Review of HIV-I reverse transcriptase 3-dimentional structure: implication for drug design Perspect. Drug Discovery and Des. 1993;1:129–150. [Google Scholar]
- Harris D, Lee R, Misra HS, Pandey PK, Pandey VN. The p51 subunit of Human Immunodeficiency Virus Type 1 Reverse Transcriptase is essential in loading the p66 Subunit on the Template Primer. Biochemistry. 1998;37:5903–5908. doi: 10.1021/bi9728452. [DOI] [PubMed] [Google Scholar]
- Pandey PK, Kaushik N, Talele TT, Yadav PNS, Pandey VN. The β7-β8 loop of the p51 Subunit in the Heterodimeric (p66/p51) Human Immunodeficiency Virus Type 1 Reverse Transcriptase is Essential for the Catalytic Function of the p66 subunit. Biochemistry. 2001;40:9505–9512. doi: 10.1021/bi002872j. [DOI] [PubMed] [Google Scholar]
- Hostomsky Z, Hostomska Z, Fu TB, Taylor J. Reverse transcriptase of human immunodeficiency virus type 1: functionality of subunits of the heterodimer in DNA synthesis. J Virol. 1992;66:3179–3182. doi: 10.1128/jvi.66.5.3179-3182.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeGrice SFJ, Nass T, Wohlgensinger N, Schatz O. Subunit-selective mutagenesis indicates minimal polymerase activity in heterodimer-associated p51 HIV-1 reverse transcriptase. EMBO J. 1991;10:3905–3911. doi: 10.1002/j.1460-2075.1991.tb04960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amacker M, Hubscher U. Chimeric HIV-1 and Feline Immunodeficiency Virus Reverse Transcriptases: Critical Role of the p51 Subunit in the Structural Integrity of Heterodimeric Lentiviral DNA Polymerases. J Mol Biol. 1998;278:757–765. doi: 10.1006/jmbi.1998.1739. [DOI] [PubMed] [Google Scholar]
- Amacker M, Hottiger M, Hubscher U. Feline immunodeficiency virus reverse transcriptase: expression, functional characterization, and reconstitution of the 66- and 51-kilodalton subunits. J Virol. 1995;69:6273–6279. doi: 10.1128/jvi.69.10.6273-6279.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M, Podusr VN, Thimmig RL, McHenry C, Hubscher U. Strand displacement activity of the human immunodeficiency virus type 1 reverse transcriptase heterodimer and its individual subunits. J Biol Chem. 1994;269:986–991. [PubMed] [Google Scholar]
- Haung SC, Smith JR, Moen LK. Contribution of the p51 subunit of HIV-1 reverse transcriptase to enzyme processivity. Biochem Biophys Res Commun. 1992;184:986–992. doi: 10.1016/0006-291x(92)90688-h. [DOI] [PubMed] [Google Scholar]
- Loya S, Gao HQ, Avidan O, Boyer PL, Hughes SH, Hizi A. Subunit-specific mutagenesis of the cysteine 280 residue of the reverse transcriptase of human immunodeficiency virus type 1: effects on sensitivity to a specific inhibitor of the RNase H activity. J Virol. 1997;71:5668–5672. doi: 10.1128/jvi.71.7.5668-5672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere H, Taymans JM, Balzarini J, Velazquez S, Camarasa MJ, Desmyter J, De Clercq E, Anne J. Resistance of HIV-1 Reverse Transcriptase against [2', 5'-bis-O-(tert-butyldimethylsilyl)-3'spiro-5"-(4"-amino-1",2"-oxathiole-2",2"-dioxide)] (TSAO) Derivatives Is Determined by the Mutation Glu 138→Lys on the p51 Subunit. J Biol Chem. 1994;269:25255–25258. [PubMed] [Google Scholar]
- Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a Covalently trapped catalytic complex of HIV-1RT: Implication for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- Tong WW, Lu C, Sharma SK, Matsuura S, So AG, Scott WA. Nucleotide-induced stable complex formation by HIV-1 reverse transcriptase. Biochemistry. 1997;36:5749–5757. doi: 10.1021/bi962410z. [DOI] [PubMed] [Google Scholar]
- Smerdon SJ, Jager J, Wang J, Kohlstaedt LA, Chirino AJ, Friedman JM, Rice PA, Steitz TA. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1994;91:3911–3915. doi: 10.1073/pnas.91.9.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel R, Beard WA, Kumar A, Casas-Finet JR, Strub M, Stahl SJ, Lewis MS, Bebenek K, Becerra P, Kunkel TA, Wilson SH. Structure/function studies of HIV-1(1) reverse transcriptase: dimerization-defective mutant L289K. Biochemistry. 1993;32:13012–13018. doi: 10.1021/bi00211a009. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Jacques PS, Rodgers DW, Ottman M, Darlix JL, LeGrice SF. Alterations to the primer grip of p66 HIV-1 reverse transcriptase and their consequences for template-primer utilization. Biochemistry. 1996;35:8553–8562. doi: 10.1021/bi952773j. [DOI] [PubMed] [Google Scholar]
- Pandey VN, Kaushik N, Rege N, Sarafianos SG, Yadav PNS, Modak MJ. Role of Methionine 184 of Human Immunodeficiency Virus Type-Reverse Transcriptase in the Polymerase function and fidelity of DNA Synthesis. Biochemistry. 1996;35:2168–2179. doi: 10.1021/bi9516642. [DOI] [PubMed] [Google Scholar]
- Kaushik N, Harris D, Rege N, Modak MJ, Yadav PNS, Pandey VN. Role of Glutamine-151 of Human Immunodeficiency virus Type-1 Reverse Transcriptase in RNA-Directed DNA Synthesis. Biochemistry. 1997;36:14430–14438. doi: 10.1021/bi970645k. [DOI] [PubMed] [Google Scholar]
- Lee R, Kaushik N, Modak MJ, Vinayak R, Pandey VN. Polyamide Nucleic Acid Targeted to the Primer Binding Site of the HIV-1 RNA Genome Blocks in Vitro HIV-1 Reverse Transcription. Biochemistry. 1998;37:900–910. doi: 10.1021/bi972197m. [DOI] [PubMed] [Google Scholar]
- Arts EJ, Li X, Gu Z, Kleiman L, Parniak M, Wainberg MA. Comparison of Deoxyoligonucleotide and t-RNAlys-3 as Primers in an Endogenous Human Immunodeficiency Virus-1 in vitro Reverse transcription/Template-switching reaction. J Biol Chem. 1994;269:14672–14680. [PubMed] [Google Scholar]
- Kaushik N, Rege N, Sarafianos SG, Yadav PNS, Modak MJ, Pandey VN. Biochemical Analysis of Catalytically Crucial Aspartate Mutants of Human Immunodeficiency Virus Type 1 Reverse Transcriptase. Biochemistry. 1996;35:11536–11546. doi: 10.1021/bi960364x. [DOI] [PubMed] [Google Scholar]
- Hsieh JC, Zinnen S, Modrich P. Kinetic Mechanism of the DNA-dependent DNA polymerase Activity of human Immunodeficiency Virus-1 Reverse Transcriptase. J Biol Chem. 1993;268:24607–24613. [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D, Kaushik N, Pandey PK, Yadav PNS, Pandey VN. Functional Analysis of Amino Acid Residues Constituting the dNTP Binding Pocket of HIV-1 Reverse Transcriptase. J Biol Chem. 1998;273:33624–33634. doi: 10.1074/jbc.273.50.33624. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JS, Smith JA, Struhl K. Current protocols in molecular Biology. Greene publishing Associates and Wiley-Intersciences, John Wiley & Sons, New York. 1987.