

Abstract
Tumour content plays a pivotal role in directing the bioinformatic analysis of molecular profiles such as copy number variation (CNV). In clinical application, tumour purity estimation (TPE) is achieved either through visual pathological review [conventional pathology (CP)] or the deconvolution of molecular data. While CP provides a direct measurement, it demonstrates modest reproducibility and lacks standardisation. Conversely, deconvolution methods offer an indirect assessment with uncertain accuracy, underscoring the necessity for innovative approaches. SoftCTM is an open‐source, multiorgan deep‐learning (DL) model for the detection of tumour and non‐tumour cells in H&E‐stained slides, developed within the Overlapped Cell on Tissue Dataset for Histopathology (OCELOT) Challenge 2023. Here, using three large multicentre colorectal cancer (CRC) cohorts (N = 1,097 patients) with digital pathology and multi‐omic data, we compare the utility and accuracy of TPE with SoftCTM versus CP and bioinformatic deconvolution methods (RNA expression, DNA methylation) for downstream molecular analysis, including CNV profiling. SoftCTM showed technical repeatability when applied twice on the same slide (r = 1.0) and excellent correlations in paired H&E slides (r > 0.9). TPEs profiled by SoftCTM correlated highly with RNA expression (r = 0.59) and DNA methylation (r = 0.40), while TPEs by CP showed a lower correlation with RNA expression (r = 0.41) and DNA methylation (r = 0.29). We show that CP and deconvolution methods respectively underestimate and overestimate tumour content compared to SoftCTM, resulting in 6–13% differing CNV calls. In summary, TPE with SoftCTM enables reproducibility, automation, and standardisation at single‐cell resolution. SoftCTM estimates (M = 58.9%, SD ±16.3%) reconcile the overestimation by molecular data extrapolation (RNA expression: M = 79.2%, SD ±10.5, DNA methylation: M = 62.7%, SD ±11.8%) and underestimation by CP (M = 35.9%, SD ±13.1%), providing a more reliable middle ground. A fully integrated computational pathology solution could therefore be used to improve downstream molecular analyses for research and clinics. © 2024 The Author(s). The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: pathology, artificial intelligence, colorectal cancer, diagnostic molecular pathology, personalised medicine
Introduction
The assessment of the cancer microenvironment plays a pivotal role in informing the interpretation of transcriptional signatures and copy number variation (CNV) calls within molecular pathology workflows [1, 2]. Semi‐quantitative visual evaluations of tumour purity (TP) are commonly employed to gauge sample adequacy prior to omic profiling. However, these estimations suffer from a lack of standardisation and exhibit poor reproducibility, potentially introducing bias into genomic analyses [3, 4]. Bioinformatic deconvolution techniques for estimating tumour content from genomic data offer a potential solution but are costly to implement, lack spatial preservation, and demonstrate relevant failure rates [5, 6, 7]. In contrast, computational pathology (CPATH) has emerged as a robust and non‐destructive approach to identifying and segmenting neoplastic and non‐neoplastic cell populations within clinical pathology samples, seamlessly integrating into existing laboratory workflows. In this study, we explored the potential of CPATH to enhance diagnostic molecular pathology, comparing it directly with expert pathologist assessment and established deconvolution methods using transcriptional and DNA methylation data. The findings of this investigation carry significant implications for selecting the most suitable method for both clinical and research purposes. Given the importance of TP estimations (TPEs) in interpreting CNV calls and next‐generation sequencing (NGS) readouts in molecular pathology, we postulate that a more precise approach to cell deconvolution could greatly benefit colorectal cancer (CRC) molecular pathology workflows and cancer genomics in general.
DNA aneuploidy has been associated with a shorter disease‐free and overall survival in CRC patients [8, 9]. NGS data can be utilised to derive information on copy number variations following TPE from deconvolution methods. However, the accuracy of this approach remains uncertain due to the lack of correlation with conventional pathology (CP) TPEs [4, 10]. Conventional methods to measure CNVs involve tissue dissociation and assessment of DNA content using flow cytometry or microscopic imaging [9]. Since copy number alterations are diluted by an increase in normal diploid cells, accurately determining TP is crucial for bioinformatic correction of CNV calls. By extracting precise measures of TP, CPATH has the potential to improve the identification of CRC cases harbouring aneuploid neoplastic populations.
Here we perform a comprehensive analysis of TP in three cohorts with a total of 1,097 CRC patients from the Stratification in COloRecTal cancer (S:CORT) programme and The Cancer Genome Atlas (TCGA) with full molecular information and available whole slide images (WSIs). We compare the impact and utility of (1) a gold‐standard molecular deconvolution method (ESTIMATE) with (2) CP, (3) a DNA‐methylation‐based deconvolution method (InfiniumPurify), and (4) a novel open‐source, multiorgan CPATH algorithm (SoftCTM) on TPEs, including downstream CNV analysis and biological stratification.
Our systematic analysis reveals that SoftCTM‐based TPE surpasses the accuracy of CP, ESTIMATE, and InfiniumPurify. These currently established methods tend to respectively underestimate and overestimate TP. TPE using SoftCTM offers analytical robustness, automation, and standardisation, resulting in remarkably high reproducibility at the single‐cell level. Leveraging CPATH approaches could therefore enhance the planning and evaluation of subsequent molecular analyses.
Materials and methods
Cohorts
This study included a total of 1,097 CRC cases from three independent cohorts (FOCUS, GRAMPIAN, TCGA) with complete digital pathology (DP) and multi‐omic datasets. Cases from FOCUS and GRAMPIAN were characterised as part of the Medical Research Council (MRC) Cancer Research UK (CRUK) S:CORT programme, the S:CORT case IDs considered in this study are detailed in supplementary material, Table S1. Figure 1A provides an overview of the three datasets' specifications, Figure 1B indicates the available data types and applied TPE methods, and Figure 1C shows their sample collection strategy. Further details on data and resource availability can be found in supplementary material, Figure S1.
Figure 1.
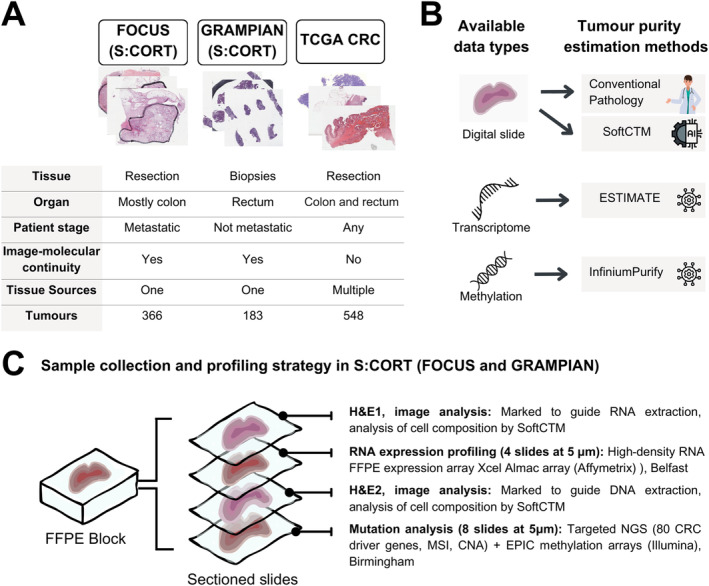
Experimental study design. (A) Specifications and data summary of the three independent datasets (FOCUS, TCGA, and GRAMPIAN) used in this study. (B) Available data types and tumour estimation methods applied on each data type. (C) Sample collection and profiling strategy in FOCUS and GRAMPIAN cohorts. Created with canva.com.
Cohort 1: FOCUS
As part of the Stratification in the S:CORT programme, 385 patients with available formalin‐fixed paraffin embedded (FFPE) blocks of the primary CRC were selected from the MRC FOCUS randomised clinical trial (RCT) that tested different strategies of sequential and combination chemotherapy for patients with advanced CRC [11]. Serial sections were cut from one representative block for H&E staining followed by four unstained sections for RNA extraction, a second H&E‐stained section, and eight unstained sections for DNA extraction (Figure 1). Glass H&E slides were re‐reviewed by an expert gastrointestinal pathologist and tumour tissue with the associated intra‐tumoural stroma was annotated in the first and second H&E‐stained section respectively to guide RNA and DNA extractions. No tumour microdissection was performed. Regions of extensive necrosis and non‐tumour tissue were excluded according to standard practice for downstream molecular tumour profiling. RNA expression microarrays (Xcel array, Affymetrix, Santa Clara, CA, USA), DNA target capture (SureSelect, Agilent, Santa Clara, CA, USA) followed by NGS sequencing (Illumina, San Diego, CA, USA), and DNA methylation arrays (EPIC arrays, Illumina) were applied in that order [12]. All H&E slides were scanned on an Aperio scanner at 20×. Digital slides were re‐reviewed by a second gastrointestinal pathologist and tumour region annotations for deep‐learning (DL) classification were generated. Areas containing folds or debris were excluded by digital annotation. Samples were excluded if they contained biopsies instead of resections and/or metastatic tissue instead of primary lesions or extensive ulceration/necrosis. Slides with irrecoverable failure of the staining or scanning procedure were excluded for technical reasons. The final set was composed of 702 primary tumour H&E slides from 366 cases.
Cohort 2: TCGA
A total of 624 digital slides from 615 cases of colon and rectal adenocarcinoma were downloaded from the TCGA Data Portal (COAD and READ datasets, https://www.cancer.gov/tcga, last accessed: 2 August 2018). All digital slides were re‐reviewed, and tumour tissue was annotated. Slides were excluded based on the same quality control (QC) criteria as described for FOCUS. Gene‐level expression data were downloaded with the R package TCGAbiolinks [13]. After excluding slides based on QC and six duplicated tumours, the final number of slides was 548, all from unique cases.
Cohort 3: GRAMPIAN
A total of 334 slides from 184 pretreatment biopsy FFPE blocks from rectal cancer patients were analysed for this study as part of the S:CORT programme [14, 15]. Following the initial biopsy, all patients received preoperative (chemo)radiotherapy followed by surgical resection. Slides and molecular profiling were processed as described for cohort 1 (FOCUS) but using five to nine sections for RNA extraction and nine for DNA. A total of four slides were excluded after QC for a final set of 332 slides from 183 cases.
Ethics approval
The FOCUS and GRAMPIAN cohorts of S:CORT have ethical approval (REC 15/EE/0241) from the East of England – Cambridge South Research Ethics Committee. TCGA (https://www.cancer.gov/tcga, last accessed: 2 August 2018) is an open‐source public database.
Analysis of cell composition by deep learning
We utilised the Soft Cell‐Tissue DL model (SoftCTM) [16] to detect tumour and non‐tumour cells in the test cohorts. The model was developed on the Overlapped Cell on Tissue Dataset for Histopathology (OCELOT) training and validation set [17] within the OCELOT 2023 Challenge. The sets respectively comprised n = 400 and n = 137 pairs of annotated cell patches at 50× and tissue patches at 12.5× extracted from 173 and 65 TCGA slides and six distinct organs (bladder, endometrium, head and neck, kidney, prostate, and stomach). SoftCTM consists of a model for tissue segmentation and cell detection (Figure 2). The tissue segmentation prediction was provided as input to the cell detection model, allowing consideration of predicted tumour versus non‐tumour tissue regions. As our test cohort slides were at 20×, we utilised the SoftCTM 20× version and WSI inference pipeline from the public GitHub repository (https://github.com/lely475/ocelot23algo, last accessed: 27 May 2024). Contrary to [16], the WSI inference pipeline does not extract a larger field of view for tissue segmentation or apply test‐time augmentation for either model, thereby reducing computational costs. We inferred the SoftCTM model on each slide (Figure 2A) and collected the tumour and background (non‐tumour) cell counts (TC, BC) (Figure 2B). Visualisations of SoftCTM predictions (supplementary material, Figure S1) were found to be of high quality based on visual review by an experienced board‐certified pathologist (VHK).
Figure 2.
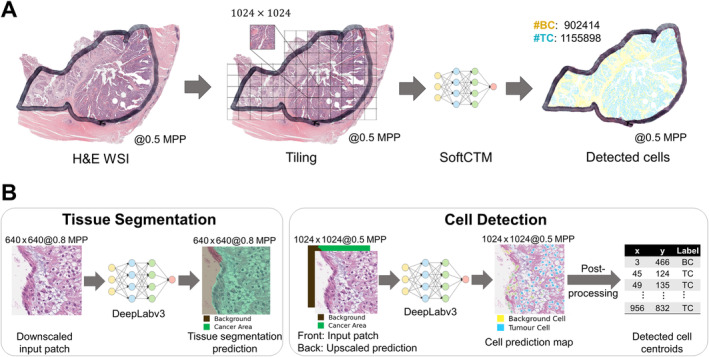
Inference workflow for tumour and background cell nuclei (TC, BC) detection by SoftCTM on a H&E‐stained WSI. (A) WSI‐level inference: Pathologist‐marked ROIs of a H&E‐stained WSI are tiled into patches (1,024 × 1,024 pixels at 0.5 MPP), on which SoftCTM is applied. The predictions are then recombined into a spatially resolved WSI‐level prediction of detected TC and BC. (B) Patch‐level inference: The SoftCTM algorithm consists of two stages: tissue segmentation and cell detection. Tissue segmentation is performed at 0.8 MPP, cell detection at 0.5 MPP. For cell detection, the tissue segmentation prediction is used as input along with the input patch. The output is a probability map for each cell class, from which detected cells are extracted through a postprocessing step. MPP, microns per pixel; WSI, whole slide image
Correlation of SoftCTM with pathologist‐supervised DP algorithm
To further verify the reliability of the SoftCTM algorithm, we investigated its correlation with a CPATH algorithm for cell detection, developed within the Indica Labs HALO AI™ digital image analysis platform [18]. Further referred to as HALO DP, the algorithm consisted of (1) a tissue segmentation algorithm, which was trained on >1,500 tissue areas from S:CORT, TCGA, TEM, and CORGI CRC cohorts with pathologist annotations (tumour, desmoplastic stroma, inflamed stroma, muscle, necrosis, mucin, mesenchyme, background) and (2) a visually optimised general cell segmentation algorithm implemented in the HALO image analysis platform. Algorithms 1 and 2 were combined for cell classification, where all cells within a predicted tissue area were treated as the respective tissue's cells (e.g. all cells in the tumour area are treated as tumour cells). This provides a more fine‐grained distinction between different non‐neoplastic cell classes than SoftCTM, but it is more restricted in the assumption that a specific tissue area cannot contain other cell types. As with SoftCTM, we derived TPEs for HALO DP as tumour cell counts divided by total cell counts. TPEs by SoftCTM and HALO DP showed a high correlation of 0.78 considering all cohorts (supplementary material, Figure S3), with a Pearson correlation coefficient of 0.85 and 0.79 for FOCUS and GRAMPIAN cohorts and slightly lower, 0.68, for TCGA.
Preprocessing of image data and exclusion criteria
Digital slides were re‐reviewed and invasive cancer regions annotated by an experienced board‐certified pathologist (VHK) using the HALO™ software version 2.3.2089.52 (Indica Labs, Corrales, NM, USA). The SoftCTM algorithm was applied within the annotated regions.
Tumour purity estimations
All TPEs were harmonised to scale 0% (e.g. no tumour) to 100% (pure tumour). CPATH estimations were determined using cell counts by SoftCTM (tumour cell counts divided by total cell counts). CP estimations were derived in TCGA and GRAMPIAN by one expert pathologist visually estimating the proportion of viable tumour versus all cells at fractions of 5%. In FOCUS, scores from two different pathologists were available showing modest correlation [r = 0.59, CI = (0.52, 0.65)]. One of them was randomly selected for all further analyses. Estimations from multi‐omic data were derived from transcriptome with ESTIMATE [19] and from methylation with InfiniumPurify [20] using their original R packages in FOCUS and GRAMPIAN. For TCGA, purity based on ESTIMATE and InfiniumPurify were retrieved from previous publications [10, 21].
Gene copy number estimation
The targeted NGS panel applied to FOCUS and GRAMPIAN cohorts contains probes spanning SNPs evenly distributed along the human genome (average of one SNP per 3 Mb) and 66 chromosomal regions recurrently gained or lost in CRC. This design allows for the generation of copy number estimations from targeted NGS at row resolution, acknowledging that such estimations may not be directly comparable to techniques that examine the entire genome. Furthermore, CNVkit [22], a tool specifically designed to enhance targeted NGS data by the analysis of both targeted and off‐target reads, was used on both cohorts adjusting by TPEs from different methods. Copy number segments with estimations ≥3 were classified as gain, 2 as neutral, and ≤1 as loss. The Whole Genome Instability Index (WGII) measuring the proportion of the genome with an aberrant copy number was calculated as the sum of the lengths of calls for either loss or gain divided by the whole length.
Consensus molecular subtype (CMS) classification
CMS was derived as described previously [14]. In brief, the R library CMSclassifier [23] was used to compute both single sample predictions after row‐centring the expression data and random forest in each of the three cohorts separately. CMS calls were generated by matching both methods without applying any cut‐off.
Statistics
Correlations were analysed using Pearson's correlation coefficient, with confidence intervals provided at 95%. Statistical differences between TPE methods were evaluated using paired‐samples t‐tests. We used the SciPy [24] statistics Python package for Pearson correlation analysis, paired‐samples t‐test, generation of boxplots and histograms and Pingouin [25] for generating Bland–Altman plots.
Results
Assessment of cell composition by DL is accurate, robust, and reproducible
The objective of this study was to evaluate the efficacy of a reliable and openly accessible CPATH technique for estimating TP on CRC histology slides and to conduct a comprehensive comparison with CP and molecular deconvolution methods (ESTIMATE, InfiniumPurify). Test cohorts including a total of 1,097 patients were selected to represent relevant clinical scenarios in the management of CRC patients including postoperative resection specimens (FOCUS, n = 702 slides from 366 patients; TCGA, n = 548 slides from 548 patients) and endoscopic biopsy material (GRAMPIAN, n = 332 slides from 183 patients) (Figure 1A). Tumour areas on each slide were annotated by a pathologist, and molecular analysis was performed on material obtained from strict serial sections (FOCUS and GRAMPIAN). Please refer to Sirinukunwattana et al [14] (https://gut.bmj.com/content/gutjnl/70/3/544/DC2/embed/inline-supplementary-material-2.pdf, last accessed: 29 October 2024) for clinical and molecular data characterisation and a summary of the GRAMPIAN and FOCUS cohorts. Technical reproducibility was checked by rerunning SoftCTM on 50 H&E slides from FOCUS, comparing cell counts and TPEs in each paired output. The correlation was excellent with an r‐coefficient equalling 1.0, showing technical repeatability and stability of SoftCTM predictions. In contrast, CP estimates by two different pathologists in the FOCUS cohort showed only mild correlation [r = 0.59, CI = (0.52, 0.65)]. As SoftCTM was not trained on CRC histology, we further investigated its agreement with a pathologist‐supervised CPATH algorithm that was trained utilising parts of the test cohort data and report a high level of agreement in TPE between the methods (supplementary material, Figure S3). To investigate intra‐sample variance, we then compared TPE by SoftCTM between the first and second H&E slides in all available cases from FOCUS and GRAMPIAN (334 and 149 pairs respectively) (Table 1 and supplementary material, Figure S4). Importantly, these slides represent serial sections with approximately 20 μm Z‐axis distance for FOCUS and 40 μm for GRAMPIAN, with additional material taken for RNA profiling between the sectioning planes of interest. Across serial sections, both the TPE and total cell counts showed excellent correlations (all r > 0.8, Table 1). We further verified that the number of detected cells correlated with the size of the invasive cancer region [supplementary material, Figure S5, r = 0.925, CI = (0.916, 0.933)]. For this comparison, only tissue regions inside the expert pathologist annotation were considered. The mean cell density in resection specimens from the FOCUS and TCGA cohorts was approximately 19,000 cells/mm2, whereas GRAMPIAN biopsy specimens exhibited a cellular density of approximately 26,000 cells/mm2 indicating increased tissue compression in biopsy samples. This difference may be attributed to the sampling technique involving compression by biopsy forceps and enhanced fixation of smaller tissue samples, resulting in greater shrinkage.
Table 1.
Pearson correlation of tumour purity estimation, tumour, and background cell counts by digital pathology for H&E sections 1 and 2, which gives insight into the reproducibility and dependency of the method on the selected tissue section (all p < 0.001).
| TPE | Tumour cells | Background cells | |
|---|---|---|---|
| FOCUS (N = 334) | r = 0.949, CI = (0.937, 0.959) | r = 0.976, CI = (0.970, 0.981) | r = 0.908, CI = (0.887, 0.925) |
| GRAMPIAN (N = 149) | r = 0.818, CI = (0.757, 0.865) | r = 0.976, CI = (0.967, 0.982) | r = 0.946, CI = (0.927, 0.961) |
| Total (N = 485) | r = 0.916, CI = (0.901, 0.929) | r = 0.983, CI = (0.980, 0.989) | r = 0.945, CI = (0.934, 0.954) |
Abbreviations: CI, confidence interval; r, Pearson correlation coefficient.
Tumour purity assessed by different methods
Next, we compared TPEs derived from cell counts of SoftCTM with visual estimates determined by expert pathologists and TPEs derived from bioinformatic deconvolution from RNA expression using ESTIMATE [19] and DNA methylation using InfiniumPurify [20]. We first conducted a statistical comparison of all TPE methods using paired‐samples t‐tests, revealing significant differences between each method (p < 0.001) (supplementary material, Figure S6). SoftCTM displays a broad distribution of TPE predictions, aligning with biological expectations. In contrast, the other three methods demonstrate narrower distributions, and these converge at varying levels (Figure 3, and cohort‐specific in supplementary material, Figure S7). Our objective was to provide additional context to these findings by considering CRC biological subgroups. To achieve this, we employed the primary transcriptomic classifier CMS as a framework, given its significant association with TPE metrics. Notably, CMS1 and CMS4 subtypes are associated with elevated levels of immune and stromal infiltration, resulting in lower TPE, whereas the canonical and metabolic subgroups (CMS2 and CMS3) are distinguished by higher epithelial content [14, 23]. As expected, we observed lower TPEs in CMS4 followed by CMS1 and higher epithelial content in CMS2/3 (supplementary material, Figure S8). These results suggest that all four methods reliably capture the expected associations across biological subgroups.
Figure 3.
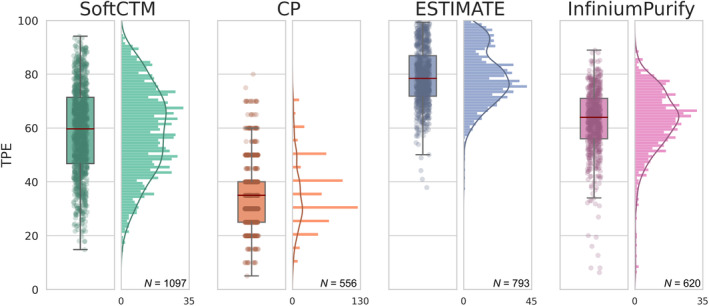
Boxplot and histogram comparing distribution of TP estimated by different methods for the combined test cohorts. We only consider samples with TPE available for all methods. The box represents the IQR, encompassing 50% of the data points. Red line indicates median, and whiskers extend to ±1.5 IQR from IQR edges. TP, tumour purity; IQR, interquartile range
We further examined the direct correlation between SoftCTM, CP, and molecular TPE methods (ESTIMATE, InfiniumPurify) (Figure 4, and cohort‐specific in supplementary material, Figure S9). The correlation coefficients (r) comparing SoftCTM with ESTIMATE and InfiniumPurify exhibited strong agreement in FOCUS and GRAMPIAN cohorts (all r in 0.49–0.70). Conversely, in TCGA, where spatial continuity between molecular and pathological profiles is lacking, the correlation was notably lower (all r in 0.29–0.32). The correlations of SoftCTM with CP were generally strong (all r in 0.53–0.61). Notably, tumour content tended to be overestimated when analysed by ESTIMATE and, to a lesser degree, by InfiniumPurify, compared to SoftCTM, while we observed underestimation by CP. When comparing CP with ESTIMATE and InfiniumPurify TPEs, we observed a mild correlation in FOCUS and GRAMPIAN (all r in 0.31–0.56) and note an overestimation by the molecular deconvolution methods compared to CP. In TCGA, there was no correlation between CP and molecular methods (all r ≈0.0).
Figure 4.
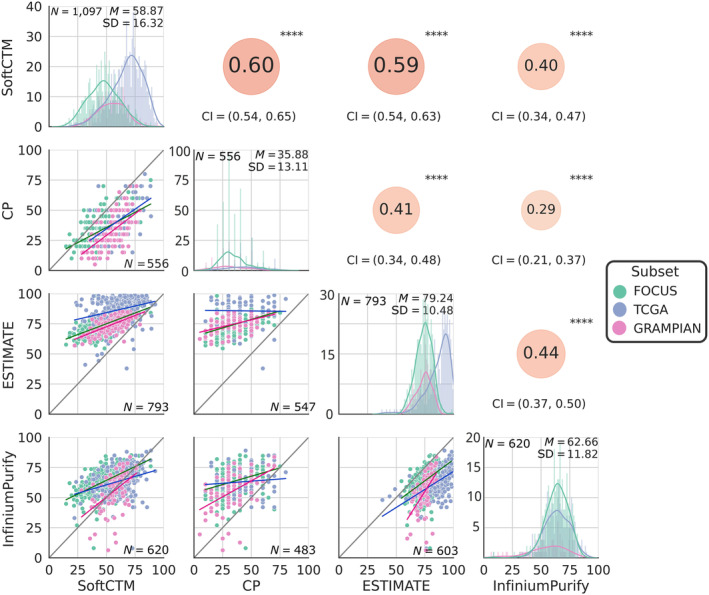
Comparison of TPE method results for test cohorts. Below diagonal: scatter plots comparing respective TPE method results. Diagonal: histogram for each TPE method with mean (M) and standard deviation (SD) in top right. Above diagonal: Pearson correlation coefficient between respective TPE methods; ****p < 0.0001. CI, confidence interval; TPE, tumour purity estimation.
Copy number adjusted using TPEs from CP, DP, and bioinformatic deconvolution methods
Our findings revealed variation in TP distribution depending on the assessment method. This variability can significantly impact the correction of downstream metrics in molecular pathology analysis. One common application involves using TPEs to normalise copy number data, which may be affected by increasing proportions of non‐tumour, diploid cells in DNA extractions. Here, we quantify the impact of utilising TPEs derived from different methods on copy number analysis. Specifically, we assessed the WGII, which measures the proportion of the genome that deviates from diploidy, within the FOCUS and GRAMPIAN cohorts (Figure 5). The TCGA cohort was excluded for this analysis, as we compared copy number adjustment by TPE of SoftCTM in H&E1 with H&E2, which is not feasible in TCGA with only a single H&E slide available.
Figure 5.
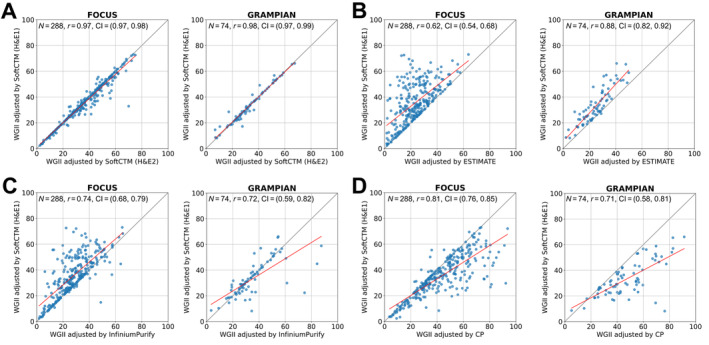
Comparison of Whole Genome Instability Index (WGII) adjusted by (A) SoftCTM, (B) ESTIMATE, (C) InfiniumPurify, and (D) CP; all p < 0.001. CI, confidence interval.
The correlation of WGII following copy number adjustment using TPEs from SoftCTM for H&E1 and H&E2 was excellent [FOCUS: r = 0.973, CI = (0.966, 0.979), GRAMPIAN: r = 0.978, CI = (0.965, 0.986)]. In contrast, comparison of TPEs by SoftCTM with TPEs from RNA (ESTIMATE) and DNA methylation (InfiniumPurify) revealed lower correlations [FOCUS: r = 0.617, CI = (0.540, 0.683), GRAMPIAN: r = 0.881, CI = (0.818, 0.924) and FOCUS: r = 0.741, CI = (0.684, 0.789), GRAMPIAN: r = 0.724, CI = (0.594, 0.818)]. Compared to SoftCTM, ESTIMATE clearly underestimated WGII, while InfiniumPurify underestimated WGII for FOCUS but showed mixed patterns with a tendency towards overestimation for GRAMPIAN. Similarly, a lack of correlation was observed in comparisons of SoftCTM TPEs with CP [FOCUS: r = 0.809, CI = (0.765, 0.845), GRAMPIAN: r = 0.714, CI = (0.581, 0.811)], where CP tends to overestimate WGII.
We then measured changes in copy number at the chromosome arm level (Table 2). When comparing SoftCTM for H&E1 and H&E2, only ~1.5% of copy number segments differed between both cohorts, with a slight tendency towards overcalling in FOCUS (76.06%) and a balanced distribution in GRAMPIAN. However, following adjustment with TPEs from ESTIMATE, 9–13% of calls differed, nearly all of them being underestimations. Conversely, when compared to following adjustment with TPEs from CP, the difference was also 8–14%, but most of them were overestimations. For adjustment with InfiniumPurify TPEs we note 6–9% differing calls with primarily underestimations in FOCUS and a tendency (67.2%) to overcalling in GRAMPIAN.
Table 2.
Comparison of copy number alteration calls in 39 chromosomal arms adjusted by different tumour purity estimation (TPE) methods for FOCUS (N = 288) and GRAMPIAN (N = 74): TPE from SoftCTM applied on H&E1, SoftCTM applied on H&E2, transcriptome‐based ESTIMATE, DNA‐methylation‐based InfiniumPurify, and conventional pathology. Undercalling refers to copy number alterations (CNAs, both losses and gains) that were detected by SoftCTM (H&E1), but not by the respective other method, and the opposite for overcalling.
| CNA calls in Chr arms | SoftCTM (H&E2) | Estimate | InfiniumPurify | CP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Loss | Neutral | Gain | Loss | Neutral | Gain | Loss | Neutral | Gain | Loss | Neutral | Gain | |||
| FOCUS | SoftCTM (H&E1) | Loss | 2,066 | 23 | 1 | 1,340 | 749 | 1 | 1,622 | 466 | 2 | 2,034 | 55 | 1 |
| Neutral | 72 | 6,662 | 74 | 0 | 6,808 | 0 | 19 | 6,761 | 28 | 416 | 6,008 | 384 | ||
| Gain | 0 | 22 | 2,312 | 0 | 675 | 1,659 | 0 | 458 | 1,876 | 0 | 43 | 2,291 | ||
| Different calls | 192/11,232 (1.7%) | 1,425/11,232 (12.7%) | 973/11,232 (8.7%) | 899/11,232 (8.0%) | ||||||||||
| Undercalling | 51/192 (23.4%) | 1,424/1,425 (99.9%) | 103/973 (95.0%) | 98/899 (10.9%) | ||||||||||
| Overcalling | 147/192 (76.6%) | 1/1,425 (0.1%) | 906/973 (5.0%) | 801/899 (89.1%) | ||||||||||
| GRAMPIAN | SoftCTM (H&E1) | Loss | 474 | 15 | 0 | 363 | 126 | 0 | 456 | 33 | 0 | 489 | 0 | 0 |
| Neutral | 6 | 1,865 | 8 | 0 | 1,878 | 1 | 56 | 1,760 | 63 | 197 | 1,455 | 227 | ||
| Gain | 0 | 14 | 504 | 1 | 118 | 399 | 2 | 26 | 490 | 3 | 2 | 513 | ||
| Different calls | 43/2,886 (1.5%) | 246/2,886 (8.5%) | 180/2,886 (6.2%) | 429/2,886 (14.9%) | ||||||||||
| Undercalling | 29/43 (67.4%) | 244/246 (99.2%) | 59/180 (32.8%) | 2/429 (0.5%) | ||||||||||
| Overcalling | 14/43 (32.6%) | 2/246 (0.8%) | 121/180 (67.2%) | 427/429 (99.5%) | ||||||||||
Discussion
H&E slides are routinely prepared in the work‐up of CRC tissue samples in pathology laboratories. TPE serves as an important QC metric for selecting tissue material suitable for molecular tumour profiling [26] and is essential for the correct interpretation of molecular diagnostic tests [27]. Here, we propose a DL‐based cell‐level TPE method and demonstrate its high reproducibility, correlation to other TPE methods, and implications for downstream molecular analyses on a large CRC dataset, including three separate cohorts with complete DP and multi‐omic data.
While the assessment of tumour percentage on a given tissue slide may appear to be a straightforward task, it is more complex than initial observation might suggest [28]. Interobserver reproducibility of CP between domain experts is low to moderate [4, 29]. Previous research associated this variability with insufficiently defined cellularity criteria and underestimation of the non‐linear correlations between area‐ and cell‐level assessments [30, 31]. This effect is particularly pronounced in cases with very high or low cellular density in regions of interest (ROIs) [30]. These limitations reflect negatively on the quality and reliability of tumour molecular profiling in clinical practice [4, 29]. Manual cell counting could be a more accurate methodology but is not routinely carried out in diagnostic practice due to prohibitive demands on time [28]. DL algorithms for automated TPE address the need for higher robustness, reproducibility, and standardisation at low cost [3, 32]. Computer‐aided diagnostic decisions can offer valuable support to pathologists in clinical practice as described elsewhere [33, 34].
We distinguish DL‐based approaches to TPE into (1) WSI‐level methods, where TP is directly predicted as a WSI‐level score, (2) tile‐level methods, where WSIs are tessellated and each tile is classified into tumour and non‐tumour or TP is directly predicted for each tile, and (3) cell‐level methods, where cells are detected and classified into tumour and non‐tumour cells. Supplementary material, Table S2 provides an overview from the literature for each approach. From approaches 1–3 we note an increase in detail and interpretability. From a pathology standpoint, the cell counting methodology represents the most direct measurement of tumour DNA content and is robust with regard to cellular compression, so it is our chosen methodology. Several applications split this task into tissue and cell segmentation, with cells classified into tumour and non‐tumour classes based on their localisation within a predicted tissue compartment, but this is not an exact representation of biological reality. Many published methods were developed for a specific cancer indication and lacked broad applicability across different cancer types. Further, accessibility is limited, with seven out of ten considered cell‐level methods not available for public access and one requiring further fine‐tuning by experts (supplementary material, Table S2). Lastly, validation and comparison against other state‐of‐the‐art approaches are often incomplete. Our chosen method, SoftCTM [16], addresses these concerns. SoftCTM is a multiorgan, open‐source (https://github.com/lely475/ocelot23algo, last accessed: 27 May 2024) solution for detecting tumour and non‐tumour cells, unrestricted by tissue segmentation prediction, developed as part of the OCELOT challenge (https://ocelot2023.grand-challenge.org, last accessed: 27 May 2024). It achieved third place in mean F1 on the OCELOT multiorgan test set [17] compared to 14 other methods, with a difference of only 0.69% mean F1 to the first place method (Test Leaderboard: https://ocelot2023.grand-challenge.org/evaluation/test/leaderboard/, last accessed: 27 May, 2024).
Here, we apply SoftCTM on three large and diverse CRC cohorts, consisting of a randomised clinical trial for patients with advanced disease [11], the CRC TCGA dataset, and a cohort of preoperative rectal biopsies, for a total of over 1,000 samples, including serial sections taken in alternation with tissue material sampled for RNA and DNA profiling within the S:CORT programme. This design ensures continuity of tissue sections extracted for molecular and imaging purposes and is a unique setting to compare the accuracy of CPATH and genomic methods for the determination of TPE within the same sample. For all samples, SoftCTM TPEs were compared with three established approaches (CP, ESTIMATE, InfiniumPurify) widely used in both research and clinical workflows. TPEs by these three methods showed very different distributions. Specifically, the average TPE by CP stood at 35.88% (SD ±13.11), contrasting sharply with InfiniumPurify at 62.66% (SD ±11.82), and even more so with ESTIMATE at 79.24% (SD ±10.48). However, these variations correspond to underlying biology as they all show expected associations with CMS classification. Notably, SoftCTM presents a middle ground, with a mean of 58.87 and broader distribution (SD ±16.32), indicative of potentially more accurate estimations due to its direct cell count measurement. Overestimation can occur in ESTIMATE due to its exclusive measurement of stromal and immune components as the non‐tumour fraction, neglecting the contribution of other normal cell types. In InfiniumPurify, normal tissue, and tumour cells are distinguished, but methylation specific to stromal/immune cells is overlooked. For both methods, this leads to overestimation of tumour cell content, particularly in cases of low purity where the impact of uncaptured non‐tumour biology is strongest. Further, underestimation of TP by CP compared to CPATH or molecular methods is uncommon, as many studies report an overestimation [33, 35, 36, 37, 38, 39]. We speculate that this discrepancy may stem from visual pathologist assessment influenced by variability in tissue appearance and cell compression across different cancer types and pathology workflows. Additionally, pathologist training plays a role, with some sources documenting both over‐ and underestimations [19, 29, 30, 40]. This underscores the necessity for more standardised and strictly quantitative methodologies.
As a result of the TPE underestimation by CP and overestimation by deconvolution methods, the adjusted arm‐level copy numbers result in consistent overcalls and undercalls by around 10% respectively compared to SoftCTM. In contrast, comparison between paired H&Es with SoftCTM shows only ~1.5% differing calls, considered to be expectable background noise. Hence, the 10% differing calls for intermethod comparison contrasted by 1.5% for intra‐method variability highlight the impact of TPE methods on subsequent molecular analysis. This is consistent with and expands recent observations by others [31]. Although the overall patterns of genomic profiles may not be strongly biased, this level of difference could have a tangible effect on future precision medicine pipelines and impact clinical decisions. In addition, deconvolution methods may be confounded by some assumptions such as level of molecular intra‐heterogeneity and/or ploidy, where near‐diploid or near‐tetraploid may provide similar genomic patterns. An accurate, unbiased cell count of tumour and non‐tumour cells before DNA/RNA extraction may provide more objective TPEs to consider for further downstream analyses.
The strengths of this study lie in its comprehensive comparison of TPE assessments across relevant clinical settings, including both resection and biopsy samples. The reference method SoftCTM is publicly available, fully reproducible, and robust with regards to slide selection. SoftCTM predictions are interpretable, as cell markups can be generated for pathologist review (supplementary material, Figure S2). This highlights the applicability and usability of CPATH for TPE. SoftCTM was not initially trained on CRC histology, but it still achieves high agreement of TPEs with a pathologist‐supervised CPATH algorithm for CRC trained on the test cohorts. Still further validation of SoftCTM with regards to cell detection performance for CRC and other indications beyond the OCELOT dataset is recommended.
Overall, SoftCTM showed excellent consistency across slides, was biologically sound, and showed reliable estimates of TPE that were directly interpretable by pathologists. Subsequent steps could lead to the development of image‐based methods for tumour diagnosis together with sample selection and downstream bioinformatic pipelines in research and clinical labs for accurate molecular profiling. While ambitious, recent successes in the classification of cancers at the intersection of digital and molecular pathology make this a plausible next development step [41].
Author contributions statement
ED, TM, J Rittscher and VHK jointly conceived the study. ED, TM, J Rittscher and VHK designed the study. ED, LAS, TM, J Rittscher and VHK drafted the manuscript. ED, KS, SR, KR, ABl, J Rittscher, CH, CW, IT, ABe, UMcD, GIM, LMS, MS, PQ, TM and VHK obtained and categorised image data, clinicopathological and molecular data. LAS designed and implemented the DP algorithm and performed the DP analysis. ED, LAS, KS, J Rittscher, TM and VHK performed data interpretation. KDM, CV and SL provided important intellectual input, provided critical resources or funding and critically reviewed the study design. ED, LAS, AC, KS, J Robineau and ABl performed bioinformatic and statistical analysis. All authors have read and given approval of the final manuscript.
Supporting information
Figure S1. Overall and cohort‐specific overlap of available data and resources
Figure S2. Visualisation of the SoftCTM predictions for areas from two randomly selected WSIs for each cohort
Figure S3. Correlation of TPE by SoftCTM and HALO DP, a cell detection algorithm developed within the digital image analysis platform HALO AI™ by IndicaLabs
Figure S4. Scatter plots comparing SoftCTM TPE, tumour and background cell counts for H&E section 1 and 2 in FOCUS (N = 334), GRAMPIAN (N = 149) and both combined (N = 483), to assess reproducibility and dependency of SoftCTM on the selected tissue section (all p < 0.001)
Figure S5. Scatter plot showing the total number of cells for each cohort over the ROI
Figure S6. Bland–Altman plots comparing results for each TPE method, bias is indicated as a thick black line, the levels of agreement as dotted lines (all p < 0.001)
Figure S7. Boxplot comparing distribution of TP estimated by different method for each test cohort
Figure S8. Box plots for TPE by different methods for each CMS subtype
Figure S9. Comparison of TPE method results for (A) FOCUS, (B) TCGA and (C) GRAMPIAN cohorts
Table S1. List of all S:CORT case ids used in this study and their cohort origin (FOCUS or GRAMPIAN)
Table S2. Selected examples of automated TPE methods
Acknowledgements
The S:CORT consortium is a Medical Research Council (MRC) stratified medicine consortium jointly funded by the MRC and CRUK (MR/M016587/1). This work was further supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre. Computation used the Oxford Biomedical Research Computing (BMRC) facility, a joint development between the Wellcome Centre for Human Genetics and the Big Data Institute supported by Health Data Research UK and the NIHR Oxford Biomedical Research Centre. J Rittscher is supported through the Engineering and Physical Sciences Research Council (EPSRC)‐funded Seebibyte programme (EP/M013774/1). VHK gratefully acknowledges funding by the Swiss National Science Foundation (P2SKP3_168322/1 and P2SKP3_168322/2), the Werner‐Hedy Berger Janser Foundation, and the Promedica Foundation F‐87701‐41‐01. The authors thank Claire Butler and Michael Youdell for excellent management in S:CORT and the MRC Clinical Trials Unit which provided the clinical data from the FOCUS trial with permission from the FOCUS trial steering group. We would further like to thank Indica Labs for providing the HALO™ software. The results published or shown here are based in part upon data generated by the TCGA Research Network established by the National Cancer Institute and National Human Genome Research Institute. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov. We would especially like to thank all patients who consented to take part in S:CORT and TCGA. The views expressed are those of the author(s) and not necessarily those of the National Health Service, the National Institute for Health and Care Research, or the Department of Health.
Appendix A.
S:CORT consortium
| Name | Affiliation |
| Richard Adams | University of Cardiff |
| Michael Youdell | University of Oxford |
| Viktor Koelzer | University of Oxford |
| Simon Bach | University of Birmingham |
| Andrew Beggs | University of Birmingham |
| Celina Whalley | University of Birmingham |
| Louise Brown | University College London |
| Francesca Buffa | University of Oxford |
| Peter Campbell | Wellcome Trust Sanger Institute |
| Jean‐Baptiste Cazier | University of Birmingham |
| Enric Domingo | University of Oxford |
| Andrew Blake | University of Oxford |
| Chieh‐His Wu | University of Southampton |
| Aikaterini Chatzipli | Wellcome Trust Sanger Institute |
| Claire Hardy | Wellcome Trust Sanger Institute |
| Susan Richman | University of Leeds |
| Philip Dunne | Queens University Belfast |
| Keara Redmond | Queens University Belfast |
| Paul Harkin | Almac Diagnostics |
| Steven Walker | Almac Diagnostics |
| Geoff Higgins | University of Oxford |
| Jim Hill | Christie Hospital Manchester |
| Chris Holmes | University of Oxford |
| Denis Horgan | European Association of Precision Medicine |
| Rick Kaplan | University College London |
| Richard Kennedy | Queens University Belfast |
| Mark Lawler | Queens University Belfast |
| Simon Leedham | University of Oxford |
| Tim Maughan | University of Oxford |
| Ultan McDermott | AstraZeneca |
| Gillies McKenna | University of Oxford |
| Gary Middleton | University of Birmingham |
| Dion Morton | University of Birmingham |
| Graeme Murray | University of Aberdeen |
| Phil Quirke | University of Leeds |
| Sanjay Rathee | University of Cambridge |
| James Robineau | University of Oxford |
| Manuel Salto‐Tellez | Queens University Belfast |
| Les Samuel | Grampian NHS Health Board |
| Anna Schuh | University of Oxford |
| David Sebag‐Montefiore | University of Leeds |
| Matt Seymour | University of Leeds |
| Ricky Sharma | Varian Medical Systems |
| Richard Sullivan | Kings College London |
| Ian Tomlinson | University of Oxford |
| Nicholas West | University of Leeds |
| Richard Wilson | University of Glasgow |
Conflict of interest statement: Both VHK and LAS have served as invited speakers on behalf of Indica Labs. VHK reports being an invited speaker for Sharing Progress in Cancer Care (SPCC), serving on the advisory board of Takeda, and sponsoring research agreements with Roche and IAG, all unrelated to the current study. VHK is a participant in several patent applications on the assessment of cancer immunotherapy biomarkers by digital pathology, a patent application on multimodal deep learning for the prediction of recurrence risk in cancer patients, and a patent application on predicting the efficacy of cancer treatment using deep learning. KS and J Rittscher are cofounders and equity holders of Ground Truth Labs. GIM is an editorial board member of The Journal of Pathology and Treasurer of The Pathological Society, the owners of this journal. No other conflicts of interest were declared.
Contributor Information
Tim Maughan, Email: tim.maughan@oncology.ox.ac.uk.
Enric Domingo, Email: enric.domingo@oncology.ox.ac.uk.
Viktor H Koelzer, Email: viktor.koelzer@usb.ch.
S:CORT consortium:
Richard Adams, Michael Youdell, Viktor Koelzer, Simon Bach, Andrew Beggs, Celina Whalley, Louise Brown, Francesca Buffa, Peter Campbell, Jean‐Baptiste Cazier, Enric Domingo, Andrew Blake, Chieh‐His Wu, Aikaterini Chatzipli, Claire Hardy, Susan Richman, Philip Dunne, Keara Redmond, Paul Harkin, Steven Walker, Geoff Higgins, Jim Hill, Chris Holmes, Denis Horgan, Rick Kaplan, Richard Kennedy, Mark Lawler, Simon Leedham, Tim Maughan, Ultan McDermott, Gillies McKenna, Gary Middleton, Dion Morton, Graeme Murray, Phil Quirke, Sanjay Rathee, James Robineau, Manuel Salto‐Tellez, Les Samuel, Anna Schuh, David Sebag‐Montefiore, Matt Seymour, Ricky Sharma, Richard Sullivan, Ian Tomlinson, Nicholas West, and Richard Wilson
Data availability statement
FOCUS raw expression data and molecular metadata are publicly available at GEO under reference GSE156915. The transcriptomic data from GRAMPIAN are publicly available at the following link: https://www.scort.org/sites/default/files/exports/scort_ws3_grampian_export_84m9fndk/ws3_grampian_expression_raw.zip. Sequencing data from whole S:CORT are publicly available in EGA (EGAS00001001521). Additional S:CORT data are available to all academic researchers on submission of a data request to the data access committee. For commercial agencies, the data will be made available through Cancer Research Horizons acting on behalf of the funders and consortium members. The TCGA datasets and images analysed in this study are openly and publicly available at https://portal.gdc.cancer.gov/.
References
References [42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55] are cited only in the supplementary materials.
- 1. Fisher NC, Byrne RM, Leslie H, et al. Biological misinterpretation of transcriptional signatures in tumor samples can unknowingly undermine mechanistic understanding and faithful alignment with preclinical data. Clin Cancer Res 2022; 28: 4056–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim J, Park WY, Kim NKD, et al. Good laboratory standards for clinical next‐generation sequencing cancer panel tests. J Pathol Transl Med 2017; 51: 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamilton PW, Wang Y, Boyd C, et al. Automated tumor analysis for molecular profiling in lung cancer. Oncotarget 2015; 6: 27938–27952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haider S, Tyekucheva S, Prandi D, et al. Systematic assessment of tumor purity and its clinical implications. JCO Precis Oncol 2020; 4: 995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chakravarthy A, Furness A, Joshi K, et al. Pan‐cancer deconvolution of tumour composition using DNA methylation. Nat Commun 2018; 9: 3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012; 30: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel NM, Jo H, Eberhard DA, et al. Improved tumor purity metrics in next‐generation sequencing for clinical practice: the integrated interpretation of neoplastic cellularity and sequencing results (IINCaSe) approach. Appl Immunohistochem Mol Morphol 2019; 27: 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kouri M, Pyrhönen S, Mecklin JP, et al. The prognostic value of DNA‐ploidy in colorectal carcinoma: a prospective study. Br J Cancer 1990; 62: 976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danielsen HE, Pradhan M, Novelli M. Revisiting tumour aneuploidy – the place of ploidy assessment in the molecular era. Nat Rev Clin Oncol 2016; 13: 291–304. [DOI] [PubMed] [Google Scholar]
- 10. Aran D, Sirota M, Butte AJ. Systematic pan‐cancer analysis of tumour purity. Nat Commun 2015; 6: 8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seymour MT, Maughan TS, Ledermann JA, et al. Different strategies of sequential and combination chemotherapy for patients with poor prognosis advanced colorectal cancer (MRC FOCUS): a randomised controlled trial. Lancet 2007; 370: 143–152. [DOI] [PubMed] [Google Scholar]
- 12. Malla S, Redmond K, Blake A, et al. Exploration of molecular signalling underpinning the DNA damage response deficiency (DDRD) assay in colorectal cancer; data from the S:CORT consortium (stratification in COlorRecTal cancer). Br J Cancer 2019; 121: 3.31114016 [Google Scholar]
- 13. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016; 44: e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sirinukunwattana K, Domingo E, Richman SD, et al. Image‐based consensus molecular subtype (imCMS) classification of colorectal cancer using deep learning. Gut 2021; 70: 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Craig SG, Humphries MP, Alderdice M, et al. Immune status is prognostic for poor survival in colorectal cancer patients and is associated with tumour hypoxia. Br J Cancer 2020; 123: 1280–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schoenpflug LA, Koelzer VH. SoftCTM: cell detection by soft instance segmentation and consideration of cell‐tissue interaction. In International Conference on Medical Image Computing and Computer‐Assisted Intervention. Springer Nature Switzerland: Lecture Notes in Computer Science: Cham, 2024. [Google Scholar]
- 17. Ryu J, Puche AV, Shin J, et al. OCELOT: overlapped cell on tissue dataset for histopathology. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition. IEEE/CVF, 2023; 23902–23912. [Google Scholar]
- 18. Domingo E, Chatzipli A, Richman S, et al. Assessment of tissue composition with digital pathology in colorectal cancer. Cancer Res 2019; 79: 4446. [Google Scholar]
- 19. Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013; 4: 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qin Y, Feng H, Chen M, et al. InfiniumPurify: an R package for estimating and accounting for tumor purity in cancer methylation research. Genes Dis 2018; 5: 43–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng X, Zhang N, Wu HJ, et al. Estimating and accounting for tumor purity in the analysis of DNA methylation data from cancer studies. Genome Biol 2017; 18: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Talevich E, Shain AH, Botton T, et al. CNVkit: genome‐wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 2016; 12: e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015; 21: 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Virtanen P, Gommers R, Oliphant TE, et al. SciPy 1.0: fundamental algorithms for scientific computing in python. Nat Methods 2020; 17: 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vallat R. Pingouin: statistics in python. J Open Source Softw 2018; 3: 1026. [Google Scholar]
- 26. Lyman GH, Moses HL. Biomarker tests for molecularly targeted therapies – the key to unlocking precision medicine. N Engl J Med 2016; 375: 4–6. [DOI] [PubMed] [Google Scholar]
- 27. Tinhofer I, Keilholz U, Rieke D. How to standardize molecular profiling programs for routine patient care. In Critical Issues in Head and Neck Oncology, Vermorken JB, Budach V, Leemans CR, et al. (eds). Springer: Cham, 2023. [Google Scholar]
- 28. Dufraing K, van Krieken JH, De Hertogh G, et al. Neoplastic cell percentage estimation in tissue samples for molecular oncology: recommendations from a modified Delphi study. Histopathology 2019; 75: 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smits AJ, Kummer JA, de Bruin PC, et al. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod Pathol 2014; 27: 168–174. [DOI] [PubMed] [Google Scholar]
- 30. Lhermitte B, Egele C, Weingertner N, et al. Adequately defining tumor cell proportion in tissue samples for molecular testing improves interobserver reproducibility of its assessment. Virchows Arch 2017; 470: 21–27. [DOI] [PubMed] [Google Scholar]
- 31. L'Imperio V, Cazzaniga G, Mannino M, et al. Digital counting of tissue cells for molecular analysis: the QuANTUM pipeline. Virchows Arch 2024. 10.1007/s00428-024-03794-9 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 32. Kazdal D, Rempel E, Oliveira C, et al. Conventional and semi‐automatic histopathological analysis of tumor cell content for multigene sequencing of lung adenocarcinoma. Transl Lung Cancer Res 2021; 10: 1666–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakamoto T, Furukawa T, Pham HHN, et al. A collaborative workflow between pathologists and deep learning for the evaluation of tumour cellularity in lung adenocarcinoma. Histopathology 2022; 81: 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frei AL, Oberson R, Baumann E, et al. Pathologist computer‐aided diagnostic scoring of tumor cell fraction: a Swiss National Study. Mod Pathol 2023; 36: 100335. [DOI] [PubMed] [Google Scholar]
- 35. Oner MU, Chen J, Revkov E, et al. Obtaining spatially resolved tumor purity maps using deep multiple instance learning in a pan‐cancer study. Patterns (N Y) 2022; 3: 100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dudley JC, Gurda GT, Tseng LH, et al. Tumor cellularity as a quality assurance measure for accurate clinical detection of BRAF mutations in melanoma. Mol Diagn Ther 2014; 18: 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mikubo M, Seto K, Kitamura A, et al. Calculating the tumor nuclei content for comprehensive cancer panel testing. J Thorac Oncol 2020; 15: 130–137. [DOI] [PubMed] [Google Scholar]
- 38. Sakamoto T, Furukawa T, Lami K, et al. A narrative review of digital pathology and artificial intelligence: focusing on lung cancer. Transl Lung Cancer Res 2020; 9: 2255–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Viray H, Li K, Long TA, et al. A prospective, multi‐institutional diagnostic trial to determine pathologist accuracy in estimation of percentage of malignant cells. Arch Pathol Lab Med 2013; 137: 1545–1549. [DOI] [PubMed] [Google Scholar]
- 40. Noorbakhsh J, Farahmand S, Foroughi Pour A, et al. Deep learning‐based cross‐classifications reveal conserved spatial behaviors within tumor histological images. Nat Commun 2020; 11: 6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lafarge MW, Koelzer VH. Towards computationally efficient prediction of molecular signatures from routine histology images. Lancet Digit Health 2021; 3: e752–e753. [DOI] [PubMed] [Google Scholar]
- 42. Brendel M, Getseva V, Assaad MA, et al. Weakly‐supervised tumor purity prediction from frozen H&E stained slides. EBioMedicine 2022; 80: 104067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petrick N, Akbar S, Cha KH, et al. SPIE‐AAPM‐NCI BreastPathQ challenge: an image analysis challenge for quantitative tumor cellularity assessment in breast cancer histology images following neoadjuvant treatment. J Med Imaging (Bellingham) 2021; 8: 034501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Su A, Lee H, Tan X, et al. A deep learning model for molecular label transfer that enables cancer cell identification from histopathology images. NPJ Precis Oncol 2022; 6: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Azimi V, Chang YH, Thibault G, et al. Breast cancer histopathology image analysis pipeline for tumor purity estimation. Proc IEEE Int Symp Biomed Imaging 2017; 2017: 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Graham S, Vu QD, Raza SEA, et al. Hover‐net: simultaneous segmentation and classification of nuclei in multi‐tissue histology images. Med Image Anal 2019; 58: 101563. [DOI] [PubMed] [Google Scholar]
- 47. Greene C, O'Doherty E, Sidi FA, et al. The potential of digital image analysis to determine tumor cell content in biobanked formalin‐fixed, paraffin‐embedded tissue samples. Biopreserv Biobank 2021; 19: 324–331. [DOI] [PubMed] [Google Scholar]
- 48. Kristoffersen S. Developing a Machine Learning Model for Tumor Cell Quantification in Standard Histology Images of Lung Cancer [Master Thesis]. UiT The Arctic University of Norway, 2023. [Google Scholar]
- 49. Onermustafaumit . Spatially Resolved Tumor Purity Maps (SRTPMs) [Computer Software]. 2021. Available from: https://github.com/onermustafaumit/SRTPMs/tree/v1.0.0.
- 50. Brendel M. wsPurity [Computer Software]. 2022. Available from: https://github.com/ih-lab/wsPurity.
- 51. Noorbakhsh J. HistCNN [Computer Software]. 2020. Available from: https://github.com/javadnoorb/HistCNN.
- 52. Su A. HEMnet [Computer Software]. 2022. Available from: https://github.com/BiomedicalMachineLearning/HEMnet.
- 53. Graham S. HoVer‐net [Computer Software]. 2019. Available from: https://github.com/simongraham/hovernet_inference.
- 54. L'Imperio V, Cazzaniga G. QuANTUM [Computer Software]. 2024. Available from: https://github.com/Gizmopath/QuANTUM.
- 55. Schoenpflug LA. SoftCTM [Computer Software]. 2023. Available from: https://github.com/lely475/SoftCTM.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overall and cohort‐specific overlap of available data and resources
Figure S2. Visualisation of the SoftCTM predictions for areas from two randomly selected WSIs for each cohort
Figure S3. Correlation of TPE by SoftCTM and HALO DP, a cell detection algorithm developed within the digital image analysis platform HALO AI™ by IndicaLabs
Figure S4. Scatter plots comparing SoftCTM TPE, tumour and background cell counts for H&E section 1 and 2 in FOCUS (N = 334), GRAMPIAN (N = 149) and both combined (N = 483), to assess reproducibility and dependency of SoftCTM on the selected tissue section (all p < 0.001)
Figure S5. Scatter plot showing the total number of cells for each cohort over the ROI
Figure S6. Bland–Altman plots comparing results for each TPE method, bias is indicated as a thick black line, the levels of agreement as dotted lines (all p < 0.001)
Figure S7. Boxplot comparing distribution of TP estimated by different method for each test cohort
Figure S8. Box plots for TPE by different methods for each CMS subtype
Figure S9. Comparison of TPE method results for (A) FOCUS, (B) TCGA and (C) GRAMPIAN cohorts
Table S1. List of all S:CORT case ids used in this study and their cohort origin (FOCUS or GRAMPIAN)
Table S2. Selected examples of automated TPE methods
Data Availability Statement
FOCUS raw expression data and molecular metadata are publicly available at GEO under reference GSE156915. The transcriptomic data from GRAMPIAN are publicly available at the following link: https://www.scort.org/sites/default/files/exports/scort_ws3_grampian_export_84m9fndk/ws3_grampian_expression_raw.zip. Sequencing data from whole S:CORT are publicly available in EGA (EGAS00001001521). Additional S:CORT data are available to all academic researchers on submission of a data request to the data access committee. For commercial agencies, the data will be made available through Cancer Research Horizons acting on behalf of the funders and consortium members. The TCGA datasets and images analysed in this study are openly and publicly available at https://portal.gdc.cancer.gov/.