

Graphical abstract
Highlights
-
•
Novel lineage tracing hypoxia reporter to mark hypoxic tumor cells and their descendants.
-
•
Pretreatment hypoxic cells drive tumor regrowth in spheroids and xenografts after radiotherapy.
-
•
Split-dose fractionation counteracts hypoxic cell repopulation and improves treatment outcome.
Abstract
Purpose
Tumor hypoxia imposes a main obstacle to the efficacy of anti-cancer therapy. Understanding the cellular dynamics of individual hypoxic cells before, during and post-treatment has been hampered by the technical inability to identify and trace these cells over time.
Methods and materials
Here, we present a novel lineage-tracing reporter for hypoxic cells based on the conditional expression of a HIF1a-CreERT2-UnaG biosensor that can visualize hypoxic cells in a time-dependent manner and trace the fate of hypoxic cells over time. We combine this system with multiphoton microscopy, flow cytometry, and immunofluorescence to characterize the role of hypoxic cells in tumor relapse after irradiation in H1299 tumor spheroids and in vivo xenografts.
Results
We validate the reporter in monolayer cultures and we show that tagged cells colocalize in spheroids and human tumor xenografts with the hypoxic marker pimonidazole. We found that irradiation of H1299-HIFcreUnaG spheroids leads to preferential outgrowth of cells from the hypoxic core. Similarly, in xenografts tumors, although initially UnaG-positive-cells coincide with pimonidazole-positive tumor areas and they are merely quiescent, upon irradiation UnaG-positive cells enrich in regrowing tumors and are mainly proliferative.
Conclusions
Collectively, our data provide clear evidence that the hypoxic cells drive tumor relapse after irradiation.
Introduction
The expansion of tumors is critically dependent on the co-development of a vascular network to ensure a sufficient supply of oxygen and other nutrients [1]. However, structural and functional vascular defects [2] coupled with the high oxygen demand of the rapidly proliferative tumor cells, can cause tumor sub-areas to become nutrient and oxygen-deprived, termed hypoxic. Tumor cells residing within these hypoxic regions can remain alive due to the action of multiple mechanisms that promote adaptation and survival under the harsh hypoxic environment. From those the most well-studied are hypoxia-inducible factors (HIFs), a family of transcription factors that through their function are core members driving cellular adaptation to hypoxia. During normoxic conditions, the α-subunits are hydroxylated on proline residues within the Oxygen Dependent Degradation Domain by the prolyl-hydroxylated domain (PHD) family of proteins. This event targets them for degradation through the VHL protein complex. In conditions of low oxygen, PHDs are inhibited, HIF-1α and HIF-1β dimerize and translocate to the nucleus where they bind to hypoxia-responsive elements (HREs) located in the promoter of hypoxia-activated genes to initiate the transcriptional response that drives the cellular adaptation to hypoxia [3], [4], [5], [6].
The role of tumor hypoxia has been assessed in several tumor types [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21]. Independently of the method employed to assess hypoxia, patients with more hypoxic tumors have worse therapeutic outcomes even when patients with locally advanced primary tumors are considered, indicating that hypoxic tumor areas might impose the main obstacle in the efficacy of cancer treatment at the primary site. Consistent with this notion, hypoxic tumor cells are more chemo-resistant [22], radioresistant [23], [24] and it was recently shown that tumor hypoxia leads to immunosuppression, hampering the efficacy of immunotherapy [25], [26]. The adverse effect of hypoxia in the outcome of radiotherapy was one of the earliest recognized [23], [24] and commonly referred to as the oxygen effect of radiotherapy. It is mainly attributed to the fact that ionizing radiation can induce both direct DNA damage on the DNA molecule, but also additional excessive damage in the presence of oxygen via the generation of reactive oxygen species (ROS) [24], [27].
However, the oxygen effect can be compromised by tumor tissue re-oxygenation which has been shown to occur very early in irradiated tissue of both mice xenografts [28], [29] and patient tumors [30]. Besides the oxygen effect, hypoxia also affects cell cycle progression [31], and this makes an additional contribution to the radioprotective effect of hypoxia [32]. These observations have led to the general assumption that the combination of the oxygen effect and hypoxia-induced tumor cell quiescence are critical factors for tumor relapse after radiation therapy.
Formal proof for this is still lacking, because one would need to be able to trace the fate of cells that are hypoxic at the time of treatment. Previous efforts to trace hypoxic cells within the primary tumor have been hampered by the natural properties of commonly used fluorescent proteins (which require molecular oxygen to acquire their fluorescent state), and by the fact that previous reporters were activated by hypoxia that occurred throughout the time course of the experiment, rather than uniquely in the cells that are hypoxic at the time of irradiation [33], [34], [35]. Here, we present a novel hypoxic cell lineage-tracer reporter based on the conditional expression of UnaG protein which does not require molecular oxygen to adopt its fluorescent state [36] and can be switched on only in the cells that are hypoxic at the time of irradiation. Using the HIF-1A-cre-ER2-UnaG reporter (HIF1creUnaG) in H1299 lung adenocarcinoma cells, we could follow the fate of hypoxic cells in real-time in multicellular spheroids and tumor xenografts during and after the course of radiation therapy.
Material and Methods (a more detailed description of material and methods can be found in supplementary material)
Generation of UnaG plasmid construct:Generation of EF1α-HIF-1αODDD-Cre-ERT2: The CMV-HIF-1α-eGFP-Cre-ERT2 construct, described in [34], by replacing the CMV promoter with an EF1α promoter, retrieved by PCR, from pHIV-H2BmRFP addgene #18982 t from Bryan Welm & Zena Werb [37] with primers:
5′CGGTTAACTTTTAAAAGAAAAGGGGGGATTCGTGAGGCTCCGGTGCCCGTC- 3′ and 5′-GCCGCCGGCGCCCTCCATGGTGGCGGCTCACGACACCTGAAATGGAAG-3′ using the HpaI and SgrAI digestive sites. Next, we used gBlock (IDT) encompassing a fusion of HIF-1A directly with Cre recombinase by use of the BstBI sites. The construct was sequence verified.
CMV UnaG reporter: A gBlock (IDT) DNA fragment was used which contained the HA tagged UnaG sequence. T overhangs were added using Taq polymerase and this was then sub cloned into the pCR 2.1-TOPO TA vector (Invitrogen). Phusion DNA polymerase was then used to add flanking MluI restriction sites, kozak sequence, and a STOP codon at the end using primers 5′- GGGACGCGTGCCGCCACCATGGTCGAGAAATTTGTTGGCACC-3′ & 5′-CCCACGCGTTCATCAGATCATGCGTAGTCTGGCACGTCGTATGGGTATTCCGTCGCCCTCCGGTAGC-3′. The product was gel purified and T overhangs added with Taq polymerase and then sub cloned into pCR 2.1-TOPO TA vector again. UnaG-HA and pLV(cmv)-NEO-FSF-tdTomato Cre reporter (addgene #160551). The UnaG-HA fragment and reporter were then gel purified. The construct was sequence verified.
Generation of eGFP / DsRed construct: Cre Reporter was a gift from Niels Geijsen (Addgene plasmid # 62732) (D’Astolfo et al., 2015) and was modified by a plasmid, Johan van Es kindly gifted. It was used for generating EF-1α loxP DsRed loxP DTR-EGFP-Puromycin lenti viral construct. The MARCer construct, described in [34], was adapted and the same EF1α-HIF-1αODDD-Cre-ERT2 fused protein was used as in the UnaG construct, with primers 5′ –. All constructs were sequence verified.
Generation of the H1299-HIFcreUnaG cell line: Viral particles for both the EF1a-HIF-1A-Cre-ERT2 and the CMV-UnaG plasmids were generated in Human Embryonic Kidney 293 T (HEK293T) cells as previously described [38] and subsequently, H1299 cells were transduced sequentially and selected with 10 µg/ml Blasticidin and 1000 µg/ml G418 antibiotics to generate a polyclonal cell line. Single clones were then tested for UnaG expression using Adeno cre to induce expression and a clone with minimal leakiness in normoxia and highest induction in hypoxia was selected.
Generation of 4T1-hypGFP cell line: Viral particles were produced using viral vectors and packaging plasmids in 293FT cells as previously published [38]. Cells were then transduced sequentially with the described plasmids selected with 2.5ug/ml Puromycin. Single clones were screened for for dual DsRed and eGFP positivity by FACS analysis. The selected clone was then transduced with pLenti PGK V5-LUC Neo (w623-2) (addgene #21471), to give constitutive luciferase expression.
Cell and spheroid culture conditions: H1299 and 4 T1-hypGFP cells were maintained in RPMI media (Gibco, Life Technology) supplemented with 1 % penicillin/streptomycin, 1 % sodium pyruvate, 1 % Non-essential amino acids and 10 % fetal bovine serum (FBS, Gibco, Life Technology) along with antibiotics. All cell lines were routinely checked for mycoplasma. Normoxic cell culture was performed in a standard humidified incubator (37 °C). An InvivO2500 physiological cell culture workstation (Baker) was used to maintain cells in hypoxia with 1% oxygen level. To retain hypoxic conditions during the time of transportation outside the hypoxic incubator, cells were kept in GasPakTM EZ Pouch Systems (BD) that reduce oxygen levels to 0.001 % within 2h. All hypoxia experiments were performed with substances that have been previously de-oxygenized for at least 12h.
Multicellular spheroids were generated by treating a 2D cell culture with nanoshuttle solution containing magnetic nano-particles (Nanoshuttle-PL, Greiner, Bio-One GmbH) according to manufacturers guidelines. After preparation of single cell suspension, cells were seeded in a non-adherent 24-well plate (Greiner, Bio-One GmbH) at a density of 50.000 cells/ml/well and maintained overnight on a magnetic frame (Greiner, Bio-One GmbH) before transfer to 6-well non-adherent plates (Greiner, Bio-One GmbH) (day3) and left to grow for 12 days before the addition of 4-OHT. During all times spheroids were maintained in phenol-red free Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Dialysed fetal bovine serum (D-FBS, Gibco, Life Technology) to avoid leakiness due to presence of estrogen in the FBS and in the presence of 10 µg/ml Blasticidin and 1000 µg/ml G418. For the multiphoton imaging, spheroids were cultured overnight with SPYDNA 650 (SPIROCHROME) 0,25 μM and then transferred in a 4 well glass bottom labtek (Nunc) in Leibovitz medium (Gibco, Ref. 21083027) during the time of imaging and then immediately transferred back to non-adherent plates and maintained with their standard DMEM.
Radiation treatment for 2D cells and multicellular spheroids: Cells and spheroids were irradiated with a Gammacell 40® Exactor (Theratronics) 137Cs gamma source with a dose rate of 0.92 Gy/min.
Colony forming assay (CFA): Cells were grown in either hypoxia (1 % − indicated as hypoxia – (H)) or normoxia according to the experimental plan. They were then moved to the irradiator for the conditions OOO (cell kept in oxic conditions prior (O), during (O) and post-IR for 24 h (O) or placed in GasPaks for 2 h to reach an oxygen level of 0,001 % and then moved to the irradiator inside GasPaks (condition indicated as Anoxia – (A)). Following irradiation all cells were placed in a normal incubator for 24 h before plated for CFA. Colonies were counted manually with a manual counter application of Fiji software.
Western Blots: Cells were lysed in Laemmli buffer, following protein calibration, 20 μg of protein were loaded and was subsequently separated by SDS-PAGE and transferred to a nitrocellulose membrane (Whatman), before stained with the indicated antibodies and visualized by chemiluminescence (GE Healthcare).
Flow cytometry analysis:Cell cycle Profile: Following Bromodeoxyuridine (BrdU) (10 μM) incubation for 30 min, cells in a maximum cell number of 500.000 were trypsinized, washed and fixed in 70% ethanol (stored at 4 °C until further processing) before staining with primary Rat anti-BrdU (in 2% BSA TBST) for 2 h at room temperature followed by secondary goat anti-rat Alexa 488 (1:400, A11006, Molecular Probes) (in 2% BSA TBST) for 2h and the final incubation with Propidium Iodide and RNAse at 37°C for 20–30 min before been moved to Attune NxT flow cytometer to analyze the cells and FlowJo (BD) was used for analysis and graphs preparation.
Multicellular spheroids processing: Single cell suspension from either H1299-HIFcreUnaG or 4T1-hypGFP spheroids were prepared and afterward passed through cell strainer (Corning, life science, ref: 352235) and transferred on ice to be analyzed on a Fortessa flow cytometer (Becton Dickinson)..
Generation and processing of H1299-HIFcreUnaG tumors: Animal experiments were conducted at the Netherlands Cancer Institute according to national regulations and ethical guidelines. Designs of the animal studies were approved by the Dutch Central Authority for Scientific Procedures on Animals (CCD) and the local animal experimental committee of the Netherlands Cancer Institute (AVD3010020172464 – CCD Protocol ID: 9.1.10916). H1299-HIFcreUnaG tumors were generated by injecting in the mammary fat pad 2,5 million H1299-HIFcreUnaG cells per 30/μL of basement membrane extract (BME, (R&D systems Cat. No. 3533–005-02) per mouse was prepared for the injection in NOD-Scid IL2Rgnull (NSGγ, Jackson) female mice, aged 6–8 weeks at the time of surgery. Once the tumor reached a measured volume between 150–180 mm3, tamoxifen (75 mg/kg, Sigma, Cat. No. T5648-5G) was administered by intraperitoneal injection.
For the characterization of the UnaG reporter, mice were administered with pimonidazole (5 mg/mouse; Hypoxyprobe, pimonidazole hydrochloride, HP-1000 mg) 24h after treatment with tamoxifen. 90 min after administration of pimonidazole, mice were sacrificed and tumors excised, fixed in formalin and subsequently embedded in paraffin. Tumor cross-sections were stained with anti-pimonidazole (hypoxia), anti-HA (UnaG-expression) and anti-Ki67 (proliferation) (Suppl. Fig. 3A).
For experiments assessing the response to radiation therapy, 24 h after the administration of tamoxifen, mice were treated with 0 Gy or 4 Gy, every 2 days with 4 fractions in total. Treatment with radiation was performed on an X-RAD 225Cx Small Animal Image-guided radiation therapy (IGRT). Before and during treatment, mice were anesthetized with isoflurane 2% (v/v). Tumor volume was monitored three times weekly using calipers until reaching 2000 mm3. Then, mice were administered intraperitoneally with 1 mg of 5-ethynyl-2-deoxyuridine (EdU, 200 μl in PBS); and pimonidazole (5 mg/mice).
For the purpose of the 4 T1-hypGFP tumors, animal experiments were by national guidelines for animal welfare and approved by the Animal Ethical Committee of Maastricht University (DEC 2017–018). 75,000 4 T1-MR cells resuspended in 10 µl matrigel® (Corning) were orthotopically injected directly into the mammary fat pad of Balb/c mice (Charles River). Tumour volumes were measured 3times/week in three orthogonal diameters using Vernier calipers. Tamoxifen (Sigma Aldrich) was injected i.p.. one hour before sacrifice Animals were injected i.p with Pimonidazole (Hypoxyprobe) 60 mg/kg and 1 min before 15 mg/kg i.v Hoechst H33342 (Sigma). Radiation planning was performed using SmART-ATP version 2.0. (SmART Scientific Solutions) based of acquired CT images. Radiation was delivered using the small animal irradiator at 225kVp, 12 mA (filtration of 0.3 mm copper) providing a dose rate of approximately 3 Gy/minute.
Immunofluorescence staining of spheroid and tumor cross-sections: Four consecutive 3 µm thickness cross-sections from the paraffin-embedded H1299-HIFcreUnaG tumors were subsequently stained for a) anti-Ki67 b) anti-Pimonidazole c) anti-HA (for UnaG) and d) PBS instead of primary antibody (negative control) with the Alexa Fluor™ 488 Tyramide SuperBoost™ Kit, goat anti-mouse IgG – (B40912) according to the manufacturer guidelines. In the case of spheroids double staining, the procedure was followed sequentially anti-pimonidazole followed by F6 with the Alexa Fluor™ 594 Tyramide SuperBoost™ Kit, goat anti-rabbit IgG – (B40925). As final step nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and embedded in DAKO fluorescent mounting medium.
Frozen sections of 4 T1-hypGFP tumors: 7 µm sections were cut and stored at −80˚C until staining. Sections were stained overnight at 4˚C with rat anti-GFP (Nacalai Tesque) 1:1000, rabbit anti-pimonidazole (Hypoxyprobe) 1:150 or rat anti-CD31 (BD Biosciences) 1:500 in PBS before incubation with secondary antibodies goat anti-Rat (A11006-ThermoFisher) and Goat Anti-Rabbit IgG H&L Cy5 (Abcam) for 1 h at RT and incubated with DAPI (ThermoFisher) 1:5000 in PBS for 10 mins before washing PBS and mounting in fluorescent mounting medium (Agilent).
Imaging and image analysis: 3D whole cross-section immunofluorescent images were acquired with a Zeiss AxioScan Z1 slide scanner (Jena, Germany), equipped with a Hamamatsu Orca FLASH 4.0 V3 camera. The 20x/0.8 Plan-Apochromat objective was scanned axially to acquire a 3D image with a voxel size of 0.325x0.325x0.5 µm. After acquisition, z-stack tiled images were stitched in Zen software v2.6 (Zeiss) and maximal projection was used to create a 2D whole slide image. These were loaded in the bioimage analysis software QuPath [ref: https://doi.org/10.1038/s41598-017–17204-5]. Segmentation of each nucleus was performed on the DAPI channel using Stardist [ref: https://doi.org/10.1007/978–3-030–00934-2_30]. The Warpy-plugin [ref: https://doi.org/10.3389/fcomp.2021.780026] was used to register the different stained sections to have cell-to-cell alignment. For the automatic identification and annotation of each individual staining, home-designed macros were applied (R. H., BvdB). Thresholds were applied to the mean signal intensity for each channel to identify positive/negative cells and a binary nuclei segmentation and color-code annotation was assigned to every single nucleus of each section (Suppl. Fig. 3B, 4A-4B, Fig. 3B, 4D).
Fig. 3.

UnaG + cells coincide with pimonidazole + cells in vivo tumor xenografts. A) Schematic representation of experimental design. B) Characteristic view of an H1299-HIFcreUnaG tumor cross-section (tumor #10) with all the different color annotations assigned to all nuclei of the section. The overlaid image was derived from aligning and processing the three different cross-sections stained for anti-pimonidazole, anti-HA UnaG and anti-Ki67 respectively with the use of Qu-Path software (See Suppl. Fig. 3 for the processing details). In the zoom-in view the individual color assignments for each staining are deconvolved. A characteristic area close to necrosis is shown. The scale bar corresponds to 1000 μm. C) Quantification of the percentage of Ki67+, pimonidazole + and UnaG + cells per tumor section analyzed from five different H1299-HIFcreUnaG xenografted tumors D) Zoom-in image of the double staining indicating the overlay percentage of cells that are positive for both UnaG and pimonidazole for the panel shown in the corresponding tumor section in B (tumor #10). E) Quantification of the percentage of Pimonidazole + and UnaG + cells per tumor section analyzed from five different H1299-HIFcreUnaG tumors. Overlay percentage of cells that are positive for both UnaG and pimonidazole across the different tumors analyzed F) zoom-in image of the double staining indicating the overlay percentage of cells that are positive for both Ki-67 and pimonidazole for the panel shown in the corresponding tumor section in B (tumor #10) G) Proliferation pattern in normoxic and hypoxic tumor cells. Upper Panel: Percentage of Ki67+ (Cyan part of bar) and Ki67- (Magenta part of bar) of pimonidazole + cells only across the tumors analyzed along with the corresponding tumor section in B (tumor #10). Lower Panel: Percentage of Ki67+ (orange part of bar) and Ki67- (grey part of bar) of pimonidazole- cells only across the tumors analyzed. H) Percentage of Ki67 + cells in pimonidazole positive and negative areas pooled from all the tumors analyzed (cyan and orange part of bars from G). Each point represents the measurement in one tumor. All scale bars of the zoom-in images from C, D, E and F corresponds to 250 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.

Response to irradiation and enrichment of UnaG + cells in regrowing H1299-HIFcreUnaG tumors. A) Schematic representation of the experimental design and the treatment arms. B) Tumor re-growth delay assay. On day −1 tamoxifen was administered (indicated by arrow) in the mice and the tumors were either left to grow unperturbed (blue line) or irradiated with 4 times 4 Gy (4 x 4 Gy) as indicated in A (red line) and excised at the end stage (indicated with the arrow). Five tumors were included per arm and when the tumors reached a 2000 mm3 volume were excised fixed and stained with the same parameters as in Fig. 3. Tumor regrowth delay was estimated as the time to reach half of their final volume (1000 mm3) and the two curves were subjected to hypothesis testing (F-test). C) Survival data of the mice in the treatment arm groups shown in A. Data of mice correspond to the 5 unirradiated and 5 irradiated tumors show in B. D) Distribution of the staining and the nuclear annotation across the three different sections for the different stained parameters for an unirradiated tumor. In the upper panel the original view of the final processing of each section after the implementation of the steps shown in Supp. Fig. 3A for each individual binary image and in the lower the dual color assignment. E) Distribution of the staining and the nuclear annotation across the three different sections for the different stained parameters for an irradiated tumor. In the upper panel the original view of the final processing of each section after the implementation of the steps shown in Supp. Fig. 3A for each individual binary image and in the lower the dual color assignment. Scale bar correspond to 1000 μm. F) Percentage of UnaG + cells quantified in the different tumors per condition. The red violin plot depicts the values of the UnaG + cells at the start of the treatment (from Fig. 3D), while the blue (unirradiated) and the green (irradiated) depicts the treatment arms. Each dot corresponds to one tumor analyzed and the p values depict the values of the Kruskal-Wallis test comparison. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Frozen cross-sections of 4 T1-hypGFP tumors were imaged using µManager software 2.0 on an BX51WI spinning disk confocal fluorescence microscope (Olympus, Hamburg, Germany) with a Hamamatsu EM-CCD C9100 digital camera. and analyzed using Image J software (www.imagej.nih.gov/ij/) [39]. Enrichment of eGFP + cells in 4T1 tumors was calculated by setting a GFP positive threshold and then using this calculating the GFP positive area / viable tumor area to give a percentage of tumor area that was GFP positive.
Live-imaging of multicellular spheroids with 2-photon microscopy: Imaging of H1299-HIFcreUnaG spheroids was performed on an inverted Leica TCS SP8 Dive multiphoton microscope (Mannheim, Germany, Leica‐microsystems.com) with either an inverted Leica SP8 Dive system (Leica, Mannheim, Germany) with a MaiTai eHP DeepSee laser (Spectra-Physics, Milpitas, CA, USA) and an InSight X3 laser (Spectra–Physics) or an inverted Leica SP8 Dive system (Leica, Mannheim, Germany) with only InSight X3 laser (Spectra–Physics). UnaG was excited with a wavelength of 980 nm and detected on an hybrid RLD detector non-descanned detector (NDD) set at a wavelength of 495–550 nm. SPY650 was excited with a wavelength of 1150 nm and detected on a hybrid RLD NDD detector set at a wavelength of 600–645 nm. All images were acquired with a 25× (HCX IRAPO N.A. 0.95 WD 2.5 mm) water objective. Three-dimensional tile scans of spheroids were taken with Z-steps of 5 µm.
Statistical analysis: All statistical analyses were performed with Prism 8 (version 8.1.2, GraphPad Software Inc., 2017). To assess differences between the variable distribution the Kruskal-Wallis test (one-way ANOVA on ranks) was performed. In every case, a p-value < 0.05 was considered significant. Cell survival curves were fitted with polynomial linear quadratic model based on the equation:
Surviving fraction = exp(-αD-βD2),
where D is the radiation dose and α, β parameters of the linear and the quadratic term respectively. Tumor growth was fitted exponential growth curve. Differences between cell survival curves or exponential curves of irradiated and unirradiated tumors were tested with F-test.
Results
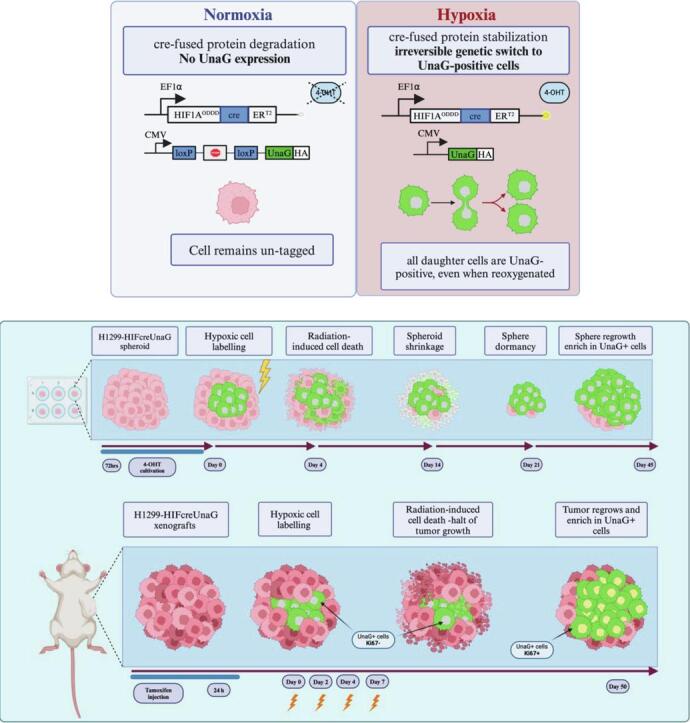
We fused the oxygen-dependent degradation domain (ODDD) of HIF-1α and the mutated Estrogen binding domain (ERT2) to Cre recombinase and placed this under control of the EF1α promoter. Temporal activity of this fusion is controlled by tamoxifen [40], allowing us to limit activation to cells that are hypoxic at the time of irradiation only. We combined this with a construct containing an HA-tagged UnaG gene preceded by the CMV promoter region and a stop codon cassette flanked by loxP sites. Due to its oxygen-independent folding, the UnaG protein will also fluoresce in hypoxic conditions [36]. Combining these constructs enables hypoxia- and 4 hydroxy-tamoxifen (4-OHT)-dependent nuclear translocation of a HIF-1A-Cre-ERT2 fusion protein. In the nucleus it elicits an irreversible recombination via excision of the loxP sites resulting in sustained expression of UnaG protein, thus irreversibly marking the cells that are hypoxic at that moment as well as their descendants, even when they are reoxygenated. We introduced this HIFcreUnaG Tracer into H1299 cells (Fig. 1A) and the stable integration of the constructs was confirmed by genomic qPCR using H1299 (Suppl. Fig. 1A). To avoid any leakiness in the system a monoclonal cell line (H1299-HIFcreUnaG) was derived. Expression of UnaG protein is induced in a 4-OHT and hypoxia-specific manner (Suppl. Fig. 1B). In the presence of 4-OHT, Cre recombinase expression parallels that of endogenous HIF-1α upon hypoxia and is lost upon reoxygenation. Cre recombinase expression can also be induced by treatment with Deferoxamine (DFO), which stabilizes HIF-1α under normoxic conditions (Suppl. Fig. 1A). We could confirm that specifically the ODDD of HIF-1α but not HIF-2α expression is required to induce UnaG expression by hypoxia or by treatment with the hypoxia mimetic CoCl2 (Suppl Fig. 1C). UnaG expression was strongly induced in > 90 % of 4-OHT-treated cells 72 h after exposure to hypoxia (1 % O2). In the absence of 4-OHT or hypoxia, UnaG-expressing cells remained below 1 % (Fig. 1B-1C). We observed significant radioresistance in cells kept in 1 % O2 and irradiated under anoxic conditions as compared to normoxic cells (Fig. 1F), consistent with the previously reported “oxygen effect”. Cells cultured in hypoxic conditions, but subsequently irradiated under hypoxic or fully oxygenated conditions displayed intermediate radioresistance (Fig. 1D). In addition, H1299-HIFcreUnaG cells grown in hypoxia (1 % O2) accumulate gradually in G1, as evidenced by flow cytometric analysis (Fig. 1E, Suppl. Fig. 1D).
Fig. 1.

Development and validation of the HIF1a-Cre-ERT2-UnaG hypoxia reporter in H1299 cells. A) Schematic representation that indicates the design of the two plasmids of the hypoxic cell reporter and how the inducible reporter expresses the UnaG protein under hypoxia and in the presence of 4-OHT. B) Flow cytometer plots from H1299-HIFcreUnaG cells cultured for 72 h in normoxia or hypoxia 1 % in the presence or absence of 500 nM 4-OHT and the respective quantification of one experiment in C. The bar graph represents the means of three independent experiments and the error bars the standard deviation of mean estimation in B. D) Colony forming assay of H1299-HIFcreUnaG cells. H1299-HIFcreUnaG cells were irradiated with graded single doses of irradiation after being for 72 h under normoxic conditions (blue curve), or for 72 h in hypoxia (1 % O2) and subsequently irradiated under almost anoxic conditions (0.001 % O2 – red curve HAO), kept under hypoxia (1 %) during irradiation, or fully reoxygenated for 3 h before been irradiated (1 %O2 – purple curve HHO, 21 % O2 green curve). All conditions reoxygenated immediately after IR The data were fitted with a linear quadratic model. The data points represent the means of three independent experiments,the error bars are the standard deviation of the means estimation and the fit of survival curves were tested with F-test hypothesis to show that are significantly different. E) Typical cell cycle distribution of H1299-HIFcreUnaG cells grown for 72 h in normoxic or hypoxic conditions (1 % O2) for different amount of days. The percentage of cells in each cell cycle phase after three independent experiments are shown in a stacked plot. Error bars represent the standard deviation of mean estimation. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Next, we generated H1299-HIFcreUnaG multicellular spheroids and we assessed the presence of hypoxia in the core of the spheroids using pimonidazole that stains cells that are under oxygen pressure below 10 mmHg (∼1.3 % O2). We could detect UnaG + cells in the core of spheroids exposed to 4-OHT (Fig. 2A).
Fig. 2.

UnaG + cells are located in hypoxic areas in unperturbed H1299-HIFcreUnaG spheroids and enrich in regrowing spheroids post-IR. A) Upper panel: Characteristic images in the middle z-plane of live H1299-HIFcreUnaG spheroid made with epifluorescent microscopy with brightfield (visualize all cells), the corresponding image with the FITC filter (depicts UnaG + cells) and the combined (upper panel − merged). Lower panel: The same spheroid fixed and stained for DAPI (visualize the nuclei) and pimonidazole in TRITC filter (to visualize pimonidazole + cells) and the combined one (lower panel − merged). B) Schematic representation of the experimental design followed to trace the fate of irradiated H1299-HIFcreUnaG spheroids. C-F) Quantification of flow cytometer plots depicting the dynamic changes in H1299-HIFcreUnaG spheroids over time post-irradiation. C) The percentage of DAPI+ (cell death) cells over the total amount of cells isolated from unirradiated and irradiated H1299-HIFcreUnaG spheroids are shown in cyan and red bars respectively. D) Quantification of spheroid areas of the mean z-plane of H1299-HIFcreUnaG spheroids. The images were made with epifluorescent microscopy (as in suppl. Fig. 2B) and correspond to the same samples of the same day that were subsequently assayed to generate the plots of C. Error bars indicate the standard deviation of the mean of at least 3 independent experiments per time-point. E) Characteristic flow cytometer plots depicting the percentage of UnaG + cells over the total amount of living cells (DAPI-negative) cells isolated from unirradiated (left panel) and irradiated (right panel) H1299-HIFcreUnaG spheroids assayed at the endpoint of the experiment, while the bars in F represent the analysis of UnaG + cells from the corresponding samples (as in C & D). The data points represent the means of three independent experiments and the error bars the standard deviation of the means estimation. G-H) Characteristic images of a Maximal intensity projection view of live H1299-HIFcreUnaG spheroids made with 2-Photon microscopy. UnaG + cells along with the total nuclei of spheroids (SPY650 − Spirochrome) and the overlay image is depicted for every occasion. The same H1299-HIFcreUnaG spheroids were followed overtime and the changes in the UnaG + cells are depicted for an unirradiated spheroid (left panel – G) and an irradiated spheroid (right panel − H). Pre-treatment day correspond to the day that 4-OHT was removed and images were made either few minutes post 4-OHT release and irradiation with 8 Gy (Upper panels). Green cytoplasmic fluorescent cells represent the UnaG + cells and magenta the total nuclei of spheroids are depicted with SPY650. Scale bars correspond to 100 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To analyze the contribution of cells that are hypoxic at the time of irradiation to sphere (pre-treatment hypoxic cells) regrowth after IR, identical size spheroids were treated with 4-OHT and 4-OHT was washed out prior to irradiation to prevent further switching (Fig. 2B). Cell death increased in irradiated spheroids, up to day 25 (Fig. 2C). Parallel to the increased cell death, we observed a gradual size decrease in irradiated spheroid, until they reached a “nadir” point at day 21, after which spheroid size started to increase again (Fig. 2D). Importantly, we observed a substantial enrichment in the fraction of UnaG + cells over time after irradiation, both in imaging (Suppl. Fig. 2A) and in flow cytometry (Fig. 2E, 2F), such that the majority of cells repopulating the spheroid were UnaG + by day 45 (Fig. 2E, 2F). The fraction of UnaG + cells is observed as early as day 7 after irradiation, coincident with the rise in cell death, and the final enrichment in regrowing spheroids displayed a clear dose-dependence (Suppl. Fig. 2C). In contrast, little fluctuation in the percentage of UnaG + cells was seen in the unirradiated spheroids. To investigate how spheroids reoxygenate post-IR, spheroids were cultured with pimonidazole for 4 h, then washed and irradiated and incubated at different time intervals following IR with F6 (another imidazole compound) and EdU to be able to monitor reoxygenation and proliferation across the spheroids post-IR (Suppl. Fig. 2A, 2B, 2C). We observed a drastic IR-induced reoxygenation in the analyzed spheroids, with complete reoxygenation achieved at 4 hr after IR, which lasted for at least 24 h post-IR (Suppl. Fig. 2D). At 48 h new hypoxic areas arise (F6 + cells) but the overall amount of hypoxia in the spheroids is substantially decreased (Suppl. Fig. 2D). Interestingly, despite full reoxygenation, proliferation at the early time-points was restricted to the outer layers of the spheroids and almost exclusively in the pimonidazole negative areas (Suppl. Fig. 2E). This implies that the hypoxic cells in the core of the spheroid, which appear to be responsible for the regrowth, remain indolent for several days after IR.
Using 2-photon microscopy, we could show that UnaG + cells in the unirradiated spheroids remained in the inner core and their fraction did not change in time, while UnaG- cells were depleted from the spheroid upon irradiation (Fig. 2G). Eventually, only a very small part of the initial spheroid structure remains, mainly containing UnaG + cells. The residual spheroid structure remains small for ∼ 3 weeks after irradiation, after which we observe the regrowth of spheroids that are mainly populated with UnaG + cells (Fig. 2H).
Next, we injected H1299-HIFcreUnaG cells in the mammary fat pads of NOD/SCID mice (NSG). To visualize the spatial distribution of UnaG+ cells, when tumors reached 180 mm3, tamoxifen was injected and mice were sacrificed 24 hr later (Fig. 3A). We observed that the fractions of proliferative and hypoxic cells were comparable in the 5 different tumors (Fig. 3C, 3D). Overall, all tumors contained between 10–25 % pimonidazole-positive cells, commonly located in the peri-necrotic areas (necrosis defined as DAPI-depleted areas in the tumors), and the percentage of UnaG-positive cells was very comparable (Fig. 3C). Importantly, UnaG + staining displayed a high degree of overlap with pimonidazole positivity (Fig. 3E). A small percentage of the UnaG-positive cells were found outside pimonidazole-positive areas, and some pimonidazole-positive cells very close to necrotic areas were not marked with UnaG (Fig. 3D, Suppl. Fig. 3B). All tumors contained a high percentage of Ki67-positive cells (Fig. 3C), but the percentage of Ki67-positive cells was very low in pimonidazole-positive areas (Fig. 3F, 3G). The vast majority of Ki67-positive cells were located in the pimonidazole-negative areas (Fig. 3F, 3G, Suppl. Fig. 3C).
To follow the changes in the irradiated tumors, H1299-HIFcreUnaG tumors were injected with a single dose of 4-OHT and left untreated, or irradiated them with 4 times 4 Gy with a 48-hour interval between fractions (Fig. 4A). As expected, irradiation delayed the outgrowth of tumors on average by 24 days, after which the tumors grew out at a similar rate as the non-irradiated tumors (Fig. 4B), resulting in prolonged survival (Fig. 4C). At the endpoint, in unirradiated tumors, the mean percentage of UnaG+ cells was low (4.9%), inferior to the initial amount of UnaG-labelled cells before treatment (15,5% in the experimental design of Fig. 3) (Fig. 4D, Suppl. Fig. 4A, 4C, 4D). In marked contrast, and consistent with our observations in the multicellular spheroids, the percentage of UnaG+ cells became significantly higher in all irradiated H1299-UnaG tumors, as compared to the unirradiated counterparts (Fig. 4E, 4F, Suppl. Fig. 4B). In addition, many of the UnaG+ cells in the irradiated tumors were Ki67-positive, indicative of enhanced proliferation of the UnaG+ cells following irradiation.
To validate our observations in another cancer model, we used murine breast carcinoma cell line 4 T1. This was combined with a construct that contains a DsRed gene flanked by loxP sites, preceded by the EF1a promoter and followed by the eGFP gene, as previously used in H1299 cells [34] (Suppl. Fig. 5A). Similar to the UnaG construct in H1299, we saw that expression of eGFP is strongly induced in a 4-OHT and hypoxia-specific manner in this 4T1-derived line (4T1-hypGFP) (Suppl. Fig. 5B). The HIFcre-ERT2 fusion protein follows (de)stabilization of endogenous HIF-1α protein upon hypoxia, DFO treatment and reoxygenation (Suppl. Fig. 5C). eGFP expression is induced in the majority of the cells exposed to hypoxia and 4-OHT (1% O2) and reoxygenated for 8 h, while minimal induction was observed in cells exposed to hypoxia without the addition of 4-OHT or in 4-OHT treated cells cultured in normoxia (Suppl. Fig. 5D, 5E). 4T1-hypGFP cells cultured under hypoxia and irradiated under anoxia displayed clear radioresistance compared to their normoxic counterparts (Suppl. Fig. 5F). We generated 3D multicellular 4 T1-hypGFP spheroids, that were subsequently treated with 4-OHT, released and irradiated with graded single doses of IR. Consistent with our observations in H1299-UnaG spheroids, we observed a significant dose-dependent enrichment of the eGFP + cells in the irradiated 4T1-hypGFP spheroids (Suppl. Fig. 5G). That occurred even though we still observed a typical sigmoid curve of Spheroid control probability (SCP) with increasing radiation dose (Suppl. Fig. 5H). Also, in xenografts of the 4T1-hypGFP cells eGFP+ staining nicely overlapped with pimonidazole staining in unirradiated tumors 3 days post-tamoxifen injection (Suppl. Fig. 5I). The fraction of eGFP+ cells significantly increased following either a single dose 15 Gy or 3x 7,25 Gy fractionated irradiation (Suppl. Fig. 5J).
Fig. 5.

An increase in duration between IR fractions allow hypoxic cell to re-enter cell cycle and opens up an opportunity to effectively eradicate them with IR. A) Schematic representation of the experimental design and the fractionation treatment arms (depicted as regimens) in H1299-HIFcreUnaG spheroids. B) Spheroid control assay. After irradiation with the indicated experimental design (A) spheroids were observed for recovery/regrowth or not (regress and never recover and a binary score (Controlled/Non-Controlled) was generated at day 45 post IR per spheroid. The bar graph indicates the percentage of controlled spheroids over total spheroid assessed in 5 different spheroid control assays (pooled together) each one containing 6 technical replicates and the p value corresponds to the unpaired t-test. C-D) Percentage of UnaG + cells in the individual spheroids that regrew assessed at day 50 (from the non-controlled spheroids of B) after single cell suspension and flow cytometry analysis of each individual spheroid assigned to a fractionation schedule. Characteristic flow cytometer plots for each individual fractionation schedule are shown in C and the quantification of three independent experiments (6 spheroid per experiment) pooled are shown in D.
Given that hypoxia promotes entry into a quiescent cellular state, whereas radiation treatment is more effective in proliferating cancer cells, we hypothesized that re-irradiating hypoxic cells at the time they re-enter the cell cycle might be a more effective strategy to kill them. To provide proof of principle for this hypothesis, we used spheroid cultures, since the time of spheroid regrowth is easily scored via microscopy. We irradiated these spheroids with a total dose of 16 Gy in 4 fractions; one set of spheroids received this dose in one week (similar to the animal fractionation protocol and referred to as regular fractionation), while in the other set a break of 20 days was introduced between the 2nd and 3rd fraction, to account for the time the UnaG+ cells take to initiate regrowth of the spheroid (Fig. 5A). This way, total dose and dose per fraction was identical; the only variable is the time between fractions. After regular fractionation, regrowth was prevented in only 3 out of 30 spheroids (SCP), while 9 out of 30 spheroids failed to regrow in the split fractionation (Fig. 5B). We again observed substantial enrichment of UnaG + cells at the endpoint (Fig. 5C). Of note, we observed significant less enrichment of UnaG + cells in the split fractionation schedule, indicating that split fractionation more effectively eliminated the UnaG + cells (Fig. 5D).
Discussion
Here we provide conclusive evidence for the cell-autonomous role of hypoxic tumor cells for tumor relapse after irradiation. Our lineage tracing confirms that pre-treatment hypoxic tumor cells in vitro and in vivo are less proliferative and more resistant to irradiation than non-hypoxic tumor cells. But more importantly, we provide the first solid evidence that tumor relapse after IR originates from the cells that are hypoxic at the time of irradiation. In addition, our data provide evidence that hypoxic tumor cells only regain proliferative potential and repopulate spheroids and tumors in vivo after approximately 2 weeks for this specific tumor model (Fig. 5E).
The hypoxic cell lineage tracing reporter presented here is based on the activation of the Hypoxia-inducible factor 1a. Our HIFcreUnaG system can specifically introduce a permanent tag in cells that are under low partial oxygen pressure. Switching occurs with high efficiency in spheroids and in tumors grown in mice, allowing us to trace their unique progeny. Previous efforts to tag and trace hypoxic cells in tumor models based on the HIF-1α expression included the incorporation of HRE elements proximal to a promoter that drives the expression of the fluorescent protein of interest based on the HIF response in hypoxia [41] and the incorporation of destabilizing enzymes or the ODDD domain to monitor the degradation of the reporter signal once the oxygen is present [36], [41]. Although such systems are very useful to visualize the cells that are hypoxic at a given time and allows one to conclude when these same cells are reoxygenated (unlike our system), they lack the ability to lineage trace in time hypoxic cells, because the tagging is reversible. Recently, the fusion of the cre recombinase with the ODDD of the HIF-1A that permanently labels hypoxic cells has been proven a powerful tool to trace in time hypoxic cells and has been used to visualize the fate of post-hypoxic cells in the development of distant metastasis [35]. The fusion of the cre with both the HIF-1AODDD and the ERT2 offers the possibility to longitudinally trace cells that have been hypoxic at a very specific point in time, e.g. at the time of irradiation. Such a reporter has been used to monitor the impact of hypoxic cells in primary tumor recurrence post-irradiation either with a luciferase-based fluorescent reporter [33] or with a dual reporter where the activation of cre permanently drives the expression of a second reporter gene [34]. However, these efforts were hampered by the physical properties of the luciferase or specific fluorophores that require molecular oxygen to acquire the emission state [36], [42], [43] resulting in lack of detection of hypoxic cells at the moment of labelling. Therefore, a significant advancement of our technology is the use of a protein that has oxygen-independent fluorescence emission properties (UnaG) into a lineage tracing tool that adequately labels cells in hypoxia [36]. Thus, while those earlier systems proved very useful to visualize the fate of post-hypoxic cells in the development of distant metastasis [35], they were unable to longitudinally trace cells that were hypoxic at a very specific point in time.
Overlap of UnaG + cells with pimonidazole + cells in vivo was less than 100 %. Possibly, cross-section overlaying generates local mismatching effects, resulting in an underestimation of the overlap. Alternatively, the difference could be due to the differential biological properties of the various hypoxia markers. Pimonidazole stains cells that are below 1.3% Oxygen level and has been reported to localize to peri-necrotic areas, indicative of very robust binding to chronic hypoxic zones and excellent diffusion properties [44], [45]. Indeed, pimonidazole was mainly located in peri-necrotic zones (Supp. Fig. 3A & 3B). HIF-1α stabilization already occurs at higher levels of oxygen partial pressure [46], so that endogenous hypoxia markers could already respond to HIF signaling occurring at higher oxygen tension (3% O2), generating inconsistencies between pimonidazole and endogenous hypoxia markers[44], [46]. Based on our 2D and 3D data, cre-dependent recombination seems to occur in the cells under low oxygen tension for a prolonged period, reflected by the high overlap of UnaG+/Pimonidazole + in our study and others [44], [46]. Thus, cells that are UnaG+/pimonidazole-, which mostly localize away from the peri-necrotic zones, most likely arise from activation of HIF signaling at higher oxygen tension, too high for bio-reduction of pimonidazole (Fig. 3D, Suppl. Fig. 3B). HIF-1α protein stabilization has been shown to occur independent of oxygen as a response to excessive ROS levels produced by mitochondria leading to oxidation of cysteine residues in the PHD2 protein resulting in their homodimerization and subsequent inactivation [47]. Additionally, increased levels of critical metabolites of the tricarboxil Acid (TCA) cycle that can arise as a consequence of mutations in succinate dehydrogenase or fumarate hydratase can also affect the stabilization of HIF-1α protein [48]. These mutations result in increased levels of succinate or fumarate, metabolites that can compete with α-ketoglutarate, a critical substrate of PHDs. Inversely, Pimo+/UnaG- cells were almost exclusively located close to peri-necrotic areas (Suppl. Fig. 3B), possibly due to diffusion limitations of tamoxifen or lack of HIF signaling. Indeed, earlier studies indicated that hypoxic cells close to necrotic areas lack HIF-1α and CA9 staining [44], [49], [50]. Additionally, in very low oxygen tensions it has been previously demonstrated that other molecular pathways play more critical roles in cellular adaptation than HIF signaling [51]. With these considerations in mind, we can conclude that the UnaG reporter is effectively tagging the majority of hypoxic cells within H1299 tumors.
We observe little proliferation in hypoxic areas of the tumors, in line with several reports indicating that hypoxia promotes cellular quiescence [31], [35], [52], [53], [54], [55]. This phenomenon is more pronounced in the peri-necrotic areas and much less evident in other pimonidazole-positive areas, supporting the notion that time and level of hypoxia play critical roles in the acquisition of the “quiescent” phenotype. Since there is limited space for reoxygenation in unirradiated tumors, the initial UnaG+ fraction will deplete over time either by being “pushed” into the necrotic zones, or by being outcompeted by highly proliferative normoxic tumor cells. In sharp contrast, in the irradiated tumors UnaG+ cells enrich, and are located outside pimonidazole-positive areas in the end-stage tumors, indicating they must have been reoxygenated. Radiation-induced reoxygenation has been demonstrated to occur in various tumor types and is a very complex phenomenon that varies in time within and across different tumor models [28], [56]. In unirradiated tumors, UnaG + cells are in their vast majority quiescent (or slow cycling) and remain in hypoxic zones. In the recurrent tumors, they acquire a more proliferative status and localize to (re)oxygenized areas. These data are in line with the increased proliferation of post-hypoxic cells in xenografts [34]. The increased proliferation of the post-hypoxic cells at the time of relapse could be also connected to the initially tagged UnaG-positive cells that are also positive for proliferation markers. The fact that this population is present in unperturbed tumors and the observation that the fraction of UnaG+ cells decreases over time if tumors are left unirradiated suggests that the impact of this population in the regrowing tumor might be limited. However, the radiation-induced changes might make the post-irradiated tumor microenvironment very different compared to un-irradiated tumors where there is little space for this population to expand. Our clonogenic data also suggest that the hypoxic cells that have a survival advantage upon irradiation, are the ones that remain quiescent at the first few days after reoxygenation, consistent with previous data from our lab indicating that hypoxia-induced quiescence is a critical component of hypoxic cell radioresistance [32]. With these considerations in mind, our findings on the relapse-stimulating role of hypoxic cells in H1299- and 4T1-derived cell lines, provide evidence for a more global role for hypoxic cells in tumor relapse.
Our data provide proof that levels of hypoxia, ranging 0–3 %, at the time of irradiation is what bestows cells with the capacity to drive tumor relapse. Based on the oxygen fixation theory, radiobiological hypoxia (severe hypoxia < 0.5 % O2) contributes to radioresistance by lowering the induction of DNA damage by IR-induced ROS. Indeed, hypoxic cells that are irradiated under nearly anoxic conditions become highly radioresistant (approximately 2.5-fold). However, irradiating previously hypoxic cells under mild hypoxia or fully reoxygenated conditions, still results in significant radioprotection, even though oxygen levels were restored. Thus, hypoxia-induced radioresistance is a more complex phenomenon than previously thought. Following IR, spheroids and xenografted tumors shrink in size and enter a latent period where macroscopically little changes are evident. At the end of this latent phase, both spheroids and tumors regrow with fast kinetics. Consistently, UnaG+ cells are dormant in xenografted tumors immediately post-irradiation, while at the end-stage, they are highly proliferative. The cells in the hypoxic cores of the spheroids regain proliferative capacity after irradiation and drive the regrowth of spheroids. Given the decrease in spheroid size and the oxygen diffusion distances (Suppl. Fig. 2A) and our data from the culturing of spheroids with two hypoxia markers, suggest that the surviving UnaG+ cells are quickly reoxygenated. However, it takes a considerable amount of time for the spheroids to grow again in size. This is consistent with previous observations that dormant cells are the key source of tumor relapse [57]. Also, the acquisition of a dormant diapause-like state has been proposed as a mechanism of chemotherapeutic drug tolerance resistance in colorectal cancer cells [58] and breast cancer cells [59]. Interestingly in both papers, there is clear evidence of a lack of pre-existing resistant clones. Cell cycle re-entry of hypoxic cells seems to be influenced by the “depth” of the quiescent state, as demonstrate by the fact that cells that have been hypoxic for 72 h take longer to re-enter the proliferative cycle than cells that have been hypoxic for 24 hr. and the reoxygenation that provides them with nutrients post-IR. We cannot yet conclude what the optimal “depth” of quiescence is to create hypoxic cells that have been optimized to drive tumor regrowth. Interestingly, relative expression of the Ki-67 marker has been previously used as an indicator of the time the cells were quiescent [60]. We observe complete absence of the Ki-67 marker in a large fraction of hypoxic cells, which indicates that they have been probably quiescent for a long time. Reoxygenation of tumors in situ could be evaluated using hypoxia PET scans at different weeks post-initiation of treatment. Indeed, reoxygenation has been shown to be a good prognostic marker for the outcome of HNSCC patients [11], [30] clearly demonstrating that reoxygenation is a key process in the radiosensitization of tumors. Our data imply, that in addition to reoxygenation, we need to be able to monitor cell cycle re-entry of pretreatment hypoxic cells, to better predict the optimal fractionation scheme. The data of the tumors and the spheroids presented here are consistent with a model that quiescence/dormancy/diapause could promote the survival and radiation resistance of hypoxic cells and indicate that some kind of cellular reprogramming is needed before the cells exit the dormant state. Currently, we do not know exactly what process underlies this slow conversion of hypoxic cells into proliferating tumor cells, but this is a matter of ongoing research in our group. Nonetheless, our data provides solid support for the conclusion that the hypoxic population has a clear survival advantage over the normoxic population following irradiation, and this is what drives tumor relapse post-radiotherapy.
The apparent discordance of reoxygenation time with the start of spheroid regrowth causes a substantial challenge in the clinical application of anti-cancer therapy. Our data in spheroid and tumor models underscore the notion that eradication of initially hypoxic cells at the start of radiation treatment will increase the efficacy of anti-cancer therapy [46], [61]. While targeting of hypoxic cells with hypoxia-activated prodrugs (HAPs) has shown promising results in preclinical models [49], [62], there is no clinically approved HAP. Our long-term experiments in spheroids indicate that the timing of delivery will be of critical importance, and that might be why these drugs have not yet translated into a clinical benefit [63]. Utilizing the acquired knowledge from our spheroid experiments and previously published observations that proliferative retina pigment epithelium (RPE) cells are more sensitive to irradiation [32], we hypothesized that a treatment break and re-irradiation at the time that the hypoxic cells acquire a proliferative state might be an interesting approach to effectively target these cells. Our data provide the first experimental evidence that such an approach might be very beneficial. Of note, these findings are supported by the outcome of a recent clinical study in lung cancer patients [64].
Taken together, we report a novel tool to tag hypoxic cells in a time-controlled manner, and subsequently trace their fate. Using this technique, we were able to follow cells that were hypoxic at the time of irradiation and demonstrate that these cells drive tumor regrowth in both spheroids and in tumors in vivo. A further understanding of the exact dynamics of this process may lead to different treatment scheduling, resulting in better long-term outcomes for cancer patients.
CRediT authorship contribution statement
Apostolos Menegakis: Writing – review & editing, Writing – original draft, Supervision, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Claire Vennin: Data curation. Jonathan Ient: Data curation. Arjan J. Groot: Data curation. Lenno Krenning: Writing – review & editing, Writing – original draft, Data curation, Conceptualization. Rob Klompmaker: Data curation. Anoek Friskes: Visualization, Data curation. Mila Ilic: Visualization, Data curation. Ala Yaromina: Software, Data curation. Rolf Harkes: Software. Bram van den Broek: Software. Jan Jakob Sonke: Resources, Conceptualization. Monique De Jong: Conceptualization. Jolanda Piepers: Data curation. Jacco van Rheenen: Resources, Conceptualization. Marc A. Vooijs: . René H. Medema: Writing – review & editing, Writing – original draft, Supervision, Project administration, Conceptualization.
Funding
This work was supported by the German Research Council (Deutche Forshungsgemeineschaft) (research fellowship ME 4924/1–1) (AM), European Research Council (ERC-CoG: 648,804 and ERc-CoG: 617060) (MV), Doctor Josef Steiner Foundation, Human Frontiers in Science Program Fellowship (CV) and the Oncode Institute (RM, JvR).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to thank the members of flowcytometry facility (Martijn van Baalen, Frank van Diepen, Anita Pfauth and Debajit Bhowmick, Guido de Roo), the members of the bioimaging facility (Lenny Brooks, Marjolijn Mertz and Bram van de broek) and all the members of the experimental animal pathology facility (JI-Ying Song, Sjoerd Klarenbeek, Ellen Riem, Joost van Ooij, Lex de Vrije, Jelrik van der Meer, Britt Janssen) of the Netherlands Cancer Institute for their excellent technical support. In addition, the authors would like to thank Professor Friedeman Kiefer for his kind donation of the original UnaG plasmid from which our construct was made.
Research data are stored in an NKI institutional repository and will be shared upon request to the corresponding author.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.radonc.2024.110592.
Contributor Information
Apostolos Menegakis, Email: a.menegakis@nki.nl.
Marc A. Vooijs, Email: marc.vooijs@maastrichtuniversity.nl.
René H. Medema, Email: r.h.medema-2@prinsesmaximacentrum.nl.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Development and validation of the HIF-1A-Cre-ERT2-UnaG hypoxia reporter in H1299 cells. A) Genomic q-PCR in H1299 wt cells and in H1299 transduced with the plasmids of the hypoxia reporter using primers against UnaG protein, Cre, and a housekeeping gene (HSPA4). B) Western blot analysis of anti-HA to visualize the UnaG protein induction. Samples were cultured for 16 hours in different oxygen levels in the presence or absence of 4-OHT. β-Actin was used as a loading control. C) Western blot analysis of endogenous HIF-1A and Cre from the same samples of H1299-HIFcreUnaG cells. H1299-HIFcreUnaG cells were cultured for 24 hours in 0,2% O2 in the presence of 4-OHT and samples were harvested in hypoxic conditions, or at different points after reoxygenation. Cells cultured in normoxia were used as negative control and in the presence of Deferoxamine as positive control. Lamin A was used as loading control. D) Western blot analysis of endogenous HIF-1A in H1299-HIFcreUnaG cells kept for 72h in normoxia or hypoxia (1%) in the presence of 500nM 4-OHT and treated for 96h with siRNAs against HIF1A, EPAS1 (HIF2A), or both. As extra controls, cells treated with siRNA against luciferase (Non targeting) and cells cultured with CoCL2 were used as positive controls. Tubulin was used as a loading control. E) Quantification of percentages of UnaG+ cells from flow cytometer analysis from the same samples as in B. F) Cell cycle distribution based on the Propidium Iodine (P.I.) staining of H1299-HIFcreUnaG cells kept either for 72h in normoxia or in 1% hypoxia for different duration (in days). The percentage of cell cycle phases are depicted in the plots.
H1299-HIFcreUnaG spheroids contain areas of hypoxia where UnaG+ cells reside and upon irradiation UnaG+ cells enrich in the spheroids. A) Schematic representation of the experimental design with the indicated times were the exogenous hypoxia markers were supplied to H1299-HIFcreUnaG spheroids. B) Representative images of a spheroid irradiated with 8 Gy and fixed 48 hours post-irradiation. The individual stainings DAPI (nuclear staining), pimonidazole (pre-treatment hypoxic cells), F6 (post-treatment hypoxic cells) and EdU (S-Phase, proliferative cells) are shown along with the image where the triple-staining is depicted. C) Nuclear segmentation and nuclear annotation process in the Qu-Path software across the three different staining parameters. The final processing of each staining parameter along with the implementation of the dual mode of each of-triple staining based on the segmented thresholds is depicted. D) Analysis of the fraction of cells that are hypoxic before irradiation (pimonidazole+ fraction) and remain hypoxic (pimo+/F6+ fraction) or reoxygenate (pimo+/F6- fraction) over different time points in un-irradiated and irradiated H1299-HIFcreUnaG spheroids. The percentage of cells in fractions are shown in a stacked plot. Error bars represent the standard deviation of mean estimation corresponding of the analysis of at least 4 spheroid cross-section per timepoint. E) Distribution of the EdU positive cells in H1299-HIFcreUnaG spheroids 48 hours post-8Gy irradiation. The mean fraction of EdU-positive cells located in pimo-positive (hypoxic prior to irradiation) or pimo-negative (normoxic at the time of irradiation) from 8 different spheroids analyzed is depicted along with the error bars showing the standard deviation of the mean. F) Characteristic images of a middle Z-plane view of different live H1299-HIFcreUnaG spheroids made with epifluorescent microscopy. The images represent the combined image of the brightfield (all cells) and the FITC channel image (UnaG+) cells and indicate the dynamic changes in percentage of UnaG+ cells and spheroid area that follow irradiation of H1299-HIFcreUnaG spheroids with 8 Gy. Such images were the basis of quantification of spheroid area that are depicted in figure 2D. G) Quantification of flow cytometer plots indicating the percentage of UnaG+ cells over the total amount of cells isolated from H1299-HIFcreUnaG spheroids (at endpoint) exposed to different radiation doses. Each dot in the plots represent the quantification of 1 spheroid and the data showed here are depicting the data from at least three independent experiments pooled together. H) Characteristic dot plots that show the fraction of UnaG+ cells isolated from H1299-HIFcreUnaG spheroids without cultivation with 4-OHT and cultured 72 hours in the presence of 4-OHT. The numbers in the plot indicate the corresponding percentages.
Nuclei segmentation steps with the Qu-Path software. A) Nuclei segmentation and color-coded annotation based on single staining by Qu-Path software. On the left panel the tumor cross-section with the original staining and imaging is shown for the three different sections/stainings. A specific area (also depicted in figure 3) is then zoomed-in to visualize the segmentation process of each nucleus with StarDist plugin on the DAPI channel of each section and the application of the threshold for each individual staining which generates a binary image (positive/negative) cells per staining/section and the filling in of the nucleus to generate the final image. Scale bars of the whole tumor cross-section in the left panels (whole tumor cross-section) correspond to 1000 μm while all the corresponding scale bars of the zoom-in images correspond to 250μm. B) Distribution of the staining and the nuclear annotation across the three different whole tumor sections for the different stained parameters. In the upper panel the original view of the stained/imaged section is shown and in the lower panels the final processing of each section after the implementation of the steps shown in A for each individual binary image and in the dual color assignment. Scale bars correspond to 1000 μm C) The total number of Ki67+ cells and their percentage distribution in pimonidazole+ (red part of the graph) and pimonidazole – cells (blue part of the graph) for all the five tumors analyzed are shown.
Microenvironmental parameters in H1299-HIFcreUnaG tumors post-IR. A) Characteristic view of an overlaid H1299-HIFcreUnaG tumor cross-section unirradiated (same as in fig. 4D). All the different color annotations assigned to all nuclei of the sections are shown after align and processing the three different cross-sections with the Qu-Path software. In the zoom-in view the individual and dual-color assignment for each staining are deconvolved. A characteristic view from a newly developed hypoxic (pimonidazole+ area) is depicted. Scale bar of the whole tumor section in the left panel (whole tumor cross-section) corresponds to 1000μm and the corresponding of the zoomed-in in the right panel to 250 μm. B) Characteristic view of an overlaid H1299-HIFcreUnaG tumor cross-section of an irradiated and re-grown tumor (same as in fig. 4E) with all the different color annotations assigned as in A. Two characteristic views are shown with a newly formed hypoxic area in the first panel (depicted as 1) and a high proliferative regrowing part in the second (depicted as 2). Scale bar of the whole tumor section in the left panel corresponds to 1000μm and the corresponding of the zoomed-in in the right panel to 250 μm.
Development and validation of the HIF-1A-Cre-ERT2-DsRed/eGFP hypoxia reporter in 4T1 cells. A) Schematic representation that indicates the design of the two plasmids of the hypoxic cell reporter and the way that the inducible reporter expresses the eGFP protein under hypoxia and in the presence of 4-OHT. B) Western blot analysis of eGFP protein induction. Samples were cultured for 16 hours in different oxygen levels in the presence or absence of 4-OHT. β-Actin was used as a loading control. C) Western blot analysis of endogenous HIF-1A and HIF-1A-Cre-ERT2 fusion protein from the same samples of 4T1-hypGFP cells. 4T1-HIF-cre-eGFP cells were cultured for 24 hours in 0,2% O2 in the presence of 4-OHT and samples were harvested in hypoxic conditions, or at different points after reoxygenation. Cells cultured in normoxia were used as negative control and in the presence of Deferoxamine as positive control. Lamin A was used as a loading control. D-E) Flow cytometer plots from 4T1- hypGFP cells cultured for 72h in normoxia or hypoxia 1% in the presence or absence of 500nM 4-OHT and reoxygenated for 8 hours. The respective quantification of one experiment in D. The bar graph represents the means of three independent experiments and the error bars the standard deviation of mean estimation in E. F) Colony forming assay of 4T1- hypGFP cells. 4T1- hypGFP cells were irradiated with graded single doses of irradiation after being for 72 hours under normoxic conditions (blue curve), or for 72 hours in hypoxia (1% O2) and subsequently irradiated under almost anoxic conditions (0.001% O2 – red curve HAO), before reoxygenated after IR. The data were fitted with a linear quadratic model. The data points represent the means of three independent experiments and the error bars are the standard deviation of the means estimation. G) Percentage of eGFP+ cells over the total amount of living cells (DAPI-negative) cells isolated from unirradiated and irradiated (with graded single doses) 4T1- hypGFP spheroids assayed at the endpoint of the experiment (45 Days post-IR). The data points represent the means of three independent experiments and the error bars are the standard deviation of the means estimation. H) Spheroid control assay for 4T1-HIF-cre-eGFP spheroids irradiated with graded single doses as in G. The sigmoid curve indicate the probability of an irradiated spheroid to be controlled and not regrow. The numbers of the controlled spheroid as a fraction of the total spheroids assessed per dose level for 3 independent experiments are depicted. I) Characteristic view of a hypoxic area of a 4T1- hypGFP xenografted tumor. In the zoomed-in image, the overlap of eGFP-positive cells in the pimonidazole staining tumor region is depicted. J) Percentage of eGFP+ cells quantified in the different tumors per condition. The blue violin plot depicts the values of the eGFP+ cells from 4T1-HIF-cre-EGFP unirradiated tumors, while the red and green violin plots depict the treatment arms. Each dot corresponds to one tumor analyzed and the p values depict the values of the Kruskal-Wallis test comparison.
References
- 1.Stapor P., Wang X., Goveia J., Moens S., Carmeliet P. Angiogenesis revisited - role and therapeutic potential of targeting endothelial metabolism. J Cell Sci. 2014;127:4331–4341. doi: 10.1242/jcs.153908. [DOI] [PubMed] [Google Scholar]
- 2.Hockel M., Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 3.Greer S.N., Metcalf J.L., Wang Y., Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31:2448–2460. doi: 10.1038/emboj.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keith B., Johnson R.S., Simon M.C. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee P., Chandel N.S., Simon M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. 2020;21:268–283. doi: 10.1038/s41580-020-0227-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza G.L. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nordsmark M., Bentzen S.M., Rudat V., Brizel D., Lartigau E., Stadler P., et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol. 2005;77:18–24. doi: 10.1016/j.radonc.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 8.Sakso M., Mortensen L.S., Primdahl H., Johansen J., Kallehauge J., Hansen C.R., et al. Influence of FAZA PET hypoxia and HPV-status for the outcome of head and neck squamous cell carcinoma (HNSCC) treated with radiotherapy: Long-term results from the DAHANCA 24 trial ( NCT01017224) Radiother Oncol. 2020;151:126–133. doi: 10.1016/j.radonc.2020.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Sanduleanu S., Hamming-Vrieze O., Wesseling F.W.R., Even A.J.G., Hoebers F.J., Hoeben A., et al. [(18)F]-HX4 PET/CT hypoxia in patients with squamous cell carcinoma of the head and neck treated with chemoradiotherapy: Prognostic results from two prospective trials. Clin Transl Radiat Oncol. 2020;23:9–15. doi: 10.1016/j.ctro.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welz S., Paulsen F., Pfannenberg C., Reimold M., Reischl G., Nikolaou K., et al. Dose escalation to hypoxic subvolumes in head and neck cancer: A randomized phase II study using dynamic [(18)F]FMISO PET/CT. Radiother Oncol. 2022;171:30–36. doi: 10.1016/j.radonc.2022.03.021. [DOI] [PubMed] [Google Scholar]
- 11.Zschaeck S., Lock S., Hofheinz F., Zips D., Sakso Mortensen L., Zophel K., et al. Individual patient data meta-analysis of FMISO and FAZA hypoxia PET scans from head and neck cancer patients undergoing definitive radio-chemotherapy. Radiother Oncol. 2020;149:189–196. doi: 10.1016/j.radonc.2020.05.022. [DOI] [PubMed] [Google Scholar]
- 12.Kim J.I., Choi K.U., Lee I.S., Choi Y.J., Kim W.T., Shin D.H., et al. Expression of hypoxic markers and their prognostic significance in soft tissue sarcoma. Oncol Lett. 2015;9:1699–1706. doi: 10.3892/ol.2015.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo D., Ren H., Zhang W., Xian H., Lian K., Liu H. Clinicopathological and prognostic value of hypoxia-inducible factor-1alpha in patients with bone tumor: a systematic review and meta-analysis. J Orthop Surg Res. 2019;14:56. doi: 10.1186/s13018-019-1101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreno Roig E., Yaromina A., Houben R., Groot A.J., Dubois L., Vooijs M. Prognostic role of hypoxia-inducible factor-2alpha tumor cell expression in cancer patients: a meta-analysis. Front Oncol. 2018;8:224. doi: 10.3389/fonc.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shamis S.A.K., McMillan D.C., Edwards J. The relationship between hypoxia-inducible factor 1alpha (HIF-1alpha) and patient survival in breast cancer: Systematic review and meta-analysis. Crit Rev Oncol Hematol. 2021;159 doi: 10.1016/j.critrevonc.2021.103231. [DOI] [PubMed] [Google Scholar]
- 16.Wang J., Wang Y., Xing P., Liu Q., Zhang C., Sui Y., et al. Development and validation of a hypoxia-related prognostic signature for breast cancer. Oncol Lett. 2020;20:1906–1914. doi: 10.3892/ol.2020.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fjeldbo C.S., Hompland T., Hillestad T., Aarnes E.K., Gunther C.C., Kristensen G.B., et al. Combining imaging- and gene-based hypoxia biomarkers in cervical cancer improves prediction of chemoradiotherapy failure independent of intratumour heterogeneity. EBioMedicine. 2020;57 doi: 10.1016/j.ebiom.2020.102841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lalonde E., Ishkanian A.S., Sykes J., Fraser M., Ross-Adams H., Erho N., et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 2014;15:1521–1532. doi: 10.1016/S1470-2045(14)71021-6. [DOI] [PubMed] [Google Scholar]
- 19.Yang L., Roberts D., Takhar M., Erho N., Bibby B.A.S., Thiruthaneeswaran N., et al. Development and validation of a 28-gene hypoxia-related prognostic signature for localized prostate cancer. EBioMedicine. 2018;31:182–189. doi: 10.1016/j.ebiom.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lane B., Khan M.T., Choudhury A., Salem A., West C.M.L. Development and validation of a hypoxia-associated signature for lung adenocarcinoma. Sci Rep. 2022;12:1290. doi: 10.1038/s41598-022-05385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linge A., Lohaus F., Lock S., Nowak A., Gudziol V., Valentini C., et al. HPV status, cancer stem cell marker expression, hypoxia gene signatures and tumour volume identify good prognosis subgroups in patients with HNSCC after primary radiochemotherapy: A multicentre retrospective study of the German Cancer Consortium Radiation Oncology Group (DKTK-ROG) Radiother Oncol. 2016;121:364–373. doi: 10.1016/j.radonc.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Cosse J.P., Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem. 2008;8:790–797. doi: 10.2174/187152008785914798. [DOI] [PubMed] [Google Scholar]
- 23.Brown J.M. The hypoxic cell: a target for selective cancer therapy–eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res. 1999;59:5863–5870. [PubMed] [Google Scholar]
- 24.Rockwell S., Dobrucki I.T., Kim E.Y., Marrison S.T., Vu V.T. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med. 2009;9:442–458. doi: 10.2174/156652409788167087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scharping N.E., Rivadeneira D.B., Menk A.V., Vignali P.D.A., Ford B.R., Rittenhouse N.L., et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. 2021;22:205–215. doi: 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson M.J., Vignali P.D.A., Mullett S.J., Overacre-Delgoffe A.E., Peralta R.M., Grebinoski S., et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. 2021;591:645–651. doi: 10.1038/s41586-020-03045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H., Jiang H., Van De Gucht M., De Ridder M. Hypoxic radioresistance: Can ROS be the key to overcome it? Cancers (Basel) 2019;11 doi: 10.3390/cancers11010112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crokart N., Jordan B.F., Baudelet C., Ansiaux R., Sonveaux P., Gregoire V., et al. Early reoxygenation in tumors after irradiation: determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int J Radiat Oncol Biol Phys. 2005;63:901–910. doi: 10.1016/j.ijrobp.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 29.Taylor E., Zhou J., Lindsay P., Foltz W., Cheung M., Siddiqui I., et al. Quantifying reoxygenation in pancreatic cancer during stereotactic body radiotherapy. Sci Rep. 2020;10:1638. doi: 10.1038/s41598-019-57364-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zips D., Zophel K., Abolmaali N., Perrin R., Abramyuk A., Haase R., et al. Exploratory prospective trial of hypoxia-specific PET imaging during radiochemotherapy in patients with locally advanced head-and-neck cancer. Radiother Oncol. 2012;105:21–28. doi: 10.1016/j.radonc.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 31.Hubbi M.E., Semenza G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am J Physiol Cell Physiol. 2015;309:C775–C782. doi: 10.1152/ajpcell.00279.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menegakis A., Klompmaker R., Vennin C., Arbusa A., Damen M., van den Broek B., et al. Resistance of hypoxic cells to ionizing radiation is mediated in part via hypoxia-induced quiescence. Cells. 2021;10 doi: 10.3390/cells10030610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harada H., Inoue M., Itasaka S., Hirota K., Morinibu A., Shinomiya K., et al. Cancer cells that survive radiation therapy acquire HIF-1 activity and translocate towards tumour blood vessels. Nat Commun. 2012;3:783. doi: 10.1038/ncomms1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vermeer J.A.F., Ient J., Markelc B., Kaeppler J., Barbeau L.M.O., Groot A.J., et al. A lineage-tracing tool to map the fate of hypoxic tumour cells. Dis Model Mech. 2020;13 doi: 10.1242/dmm.044768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Godet I., Shin Y.J., Ju J.A., Ye I.C., Wang G., Gilkes D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat Commun. 2019;10:4862. doi: 10.1038/s41467-019-12412-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erapaneedi R., Belousov V.V., Schafers M., Kiefer F. A novel family of fluorescent hypoxia sensors reveal strong heterogeneity in tumor hypoxia at the cellular level. EMBO J. 2016;35:102–113. doi: 10.15252/embj.201592775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welm B.E., Dijkgraaf G.J., Bledau A.S., Welm A.L., Werb Z. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell. 2008;2:90–102. doi: 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groot A.J., Habets R., Yahyanejad S., Hodin C.M., Reiss K., Saftig P., et al. Regulated proteolysis of NOTCH2 and NOTCH3 receptors by ADAM10 and presenilins. Mol Cell Biol. 2014;34:2822–2832. doi: 10.1128/MCB.00206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feil R., Wagner J., Metzger D., Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- 41.Le A., Stine Z.E., Nguyen C., Afzal J., Sun P., Hamaker M., et al. Tumorigenicity of hypoxic respiring cancer cells revealed by a hypoxia-cell cycle dual reporter. PNAS. 2014;111:12486–12491. doi: 10.1073/pnas.1402012111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Godet I., Doctorman S., Wu F., Gilkes D.M. Detection of hypoxia in cancer models: significance, challenges, and advances. Cells. 2022;11 doi: 10.3390/cells11040686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heim R., Prasher D.C., Tsien R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. PNAS. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rademakers S.E., Lok J., van der Kogel A.J., Bussink J., Kaanders J.H. Metabolic markers in relation to hypoxia; staining patterns and colocalization of pimonidazole, HIF-1alpha, CAIX, LDH-5, GLUT-1, MCT1 and MCT4. BMC Cancer. 2011;11:167. doi: 10.1186/1471-2407-11-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaanders J.H., Wijffels K.I., Marres H.A., Ljungkvist A.S., Pop L.A., van den Hoogen F.J., et al. Pimonidazole binding and tumor vascularity predict for treatment outcome in head and neck cancer. Cancer Res. 2002;62:7066–7074. [PubMed] [Google Scholar]
- 46.Wilson W.R., Hay M.P. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 47.Lee G., Won H.S., Lee Y.M., Choi J.W., Oh T.I., Jang J.H., et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1alpha activation. Sci Rep. 2016;6:18928. doi: 10.1038/srep18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pollard P.J., Briere J.J., Alam N.A., Barwell J., Barclay E., Wortham N.C., et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 49.Singleton D.C., Macann A., Wilson W.R. Therapeutic targeting of the hypoxic tumour microenvironment. Nat Rev Clin Oncol. 2021;18:751–772. doi: 10.1038/s41571-021-00539-4. [DOI] [PubMed] [Google Scholar]
- 50.Swartz J.E., Smits H.J.G., Philippens M.E.P., de Bree R., J HAMK, Willems SM. Correlation and colocalization of HIF-1alpha and pimonidazole staining for hypoxia in laryngeal squamous cell carcinomas: A digital, single-cell-based analysis. Oral Oncol. 2022;128 doi: 10.1016/j.oraloncology.2022.105862. [DOI] [PubMed] [Google Scholar]
- 51.Wouters B.G., Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 52.Fluegen G., Avivar-Valderas A., Wang Y., Padgen M.R., Williams J.K., Nobre A.R., et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19:120–132. doi: 10.1038/ncb3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goda N., Dozier S.J., Johnson R.S. HIF-1 in cell cycle regulation, apoptosis, and tumor progression. Antioxid Redox Signal. 2003;5:467–473. doi: 10.1089/152308603768295212. [DOI] [PubMed] [Google Scholar]
- 54.Goda N., Ryan H.E., Khadivi B., McNulty W., Rickert R.C., Johnson R.S. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23:359–369. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ortmann B., Druker J., Rocha S. Cell cycle progression in response to oxygen levels. Cell Mol Life Sci. 2014;71:3569–3582. doi: 10.1007/s00018-014-1645-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petersen C., Zips D., Krause M., Schone K., Eicheler W., Hoinkis C., et al. Repopulation of FaDu human squamous cell carcinoma during fractionated radiotherapy correlates with reoxygenation. Int J Radiat Oncol Biol Phys. 2001;51:483–493. doi: 10.1016/s0360-3016(01)01686-8. [DOI] [PubMed] [Google Scholar]
- 57.Santos-de-Frutos K., Djouder N. When dormancy fuels tumour relapse. Commun Biol. 2021;4:747. doi: 10.1038/s42003-021-02257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rehman S.K., Haynes J., Collignon E., Brown K.R., Wang Y., Nixon A.M.L., et al. Colorectal cancer cells enter a diapause-like DTP state to survive chemotherapy. Cell. 2021;184:e21. doi: 10.1016/j.cell.2020.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dhimolea E., de Matos S.R., Kansara D., Al'Khafaji A., Bouyssou J., Weng X., et al. An embryonic diapause-like adaptation with suppressed myc activity enables tumor treatment persistence. Cancer Cell. 2021;39:e11. doi: 10.1016/j.ccell.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller I., Min M., Yang C., Tian C., Gookin S., Carter D., et al. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 2018;24:e5. doi: 10.1016/j.celrep.2018.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wigerup C., Pahlman S., Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152–169. doi: 10.1016/j.pharmthera.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 62.Baran N., Konopleva M. Molecular pathways: hypoxia-activated prodrugs in cancer therapy. Clin Cancer Res. 2017;23:2382–2390. doi: 10.1158/1078-0432.CCR-16-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salem A., Asselin M.C., Reymen B., Jackson A., Lambin P., West C.M.L., et al. Targeting hypoxia to improve non-small cell lung cancer outcome. J Natl Cancer Inst. 2018;110 doi: 10.1093/jnci/djx160. [DOI] [PubMed] [Google Scholar]
- 64.Alite F., Stang K., Balasubramanian N., Adams W., Shaikh M.P., Small C., et al. Local control dependence on consecutive vs. nonconsecutive fractionation in lung stereotactic body radiation therapy. Radiother Oncol. 2016;121:9–14. doi: 10.1016/j.radonc.2016.07.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Development and validation of the HIF-1A-Cre-ERT2-UnaG hypoxia reporter in H1299 cells. A) Genomic q-PCR in H1299 wt cells and in H1299 transduced with the plasmids of the hypoxia reporter using primers against UnaG protein, Cre, and a housekeeping gene (HSPA4). B) Western blot analysis of anti-HA to visualize the UnaG protein induction. Samples were cultured for 16 hours in different oxygen levels in the presence or absence of 4-OHT. β-Actin was used as a loading control. C) Western blot analysis of endogenous HIF-1A and Cre from the same samples of H1299-HIFcreUnaG cells. H1299-HIFcreUnaG cells were cultured for 24 hours in 0,2% O2 in the presence of 4-OHT and samples were harvested in hypoxic conditions, or at different points after reoxygenation. Cells cultured in normoxia were used as negative control and in the presence of Deferoxamine as positive control. Lamin A was used as loading control. D) Western blot analysis of endogenous HIF-1A in H1299-HIFcreUnaG cells kept for 72h in normoxia or hypoxia (1%) in the presence of 500nM 4-OHT and treated for 96h with siRNAs against HIF1A, EPAS1 (HIF2A), or both. As extra controls, cells treated with siRNA against luciferase (Non targeting) and cells cultured with CoCL2 were used as positive controls. Tubulin was used as a loading control. E) Quantification of percentages of UnaG+ cells from flow cytometer analysis from the same samples as in B. F) Cell cycle distribution based on the Propidium Iodine (P.I.) staining of H1299-HIFcreUnaG cells kept either for 72h in normoxia or in 1% hypoxia for different duration (in days). The percentage of cell cycle phases are depicted in the plots.
H1299-HIFcreUnaG spheroids contain areas of hypoxia where UnaG+ cells reside and upon irradiation UnaG+ cells enrich in the spheroids. A) Schematic representation of the experimental design with the indicated times were the exogenous hypoxia markers were supplied to H1299-HIFcreUnaG spheroids. B) Representative images of a spheroid irradiated with 8 Gy and fixed 48 hours post-irradiation. The individual stainings DAPI (nuclear staining), pimonidazole (pre-treatment hypoxic cells), F6 (post-treatment hypoxic cells) and EdU (S-Phase, proliferative cells) are shown along with the image where the triple-staining is depicted. C) Nuclear segmentation and nuclear annotation process in the Qu-Path software across the three different staining parameters. The final processing of each staining parameter along with the implementation of the dual mode of each of-triple staining based on the segmented thresholds is depicted. D) Analysis of the fraction of cells that are hypoxic before irradiation (pimonidazole+ fraction) and remain hypoxic (pimo+/F6+ fraction) or reoxygenate (pimo+/F6- fraction) over different time points in un-irradiated and irradiated H1299-HIFcreUnaG spheroids. The percentage of cells in fractions are shown in a stacked plot. Error bars represent the standard deviation of mean estimation corresponding of the analysis of at least 4 spheroid cross-section per timepoint. E) Distribution of the EdU positive cells in H1299-HIFcreUnaG spheroids 48 hours post-8Gy irradiation. The mean fraction of EdU-positive cells located in pimo-positive (hypoxic prior to irradiation) or pimo-negative (normoxic at the time of irradiation) from 8 different spheroids analyzed is depicted along with the error bars showing the standard deviation of the mean. F) Characteristic images of a middle Z-plane view of different live H1299-HIFcreUnaG spheroids made with epifluorescent microscopy. The images represent the combined image of the brightfield (all cells) and the FITC channel image (UnaG+) cells and indicate the dynamic changes in percentage of UnaG+ cells and spheroid area that follow irradiation of H1299-HIFcreUnaG spheroids with 8 Gy. Such images were the basis of quantification of spheroid area that are depicted in figure 2D. G) Quantification of flow cytometer plots indicating the percentage of UnaG+ cells over the total amount of cells isolated from H1299-HIFcreUnaG spheroids (at endpoint) exposed to different radiation doses. Each dot in the plots represent the quantification of 1 spheroid and the data showed here are depicting the data from at least three independent experiments pooled together. H) Characteristic dot plots that show the fraction of UnaG+ cells isolated from H1299-HIFcreUnaG spheroids without cultivation with 4-OHT and cultured 72 hours in the presence of 4-OHT. The numbers in the plot indicate the corresponding percentages.
Nuclei segmentation steps with the Qu-Path software. A) Nuclei segmentation and color-coded annotation based on single staining by Qu-Path software. On the left panel the tumor cross-section with the original staining and imaging is shown for the three different sections/stainings. A specific area (also depicted in figure 3) is then zoomed-in to visualize the segmentation process of each nucleus with StarDist plugin on the DAPI channel of each section and the application of the threshold for each individual staining which generates a binary image (positive/negative) cells per staining/section and the filling in of the nucleus to generate the final image. Scale bars of the whole tumor cross-section in the left panels (whole tumor cross-section) correspond to 1000 μm while all the corresponding scale bars of the zoom-in images correspond to 250μm. B) Distribution of the staining and the nuclear annotation across the three different whole tumor sections for the different stained parameters. In the upper panel the original view of the stained/imaged section is shown and in the lower panels the final processing of each section after the implementation of the steps shown in A for each individual binary image and in the dual color assignment. Scale bars correspond to 1000 μm C) The total number of Ki67+ cells and their percentage distribution in pimonidazole+ (red part of the graph) and pimonidazole – cells (blue part of the graph) for all the five tumors analyzed are shown.
Microenvironmental parameters in H1299-HIFcreUnaG tumors post-IR. A) Characteristic view of an overlaid H1299-HIFcreUnaG tumor cross-section unirradiated (same as in fig. 4D). All the different color annotations assigned to all nuclei of the sections are shown after align and processing the three different cross-sections with the Qu-Path software. In the zoom-in view the individual and dual-color assignment for each staining are deconvolved. A characteristic view from a newly developed hypoxic (pimonidazole+ area) is depicted. Scale bar of the whole tumor section in the left panel (whole tumor cross-section) corresponds to 1000μm and the corresponding of the zoomed-in in the right panel to 250 μm. B) Characteristic view of an overlaid H1299-HIFcreUnaG tumor cross-section of an irradiated and re-grown tumor (same as in fig. 4E) with all the different color annotations assigned as in A. Two characteristic views are shown with a newly formed hypoxic area in the first panel (depicted as 1) and a high proliferative regrowing part in the second (depicted as 2). Scale bar of the whole tumor section in the left panel corresponds to 1000μm and the corresponding of the zoomed-in in the right panel to 250 μm.
Development and validation of the HIF-1A-Cre-ERT2-DsRed/eGFP hypoxia reporter in 4T1 cells. A) Schematic representation that indicates the design of the two plasmids of the hypoxic cell reporter and the way that the inducible reporter expresses the eGFP protein under hypoxia and in the presence of 4-OHT. B) Western blot analysis of eGFP protein induction. Samples were cultured for 16 hours in different oxygen levels in the presence or absence of 4-OHT. β-Actin was used as a loading control. C) Western blot analysis of endogenous HIF-1A and HIF-1A-Cre-ERT2 fusion protein from the same samples of 4T1-hypGFP cells. 4T1-HIF-cre-eGFP cells were cultured for 24 hours in 0,2% O2 in the presence of 4-OHT and samples were harvested in hypoxic conditions, or at different points after reoxygenation. Cells cultured in normoxia were used as negative control and in the presence of Deferoxamine as positive control. Lamin A was used as a loading control. D-E) Flow cytometer plots from 4T1- hypGFP cells cultured for 72h in normoxia or hypoxia 1% in the presence or absence of 500nM 4-OHT and reoxygenated for 8 hours. The respective quantification of one experiment in D. The bar graph represents the means of three independent experiments and the error bars the standard deviation of mean estimation in E. F) Colony forming assay of 4T1- hypGFP cells. 4T1- hypGFP cells were irradiated with graded single doses of irradiation after being for 72 hours under normoxic conditions (blue curve), or for 72 hours in hypoxia (1% O2) and subsequently irradiated under almost anoxic conditions (0.001% O2 – red curve HAO), before reoxygenated after IR. The data were fitted with a linear quadratic model. The data points represent the means of three independent experiments and the error bars are the standard deviation of the means estimation. G) Percentage of eGFP+ cells over the total amount of living cells (DAPI-negative) cells isolated from unirradiated and irradiated (with graded single doses) 4T1- hypGFP spheroids assayed at the endpoint of the experiment (45 Days post-IR). The data points represent the means of three independent experiments and the error bars are the standard deviation of the means estimation. H) Spheroid control assay for 4T1-HIF-cre-eGFP spheroids irradiated with graded single doses as in G. The sigmoid curve indicate the probability of an irradiated spheroid to be controlled and not regrow. The numbers of the controlled spheroid as a fraction of the total spheroids assessed per dose level for 3 independent experiments are depicted. I) Characteristic view of a hypoxic area of a 4T1- hypGFP xenografted tumor. In the zoomed-in image, the overlap of eGFP-positive cells in the pimonidazole staining tumor region is depicted. J) Percentage of eGFP+ cells quantified in the different tumors per condition. The blue violin plot depicts the values of the eGFP+ cells from 4T1-HIF-cre-EGFP unirradiated tumors, while the red and green violin plots depict the treatment arms. Each dot corresponds to one tumor analyzed and the p values depict the values of the Kruskal-Wallis test comparison.