

Abstract
Sarcoidosis is a multisystemic syndrome characterized by non-caseous granulomatous inflammation, although necrotizing sarcoid granulomatosis is considered part of the spectrum of the disease. Drug induced sarcoidosis-like reaction (DISR) is a systemic granulomatous reaction, which is histopathologically identical to primary sarcoidosis - mostly described after the use of biologics like tumour necrosis factor alpha antagonists but also anti-CD20 (rituximab). The authors present the very rare case of a woman with a primary Sjögren’s syndrome (pSS) started on rituximab for disease control, which evolved with a 3-year indolent progressive systemic sarcoid reaction. There has been much speculation about the potential role of B cells in sarcoidosis. Findings show a decrease of B memory cells and an increase in naïve and active subsets of regulatory B cells in sarcoidosis patients, which resembles the repopulation with naïve B cells after treatment with rituximab. Moreover, granulomatous lymphocytic interstitial lung disease associated with common variable immunodeficiency and immune reconstitution syndrome in patients wirh human immunodeficiency virus show clinical similarities to DISR and can help unveil new cytogenic and physiologic pathways. To the authors’ knowledge this is the first report of a systemic sarcoidosis-like reaction with necrotizing granulomas following an anti-CD20 therapy and also the first described in a pSS patient - underlining the importance of recognizing necrotizing sarcoid granulomatous processes in the diferential diagnosis of patients with caseous inflammation. Although this is a very rare adverse effect, the case enhances the importance of actively searching for DISR after biologics, even in patients undergoing rescue on-label therapies, such as rituximab.
LEARNING POINTS
First report of a systemic sarcoidosis-like reaction with necrotizing granulomas following an anti-CD20 therapy, in a patient with primary Sjögren’s syndrome.
Recognizing immunotherapy and biological therapies as the possible causative agents of rare and underrecognized adverse effects in patients with rare diseases in the era of biologics.
Recognizing necrotizing sarcoid granulomas in the diferential diagnosis of patients with caseous inflammation.
Keywords: Secondary sarcoidosis, drug induced sarcoid reaction, rituximab, anti-CD20, primary Sjögren’s syndrome, necrotizing sarcoid granulomatous
INTRODUCTION
Sarcoidosis is a multisystemic syndrome characterized by non-caseous granulomatous inflammation. Although caseating granulomas are more commonly associated with infectious diseases, necrotizing sarcoid granulomatosis is a rare presentation considered as part of the spectrum of sarcoidosis[1]. Recently, another sarcoid presentation has been described: the drug induced sarcoidosis-like reaction (DISR), a systemic granulomatous reaction, which is histopathologically identical to primary sarcoidosis. It usually presents with indolent and unspecific symptoms and is most commonly described after therapies with like tumour necrosis factor alpha (TNF-alpha) antagonists, interferons, checkpoint inhibitors. Rituximab, an anti-CD20 IgG monoclonal antibody, has been used in small cohorts of refractory sarcoidosis patients with mixed results, however, rarely, DISR have also been reported after this treatment. Although T cells, especially CD4 cells, have been repeatedly reported as one of the main culprits in sarcoidosis, there is a possible role for the interplay between B and T cells[2]. Primary Sjögren’s syndrome (pSS) is also a multisystemic autoimmune disease, characterized by lymphocytic glandular involvement with fibrotic progression. Extraglandular involvement is common and can be severe on presentation, which determines the treatment approach. Rituximab has shown promising outcomes in severe disease. We present the very rare case of a woman with pSS with severe kidney involvement, started on rituximab, followed by a progressive systemic sarcoid reaction.
CASE DESCRIPTION
We present the case of a 65- year-old woman followed in the Internal Medicine outpatient clinic since 2019 for pSS, who developed new progressive lung opacities first reported on June 2021 (Fig. 1).
Figure 1.

Chronogram of disease progression concerning the onset of new symptoms, invasive diagnostic procedures, treatment, imaging and function assessment.
Abbreviations: HR, high resolution; CT, computed tomography; EBUS-TBNA, endobronchial ultrasoundnography-guided transbronchial needle aspiration.
Since 1998 the patient has sicca syndrome. pSS was diagnosed in 2003 through acute interstitial nephropathy with positivity for anti-Sjögren’s syndrome A (SSA), anti-Sjögren’s syndrome B (SSB) antibodies, hypergammaglobulinemia, and an antinuclear antibody (ANA) level of 1/640. No treatment was initiated at the time of diagnosis. In 2020, the patient developed end stage kidney diseaseand was therefore started on peritoneal dialysis. The patient also developed incipient mild interstitial lung changes, first documented in August 2020, with reticulation of the basal segments of both lower lobes and pSS-interstitial lung disease (ILD) was suspected. When the patient was found to have end stage kidney disease, after multidisciplinary discussion, a double cycle of rituximab was started in August 2020. There was clinical improvement but there was no change in kidney function.
In June 2021, re-evaluation with chest computed tomography (CT) scan showed no regression of the previous ILD findings plus new increased aorto-pulmonary lymph nodes, small subsolid nodules in the posterior left upper lobe and a few diffuse subpleural micronodules (Fig. 2), with no respiratory symptoms or abnormal lung function tests (Table 1). A year after treatment the patient’s had worsening symptoms along with a systemic inflammatory response. A pSS flare with progressive ILD was assumed and she underwent a repeat double cycle of rituximab in January 2022 with symptomatic improvement, however without total resolution of arthralgia. A CT scan of the chest done in February documented enlargement of the previous left upper lobe nodule and a new subsolid nodule on right upper lobe raising concern about lung neoplasms and warranting prompt short-term reassessment. About 9 months after rituximab, clinical assessment reported new-onset fatigue, a non-tender solid nodule in patient’s right leg and analytic inflammatory response. A confront CT scan of the chest showed new nodules and increasing dimensions of the previously known nodules (Fig. 2), along with functional impairment (Table 1). Positron emission tomography (PET) CT scan documented hypermetabolic active lesions in subfissural and subpleural known nodules. A transthoracic biopsy of the right lung nodule (Fig. 3) ensued and histopathological assessment revealed a lymphohistiocytic infiltrate, ruling out neoplastic disease or amyloidosis. After rediscussion, all symptoms were assumed to be related to pSS and the patient underwent a repeat cycle of rituximab in February 2023, this time with no symptomatic improvement.
Figure 2.
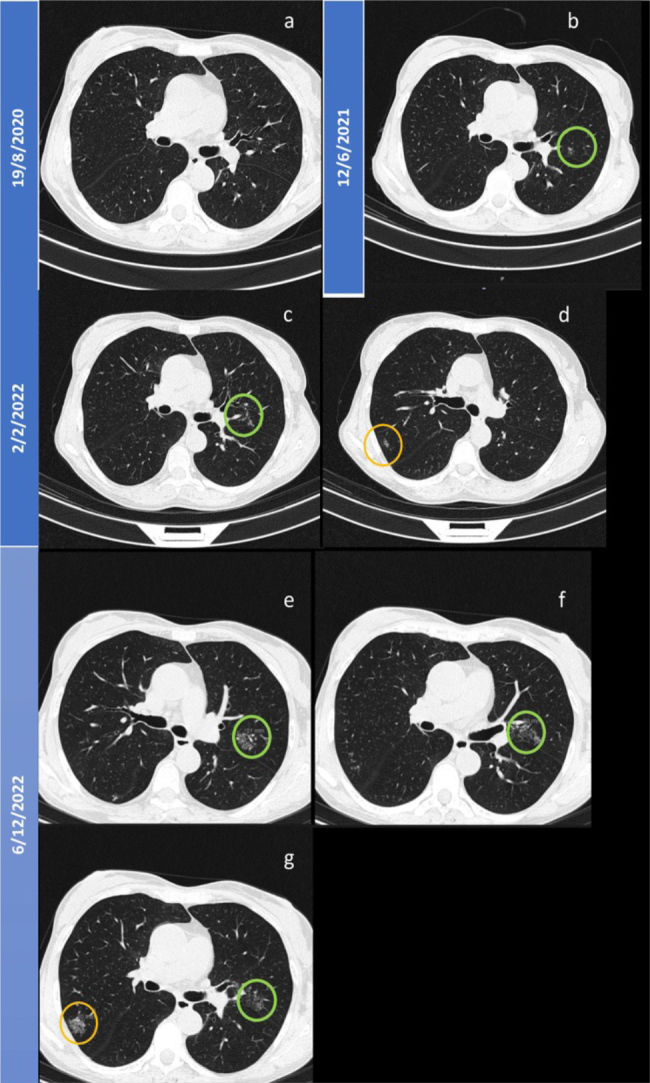
Evolution of lung micronodules until December 2022. A) Chest CT before rituximab; B) Green circle showing the first documented subfissural micronodule in the left upper lung (LUL); C, D) Small increase in dimension of LUL micronodules and new onset (orange circle) of peripheral subsolid nodules in the right upper lung (RUL); E, F and G) Progression of the known nodules in RUL and LUL.
Table 1.
Evolution of lung function tests.
| LFT measures | June 2021 | September 2023 | August 2024 | |||
|---|---|---|---|---|---|---|
| Absolute value | % value | Absolute value | % value | Absolute value | % value | |
| FVC | 2.9 | 111.7 | 2.3 | 90.9 (−20.8) | 2.23 | 89 (−0.2) |
| FEV1 | 2.14 | 105.2 | 1.7 | 86.1 | 1.73 | 88.6 |
| FEV1/FVC | 73.93 | 93.7 | 73.99 | 94.1 | 77.76 | 99 |
| DLCONA | 1.04 | 72.5 | 0.82 | 57 (−15.5) | 0.91 | 63.9 (+6.9) |
Abbreviations: LFT, lung function test; FEV, forced expiratory volume in the first minute; FVC, forced vital capacity; DLCO, diffusing capacity of the lungs for carbon monoxide; VA, alveolar volume.
Figure 3.
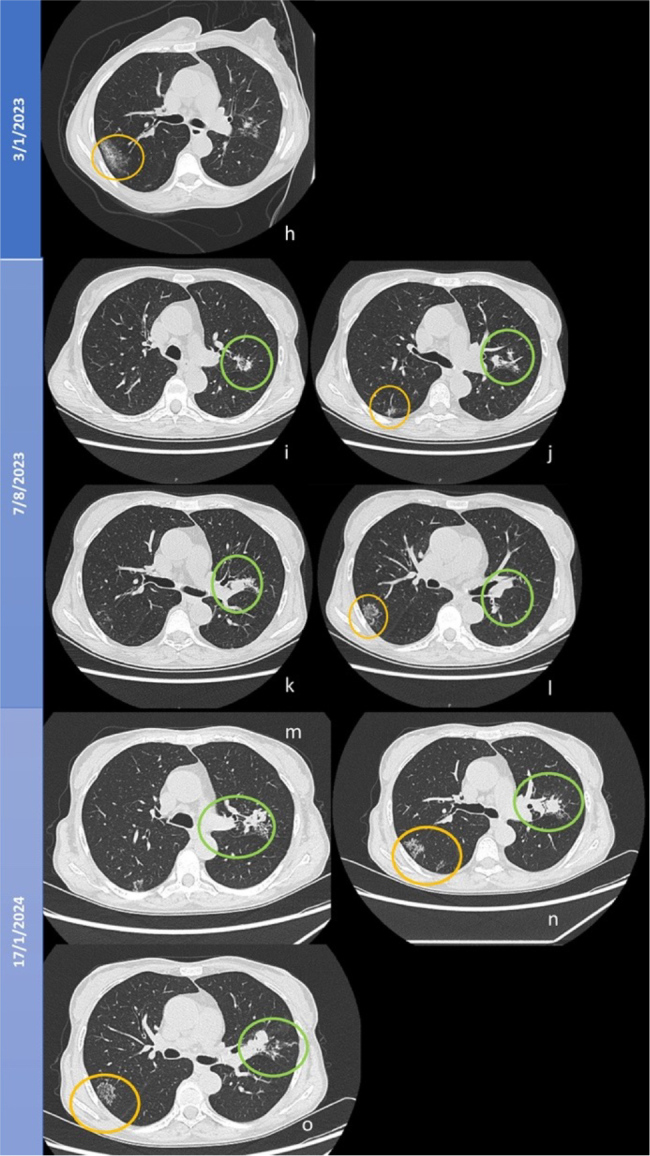
Evolution of lung micronodules until January 2024. H) Chest computed tomography scan after right upper lobe (RUL) nodule (orange circle) lung biopsy with subsequent haemorrhagic lesion; I, J, K and L) Confluence of nodules forming the galaxy sign in the left upper lobe (green circle); J) New nodule in RUL; L) regrouping of RUL nodules after biopsy resembling the galaxy sign (orange circle); M, N and O) Galaxy sign.
In May, the patient presented with non-occlusive right parotiditis with submandibular gland extension, that resolved with 1 week of anti-inflammatory therapy. In August, she presented with non-occlusive left parotitis, with a large cystic nodule and diffuse heterogenicity of both glands. A biopsy was performed, and cytometry and histopathological assessment excluded lymphoproliferative disease and revealed a necrotizing granuloma (Fig. 4). The patient started empiric antibiotics, however, when there was no improvement and necrotizing agents, including mycobacteria, were ruled out, she had to undergo maxillofacial surgery. Histolopathological assessment of the removed parotid glands and lymph nodes reaffirmed a process of necrotizing granulomatous inflammation with no signs of malignancy and negative for infectious agents (Fig. 4). As the systemic disease progressed a new CT scan of the chest (Fig. 3) and PET-scan were performed revealing the increasing number, size and metabolic activity of known nodules and new hypermetabolic activity of bilateral mediastinal hilar adenopathy. An endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) with bronchioalveolar lavage (BAL) was performed and both revealed no signs of tuberculosis, granulomas or malignancy.
Figure 4.
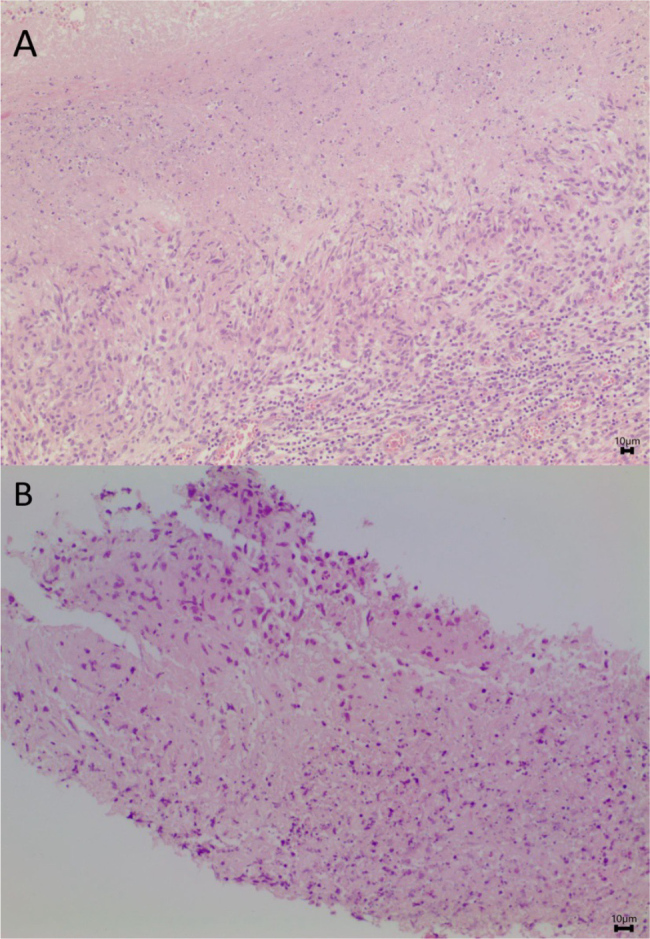
Necrotizing granulomas formed by histiocytes. A) Adenopathy (H&E 100x); B) Parotid gland (H&E 200x).
The case was discussed in the pulmonary multidisciplinary board meeting and a diagnosis of DISR following rituximab was made. Rituximab was therefore stopped and the patient was started on 0.5 mg/kg/day of prednisolone in July. Imaging and functional improvement was observed after 1 month of therapy, and after 4 months PET scan showed no hypermetabolic activity. Mycophenolate mofetil was proposed as an off-label alternative corticosteroid sparing agent.
DISCUSSION
This case describes a female patient with long-term pSS who was started on rituximab therapy, due to rapid kidney deterioration. The patient developed progressing non-responding lung micronodules and she was ultimately treated with repeat rituximab cycles over a period of 3 years, since the clinical picture was pSS. A retrospective analysis of the chest CT scan findings, shows the typical lymphangitic distributionin the upper lobes seen in sarcoidosis and it is possible to recognize a slow formation of the specific galaxy sign, non-existant in SS-ILD. Lung biopsy also revealed a lymphohistiocytic infiltrate, a rare histologic finding, that is not typical of SS and might represent an early stage of granuloma formation[3]. The patient presented throughout with non-tender leg nodules, resembling erythema nodosum, a rare manifestation in SS, but very common in Löfgren syndrome. Also, she had asynchronous bilateral parotid gland swealing, which is typical for both diseases, however the parotid tissue revealed a sterile necrotizing granulomatous inflammation, which is atypical of SS but, despite being rare, has been documented in some cases of sarcoidosis[1].
After ruling out malignancy and infectious diseases, the findings along with the chronological link to the rituximab cycles, which are in agreement with the time-to-symptoms reported in the few known rituximab DISR cases[4,5], strongly point to the diagnosis of rituximab induced sarcoidosis-like reaction. Moreover, the calculated Naranjo adverse drug reaction probability scale score was 9 points[6]. Additionally, DISR usually present a very goodresponse to corticosteroids and, in some cases, the discontinuation of the offending agent is reported as sufficient. In this case, since the patient presented with a systemic progressing disease with functional impairment the authors chose to start corticosteroids. However, after 1 month of high dose prednisone the patient developed important adverse effects related to corticotherapy, which necessitated faster down-titration. Despite that, PET scan still showed no signs of active disease, supporting the aforementioned diagnosis.
The role of B cells in the pathophysiology of sarcoidosis has been investigated. Recent findings report a decrease in B-memory cells and increased naïve and active subsets of regulatory B cells and B cell activating factor (BAFF) producers, in sarcoidosis patients. It is known that rituximab causes complete depletion of B cells approximately 21 days after treatment, and repopulating naïve B cells rise approximately 6 to 9 months later[2,5]. Therefore, a comparison between post-rituximab repopulation and cell populations found in sarcoidosis patients can be made. Findings in other granulomatous diseases, such as the granulomatous lymphocytic ILD (GLILD) associated to common variable immunodeficiency (CVID) suggest that GLILD also depends on the interaction between B and T cells in which B naïve cells invade B tolerance checkpoints causing dysregulation and increasing autoreactive B cells. Also BAFF is overproduced as a negative response to typically high IFN-gama in CVID[7]. The authors hypothesize that in some patients, B cell depletion and repopulation following anti-CD20 therapy can dysregulate the tolerance process of B-cells and trigger a granulomatous process. Moreover, sarcoidosis after highly active anti-retroviral therapy (HAART) in human immunodeficiency virus patients is described as an immune reconstitution syndrome following the rapid rise of CD4+ T cells[8]. The authors also hypothesize that this case can represent a form of latent sarcoidosis, which could have been triggered by the depletion of B-cells and disturbance of the interplay between B and T cells - resembling the immune reconstitution syndrome. Although there are 32 cases of rituximab DISR reported in the World Heath Organisation database[9], only a few cases have been published, especially cases focused on single organ sarcoid-reaction or Löfgren syndrome[10]. To the best of the authors’ knowledge this is the first report of a systemic sarcoidosis-like reaction with necrotizing granulomas following anti-CD20 therapy and also the first case described in pSS. This case also had an extraordinarily indolent presentation, especially concerning the late appearance of typical bilateral mediastinal hilar adenopathy. Currently it is not yet possible to fully understand the etiology of rituximab DISR. Moreover, the authors want to underline the importance of multidisciplinary board discussions to mitigate erroneous assumptions and also reaffirm the implication of recognizing necrotizing sarcoid granulomatous reactions as a diagnostic hypothesis in patients with caseous inflammation. Although this is a very rare adverse event, this case enhances the importance of actively searching for DISR after biologics.
Footnotes
Conflicts of Interests: The Authors declare that there are no competing interests.
Patient Consent: The patient consented the use of the information used in this publication.
REFERENCES
- 1.Makimoto G, Kawakado K, Nakanishi M, Tamura T, Sato Y, Kuyama S. Successful corticosteroid treatment of necrotizing sarcoid granulomatosis associated with tracheal lesion recurred after a surgical lung biopsy. Respir Med Case Rep. 2021;33:101402. doi: 10.1016/j.rmcr.2021.101402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miedema J, Cinetto F, Smed-Sörensen A, Spagnolo P. The immunopathogenesis of sarcoidosis. J Autoimmun. 2024;149:103247. doi: 10.1016/j.jaut.2024.103247. [DOI] [PubMed] [Google Scholar]
- 3.Yousem SA, Dacic S. Pulmonary lymphohistiocytic reactions temporally related to etanercept therapy. Mod Pathol. 2005;18:651–655. doi: 10.1038/modpathol.3800333. [DOI] [PubMed] [Google Scholar]
- 4.Mrabet S, Dahmene R, Fradi A, Jaziri A, Boukadida R, Azzebi A, et al. Sarcoid-Like Reaction in the Kidney Following Rituximab for Mantle Lymphoma in a 60-Year-Old Man. Am J Mens Health. 2023;17:15579883231159343. doi: 10.1177/15579883231159343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galimberti F, Fernandez AP. Sarcoidosis following successful treatment of pemphigus vulgaris with rituximab: a rituximab-induced reaction further supporting B-cell contribution to sarcoidosis pathogenesis? Clin Exp Dermatol. 2016;41:413–416. doi: 10.1111/ced.12793. [DOI] [PubMed] [Google Scholar]
- 6.Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239–245. doi: 10.1038/clpt.1981.154. [DOI] [PubMed] [Google Scholar]
- 7.Galant-Swafford J, Catanzaro J, Achcar RD, Cool C, Koelsch T, Bang TJ, et al. Approach to diagnosing and managing granulomatous-lymphocytic interstitial lung disease. EClinicalMedicine. 2024;75:102749. doi: 10.1016/j.eclinm.2024.102749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grunewald J, Grutters JC, Arkema EV, Saketkoo LA, Moller DR, Müller-Quernheim J. Sarcoidosis. Nat Rev Dis Primers. 2019;5:45. doi: 10.1038/s41572-019-0096-x. [DOI] [PubMed] [Google Scholar]
- 9.Cohen Aubart F, Lhote R, Amoura A, Valeyre D, Haroche J, Amoura Z, et al. Drug-induced sarcoidosis: an overview of the WHO pharmacovigilance database. J Intern Med. 2020;288:356–362. doi: 10.1111/joim.12991. [DOI] [PubMed] [Google Scholar]
- 10.Fakih O, Verhoeven F, Prati C, Wendling D. Paradoxical Löfgren’s syndrome in a patient treated with rituximab: interferon is not the key. Rheumatology (Oxford) 2020;59:1181–1182. doi: 10.1093/rheumatology/kez463. [DOI] [PubMed] [Google Scholar]