

ABSTRACT
Process intensification has become an important strategy to lower production costs and increase manufacturing capacities for biopharmaceutical products. In particular for the production of viral vectors like lentiviruses (LVs), the transition from (fed‐)batch to perfusion processes is a key strategy to meet the increasing demands for cell and gene therapy applications. However, perfusion processes are associated with higher medium consumption. Therefore, it is necessary to develop appropriate small‐scale models to reduce development costs. In this work, we present the use of the acoustic wave separation technology in combination with the Ambr 250 high throughput bioreactor system for intensified perfusion process development using stable LV producer cells. The intensified perfusion process developed in the Ambr 250 model, performed at a harvest rate of 3 vessel volumes per day (VVD) and high cell densities, resulted in a 1.4‐fold higher cell‐specific functional virus yield and 2.8‐fold higher volumetric virus yield compared to the control process at a harvest rate of 1 VVD. The findings were verified at bench scale after optimizing the bioreactor set‐up, resulting in a 1.4‐fold higher cell‐specific functional virus yield and 3.1‐fold higher volumetric virus yield.
Keywords: gene and cell therapy, lentiviral vector, lentiviral vector production, perfusion process development, small‐scale model, stable suspension producer cell line
A scale‐down model of the acoustic wave‐mediated cell retention technology was used in combination with a mini‐bioreactor system to develop an intensified perfusion process for the production of recombinant lentiviruses. The enhanced process was scaled up to bench‐top bioreactor scale, resulting in a threefold increase in functional vector yield.
1. Introduction
Continuous biomanufacturing becomes increasingly important in the pharmaceutical industry (Bielser et al. 2018; MacDonald et al. 2021; Matanguihan and Wu 2022). The development of cell culture perfusion processes for the production of recombinant molecules offers several advantages over (fed‐)batch processes, such as reduced product heterogeneity and increased volumetric productivity by increasing biomass concentration, which allows for a reduction in the footprint of production facilities (Bielser et al. 2018; Karst et al. 2017; Karst, Steinebach, and Morbidelli 2018; Walther et al. 2018). More recently, perfusion processing has become an important method to maximize production yields of vector‐based vaccines or viral vectors used for gene and cell therapy applications, such as vesicular stomatitis viruses, influenza A virus, modified vaccinia virus Ankara, adenoviruses, adeno‐associated viruses and lentiviruses (LV) (Ansorge et al. 2009; Brühlmann and Göbel 2024; Coronel, Gränicher, et al. 2020; Coronel et al. 2021; Göbel et al. 2023; Gränicher et al. 2021; Karst, Steinebach, and Morbidelli 2018; Klimpel et al. 2023; Manceur et al. 2017; Matanguihan and Wu 2022; Mendes et al. 2022; Nie et al. 2022; Silva, Kamen, and Henry 2023; Todesco et al. 2024; Tona et al. 2023; Tran and Kamen 2022; Vázquez‐Ramírez et al. 2019; Wu et al. 2021). This is particularly important as the cost of producing these vectors is relatively high, due to the production technologies used and low process yields. Process intensification through vector production in perfusion mode can reduce the manufacturing cost per dose, ultimately lowering the cost of cell and gene therapies and enabling better accessibility to these treatments (Comisel et al. 2021; van der Loo and Wright 2016; Milone and O'Doherty 2018).
The increasing importance of continuous biomanufacturing requires the development of suitable scale‐down models to accelerate process development and reduce the development costs, especially in view of the significantly higher media consumption per run (Bareither and Pollard 2010; Bielser et al. 2018). Most of the available scale‐down models are based on different types of well plates, shake tubes, shake flasks or microbioreactor systems such as the Ambr 15 and Mobius Breez (Dorn, Klottrup‐Rees, et al. 2024; Dorn, Lucas, et al. 2024; Gagliardi et al. 2019; Gomez et al. 2017; Janoschek et al. 2018; Jin et al. 2020; Kreye et al. 2019; Mayrhofer, Castan, and Kunert 2021; Schwarz et al. 2023; Tregidgo, Lucas, Dorn, et al. 2023; Villiger‐Oberbek et al. 2015). Well plates, shake tubes, and shake flasks are inexpensive options for implementation. However, cultivation in semi‐perfusion mode requires manual manipulation steps, such as centrifugation and replacing the supernatant with fresh medium, making the process labor intensive. In contrast, the use of the microbioreactor system Ambr 15 enables a certain degree of automation, and process parameters such as dissolved oxygen (DO) and pH can be monitored and controlled. However, cell separation for medium exchange is achieved through cell sedimentation or manual centrifugation of the microbioreactors (Gagliardi et al. 2019; Janoschek et al. 2018; Kreye et al. 2019), which may impact the metabolism of the cell culture due to the absence of process controls during the cell separation process. Additionally, the medium exchange is only carried out in semi‐continuous mode. The Mobius Breez microbioreactor system allows bubble‐free cell cultivation at 2 mL working volume in perfusion mode mediated by a 1.2 µm membrane for cell separation, while process relevant parameters can be monitored (Schwarz et al. 2023). A drawback is that the stirring and sparging mechanism of a stirred‐tank bioreactor isn't mimicked.
Therefore, recent developments to establish perfusion scale‐down models have focused on the use of mini‐bioreactors to mimic stirred‐tank bioreactor‐specific properties and cell retention methods commonly used at larger scales. A novel 250 mL perfusion reactor system has been recently described, allowing tangential flow filtration (TFF) mediated perfusion processing at high viable cell densities (VCDs) (Tregidgo, Dorn, Lucas, et al. 2023). The Ambr 250 high throughput bioreactor system is a widely used tool for process development and clone screening, allowing the parallel, automated cultivation of up to 24 vessels at a working volume of up to 250 mL (Bareither et al. 2013; Manahan et al. 2019; Xu et al. 2017). Sartorius has recently launched a perfusion‐ready version of the Ambr 250 system, in which perfusion can be mediated using TFF or alternating tangential flow filtration (ATF). Joe et al. (2023) have demonstrated the use of the Ambr 250 ATF perfusion system for accelerated manufacturing of an adenovirus‐vectored vaccine. However, the small pore size of most available ATF membranes can lead to a retention of larger molecules such as viruses during the production, which can reduce virus yields in the case of unstable products such as influenza A virus or LVs (Genzel et al. 2014; Gränicher et al. 2020; Tona et al. 2023). A few cell retention technologies have been described that allow continuous harvesting of larger virus‐associated products. These include the use of alternative ATF membranes with larger cutoffs (Hein et al. 2021), the tangential flow depth filtration (TFDF) technology (Göbel et al. 2024; Silva, Kamen, and Henry 2023; Tona et al. 2023; Tran and Kamen 2022; Williams et al. 2020), and the membrane‐free acoustic wave‐mediated cell separation technology (Ansorge et al. 2009; Gränicher et al. 2020; Klimpel et al. 2023; Klimpel et al. 2024; Manceur et al. 2017). However, none of these technologies have been translated into suitable perfusion scale‐down models.
In this manuscript, we have evaluated the acoustic wave‐mediated cell separation technology for LV perfusion process development in the Ambr 250 high throughput bioreactor system. We have used a stable inducible LV producer cell pool as a model, expressing LVs pseudotyped with the vesicular stomatitis virus envelope glycoprotein (VSV‐G) upon induction, which is widely used for gene and cell therapies due to its broad tropism (Finkelshtein et al. 2013). We have evaluated the scale‐down model to investigate the effect of higher perfusion rates and cell densities on process yield compared to a previously established production process (Klimpel et al. 2023). The optimized process was then scaled up to bench‐top bioreactor scale. After refining the technical set‐up and media composition, the intensified process resulted in a 3.1‐fold higher functional vector yield per bioreactor volume.
2. Results and Discussion
To increase the LV process yield of our previously developed perfusion process (Klimpel et al. 2023), we aimed to increase the viable cell density (VCD) during virus production. This is commonly achieved by applying higher perfusion rates to provide sufficient nutrient supply and removal of cellular metabolites. For the production of instable products like LVs, increased perfusion rates may also impact process yields and product quality by reducing the residence time at bioreactor conditions.
VSV‐G pseudotyped LVs have a temperature‐dependent finite half‐life, measurable by gradual loss of infectivity over time, that can be approximated using an exponential decay function. Different LV half‐lives have been previously described with a range of 34–200 h at 4°C (Higashikawa and Chang 2001; Jiang et al. 2015; Rahman et al. 2013), and 3–35 h at 37°C (Ansorge, Henry, and Kamen 2010; Dautzenberg, Rabelink, and Hoeben 2020; Higashikawa and Chang 2001). The exact mechanism for the loss of infectivity is unknown, but the envelope protein appears to play a major role, as the type of envelope affects the stability of the vector (Dautzenberg, Rabelink, and Hoeben 2020). In addition, VSV‐G variants with a superior thermostability profile have been developed through directed evolution (Hwang and Schaffer 2013). Furthermore, the reverse transcription process has been described as thermosensitive, which may be influenced by the envelope used (Carmo et al. 2009). The stability of retroviruses including lentiviruses is also affected by physical parameters like osmolarity, ionic strength and the pH (Holic et al. 2014; Moreira et al. 2020; Rodrigues et al. 2007; de las Mercedes Segura et al. 2005). Thus, the wide range of half‐lives described for VSV‐G pseudotyped LVs is most likely due to differences in the producer cell line, genetic constructs, VSV‐G variants, media, supplements, and buffers used in the production process. Therefore, the LV stability profile should be characterized for each individual expression system and production process. For LVs carrying a WAS‐T2A‐GFP construct produced using GPRTGs‐derived stable producer cells in stirred‐tank perfusion bioreactors (Klimpel et al. 2023), we found a half‐life of 153 h at 4°C and a half‐life of 6 h at 37°C in the unprocessed cell‐free harvest matrix (Figure 1), which is in the range that was previously described by other groups. The current established production process is applying a harvest rate of 1 VVD with a variable cell bleed of up to 0.33 VVD, resulting in a maximum perfusion rate of 1.33 VVD. It should be noted that in a perfusion bioreactor operated with acoustic wave‐mediated cell separation, the virus‐containing supernatant is continuously diluted out of the vessel, which can be approximated by assuming that a perfusion bioreactor behaves as an ideally mixed continuous stirred‐tank reactor (Equation 5). For example, for virus produced at a given time at a perfusion rate of 1 VVD, approximately 37% of the virus remains in the bioreactor after 24 h. As the infectious titer in the cell‐free cell culture supernatant was dropping below the quantifiable assay limit after an incubation for 21 h at 37°C (Figure 1), we hypothesized that higher perfusion rates would increase LV yields by reducing the average residence time of produced vector under bioreactor conditions and a faster vector delivery to the 4°C environment.
Figure 1.
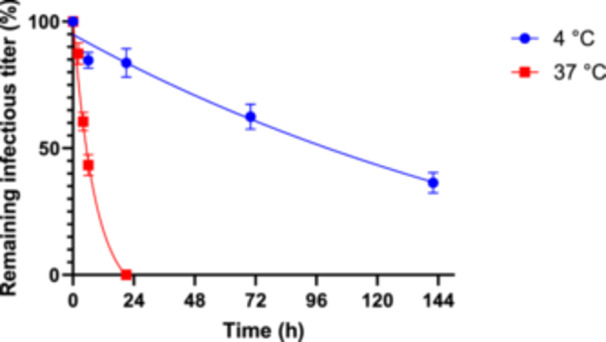
Temperature‐ and time‐dependent inactivation of WAS‐T2A‐GFP lentivirus. The cell‐free vector harvest, produced using stable GPRTGs producer cells in perfusion bioreactors, was incubated at both 4°C in a fridge and at 37°C in an incubator at a 5% CO2 atmosphere, and subsequently frozen at −80°C at different time points. A half‐life of 6 h at 37°C and 153 h at 4°C was determined by a nonlinear regression using a one phase decay equation with a robust fit.
A further potential mechanism that we previously investigated was a temperature reduction during the vector production process, with the aim of slowing down LV inactivation, given that LV inactivation is temperature dependent. A reduction in temperature may also enhance productivity by reducing the activity or secretion of proteolytic enzymes, a reduced cell proliferation and changes in cellular pathways. A mild reduction in temperature has been shown to be beneficial to produce different viruses like influenza A viruses (Petiot et al. 2011; Wu et al. 2021), recombinant adenoviruses (Cortin et al. 2004; Jardon and Garnier 2003) and herpes‐based viral vectors (Wechuck et al. 2002) as well as recombinant proteins (Furukawa and Ohsuye 2006, Kumar et al. 2008; Yoon et al. 2004). However, for GPRTGs‐derived producer cell lines, LV titers were significantly reduced when the temperature was lowered from 37.0°C to 35.5°C (p = 0.0004) (Supporting Information: Figure 1 C). The data indicate that a lower production temperature negatively affects the cellular metabolism for GPRTGs‐derived cell lines in terms of viral productivity. The finding was also verified with an adherent GPRTG‐derived producer cell line (data not shown) and is likely transferable to all GPRTG‐derived producer cell lines. Contrary to the results obtained for a decrease in temperature, an increase in temperature from 37.0°C to 38.5°C did not result in a significant change in LV titers (Supporting Information: Figure 1 C). It is worth noting that cell‐specific productivity at 38.5°C was even higher, although not significantly (p = 0.0506), due to lower average VCDs during production (Supporting Information: Figure 1 A). This finding is surprising as higher temperatures result in faster inactivation of LVs, which may indicate an even higher cellular productivity. As no significant increase in LV titers was observed at the temperatures investigated, all subsequent experiments were performed at 37.0°C. Further investigation is required to understand the effect of different production temperatures on virus quality.
To develop an intensified perfusion process, a novel small‐scale perfusion system was used, consisting of an acoustic separation chamber (ASC) for cell separation that can be connected to the Ambr 250 high throughput bioreactor system (Figure 2). Although the use of the Ambr 250 system has been previously described for intensified ATF‐mediated perfusion process development to produce an adenovirus‐vectored vaccine (Joe et al. 2023), this is the first work that shows the use of the acoustic wave separation mediated perfusion in a high throughput bioreactor system. All bioreactor productions were performed with stable GPRTGs WAS‐T2A‐GFP producer cells based on a Tet‐off inducible expression system, and LV production was initiated by a previously developed dilution method (Klimpel et al. 2023).
Figure 2.
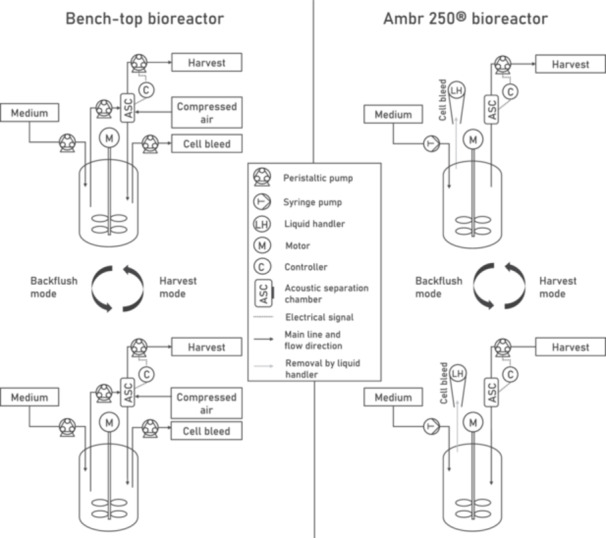
Schematic drawing of the acoustic wave‐mediated perfusion set‐up for bench‐top bioreactors and for Ambr 250 bioreactors. In the harvest mode, the acoustic field in the cell separation chamber allows to continuously collect cell‐free cell culture supernatant. In the backflush mode, the acoustic field is switched off and the flow direction of the harvest pump is reversed to return the separated cells into the bioreactor. For the operation in the Ambr 250 system, six vessels were equipped individually with an acoustic separation chamber (ASC).
Six Ambr 250 bioreactors were inoculated at a VCD of 1.50 ± 0.09 × 106 cells mL−1 and perfusion was initiated after a 3‐day batch phase for all vessels (Figure 3A). The previously established standard LV production process (Klimpel et al. 2023) applies a harvest rate of 1 VVD with a variable cell bleed to maintain a VCD of approximately 2 × 107 cells mL−1 (Figure 3A,B). To increase cell densities during LV production, the harvest rate was gradually increased to 3 VVD at 6 days post induction (dpi) for three bioreactors and cells were grown to an average VCD of 3.99 ± 0.34 × 107 cells mL−1 at 7 dpi before cell bleeding was initiated (Figure 3A,B). We found that a VCD of 4–5 × 107 cells mL−1 seems to be the limiting cell density that can be applied with the used technical set‐up to maintain a separation efficiency of > 90%. A similar cell viability profile was found between a process with a harvest rate of 1 VVD and 3 VVD, with average cell viabilities of 80.5 ± 2.8% and 79.0 ± 2.1% at 11 dpi, respectively (Figure 3C). A similar profile of infectious titers was found for both processes, reaching maximum values of 7.27 ± 1.74 × 107 TU mL−1 at a harvest rate of 1 VVD and 8.47 ± 5.26 × 107 TUmL−1 at a harvest rate of 3 VVD, both at 10 dpi (Figure 3D). As the flow rates in the harvest line are rather low due to the small bioreactor volume of the Ambr vessels, the increased residence time of the virus in the harvest line at room temperature may lead to increased virus degradation. The different flow rates for an operation at different harvest rates may distort the results for a comparison due to different average residence times in the harvest line, which is not temperature controlled. Thus, infectious titers determined in the bioreactors were used to compare the processes in terms of volumetric and cell‐specific vector productivities and yields. The cumulative virus yield per bioreactor volume was found to be significantly higher at a harvest rate of 3 VVD at 11 dpi (2.27 ± 0.28 × 108 TU mLBioreactor −1 vs. 6.91 ± 2.54 × 108 TU mLBioreactor −1; p = 0.0396) (Figure 3E). To investigate the effect of higher perfusion rates on virus recovery, the virus productivity is normalized per cell. Figure 3F shows that the mean cell‐specific virus productivity increased for the 3 VVD process at 6 dpi, which is the first sampling timepoint after the perfusion rate was higher compared to the standard process (Figure 3A). We determined significant differences when comparing cumulative cell‐specific virus yields at 6–9 dpi (all p values < 0.002), supporting the hypothesis that increased perfusion rates are increasing functional LV recoveries. The cumulative cell‐specific yield at the end of the process was 17.1 ± 1.2 TU cell−1 at a harvest rate of 1 VVD and 24.2 ± 5.0 TU cell−1 at a harvest rate of 3VVD (p = 0.0745; Figure 3G). It should be noted that we observed increasing volume fluctuations in the bioreactors starting from 9 dpi, particularly at a harvest rate of 3 VVD, which required daily readjustment of the pump speed to control the working volume. This can explain the increasing standard deviation for the VCD and titer‐related parameters at later process times. The main reason is an increasing blockage of the inlet of the integrated dip tube, which has an inner diameter of < 1 mm. Glucose concentrations of > 9 mmol L−1 were maintained for both processes, suggesting that no glucose limitation occurred (Supporting Information: Figure 3). Similar glucose consumption and lactate production rates were determined for both processes after reaching the target VCD, indicating a comparable cellular metabolic state between the processes (Supporting Information: Figure 4).
Figure 3.
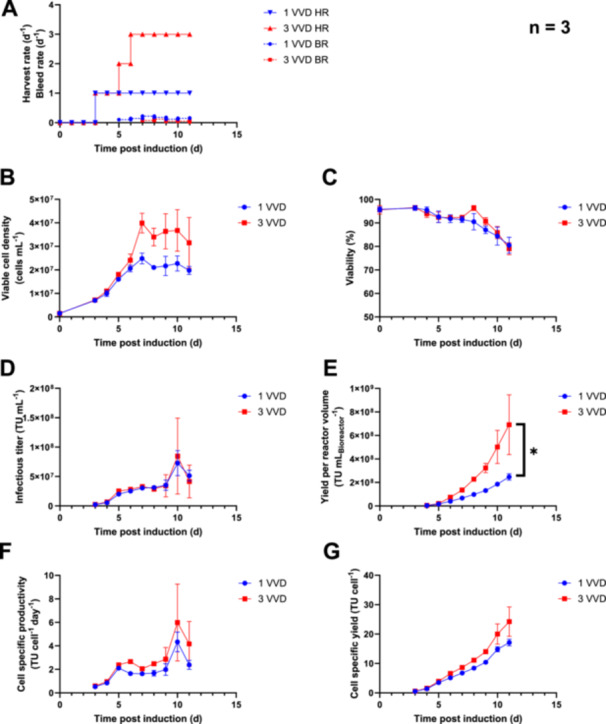
Investigation of an intensified perfusion process for stable lentivirus production using acoustic wave cell separation in the Ambr 250 high throughput bioreactor system. The standard production process applying a harvest rate of 1 VVD was compared with an intensified perfusion process at a harvest rate of 3 VVD and higher viable cell densities. The data were generated using three vessels per condition (n = 3). All six vessels were inoculated using the same preculture. Values are shown as mean for A and as mean ± SD for B–G. BR = bleed rate, HR = harvest rate, VVD = vessel volumes per day. (A) Harvest rate and bleed rate. (B) Viable cell density. (C) Cell viability. (D) Infectious virus titer. (E) Yield of infectious titer per reactor volume or volumetric yield. p = 0.0396 by an unpaired t‐test. (F) Cell‐specific productivity of infectious virus. (G) Cell‐specific infectious virus yield.
To verify the findings from the small‐scale model, the effect of higher perfusion rates was tested at bench‐top bioreactor scale. Higher perfusion rates require higher recirculation rates, which result in higher chamber turbulence and decreased cell separation efficiencies (Gorenflo et al. 2002, 2003). In addition, the flow rates applied may influence the heat dissipation in the ASC, which can affect the cellular metabolism and virus stability (Drouin et al. 2007; Gränicher et al. 2020). Therefore, the bioreactor was proportionally scaled down from 4.5 L working volume for a harvest rate of 1 VVD to 1.5L working volume for a harvest rate of 3 VVD. The set‐up allows to use the same technical settings and flow rates for the acoustic perfusion system between both processes, which reduces the impact of technical differences and facilitates the investigation of different harvest rates on viral productivity. For LV production at higher perfusion rates, a similar process strategy like in the Ambr 250 was applied by gradually increasing the perfusion rate after an initial batch phase (Figure 4A). For the process at 1 VVD, a continuous cell bleed was applied to control the cell density after reaching a VCD of 2.27 × 107 cells mL−1 at 6 dpi (Figure 4A,B). For the process at 3 VVD, the cell bleed was initiated after reaching a VCD of 4.29 × 107 cells mL−1 at 7 dpi. As the cell growth was slowing down at the end of both processes, the cell bleed was turned off at 16 dpi for the 1 VVD process and at 15 dpi for the 3 VVD process. A similar viability profile was obtained between a harvest rate of 1 VVD and 3 VVD. A viability decline occurred at the end of the process, reaching 71.4% for 1 VVD at 17 dpi and 57.7% for 3 VVD at 16 dpi (Figure 4C). At the same time, a turbidity increase of the cell free harvest was observed (data not shown). This may be due to the persistent expression of cytostatic and cytotoxic vector components like VSV‐G and the viral protease (Ferreira, Cabral, and Coroadinha 2020; Hoffmann et al. 2010), and may be specific for individual producer clones (Klimpel et al. 2024). We observed a similar infectious titer profile between a harvest rate of 1 VVD and 3 VVD, with slightly higher titers for the process at 3 VVD (Figure 4D). The highest infectious titer determined was 1.24 × 108 TU mL−1 at 12 dpi at 1 VVD and 1.74 × 108 TU mL−1 at 14 dpi at 3 VVD. The final cumulative yield at 3 VVD was 3.1‐fold higher, reaching a value of 3.31 × 109 TU mLBioreactor −1 compared to 1.08 × 109 TU mLBioreactor −1 at 1 VVD (Figure 4E). The higher functional virus yields obtained by the higher perfusion rate were also verified by determining the titers in the collected harvest. It should be noted that titers in the final harvest collected at 4°C were on average 25% lower compared to the average functional titers determined in the bioreactor (Supporting Information: Figure 5), as the virus degradation is only slowed down but not prevented at 4°C (Figure 1). Cell‐specific productivities were higher at a harvest rate of 3 VVD, except for the last day of the cultivation (Figure 4F). Highest cell‐specific productivities were obtained at 14 dpi in both bioreactors, showing values of 7.7TU cell−1 day−1 at 1 VVD and 11.1 TU cell−1 day−1 at 3 VVD. The final cumulative cell‐specific virus yield was 61.9 TU cell−1 at 1 VVD and 89.1 TU cell−1 at 3 VVD (Figure 4G). Higher cell‐specific physical vector RNA productivities were obtained at 3 VVD throughout the process (Figure 4H), resulting in a 3.0‐fold higher final cumulative yield of 5.02 × 1012 RNA copies mLBioreactor −1 compared to 1.66 × 1012 RNAcopies mLBioreactor −1 at 1 VVD. DNA concentrations in the supernatant were increasing over process time, reaching a peak concentration of 9452 ng mL−1 at 12 dpi for the 1 VVD process and 9522 ngmL−1 at 16 dpi for the 3 VVD process (Figure 5A), indicating a similar impurity profile between both processes.
Figure 4.
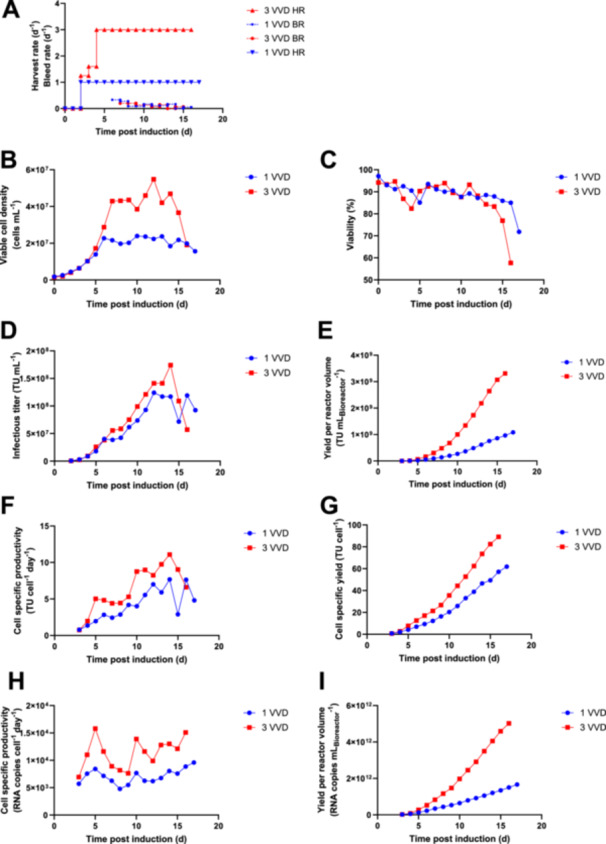
Bench‐top bioreactor perfusion processes for stable lentivirus production using acoustic wave cell separation with optimized set‐up. The standard process at a harvest rate of 1 VVD was performed in a 5 L bioreactor. The intensified process was performed at a harvest rate of 3 VVD in a 2 L bioreactor. A PharmaPure low spallation pump tubing size 17 was used to recirculate the cell suspension for operation in perfusion mode. The medium was supplemented with 0.5% poloxamer 188 and 0.4% cholesterol lipid concentrate. The run was performed with one vessel per condition. BR = bleed rate, HR = harvest rate, VVD = vessel volumes per day. (A) Harvest rate and bleed rate. (B) Viable cell density. (C) Cell viability. (D) Infectious virus titer. (E) Yield of infectious titer per reactor volume or volumetric yield. (F) Cell‐specific productivity of infectious virus. (G) Cell‐specific infectious virus yield. (H) Cell‐specific productivity of vector RNA genome. (I) Cell‐specific yield of vector RNA genome.
Figure 5.
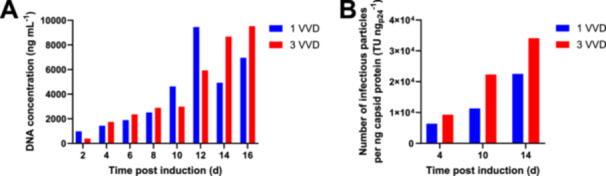
DNA concentration and number of infectious particles per ng p24 capsid protein determined for cell culture supernatant sampled from bench‐top bioreactors with optimized set‐up. (A) DNA concentration. (B) Number of infectious particles per ng p24 capsid protein.
Productivities and yields of infectious virus seem higher for a production in bench‐top bioreactors when comparing values for the same process duration in the Ambr 250 (Table 1). These results are unexpected as the shear stress introduced by recirculation pumps is described to negatively affect cellular transcription and lower cell‐specific productivities (Merten 2000; Nie et al. 2022; Zhan et al. 2020), which is only part of the bench‐scale set‐up (Figure 2). However, Gränicher et al. (2020) compared different recirculation strategies for influenza virus A production in perfusion mode using acoustic wave separation. The authors reported higher cellular productivity for a pump‐based recirculation with higher mechanical shear stress but lower maximum temperatures in the chamber compared to a valve‐based recirculation. The effect of higher chamber temperatures might be the underlying mechanism for lower productivities in the Ambr 250, as it can affect the cellular metabolism and viral inactivation kinetics. The recirculation at bench scale may contribute to a more efficient heat dissipation in the ASC and reduced DO fluctuations in the chamber. In addition, ASCs connected to the Ambr 250 system are only cooled by the air flow of the safety cabinet, while ASCs at bench‐scale are cooled by compressed air. Although the power input of the small‐scale ASC is approximately 10 times lower (Table 1) and the surface to volume ratio is higher than that of the bench‐scale ASC, which facilitates heat dissipation, potential differences in temperature distribution and dissipation between the scales must be taken into account. However, the fold increase of cell‐specific and volumetric infectious vector yields was demonstrated across both scales. The intensified perfusion process at a harvest rate of 3 VVD resulted in a 1.4‐fold higher cell‐specific virus yield in both the Ambr 250 and the bench‐top scale. The volumetric yield was increased by 2.8‐fold in the Ambr 250 and by 3.1‐fold in the bench‐top bioreactor scale.
Table 1.
Comparison of bioreactor conditions, settings for the acoustic separation chamber (ASC) and process results for production in AMBR 250 and bench‐top bioreactors. Italic font for the bench‐top bioreactor process results shows values obtained after a process duration of 11 days to allow a better comparison to the results obtained in the AMBR 250.
| AMBR 250 with 1 mL ASC | Bench‐top bioreactor with 30 mL ASC | ||||
|---|---|---|---|---|---|
| Bioreactor conditions | Harvest rate (d−1) | 1 | 3 | 1 | 3 |
| Working volume (mL) | 200 | 200 | 4500 | 1500 | |
| Max. stirring speed (rpm) | 250 | 250 | 140 | 160 | |
| Dissolved oxygen (%) | 50 | 50 | 50 | 50 | |
| pH | 6.95 ± 0.15 | 6.95 ± 0.15 | 6.95 ± 0.15 | 6.95 ± 0.15 | |
| Settings ASC | Power (W) | 0.3 | 0.4 | 3.0 | 3.0 |
| Forward time (min) | 5 | 2 | 5 | 5 | |
| Settling time (s) | 3 | 3 | 3 | 3 | |
| Reverse time (s) | 7 | 12 | 10 | 10 | |
| Process results | Process duration (d) | 11 | 11 | 17 (11) | 16 (11) |
| Total media consumption per vessel (L) | 2.0 | 4.1 | 78.3 (49.3) | 61.1 (38.2) | |
| Max. volume flow recirculation (L d−1) | n/a | n/a | 10 (10) | 10 (10) | |
| Max. viable cell density (cells mL−1) | 2.48 ± 0.18 × 107 | 3.99 ± 0.34 × 107 | 2.39 × 107 (2.39 × 107 ) | 5.48 × 107 (4.59 × 107 ) | |
| Max. infectious titer (TU mL−1) | 7.27 ± 1.74 × 107 | 8.47 ± 5.26 × 107 | 1.24 × 108 (9.27 × 107 ) | 1.74 × 108 (1.21 × 108 ) | |
| Max. cell‐specific productivity (TU cell−1 day−1) | 4.3 ± 0.7 | 6.0 ± 2.7 | 7.7 (5.6) | 11.1 (9.0) | |
| Volumetric yield (TU mLBioreactor −1) | 2.27 ± 0.28 × 108 | 6.91 ± 2.54 × 108 | 1.08 × 109 (3.66 × 108 ) | 3.31 × 109 (1.34 × 109 ) | |
| Cell‐specific yield (TU cell−1) | 17.1 ± 1.2 | 24.2 ± 5.0 | 61.9 (25.9) | 89.1 (44.4) | |
Abbreviation: n/a = not applicable.
Higher volumetric LV yields obtained by the newly developed process demonstrate the importance of perfusion process intensification for LV production. The process allows to increase the cumulative LV yields per bioreactor volume in the same process time (Figure 4E), resulting in tripled space‐time yields. This allows the footprint of the manufacturing facility to be reduced for the same output, or the output to be increased for the same footprint, ultimately reducing manufacturing costs. An important factor for increased space‐time yields is the application of higher cell densities. Even higher LV yields can potentially be obtained by further increasing target VCDs, which might also reduce the product loss by reducing the overall cell bleed. However, it is possible that the application of even higher VCDs could have an impact on process robustness. As the acoustic wave separation technology does not have a physical cell barrier, uncontrolled increases in cell density can affect separation efficiency and result in cell loss through the harvest stream when the technical limits of the cell retention device are reached. This can affect the harvest characteristics and may affect subsequent downstream processing steps. Therefore, the use of a cell bleed may be beneficial to control for a specific target VCD.
Another factor contributing to the increased space‐time yields from the enhanced perfusion process is the increased cell‐specific LV yields achieved (Figures 3G and 4G). The results obtained at higher harvest rates support the hypothesis of increasing LV recovery by decreasing the LV residence time in the bioreactor due to a low vector half‐life of 6 h at 37°C (Figure 1). Similar findings were previously reported for the production of retroviral vectors pseudotyped with the GALV envelope due to a low vector half‐life of 2–8 h at 37°C (Kotani et al. 1994; Le Doux et al. 1999; G. M. Lee et al. 1998; S.‐G. Lee et al. 1996), suggesting that a perfusion rate of three to four volume exchanges might be optimal for cell growth and vector production (Merten et al. 2001, 2004). As a half‐life range of 3–35 h at 37°C is described for LVs pseudotyped with VSV‐G (Ansorge, Henry, and Kamen 2010; Dautzenberg, Rabelink, and Hoeben 2020; Higashikawa and Chang 2001), the optimal applied perfusion rate may need to be determined for each individual production system. The reduction in virus degradation due to higher harvest rates could affect not only the yield of the LV process but also the quality of the LV produced. The number of infectious particles per ng capsid protein p24 was determined at selected time points as an indicator for LV quality (Figure 5B). When comparing the values at 10 dpi and 14 dpi, at which each respective target harvest rate was reached, higher values of 2.23 × 104 TU ngp24 −1 and 3.41 × 104 TU ngp24 −1 were obtained at 3 VVD compared to 1.13 × 104 TU ngp24 −1 and 2.25 × 104 TU ngp24 −1 at 1 VVD. The results indicate that higher perfusion rates result in higher LV quality. Although the reduced LV inactivation by reducing the bioreactor residence time is the most likely reason for increased cell‐specific productivities, other contributing factors should be considered. The average cell‐specific perfusion rate for the intensified process is slightly higher. No significant differences in glucose consumption rates were found in the Ambr 250 model between both processes and only slightly higher consumption rates at bench‐top scale after reaching steady‐state conditions (Figure 4A,E). However, it is possible that the availability and cellular consumption rate of other media components may impact cellular productivity, as well as an improved removal of potential cellular metabolites by higher perfusion rates. Another potential factor for increased cell‐specific productivities by the intensified process could be improved cooling of the ASC by higher flow rates. Other groups have reported increased temperatures up to 40°C in the top part of the chamber (Drouin et al. 2007; Gränicher et al. 2020), which could affect the cell culture metabolism and virus stability. Higher flow rates in the ASC can improve heat dissipation and reduce the time of exposure to higher temperatures. However, for the bench‐scale comparison, the bioreactor working volume for production at the higher perfusion rate of 3 VVD was proportionally reduced to 1.5 L compared to the standard process at 1 VVD with 4.5 L working volume. In fact, the flow rates between the processes and also the heat dissipation is comparable.
It is important to mention that the perfusion bench‐top bioreactor set‐up was previously optimized to support lentivirus production at higher perfusion rates. When a non‐optimized set‐up was used at a harvest rate of 3 VVD, the cell‐specific and volumetric cumulative functional vector yield showed an increasing trend at the beginning of the process compared to the control process at 1 VVD (Supporting Information: Figure 2E,G). However, the recirculation pump, as a specific part of the bench‐scale setup (Figure 2), introduced cellular shear stress at higher harvest rates, resulting in stagnation of cell growth followed by a decrease in VCD (Supporting Information: Figure 2B). The negative effect of the cell recirculation was reduced to a tolerable level by using a specific low spallation pump tubing with a smooth inner surface and a larger inner diameter to lower the pump speed at the same flow rate. Similar findings were reported by other groups for the production of recombinant proteins in perfusion mode using Chinese hamster ovary (CHO) or HEK293‐derived cell lines (Wang et al. 2017; Zhan et al. 2020). It should be noted that the LV producer cells are expressing some vector components, like VSV‐G and the human immunodeficiency virus 1 protease encoded by the gag‐pol gene, which are described to have cytostatic or cytotoxic effects (Ferreira, Cabral, and Coroadinha 2020; Hoffmann et al. 2010). This may be a factor contributing to an increased shear sensitivity of the cell line used in this study. Therefore, we increased the poloxamer 188 concentration in the medium from 0.1% to 0.5%. Poloxamer 188 is a widely used nonionic surfactant to protect cell cultures from hydrodynamic and bubble‐induced shear in bioreactors (Guzniczak et al. 2018; Tharmalingam et al. 2008; Tharmalingam and Goudar 2014; Wei et al. 2023; Xu et al. 2017; Zhang, Al‐Rubeai, and Thomas 1992). An increased poloxamer 188 concentration is also described to protect HEK293 cells against shear forces generated by peristaltic pumps for the production of adenoviruses in perfusion processes (Nie et al. 2022).
In this context, it should be noted that the production of infectious enveloped viruses like LVs relies on a budding process from the cells surface, in which the viral envelope is formed (Welsch, Müller, and Kräusslich 2007). The organization and composition of the cellular membrane is a crucial factor for the production of functional HIV particles, which are budding selectively from cholesterol‐rich lipid rafts with a specific composition (Aloia, Tian, and Jensen 1993; Nguyen and Hildreth 2000; Ono and Freed 2001). On the same note, the presence of cholesterol is a critical factor for productivity and infectivity of retroviral vectors, including HIV‐1‐derived LVs pseudotyped with VSV‐G (Chen et al. 2009; Liu, Huang, and Yu 2021; Nieto‐Garai et al. 2020; Ono and Freed 2001; Rodrigues et al. 2009). However, some cell models have shown that shear stress increases the cell membrane fluidity and decreases the membrane lipid order (Espina et al. 2023; Haidekker, L'Heureux, and Frangos 2000; Yamamoto and Ando 2015). Furthermore, high shear stress in perfusion processes using HEK293 cells downregulated lipid biosynthetic processes (Zhan et al. 2020). Zhan et al. (2020) also described increased lactate production rates induced by shear stress, which correlates with our findings (Supporting Information: Figure 4D). In consequence, increased shear stress may impact the infectivity of produced enveloped viruses. Although we did not find a significant effect of cholesterol‐lipid supplementation on LV productivity in shake flask experiments (data not shown), supplementation under shear stress conditions may be useful, as it has been described to block the negative effects, such as reduced plasma membrane cholesterol levels (Yamamoto et al. 2020).
Our developed intensified perfusion process at 3 VVD resulted in a cumulative functional yield of 3.31 × 1012 TU LBioreactor −1 calculated using infectious titers determined in the bioreactor. Considering a recovery of 75% in the collected bulk harvest (Supporting Information: Figure 5), it can be assumed that a cumulative functional yield of 2.48 × 1012 TU LBioreactor −1 can be obtained with the performed harvest strategy. We have previously reported a cumulative functional yield of 2.4 × 1011 TU LBioreactor −1 in a bioreactor production process for a WAS‐T2A‐GFP LV (Klimpel et al. 2023) like produced in the present study, indicating that the cumulative functional yield was improved by more than 10‐fold. Other groups have previously reported intensified perfusion processing for LV production using the TFDF technology (Tona et al. 2023; Tran and Kamen 2022; Williams et al. 2020). Tran and Kamen (2022) reported a cumulative functional yield of up to 3.9 × 1011 transducing units at a working volume of 2 L, which corresponds to a volumetric yield of 1.95 × 1011 TU LBioreactor −1. Tona et al. (2023) reported an average cumulative functional yield of 122 × 1011 transducing units per batch by three perfusion runs performed at 2 L working volume, which corresponds to a volumetric yield of 6.10 × 1012 TU LBioreactor −1. Although the results cannot be directly compared since different packaging constructs, stable producer cells and methods for titer quantification were used, the data indicate that comparable functional vector yields can be obtained by our developed process. Tran and Kamen (2022) have also reported cell‐specific productivities using the TFDF technology, reaching peak values of 7 TU cell−1. Manceur et al. (2017) reported peak values of 11.5 TU cell−1 using the same cell line like Tran and Kamen, but acoustic wave‐mediated cell separation for operation in perfusion. We obtained comparable peak cell‐specific productivities of up to 11.1 TU cell−1 for the process using a harvest rate of 3 VVD. Tran and Kamen also determined residual DNA concentrations in the harvest and reported values of approximately 5000–8000 ng mL−1 up to 72 h post induction and up to 20,000–30,000 ng mL−1 at 96 h post induction, whereas our maximum DNA concentration determined was 9522 ng mL−1, with lower concentrations at earlier process times.
The most suitable perfusion technologies for the production of larger enveloped viruses include ATF using membranes with larger cutoffs (Genzel et al. 2014; Hein et al. 2021), TFDF (Göbel et al. 2024; Silva, Kamen, and Henry 2023; Tona et al. 2023; Tran and Kamen 2022; Williams et al. 2020) and acoustic wave‐mediated cell separation as used in the present work (Ansorge et al. 2009; Gränicher et al. 2020; Klimpel et al. 2023; Klimpel et al. 2024; Manceur et al. 2017). Membrane based methods facilitate harvest pre‐clarification, particularly the TFDF technology due to the integrated depth filtration function. In addition, ATF and TFDF technology are considered to be scalable to industrial scale. A drawback of membrane‐based cell retention technologies is filter fouling and clogging that can occur with increased process times, which can affect virus recovery and limit the overall process time. Tona et al. (2023) reported noticeably lower LV recoveries at later harvest time points using TFDF along with increasing transmembrane pressure after 3.5 days of operation. Given that GPRTGs‐derived cell lines are capable of producing infectious virus at high titer for up to several weeks (Klimpel et al. 2023, 2024), the acoustic wave separation technology is advantageous as no membrane clogging can occur, allowing process time and ultimately process yields to be increased. One disadvantage of the acoustic wave separation technology is the generation and dissipation of heat during the process of scale‐up. Nevertheless, the technology can be scaled up to a volumetric perfusion rate of 200–1000 L d−1 (Gorenflo et al. 2002; Gränicher et al. 2020), indicating that the developed process can be scaled up to industrial‐scale bioreactors. Moreover, the manufacturer introduced a novel multi‐chamber configuration with the objective of circumventing issues associated with heat dissipation. However, this configuration has yet to be demonstrated at a scale exceeding that described in earlier studies.
The intensified process of this work was initially developed using the acoustic wave separation technology in combination with the Ambr 250 high throughput bioreactor system. The development of small‐scale models is critical to reducing development costs, particularly for perfusion processes, which typically have higher media consumption and longer process times compared to batch or fed‐batch processes. The small‐scale model allowed media consumption to be reduced by at least tenfold compared to an operation of an equivalent number of bench‐top bioreactors (Table 1). While only six Ambr 250 vessels were operated in parallel in this study, the system can be expanded to use of up to 24 vessels in parallel, with the potential to accelerate perfusion process development. In addition, the ASC can potentially be used in combination with the Ambr 250 modular, which may be a more affordable alternative for studies in academic settings. Our data show that the small‐scale model can predict the effect of increased perfusion rate on cumulative functional and cell‐specific LV yields. The cumulative LV yield per bioreactor volume increased 2.8‐fold and the cell‐specific LV yield 1.4‐fold at a harvest rate of 3 VVD compared to a harvest rate of 1 VVD using the Ambr 250 small‐scale model. A 3.1‐fold increase of the cumulative LV yield per bioreactor volume and a 1.4‐fold increase of cell‐specific LV yields was found after successfully implementing higher harvest rates at bench‐top bioreactor scale. The obtained data demonstrate the function of the model as a predictive small‐scale model.
One limitation of the technical set‐up used is the small inlet diameter of the pre‐integrated dip tube. This can cause volume fluctuations over prolonged cultivation periods, particularly in processes that use high perfusion rates and HEK293T cells, which tend to aggregate in suspension. This limitation could be circumvented by using a different Ambr 250 vessel design with a larger inner diameter of the line connected to the ASC. Alternatively, a higher concentration of anticlumping reagent might be beneficial to reduce cell aggregate formation. For the implementation of higher perfusion rates at bench‐top scale, the shear stress induced by the recirculation pump must be considered. We found that the HEK293T‐based LV producer cells used in this work show a much lower tolerance for higher pump rates compared to CHO‐derived cell lines used for the production of recombinant proteins (unpublished data). Our introduced optimizations for the cell recirculation reduced the shear stress for the producer cells used to a tolerable level, which allowed to successfully scale up the intensified perfusion process at bench scale.
3. Conclusion
Here, we described a novel small‐scale perfusion system based on acoustic wave‐mediated cell separation that can be used in combination with high‐throughput bioreactor systems. The system is a useful tool for perfusion process development to reduce media costs and was used to develop an intensified LV production process using stable producer cells. A higher perfusion rate and higher cell densities resulted in a 1.4‐fold increased cell‐specific LV yield and a 2.8‐fold higher volumetric LV yield. The findings from the small‐scale model were successfully verified at bench‐scale using an optimized technical set‐up. Our data demonstrate the importance of process intensification to maximize LV yields and potentially optimize LV quality.
4. Material and Methods
4.1. Cell Culture
A Tet‐off inducible polyclonal GPRTGs suspension producer cell line expressing a WAS‐T2A‐GFP LV upon induction (Klimpel et al. 2023) was cultivated in TransFx‐H medium (Cytiva) supplemented with 6 mM GlutaMAX (Thermo Fisher), 0.1% poloxamer 188 (Merck), 5% Cell Boost 5 (Cytiva) and 0.01% Anticlumping Agent (Thermo Fisher), hereafter called complete TransFx‐H medium. The cell line originates from the stable adherent packaging cell line GPRTG, which was developed based on the HEK293T/17 clone (Bonner et al. 2015; Throm et al. 2009). Cell cultivation was performed in plain shake flasks (Corning) at a maximum relative working volume of 32% using a shaker incubator (Infors HT, Multitron) set at 37°C, 5% CO2 and 70% relative humidity. Cell counts were performed using Nucleocounter NC‐200 (Chemometec). Before counting, a cell dissociation step was performed by incubating 100 µL of cell suspension with 300 µL TrypleLE (Thermo Fisher) for 15 min at 37°C.
For seed expansion, cells were seeded at a VCD of 4 × 105 cells mL−1 for a 3‐day split and at 3 × 105 cells mL−1 for a 4‐day split in complete TransFx‐H medium supplemented with 2.5 ng mL−1 doxycycline (MP Biomedicals) to suppress the virus expression by Tet‐controlled constructs like the vector genomic RNA expression cassette, tat, rev, and VSV‐G (Bonner et al. 2015; Throm et al. 2009). Four days before inoculation for virus production, cells were seeded at a VCD of 1 × 106 cells mL−1 at a doxycycline concentration of 1 ng mL−1. 24 h before inoculation for virus production, a medium exchange was performed after centrifuging the cell suspension at 100g for 10 min and replacing 66% of the spent medium with complete TransFx‐H medium supplemented with 1 ng mL−1 doxycycline. Induction of LV production in all bioreactors was performed by direct inoculation into doxycycline‐free medium according to a previously developed dilution method (Klimpel et al. 2023). The method consists of an N‐1 perfusion seed expansion to grow cells to high VCD at a reduced doxycycline concentration, which allows efficient induction of virus production by reaching an approximately sevenfold dilution after inoculation in the production reactor.
4.2. Comparison of Different Medium Exchange Rates in Ambr 250 Bioreactors
A 24‐way Ambr 250 high throughput bioreactor system (Sartorius) was used to investigate the effect of higher perfusion rates on LV production. Cultivation was performed in six mammalian vessels with a dual pitch‐blade impeller. Each vessel was equipped with an APS‐2402 system (SonoSep Technologies) consisting of a 1 mL acoustic separation chamber (ASC) (part number: SS‐01) and a controller (part number: SC‐2402), enabling LV production in perfusion mode by acoustic wave‐mediated cell separation (Figure 2). The ASC has a tubing connection at the bottom part and at the top part, respectively. The bottom tubing connection is connected to the Ambr vessel via one of the four feed lines, which are linked to the integrated dip tube of the vessel. The top tubing connection is connected to a harvest line, enabling to pump cells into the ASC using a separate peristaltic harvest pump for each vessel. The peristaltic pumps are connected to the APS controllers, allowing to control the power, pump speeds and time intervals for the cell separation.
The bioreactors were operated at a stirring speed of 250 rotations per minute (rpm), a temperature of 37°C, a DO level of 50% and a pH of 6.95 ± 0.15 for cell cultivation. CO2 was sparged to control the upper pH limit. The overlay was set to a fixed air volume flow of 10 mL min−1. On the day before inoculation, the vessels were preconditioned with complete doxycycline‐free TransFx‐H medium. Six vessels were inoculated at a VCD of 1.5 × 106 cells mL−1 at a final working volume of 200 mL using cells from the same preculture. 200 µL of EX‐CELL antifoam (Sigma‐Aldrich) were added per vessel after inoculation using the liquid handler. After a 3‐day batch cultivation, perfusion was initiated at a harvest rate of 1 VVD for all vessels and 200 µL of antifoam was regularly added in an interval of 6 h. The liquid levels were controlled manually by adjusting the flow rates of the harvest pumps. For the investigation of a higher perfusion rate, the harvest rate was increased to 2 VVD at 5 dpi and to 3 VVD at 6 dpi for three of the six vessels (Figure 3A). As the volume of concentrated cell culture in the ASC is continuously increasing during every harvest cycle, the set‐up requires a repetitive back flush into the bioreactor to avoid cell loss into the harvest stream. The capacity of the system is mainly determined by (1) the cell density in the bioreactor and (2) the target harvest rate. If the cell densities or harvest rates are increasing, the volume of concentrated cell suspension in the ASC is increasing more quickly, requiring more frequent back‐flush cycles. Detailed settings for the acoustic separation system are summarized in Table 1. Cell densities were controlled by variable removal of cell suspension at 6 h intervals using the liquid handler and subsequent bolus addition of media to reach the initial working volume. All bioreactors were sampled at 0 dpi and daily starting from 3 dpi by removing 7 mL of cell suspension using the liquid handler and subsequent addition of 7 mL fresh medium to reach the initial working volume. 100 µL of the cell suspension were used to perform a cell count. The remaining volume was centrifuged for 5 min at 336 g and the supernatant frozen at ‐80°C in 1 mL aliquots for quantification of infectious titers and metabolites. Lactate and glucose concentrations in the supernatant were determined using EPOC Blood Analysis System (Siemens Healthcare).
4.3. Scale‐Up in Bench‐Top Bioreactors
A Biostat B‐DCU system (Sartorius) was used for LV productions at bench‐top bioreactor scale. All cultivations were performed at a temperature of 37°C, a DO level of 50% and a pH of 6.95 ± 0.15. CO2 was sparged to control the upper pH limit. The bioreactors were equipped with an APS‐107 system (SonoSep Technologies) consisting of a controller (part number: SC‐107) and a 30 mL ASC (part number: SS‐30), enabling LV production in perfusion mode by acoustic wave‐mediated cell separation. The investigation of a harvest rate of 1 VVD was performed in a 5 L glass bioreactor (Sartorius) at a final working volume of 4.5 L. Cultivation was performed at a stirring speed of 100–140 rpm, and the overlay was set to a fixed air volume flow of 0.2 L min−1. The investigation of a harvest rate of 3 VVD was performed in a 2 L glass bioreactor (Sartorius) at a final working volume of 1.5 L. Cultivation was performed at a stirring speed of 114–160 rpm, and the overlay was set to a fixed air volume flow of 0.08 L min−1.
Complete TransFx‐H medium was supplemented with 0.4% cholesterol lipid concentrate (Thermo Fisher) and 0.5% poloxamer 188. A size 17 PharmaPure low spallation pump tubing (Saint‐Gobain) was used for cell recirculation. The bioreactors were preconditioned with doxycycline‐free medium and inoculated at a VCD of 1.5 × 106 cells mL−1. After cultivation in batch mode for 2 days, perfusion was initiated at a harvest rate of approximately 1 VVD and the harvest was continuously collected at 4°C. For the 2 L bioreactor, the harvest rate was gradually increased to 3 VVD (Figure 4A). The VCD was controlled by a continuous cell bleed based on the daily determined VCD. The volume of cell culture removed for bleeding was replaced by adding the same volume of fresh medium. EX‐CELL antifoam was added manually on demand. The bioreactors were sampled daily. 100 µL of the sampled cell suspension was used to perform a cell count. The remaining cell suspension was centrifuged for 5 min at 336 g. The supernatant was separated from the cell pellet and frozen at −80°C in 1 mL aliquots for quantification of infectious titers and metabolites. Lactate and glucose concentrations in the supernatant were determined using EPOC Blood Analysis System (Siemens Healthcare).
4.4. LV Stability Study
LV containing cell suspension was collected from stirred‐tank perfusion bioreactors by sampling using a syringe. The samples were centrifuged for 5 min at 336g for cell separation. The supernatants were aliquoted into 1.5 mL screwed‐cap sample tubes (VWR) and incubated at 4°C in a fridge or at 37°C in a 5% CO2 atmosphere in an incubator. The samples were frozen at ‐80°C at different time points for infectious titer determination. Aliquots that were incubated at 4°C were frozen after 0, 6, 21, 70, and 142 h, respectively. Aliquots that were incubated at 37°C were frozen after 0, 2, 4, 6, and 21 h, respectively. For representation, all determined titers which originated from the same sampled supernatant were normalized to the respective titer determined at 0 h.
4.5. Infectious Titer Determination
Adherent HEK293T/17 cells (ATCC) were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin‐Streptomycin (all Thermo Fisher) at 5% CO2 using a static incubator (Thermo HERAcell 250i). For the quantification of transducing units (TU), 2 × 104 cells per well were seeded in 96‐well plates in a volume of 80 µL per well using DMEM medium supplemented with 10% FBS and 10 µg mL−1 polybrene (Merck), hereafter called transduction medium. Virus containing supernatants were thawed quickly and fivefold serial dilutions were performed in duplicates using transduction medium, starting from a 50‐fold dilution. 20 µL of each dilution was added to seeded cells, resulting in a final volume of 100 µL per well. Tracking controls were included by diluting a GFP LV preparation with a known virus concentration 25,000‐fold using transduction medium. Negative controls were included by adding 20 µL DMEM medium with 10% FBS instead of virus containing supernatants. Cells were trypsinized 4 days post transduction, washed with PBS (Thermo Fisher) and resuspended in 100 µL cold MilliQ water supplemented with 0.1% Fixation/Permeabilization solution (BD Biosciences), hereafter called fixation solution. The samples were incubated for 20 min at 4°C, and 100 µL cold fixation solution were added. Samples were centrifuged for 2 min at 800 g, the supernatant discarded, and washed with 200 µL cold fixation solution. Samples were centrifuged for 2 min at 800g, and the cell pellet resuspended in 200 µL autoMACS Running Buffer (Miltenyi Biotec). The percentage of GFP+ cells was determined by analyzing 10,000 events using MACSQuant Analyzer 16 Flow Cytometer (Miltenyi Biotec). Infectious titers were calculated in TU mL−1 using dilutions that led to a proportion of 5% to 30% GFP+ cells in the sample.
4.6. Digital Droplet PCR for Determination of Vector RNA Concentration
For the determination of the WAS‐T2A‐GFP LV RNA concentration, all samples were spiked with a β‐globin LV with a known vector concentration before the extraction process. Samples were then treated with DNAse (Qiagen) and incubated for 10 min at room temperature to digest host cell DNA. Vector RNA extraction was performed using QIAamp Viral RNA Mini Kit and QIAcube connect (Qiagen) according to manufacturer's instructions. The RT‐PCR step was done using the High‐Capacity cDNA Reverse Transcription Kit (Thermo Scientific). LV RNA concentration was determined by ddPCR using QX200™ Droplet Digital PCR (Bio‐Rad Laboratories) according to manufacturer's instructions. In brief, 20 µL of the reaction mixture containing 50 ng template DNA, 1x ddPCR Supermix for Probes (No dUTPs) (Bio‐Rad), 50 units μL−1 of HaeIII restriction enzyme (New England Biolabs), 900 nM of each primer, and 250 nM of each probe (Bio‐Rad) was loaded into the sample wells in the QX100 Droplet Generator (Bio‐Rad). A total of 40 μL of oil‐water emulsion, containing approximately 20,000 droplets, was generated with the droplet generator, and transferred into a separate well of a 96‐well PCR plate. PCR was performed under the following thermocycling conditions: enzyme activation at 95°C for 10 min, 40 cycles of denaturation at 94°C for 30 s and annealing/extension at 60°C for 1 min, and final enzyme deactivation at 98°C for 10 min. After PCR amplification, positive and negative droplets were counted using QX200™ Droplet Reader and QuantaSoft software (Bio‐Rad). The calculated WAS‐T2A‐GFP LV copy number was corrected using the normalization factor calculated by the calculated β‐globin LV copy number.
The following primer‐probe sets were used to amplify the GFP gene and the β‐globin gene (rGbG) as a control for normalization (GFP: fwd 5′‐CTGCTGCCCGACAACCA‐3′, rev 5′‐TGTGATCGCGCTTCTCGTT‐3′ and probe 5′‐HEX‐TACCTGAGC ACCCAGTCCGCCCT‐3′; rGbG: fwd 5′ CCCCATACCATCAGTACAAATTGCT‐3′, rev 5′‐TGTTAGAGGACACATGCTCACATACAT‐3′ and probe 5′‐FAM‐CCTCCTTTGCAAGTGTATTTACGACGGT‐3′).
4.7. Lentivirus‐Associated HIV p24 ELISA
Determination of lentivirus‐associated HIV p24 core protein was performed using the commercially available QuickTiter™ Lentivirus Titer Kit (Cell Biolabs) according to manufacturer's instructions.
4.8. Quantification of Total DNA Concentration
Total DNA was determined using the Qubit™ double‐stranded (ds) DNA Assay‐Kit (Thermo Fisher). Samples and controls were diluted under light protection using the HS buffer and reagent dilutions supplied with the kit. Samples were incubated for a maximum of 15 min and read using a SPARK microplate reader (Tecan) at 485 nm excitation and 530 nm emission.
4.9. Equations for Process Comparison
The perfusion rate is defined as the sum of the harvest and bleed rate:
| (1) |
P = perfusion rate [d−1]; H = harvest rate [d−1]; B = bleed rate [d−1].
The equation for the cell‐specific virus productivity qv was adapted from Coronel, Heinrich, et al. (2020). The calculation at a timepoint t i was estimated by using determined infectious titers in the bioreactor:
| (2) |
q v = cell‐specific virus productivity [TU cell−1 d], c v −1 = infectious titer [TU mL−1], t = time post induction [h], X = cells density [cells mL−1]
The cell‐specific virus yield Q v,k is defined as the total lentivirus amount produced per cell at a given time point t k:
| (3) |
Q v = cell‐specific virus yield [TU cell−1].
The total virus yield produced was normalized per reactor volume to allow a comparison between different bioreactor sizes. The arithmetic mean of infectious titers measured in the bioreactor was used for an approximation:
| (4) |
VYv = volumetric yield or yield per reactor volume [TU mLBioreactor −1], VBioreactor = working volume bioreactor [mL].
The following equation was used to describe the continuous dilution of a substance in an ideally mixed continuous stirred‐tank reactor:
| (5) |
c i = relative concentration at t i [%], c 0 = relative concentration at t 0 [%], t i = time after t 0 [d], P = perfusion rate [d −1].
4.10. Statistical Analysis
Statistical analysis was performed using GraphPad Prism 10. The LV half‐life was determined by a nonlinear regression using a one phase decay equation with a robust fit. Multiple unpaired t‐tests were performed to compare virus production using the Ambr 250 between a harvest rate of 1 VVD and 3 VVD with respect to cell‐specific yields and volumetric yields. Differences were considered significant when *p < 0.05.
Author Contributions
Maximilian Klimpel made substantial contributions to the conception of the research idea, the design of the experiments, the experimental execution, data collection, and data analysis. Maximilian Klimpel drafted the original manuscript and is responsible for revisions based on reviewer feedback. Beatrice Pflüger‐Müller and Sarah Schwingal made substantial contributions to the development of the analytical assays and to the acquisition and interpretation of the analytical data. Marta Arrizabalaga Cascallana assisted in the design and execution of the experiments. Nikki Indresh Lal assisted in the execution of the experiments. Holger Laux contributed to the conception of the research idea and the design of the experiments. Thomas Noll and Vicky Pirzas supported with the review of the experimental designs and review of the manuscript.
Conflicts of Interest
The authors have read the journal's policy and the authors of this manuscript have the following competing interests: Some of the authors own CSL Limited stocks. Maximilian Klimpel and Holger Laux are inventors of a related patent application.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Acknowledgments
The authors gratefully acknowledge the support by team members of the VVPD, VVB and Research department of CSL Innovation GmbH. This research was funded by CSL Limited and its subsidiaries.
Data Availability Statement
All relevant data are within the paper. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Aloia, R. C. , Tian H., and Jensen F. C.. 1993. “Lipid Composition and Fluidity of the Human Immunodeficiency Virus Envelope and Host Cell Plasma Membranes.” Proceedings of the National Academy of Sciences of the United States of America 90, no. 11: 5181–5185. 10.1073/pnas.90.11.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge, S. , Henry O., and Kamen A.. 2010. “Recent Progress in Lentiviral Vector Mass Production.” Biochemical Engineering Journal 48, no. 3: 362–377. 10.1016/j.bej.2009.10.017. [DOI] [Google Scholar]
- Ansorge, S. , Lanthier S., Transfiguracion J., Durocher Y., Henry O., and Kamen A.. 2009. “Development of a Scalable Process for High‐Yield Lentiviral Vector Production by Transient Transfection of HEK293 Suspension Cultures.” The Journal of Gene Medicine 11, no. 10: 868–876. 10.1002/jgm.1370. [DOI] [PubMed] [Google Scholar]
- Bareither, R. , Bargh N., Oakeshott R., Watts K., and Pollard D.. 2013. “Automated Disposable Small Scale Reactor for High Throughput Bioprocess Development: A Proof of Concept Study.” Biotechnology and Bioengineering 110, no. 12: 3126–3138. 10.1002/bit.24978. [DOI] [PubMed] [Google Scholar]
- Bareither, R. , and Pollard D.. 2010. “A Review of Advanced Small‐Scale Parallel Bioreactor Technology for Accelerated Process Development: Current State and Future Need.” Biotechnology Progress 27, no. 1: 2–14. 10.1002/btpr.522. [DOI] [PubMed] [Google Scholar]
- Bielser, J. M. , Wolf M., Souquet J., Broly H., and Morbidelli M.. 2018. “Perfusion Mammalian Cell Culture for Recombinant Protein Manufacturing – A Critical Review.” Biotechnology Advances 36, no. 4: 1328–1340. 10.1016/j.biotechadv.2018.04.011. [DOI] [PubMed] [Google Scholar]
- Bonner, M. , Ma Z., Zhou S., et al. 2015. “81. Development of a Second Generation Stable Lentiviral Packaging Cell Line in Support of Clinical Gene Transfer Protocols.” Molecular Therapy 23: S35. 10.1016/S1525-0016(16)33686-3. [DOI] [Google Scholar]
- Brühlmann, B. , and Göbel S.. 2024. “Intensified Production of Recombinant Vesicular Stomatitis Virus‐Based Vectors by Tangential Flow Depth Filtration.” Cell and Gene Therapy Insights 10, no. 04: 240. 10.18609/cgti.2024.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo, M. , Dias J. D., Panet A., et al. 2009. “Thermosensitivity of the Reverse Transcription Process as an Inactivation Mechanism of Lentiviral Vectors.” Human Gene Therapy 20, no. 10: 1168–1176. 10.1089/hum.2009.068. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Ott C. J., Townsend K., Subbaiah P., Aiyar A., and Miller W. M.. 2009. “Cholesterol Supplementation During Production Increases the Infectivity of Retroviral and Lentiviral Vectors Pseudotyped With the Vesicular Stomatitis Virus Glycoprotein (VSV‐G).” Biochemical Engineering Journal 44, no. 2–3: 199–207. 10.1016/j.bej.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comisel, R.‐M. , Kara B., Fiesser F. H., and Farid S. S.. 2021. “Lentiviral Vector Bioprocess Economics for Cell and Gene Therapy Commercialization.” Biochemical Engineering Journal 167: 107868. 10.1016/j.bej.2020.107868. [DOI] [Google Scholar]
- Coronel, J. , Gränicher G., Sandig V., Noll T., Genzel Y., and Reichl U.. 2020. “Application of an Inclined Settler for Cell Culture‐Based Influenza A Virus Production in Perfusion Mode.” Frontiers in Bioengineering and Biotechnology 8: 00672. 10.3389/fbioe.2020.00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronel, J. , Heinrich C., Klausing S., Noll T., Figueredo‐Cardero Alvio, and Castilho L. R.. 2020. “Perfusion Process Combining Low Temperature and Valeric Acid for Enhanced Recombinant Factor VIII Production.” Biotechnology Progress 36, no. 1: e2915. 10.1002/btpr.2915. [DOI] [PubMed] [Google Scholar]
- Coronel, J. , Patil A., Al‐Dali A., Braβ T., Faust N., and Wissing S.. 2021. “Efficient Production of Raav in a Perfusion Bioreactor Using an ELEVECTA Stable Producer Cell Line.” Genetic Engineering & Biotechnology News 41, no. S2: S22–S33. [Google Scholar]
- Cortin, V. , Thibault J., Jacob D., and Garnier A.. 2004. “High‐Titer Adenovirus Vector Production in 293S Cell Perfusion Culture.” Biotechnology Progress 20, no. 3: 858–863. 10.1021/bp034237l. [DOI] [PubMed] [Google Scholar]
- Dautzenberg, I. J. C. , Rabelink M. J. W. E., and Hoeben R. C.. 2020. “The Stability of Envelope‐Pseudotyped Lentiviral Vectors.” Gene Therapy 28, no. 1–2: 89–104. 10.1038/s41434-020-00193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn, M. , Klottrup‐Rees K., Lee K., and Micheletti M.. 2024. “Platform Development for High‐Throughput Optimization of Perfusion Processes: Part I: Implementation of Cell Bleeds in Microwell Plates.” Biotechnology and Bioengineering 121, no. 6: 1759–1773. 10.1002/bit.28682. [DOI] [PubMed] [Google Scholar]
- Dorn, M. , Lucas C., Klottrup‐Rees K., Lee K., and Micheletti M.. 2024. “Platform Development for High‐Throughput Optimization of Perfusion Processes—Part II: Variation of Perfusion Rate Strategies in Microwell Plates.” Biotechnology and Bioengineering 121, no. 6: 1774–1788. 10.1002/bit.28685. [DOI] [PubMed] [Google Scholar]
- Le Doux, J. M. , Davis H. E., Morgan J. R., and Yarmush M. L.. 1999. “Kinetics of Retrovirus Production and Decay.” Biotechnology and Bioengineering 63, no. 6: 654–662. . [DOI] [PubMed] [Google Scholar]
- Drouin, H. , Ritter J. B., Gorenflo V. M., Bowen B. D., and Piret J. M.. 2007. “Cell Separator Operation Within Temperature Ranges to Minimize Effects on Chinese Hamster Ovary Cell Perfusion Culture.” Biotechnology Progress 23, no. 6: 1473–1484. 10.1021/bp070276b. [DOI] [PubMed] [Google Scholar]
- Espina, J. A. , Cordeiro M. H., Milivojevic M., Pajić‐Lijaković I., and Barriga E. H.. 2023. “Response of Cells and Tissues to Shear Stress.” Journal of Cell Science 136, no. 18. 10.1242/jcs.260985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, M. V. , Cabral E. T., and Coroadinha A. S.. 2020. “Progress and Perspectives in the Development of Lentiviral Vector Producer Cells.” Biotechnology Journal 16, no. 1: 2000017. 10.1002/biot.202000017. [DOI] [PubMed] [Google Scholar]
- Finkelshtein, D. , Werman A., Novick D., Barak S., and Rubinstein M.. 2013. “LDL Receptor and Its Family Members Serve as the Cellular Receptors for Vesicular Stomatitis Virus.” Proceedings of the National Academy of Sciences of the United States of America 110, no. 18: 7306–7311. 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa, K. , and Ohsuye K.. 2006. Enhancement of Productivity of Recombinant α‐Amidating Enzyme by Low Temperature Culture. Dordrecht: Kluwer Academic Publishers eBooks, 153–157. 10.1007/0-306-46865-4_27. [DOI] [Google Scholar]
- Gagliardi, T. M. , Chelikani R., Yang Y., Tuozzolo G., and Yuan H.. 2019. “Development of a Novel, High‐Throughput Screening Tool for Efficient Perfusion‐Based Cell Culture Process Development.” Biotechnology Progress 35, no. 4: e2811. 10.1002/btpr.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzel, Y. , Vogel T., Buck J., et al. 2014. “High Cell Density Cultivations By Alternating Tangential Flow (ATF) Perfusion for Influenza A Virus Production Using Suspension Cells.” Vaccine 32, no. 24: 2770–2781. 10.1016/j.vaccine.2014.02.016. [DOI] [PubMed] [Google Scholar]
- Göbel, S. , Jaén K. E., Dorn M., et al. 2023. “Process Intensification Strategies Toward Cell Culture‐Based High‐Yield Production of a Fusogenic Oncolytic Virus.” Biotechnology and Bioengineering 120, no. 9: 2639–2657. 10.1002/bit.28353. [DOI] [PubMed] [Google Scholar]
- Göbel, S. , Pelz L., Silva C. A. T., et al. 2024. “Production of Recombinant Vesicular Stomatitis Virus‐Based Vectors By Tangential Flow Depth Filtration.” Applied Microbiology and Biotechnology 108, no. 1: 240. 10.1007/s00253-024-13078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, N. , Ambhaikar M., Zhang L., et al. 2017. “Analysis of Tubespins As a Suitable Scale‐Down Model of Bioreactors for High Cell Density Cho Cell Culture.” Biotechnology Progress 33, no. 2: 490–499. 10.1002/btpr.2418. [DOI] [PubMed] [Google Scholar]
- Gorenflo, V. M. , Angepat S., Bowen B. D., and Piret J. M.. 2003. “Optimization of an Acoustic Cell Filter With a Novel Air‐Backflush System.” Biotechnology Progress 19, no. 1: 30–36. 10.1021/bp025625a. [DOI] [PubMed] [Google Scholar]
- Gorenflo, V. M. , Smith L., Dedinsky B., Persson B., and Piret J. M.. 2002. “Scale‐Up and Optimization of an Acoustic Filter for 200 L/Day Perfusion of a CHO Cell Culture.” Biotechnology and Bioengineering 80, no. 4: 438–444. 10.1002/bit.10386. [DOI] [PubMed] [Google Scholar]
- Gränicher, G. , Coronel J., Trampler F., Jordan I., Genzel Y., and Reichl U.. 2020. “Performance of an Acoustic Settler Versus a Hollow Fiber–Based Atf Technology for Influenza Virus Production in Perfusion.” Applied Microbiology and Biotechnology 104, no. 11: 4877–4888. 10.1007/s00253-020-10596-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gränicher, G. , Tapia F., Behrendt I., Jordan I., Genzel Y., and Reichl U.. 2021. “Production of Modified Vaccinia Ankara Virus by Intensified Cell Cultures: A Comparison of Platform Technologies for Viral Vector Production.” Biotechnology Journal 16, no. 1: e2000024. 10.1002/biot.202000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzniczak, E. , Jimenez M., Irwin M., Otto O., Willoughby N., and Bridle H.. 2018. “Impact of Poloxamer 188 (Pluronic F‐68) Additive on Cell Mechanical Properties, Quantification by Real‐Time Deformability Cytometry.” Biomicrofluidics 12, no. 4: 044118. 10.1063/1.5040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidekker, M. A. , L'Heureux N., and Frangos J. A.. 2000. “Fluid Shear Stress Increases Membrane Fluidity in Endothelial Cells: A Study with DCVJ Fluorescence.” American Journal of Physiology‐Heart and Circulatory Physiology 278, no. 4: H1401–H1406. 10.1152/ajpheart.2000.278.4.h1401. [DOI] [PubMed] [Google Scholar]
- Hein, M. D. , Chawla A., Cattaneo M., Kupke S. Y., Genzel Y., and Reichl U.. 2021. “Cell Culture–Based Production of Defective Interfering Influenza A Virus Particles in Perfusion Mode Using an Alternating Tangential Flow Filtration System.” Applied Microbiology and Biotechnology 105, no. 19: 7251–7264. 10.1007/s00253-021-11561-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashikawa, F. , and Chang L. J.. 2001. “Kinetic Analyses of Stability of Simple and Complex Retroviral Vectors.” Virology 280, no. 1: 124–131. 10.1006/viro.2000.0743. [DOI] [PubMed] [Google Scholar]
- Hoffmann, M. , Wu Y.‐J., Gerber M., et al. 2010. “Fusion‐Active Glycoprotein G Mediates the Cytotoxicity of Vesicular Stomatitis Virus M Mutants Lacking Host Shut‐Off Activity.” Journal of General Virology 91, no. 11: 2782–2793. 10.1099/vir.0.023978-0. [DOI] [PubMed] [Google Scholar]
- Holic, N. , Seye A. K., Majdoul S., et al. 2014. “Influence of Mildly Acidic pH Conditions on the Production of Lentiviral and Retroviral Vectors.” Human Gene Therapy Clinical Development 25, no. 3: 178–185. 10.1089/humc.2014.027. [DOI] [PubMed] [Google Scholar]
- Hwang, B. Y. , and Schaffer D. V.. 2013. “Engineering a Serum‐Resistant and Thermostable Vesicular Stomatitis Virus G Glycoprotein for Pseudotyping Retroviral and Lentiviral Vectors.” Gene Therapy 20, no. 8: 807–815. 10.1038/gt.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoschek, S. , Schulze M., Zijlstra G., Greller G., and Matuszczyk J.. 2018. “A Protocol to Transfer a Fed‐Batch Platform Process into Semi‐Perfusion Mode: The Benefit of Automated Small‐Scale Bioreactors Compared to Shake Flasks As Scale‐Down Model.” Biotechnology Progress 35, no. 2: e2757. 10.1002/btpr.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardon, M. , and Garnier A.. 2003. “pH, PCO2, and Temperature Effect on R‐Adenovirus Production.” Biotechnology Progress 19, no. 1: 202–208. 10.1021/bp025585a. [DOI] [PubMed] [Google Scholar]
- Jiang, W. , Hua R., and Wei M., et al. 2015. “An Optimized Method for High‐Titer Lentivirus Preparations Without Ultracentrifugation.” Scientific Reports 5, no. 1: 13875. 10.1038/srep13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, L. , Wang Z.‐S., Cao Y., Sun R.‐Q., Zhou H., and Cao R.‐Y.. 2020. “Establishment and Optimization of a High‐Throughput Mimic Perfusion Model in Ambr 15.” Biotechnology Letters 43, no. 2: 423–433. 10.1007/s10529-020-03026-5. [DOI] [PubMed] [Google Scholar]
- Joe, C. C. D. , Segireddy R. R., Oliveira C., et al. 2023. “Accelerated and Intensified Manufacturing of an Adenovirus‐Vectored Vaccine to Enable Rapid Outbreak Response.” Biotechnology and Bioengineering 121, no. 1: 176–191. 10.1002/bit.28553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst, D. J. , Scibona E., Serra E., et al. 2017. “Modulation and Modeling of Monoclonal Antibody N‐Linked Glycosylation in Mammalian Cell Perfusion Reactors.” Biotechnology and Bioengineering 114, no. 9: 1978–1990. 10.1002/bit.26315. [DOI] [PubMed] [Google Scholar]
- Karst, D. J. , Steinebach F., and Morbidelli M.. 2018. “Continuous Integrated Manufacturing of Therapeutic Proteins.” Current Opinion in Biotechnology 53: 76–84. 10.1016/j.copbio.2017.12.015. [DOI] [PubMed] [Google Scholar]
- Klimpel, M. , Terrao M., and Bräuer M., et al. 2024. “Generation of Stable Suspension Producer Cell Lines for Serum‐Free Lentivirus Production.” Biotechnology Journal 19, no. 5: e2400090. 10.1002/biot.202400090. [DOI] [PubMed] [Google Scholar]
- Klimpel, M. , Terrao M., Ching N., et al. 2023. “Development of a Perfusion Process for Continuous Lentivirus Production Using Stable Suspension Producer Cell Lines.” Biotechnology and Bioengineering 120, no. 9: 2622–2638. 10.1002/bit.28413. [DOI] [PubMed] [Google Scholar]
- Kotani, H. , Newton P. B., Zhang S., et al. 1994. “Improved Methods of Retroviral Vector Transduction and Production for Gene Therapy.” Human Gene Therapy 5, no. 1: 19–28. 10.1089/hum.1994.5.1-19. [DOI] [PubMed] [Google Scholar]
- Kreye, S. , Stahn R., Nawrath K., Goralczyk V., Zoro B., and Goletz S.. 2019. “A Novel Scale‐Down Mimic of Perfusion Cell Culture Using Sedimentation in an Automated Microbioreactor (Sam).” Biotechnology Progress 35, no. 5: e2832. 10.1002/btpr.2832. [DOI] [PubMed] [Google Scholar]
- Kumar, N. , Gammell P., Meleady P., Henry M., and Clynes M.. 2008. “Differential Protein Expression Following Low Temperature Culture of Suspension CHO‐K1 Cells.” BMC Biotechnology 8, no. 1: 42. 10.1186/1472-6750-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Mercedes Segura, M. , Kamen A., Trudel P., and Garnier A.. 2005. “A Novel Purification Strategy for Retrovirus Gene Therapy Vectors Using Heparin Affinity Chromatography.” Biotechnology and Bioengineering 90, no. 4: 391–404. 10.1002/bit.20301. [DOI] [PubMed] [Google Scholar]
- Lee, G. M. , Choi J. H., Jun S. C., and Palsson B. O.. 1998. “Temperature Significantly Affects Retroviral Vector Production and Deactivation Rates, and Thereby Determines Retroviral Titers.” Bioprocess Engineering 19, no. 5: 343. 10.1007/s004490050530. [DOI] [Google Scholar]
- Lee, S.‐G. , Kim S., Robbins P. D., and Kim B.‐G.. 1996. “Optimization of Environmental Factors for the Production and Handling of Recombinant Retrovirus.” Applied Microbiology and Biotechnology 45, no. 4: 477–483. 10.1007/bf00578459. [DOI] [PubMed] [Google Scholar]
- Liu, C.‐H. , Huang S. J., and Yu T. Y.. 2021. “Cholesterol Modulates the Interaction between HIV‐1 Viral Protein R and Membrane.” Membranes 11, no. 10: 784. 10.3390/membranes11100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Loo, J. C. M. , and Wright J. F.. 2016. “Progress and Challenges in Viral Vector Manufacturing.” Human Molecular Genetics 25, no. R1: R42–R52. 10.1093/hmg/ddv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald, M. A. , Nöbel M., Roche Recinos D., et al. 2021. “Perfusion Culture of Chinese Hamster Ovary Cells for Bioprocessing Applications.” Critical Reviews in Biotechnology 42, no. 7: 1099–1115. 10.1080/07388551.2021.1998821. [DOI] [PubMed] [Google Scholar]
- Manahan, M. , Nelson M., Cacciatore J. J., Weng J., Xu S., and Pollard J.. 2019. “Scale‐Down Model Qualification of Ambr 250 High‐Throughput Mini‐Bioreactor System for Two Commercial‐Scale mAb Processes.” Biotechnology Progress 35, no. 6: e2870. 10.1002/btpr.2870. [DOI] [PubMed] [Google Scholar]
- Manceur, A. P. , Kim H., Misic V., et al. 2017. “Scalable Lentiviral Vector Production Using Stable HEK293SF Producer Cell Lines.” Human Gene Therapy Methods 28, no. 6: 330–339. 10.1089/hgtb.2017.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matanguihan, C. , and Wu P.. 2022. “Upstream Continuous Processing: Recent Advances in Production of Biopharmaceuticals and Challenges in Manufacturing.” Current Opinion in Biotechnology 78: 102828. 10.1016/j.copbio.2022.102828. [DOI] [PubMed] [Google Scholar]
- Mayrhofer, P. , Castan A., and Kunert R.. 2021. “Shake Tube Perfusion Cell Cultures Are Suitable Tools for the Prediction of Limiting Substrate, CSPR, Bleeding Strategy, Growth and Productivity Behavior.” Journal of Chemical Technology & Biotechnology 96, no. 10: 2930–2939. 10.1002/jctb.6848. [DOI] [Google Scholar]
- Mendes, J. P. , Fernandes B., Pineda E., et al. 2022. “AAV Process Intensification by Perfusion Bioreaction and Integrated Clarification.” Frontiers in Bioengineering and Biotechnology 10: 1020174. 10.3389/fbioe.2022.1020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merten, O. W. 2000. “Constructive Improvement of the Ultrasonic Separation Device Adi 1015.” Cytotechnology 34, no. 1–2: 175–179. 10.1023/A:1008147822625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merten, O.‐W. 2004. “State‐of‐the‐Art of the Production of Retroviral Vectors.” The Journal of Gene Medicine 6 Suppl 1, no. S1: 105–124. 10.1002/jgm.499. [DOI] [PubMed] [Google Scholar]
- Merten, O.‐W. , Cruz P. E., Rochette C., et al. 2001. “Comparison of Different Bioreactor Systems for the Production of High Titer Retroviral Vectors.” Biotechnology Progress 17, no. 2: 326–335. 10.1021/bp000162z. [DOI] [PubMed] [Google Scholar]
- Milone, M. C. , and O'Doherty U.. 2018. “Clinical Use of Lentiviral Vectors.” Leukemia 32, no. 7: 1529–1541. 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira, A. S. , Cavaco D. G., Faria T. Q., Alves P. M., Carrondo M. J. T., and Peixoto C.. 2020. “Advances in Lentivirus Purification.” Biotechnology Journal 16, no. 1: 2000019. 10.1002/biot.202000019. [DOI] [PubMed] [Google Scholar]
- Nguyen, D. H. , and Hildreth J. E. K.. 2000. “Evidence for Budding of Human Immunodeficiency Virus Type 1 Selectively from Glycolipid‐Enriched Membrane Lipid Rafts.” Journal of Virology 74, no. 7: 3264–3272. 10.1128/jvi.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie, J. , Sun Y., Ren H., et al. 2022. “Optimization of an Adenovirus‐Vectored Zoster Vaccine Production Process with Chemically Defined Medium and a Perfusion System.” Biotechnology Letters 44, no. 11: 1347–1358. 10.1007/s10529-022-03302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto‐Garai, J. A. , Arboleya A., Otaegi S., et al. 2020. “Cholesterol in the Viral Membrane Is a Molecular Switch Governing HIV‐1 Env Clustering.” Advanced Science 8, no. 3: 2003468. 10.1002/advs.202003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, A. , and Freed E. O.. 2001. “Plasma Membrane Rafts Play a Critical Role in HIV‐1 Assembly and Release.” Proceedings of the National Academy of Sciences of the United States of America 98, no. 24: 13925–13930. 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot, E. , Jacob D., Lanthier S., Lohr V., Ansorge S., and Kamen A. A.. 2011. “Metabolic and Kinetic Analyses of Influenza Production in Perfusion HEK293 Cell Culture.” BMC Biotechnology 11, no. 1: 84. 10.1186/1472-6750-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, H. , Taylor J., Clack B., Stewart R. S., and Canterberry S. C.. 2013. “Effects of Storage Conditions on the Morphology and Titer of Lentiviral Vectors”.” Faculty Publications 94: 1–9. https://scholarworks.sfasu.edu/biology/94. [Google Scholar]
- Rodrigues, A. F. , Carmo M., Alves P. M., and Coroadinha A. S.. 2009. “Retroviral Vector Production Under Serum Deprivation: The Role of Lipids.” Biotechnology and Bioengineering 104, no. 6: 1171–1181. 10.1002/bit.22499. [DOI] [PubMed] [Google Scholar]
- Rodrigues, T. , Carrondo M. J. T., Alves P. M., and Cruz P. E.. 2007. “Purification of Retroviral Vectors for Clinical Application: Biological Implications and Technological Challenges.” Journal of Biotechnology 127, no. 3: 520–541. 10.1016/j.jbiotec.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Schwarz, H. , Lee K., Castan A., and Chotteau V.. 2023. “Optimization of Medium with Perfusion Microbioreactors for High Density CHO Cell Cultures at Very Low Renewal Rate Aided by Design of Experiments.” Biotechnology and Bioengineering 120, no. 9: 2523–2541. 10.1002/bit.28397. [DOI] [PubMed] [Google Scholar]
- Silva, C. A. T. , Kamen A. A., and Henry O.. 2023. “Intensified Influenza Virus Production in Suspension HEK293SF Cell Cultures Operated in Fed‐Batch or Perfusion with Continuous Harvest.” Vaccines 11, no. 12: 1819. 10.3390/vaccines11121819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharmalingam, T. , Ghebeh H., Wuerz T., and Butler M.. 2008. “Pluronic Enhances the Robustness and Reduces the Cell Attachment of Mammalian Cells.” Molecular Biotechnology 39, no. 2: 167–177. 10.1007/s12033-008-9045-8. [DOI] [PubMed] [Google Scholar]
- Tharmalingam, T. , and Goudar C. T.. 2014. “Evaluating the Impact of High PluronicF68 Concentrations on Antibody Producing CHO Cell Lines.” Biotechnology and Bioengineering 112, no. 4: 832–837. 10.1002/bit.25491. [DOI] [PubMed] [Google Scholar]
- Throm, R. E. , Ouma A. A., Zhou S., et al. 2009. “Efficient Construction of Producer Cell Lines for a SIN Lentiviral Vector for SCID‐X1 Gene Therapy by Concatemeric Array Transfection.” Blood 113, no. 21: 5104–5110. 10.1182/blood-2008-11-191049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todesco, H. M. , Gafuik C., John C. M., et al. 2024. “High‐Titer Manufacturing of SARS‐CoV‐2 Spike‐Pseudotyped VSV in Stirred‐Tank Bioreactors.” Molecular Therapy. Methods & Clinical Development 32, no. 1: 101189. 10.1016/j.omtm.2024.101189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tona, R. M. , Shah R., Middaugh K., et al. 2023. “Process Intensification for Lentiviral Vector Manufacturing Using Tangential Flow Depth Filtration.” Molecular Therapy. Methods & Clinical Development 29: 93–107. 10.1016/j.omtm.2023.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, M. Y. , and Kamen A. A.. 2022. “Production of Lentiviral Vectors Using a HEK‐293 Producer Cell Line and Advanced Perfusion Processing.” Frontiers in Bioengineering and Biotechnology 10: 887716. 10.3389/fbioe.2022.887716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregidgo, M. , Dorn M., Lucas C., and Micheletti M.. 2023. “Design and Characterization of a Novel Perfusion Reactor for Biopharmaceuticals Production.” Chemical Engineering Research and Design 194: 344–357. 10.1016/j.cherd.2023.04.066. [DOI] [Google Scholar]
- Tregidgo, M. , Lucas C., Dorn M., and Martina M.. 2023. “Development of mL‐Scale Pseudo‐Perfusion Methodologies for High‐Throughput Early Phase Development Studies.” Biochemical Engineering Journal 195: 108906. 10.1016/j.bej.2023.108906. [DOI] [Google Scholar]
- Vázquez‐Ramírez, D. , Jordan I., Sandig V., Genzel Y., and Reichl U.. 2019. “High Titer MVA and Influenza A Virus Production Using a Hybrid Fed‐Batch/Perfusion Strategy With an Atf System.” Applied Microbiology and Biotechnology 103, no. 7: 3025–3035. 10.1007/s00253-019-09694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villiger‐Oberbek, A. , Yang Y., Zhou W., and Yang J.. 2015. “Development and Application of a High‐Throughput Platform for Perfusion‐Based Cell Culture Processes.” Journal of Biotechnology 212: 21–29. 10.1016/j.jbiotec.2015.06.428. [DOI] [PubMed] [Google Scholar]
- Walther, J. , Lu J., Hollenbach M., et al. 2018. “Perfusion Cell Culture Decreases Process and Product Heterogeneity in a Head‐To‐Head Comparison With Fed‐Batch.” Biotechnology Journal 14, no. 2: 1700733. 10.1002/biot.201700733. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Godfrey S., Ravikrishnan J., Lin H., Vogel J., and Coffman J.. 2017. “Shear Contributions to Cell Culture Performance and Product Recovery in ATF and TFF Perfusion Systems.” Journal of Biotechnology 246: 52–60. 10.1016/j.jbiotec.2017.01.020. [DOI] [PubMed] [Google Scholar]
- Wechuck, J. B. , Ozuer A., Goins W. F., et al. 2002. “Effect of Temperature, Medium Composition, and Cell Passage on Production of Herpes‐Based Viral Vectors.” Biotechnology and Bioengineering 79, no. 1: 112–119. 10.1002/bit.10310. [DOI] [PubMed] [Google Scholar]
- Wei, Z. , Xia Y., and Su Y., et al. 2023. “Modulating and Optimizing Pluronic F‐68 Concentrations and Feeding for Intensified Perfusion Chinese Hamster Ovary Cell Cultures.” Biotechnology Progress 39, no. 4: e3340. 10.1002/btpr.3340. [DOI] [PubMed] [Google Scholar]
- Welsch, S. , Müller B., and Kräusslich H.‐G.. 2007. “More Than One Door ‐ Budding of Enveloped Viruses through Cellular Membranes.” FEBS Letters 581, no. 11: 2089–2097. 10.1016/j.febslet.2007.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, T. , Goodyear O., Davies L., et al. 2020. “Lentiviral Vector Manufacturing Process Enhancement Utilizing TFDF™ Technology.” Cell and Gene Therapy Insights 6, no. 3: 455–467. 10.18609/cgti.2020.053. [DOI] [Google Scholar]
- Wu, Y. , Bissinger T., Genzel Y., Liu X., Reichl U., and Tan W. S.. 2021. “High Cell Density Perfusion Process for High Yield of Influenza A Virus Production Using Mdck Suspension Cells.” Applied Microbiology and Biotechnology 105, no. 4: 1421–1434. 10.1007/s00253-020-11050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P. , Clark C., Ryder T., et al. 2017. “Characterization of TAP Ambr 250 Disposable Bioreactors, as a Reliable Scale‐Down Model for Biologics Process Development.” Biotechnology Progress 33, no. 2: 478–489. 10.1002/btpr.2417. [DOI] [PubMed] [Google Scholar]
- Xu, S. , Jiang R., Chen Y., Wang F., and Chen H.. 2017. “Impact of Pluronic F68 on Hollow Fiber Filter‐Based Perfusion Culture Performance.” Bioprocess and Biosystems Engineering 40, no. 9: 1317–1326. 10.1007/s00449-017-1790-2. [DOI] [PubMed] [Google Scholar]
- Yamamoto, K. , and Ando J.. 2015. “Vascular Endothelial Cell Membranes Differentiate between Stretch and Shear Stress Through Transitions in Their Lipid Phases.” American Journal of Physiology‐Heart and Circulatory Physiology 309, no. 7: H1178–H1185. 10.1152/ajpheart.00241.2015. [DOI] [PubMed] [Google Scholar]
- Yamamoto, K. , Nogimori Y., Imamura H., and Ando J.. 2020. “Shear Stress Activates Mitochondrial Oxidative Phosphorylation by Reducing Plasma Membrane Cholesterol in Vascular Endothelial Cells.” Proceedings of the National Academy of Sciences of the United States of America 117, no. 52: 33660–33667. 10.1073/pnas.2014029117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, S. K. , Choi S. L., Song J. Y., and Lee G. M.. 2004. “Effect of Culture pH on Erythropoietin Production by Chinese Hamster Ovary Cells Grown in Suspension at 32.5 and 37.0°C.” Biotechnology and Bioengineering 89, no. 3: 345–356. 10.1002/bit.20353. [DOI] [PubMed] [Google Scholar]
- Zhan, C. , Bidkhori G., Schwarz H., et al. 2020. “Low Shear Stress Increases Recombinant Protein Production and High Shear Stress Increases Apoptosis in Human Cells.” IScience 23, no. 11: 101653. 10.1016/j.isci.2020.101653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Al‐Rubeai M., and Thomas C. R.. 1992. “Effect of Pluronic F‐68 on the Mechanical Properties of Mammalian Cells.” Enzyme and Microbial Technology 14, no. 12: 980–983. 10.1016/0141-0229(92)90081-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
All relevant data are within the paper. The data that support the findings of this study are available from the corresponding author upon reasonable request.