


Abstract
The role of the 50S particle of Escherichia coli ribosome and its 23S rRNA in the refolding and subunit association of dimeric porcine heart cytoplasmic malate dehydrogenase (s-MDH) has been investigated. The self-reconstitution of s-MDH is governed by two parallel pathways representing the folding of the inactive monomeric and the dimeric intermediates. However, in the presence of these folding modulators, only one first order kinetics was observed. To understand whether this involved the folding of the monomers or the dimers, subunit association of s-MDH was studied using fluorescein-5-isothiocyanate–rhodamine-isothiocyanate (FITC–RITC) fluorescence energy transfer and chemical cross-linking with gluteraldehyde. The observation suggests that during refolding the interaction of the unstructured monomers of s-MDH with these ribosomal folding modulators leads to very fast formation of structured monomers that immediately dimerise. These inactive dimers then fold to the native ones, which is the rate limiting step in 23S or 50S assisted refolding of s-MDH. Furthermore, the sequential action of the two fragments of domain V of 23S rRNA has been investigated in order to elucidate the mechanism. The central loop of domain V of 23S rRNA (RNA1) traps the monomeric intermediates, and when they are released by the upper stem–loop region of the domain V of 23S rRNA (RNA2) they are already structured enough to form dimeric intermediates which are directed towards the proper folding pathway.
INTRODUCTION
For proteins active as rigid oligomers, ‘subunit association’ is as important as chain ‘folding’ and these two coupled processes occur so uniquely in different oligomeric systems that no general model predicting the sequence of folding and association could be given. The suggested model for oligomerisation emphasises two processes. First, the size of the associating monomers should be comparable, preferably full length, or if not so at least grown enough to exceed the subdomain size so that solvent interactions enforce a certain intrinsic backbone structure to the entities undergoing association. Secondly, the formation of the ‘structured monomer’, with well-constructed secondary structure, must precede the association reaction because otherwise ‘wrong aggregates’ will be formed due to incomplete folding or incorrect association. In vitro, the structured oligomers are formed very fast, almost at the nanosecond level, by hydrophobic collapse immediately after dilution of the denaturant. Within the cell, the growing polypeptide chain most probably acquires that secondary structure co-translationally and thereby fulfils the prerequisite for oligomerisation.
Dimeric proteins represent the simplest oligomeric system. For these proteins ‘in vitro’ reconstitution consists of only one association step apart from two possible folding reactions at the level of the monomer and the dimer. Which of these two reactions is rate limiting cannot be predicted a priori. Generally, fast folding steps preceding the formation of the ‘structured monomer’ are not included in the folding pathway because they are not rate limiting. Depending on whether the ‘folding and assembly of the structured monomers’ or the ‘reshuffling of the incompletely folded dimer’ is rate limiting, a uni-bimolecular or a simple unimolecular mechanism is expected to hold.
Porcine heart cytoplasmic malate dehydrogenase (s-MDH) is a homodimer (2 × 35 kDa). The self-folding of s-MDH confirms both of the two possible alternatives by exhibiting two parallel folding pathways. The kinetic analysis of self-folding and association of s-MDH shows that for ∼70% of molecules formation of the native dimer is determined by a slow folding (isomerisation) step at the monomer level (K1 = 1.3 × 10–3 s–1 ), followed by a very fast association reaction with rate constant like a diffusion controlled process (K > 106 mol–1 s–1). The remaining 30% of chains rapidly form an inactive dimeric intermediate which is rearranged to form the native dimer by a very slow folding reaction (K2 of the order of 10–5 s–1) (1). The kinetics and yield of folding of s-MDH remain essentially unchanged even in the presence of chaperonins GroEL/GroES (2).
There are previous reports that ribosomes assist in protein folding (3–7). This activity was first identified in the large subunit of the ribosome and finally traced to domain V of 23S rRNA, both in vitro and in vivo (8,9). It was further demonstrated that the in vitro synthesised domain V of 23S rRNA also had refolding activity (10). When refolding of s-MDH was studied in the presence of 70S ribosome or its components having protein folding activity (50S subunit or 23S rRNA or in vitro synthesised domain V of 23S rRNA), not only were the folding yields consistently higher than for self-folding but it also showed completely different folding kinetics. Instead of two parallel folding pathways as was in the case for s-MDH self-folding, it showed only one with first order kinetics (K1 = 2.6 × 10–3 s–1 in 23S rRNA assisted folding). Whether this was due to the isomerisation at the monomer level or at the dimer level could not be concluded from reactivation experiments.
To answer this question, the reassociation or dimerisation of s-MDH was studied by two different approaches: (i) fluorescence energy transfer and (ii) chemical cross-linking. The fluorescence energy transfer method has been successfully used in many systems to study association of the macromolecules. The strategy used to study dimerisation of s-MDH was to label it with either fluorescein-5-isothiocyanate (FITC, donor) or rhodamine-isothiocyanate (RITC, acceptor). The Förster distance for this donor acceptor pair is 40 Å, at which the efficiency of energy transfer is 50% (11). Upon mixing the FITC labelled s-MDH and RITC labelled s-MDH no energy transfer was found. However when these mixed samples were denatured and refolding was studied upon dilution of the denaturant, energy transfer was seen. The extent of energy transfer was greater in the case of 50S and 23S rRNA assisted folding than for self-folding. By calculating the efficiency of energy transfer the distance between the donor and acceptor labels could be measured (34.7 Å) and the extent of dimerisation could also be predicted.
The chemical cross-linking method, using gluteraldehyde followed by NaDodSO4–polyacrylamide gel electrophoresis, had been previously employed to elucidate individual steps in the complex association reaction of oligomeric proteins (12,13), including s-MDH (1). This method allows quantification of the different molecular species formed at any time during the process of reconstitution. So from this experiment the 23S rRNA assisted refolding of s-MDH could be fully traced and the rate constants of the individual association and isomerisation steps could be determined.
We have reported earlier that two fragments of domain V of 23S rRNA play complementary roles in the protein folding process. The putative secondary structure of these in vitro transcribed RNA fragments, namely RNA1 (337 nt, covering the central loop and the lower portions of the domain V of Bacillus subtilis 23S rRNA) and RNA2 (425 nt, covering the upper stem–loop region of domain V) are shown in Figure 1. These two fragments together form the whole domain V. Using monomeric carbonic anhydrase as the refolding study system, it has been shown that RNA1 binds and interacts with some protein folding intermediate and RNA2 releases it, which then folds to the native form (14). In the case of dimeric s-MDH the observation is similar, which leads to further insight into the process. The sequential binding and releasing actions (by RNA1 and RNA2, respectively) have been studied by gel-filtration chromatography using radioactively labelled RNA transcripts and fluorescence labelled s-MDH. Subsequently association and folding of s-MDH was studied using fluorescence energy transfer and activity assays. The observation suggests that, during refolding, the sequential interaction of the unfolded s-MDH monomers with these fragments of domain V of 23S rRNA lead to the formation of ‘structured monomers’, which dimerise immediately after release from RNA1, and that these dimeric intermediates fold to produce the native dimers. This clearly indicates that the involvement of the 23S RNA in protein folding is to drive the unfolded species to the productive folding pathway, which is very specific and this makes it different from self-folding, where the first population formed after dilution of the denaturant is rather a random ensemble.
Figure 1.
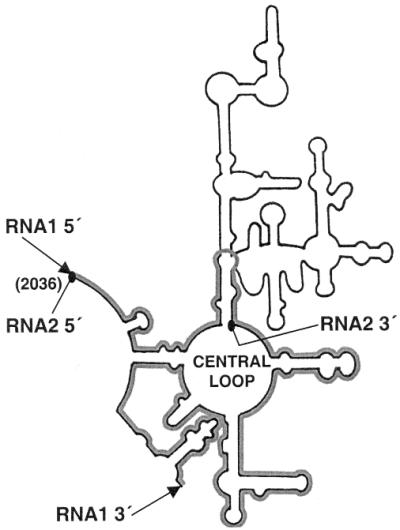
Putative secondary structure of domain V of 23S rRNA of B.subtilis. The 5′ and 3′ ends of the two RNA constructs, RNA1 [337 nt RNA fragment containing the central loop of domain V, (highlighted)] and RNA2 (425 nt fragment from the 5′ end of the domain V containing mainly the upper stem–loop regions) are marked with ‘triangular headed arrow’ and ‘oval headed arrow’ symbols, respectively. These two constructs together form the whole domain V.
MATERIALS AND METHODS
Enzymes and reagents
s-MDH (EC 1.1.1.37), bought from Sigma (St Louis, MO), was obtained as a precipitate in 3.2 M (NH4)2SO4 added as a stabilising salt during its storage. To remove this high salt, the enzyme solution was dialysed against 100 mM potassium phosphate buffer pH 7.6 containing 5 mM β-ME. After dialysis, s-MDH had a specific activity of 350 IU/mg, as determined at 25°C, pH 7.6, in the presence of 0.5 mM oxaloacetate and 0.2 mM NADH. Enzyme concentrations were determined by using an extinction coefficient of E0.1%280nm = 1.08 (15). Molar concentrations refer to a subunit molecular weight of 35 000 Da. Gdn–HCl, DEAE cellulose, SDS, glycine, NaBH4, Na deoxycholate and gluteraldehyde were purchased from Sigma. FITC and RITC were purchased from Molecular Probes Inc. All other reagents were of analytical grade.
Preparation of Escherichia coli 50S particle and its 23S rRNA and preparation of segments of domain V of 23S rRNA from B.subtilis
Purification of E.coli 50S particle and its 23S rRNA was done following the protocol described earlier (6). The purity of the 23S rRNA was checked as described in Pal et al. (14). The preparations of RNA1 and RNA2, the segments of domain V of 23S rRNA from B.subtilis, have also been described in Pal et al. (14).
Denaturation and renaturation of s-MDH
Denaturation and dissociation of s-MDH were performed by 40 min incubation at 20°C in 6 M Gdn–HCl in 0.1 M sodium phosphate buffer, pH 7.6. The enzyme concentration was varied according to the type of experiment done. Renaturation and reassociation were performed by appropriate dilution in 0.1 M sodium phosphate buffer pH 7.6 containing 5 mM β-ME (except for the cross-linking experiments), 10 mM Mg acetate and 50–150 mM NaCl at 25°C. In the case of the assisted folding, the folding modulators (50S, 23S rRNA, RNA1 and/or RNA2) were pre-incubated in the refolding buffer.
FITC and RITC labelling
s-MDH was dialysed against 100 mM sodium phosphate buffer, pH 8.0. One microgram of FITC (molecular weight 389.38 Da) was dissolved in 50 µl acetone. The fine particles were removed by centrifugation and the clear FITC solution was added to 200 µl of 4 mg/ml s-MDH. The mixture was incubated at 4°C for 30–60 min and then loaded on a Sephadex G-50 column. Fractions were collected and assayed following the standard procedure of s-MDH assay. In parallel, FITC fluorescence of the fractions at 520 nm was monitored by following excitation at 495 nm. Both the parameters were plotted and the overlapping peak fractions were collected as FITC labelled s-MDH. From the absorbance spectrum of this FITC labelled s-MDH (FITC: ɛM = 76 000 at 495 nm and s-MDH: E0.1% = 1.08 at 280 nm), the relative molar proportion of the label to the enzyme was obtained.
The procedure for RITC (molecular weight 584 Da) labelling was same as that for FITC labelling. The only difference was that it was dissolved in methanol. To check RITC fluorescence the fractions were excited at 570 nm and the fluorescence emission was monitored at 595 nm. RITC concentration was calculated from the absorbance spectrum using ɛM = 93 000 at 570 nm. In contrast to FITC, RITC was found to affect the absorbance spectrum of s-MDH. Hence, enzyme concentration was calculated from its specific activity. In both cases the incubation period and conditions were optimised to get a 1:1 labelling ratio.
Subunit reassociation studied by energy transfer
FITC labelled s-MDH (D) and RITC labelled s-MDH (A) were mixed at a ratio of 1:1. The mixture was excited at 495 nm (excitation wavelength for FITC) and the emission spectra of both FITC and RITC were recorded. This combined spectrum was further used as control.
The mixture was then denatured with 6 M Gdn–HCl at 20°C for 40 min at an enzyme concentration of 2.4 µM (monomer) in a 100 mM Na–phosphate buffer, pH 8.0. The denatured samples were diluted 80-fold in the same buffer at 20°C containing 50 mM NaCl for reconstitution. Upon random association, three types of dimers are expected to be reconstituted: DD, DA and AA. Among these, when D is excited, only DA dimers will show energy transfer. So unless dimers are formed no energy transfer will be found or, in other words, D to A energy transfer will directly reflect dimer formation. The fluorescence energy transfer was studied immediately after dilution of the denaturant. Fluorescence emission spectra of FITC and RITC were monitored and F520 (emission λmax of FITC) and F595 (emission λmax of RITC) values were recorded from time to time, exciting the samples at 495 nm (excitation λmax of FITC). The same experiments were performed in the presence of 30 nM 23S rRNA and 30 nM 50S to study their effect on dimerisation. The efficiency of energy transfer and the distance between the donor and acceptor were calculated using standard formulae (16), the Förster distance for this D–A pair being 40 Å.
Chemical cross-linking experiments
For cross-linking experiments, denaturation of native s-MDH was performed at a concentration of 4 mg/ml in 6 M Gdn–HCl at 20°C for 2 h and reconstitution was initiated by 200-fold dilution of the denaturation mixture so that in the refolding buffer the residual Gdn–HCl concentration was 30 mM, above which gluteraldehyde cross-linking could not be done. Chemical cross-linking with gluteraldehyde (1.2%) was done modifying the method of Zettlmeissl el al. (13). In the case of 23S rRNA assisted folding, RNA was removed from the mixture by passing the cross-linked product twice through small DEAE cellulose spin columns before running in the gel. The cross-linked products were then subjected to SDS–PAGE to obtain the actual particle distribution profile following Sambrook et al. (17). The gels were silver stained and individual lanes were scanned with a Bio-Rad gel documentation system and the profiles were plotted.
Gel filtration chromatography
Gel filtration was performed while refolding s-MDH (a mixture of FITC and RITC labelled s-MDH) with an excess of RNA1 and/or RNA2. 100 µl of refolding mix containing 30 nM s-MDH and a 5-fold excess of RNA1 was loaded on a 12 ml Sephadex G-100 column. In the fractions, 32P count was taken to trace the RNAs and F520 (emission λmax of FITC) and F595 (emission λmax of RITC) were recorded, setting excitation wavelength at 495 nm, to trace the protein. RITC fluorescence at 595 nm was recorded to see whether there was any energy transfer indicating dimerisation. The same gel filtration experiment was also performed after addition of 100 nM of RNA2 in the refolding mixture containing 30 nM s-MDH and 100 nM RNA1.
RESULTS
Reactivation kinetics of denatured s-MDH with 50S particle and 23S rRNA
In 1986 Rudolph et al. reported a detailed study on self-folding of s-MDH (1). It showed two parallel slow and very slow first order kinetics with rate constants K1 = 1.3 × 10–3 s–1 and K2 = 7 × 10–5 s–1 at 20°C. The two first order reactions are due to the folding at the monomer and the dimer level respectively. Our results of self-folding experiments with s-MDH agree well with their findings.
Figure 2 shows the time course of reactivation of 6 M Gdn–HCl denatured s-MDH (14 nM) with stoichiometric amounts of 50S and 23S rRNA. The kinetics follow a single first order reaction (inset Fig. 2) in both cases, with the rate constants K1 = 1.6 × 10–3 s–1 for 50S and K1 = 2.6 × 10–3 s–1 for 23S rRNA. At zero time (immediately after dilution), reactivation started from zero activity. When experiments were done with 10-fold less protein concentration, a detectable lag period of ∼1–2 min was seen at the beginning of the refolding reaction. However, the rate constants are found to be essentially independent of the s-MDH concentration during refolding (range 100 ng/ml to 10 µg/ml).
Figure 2.

Kinetics of reactivation of s-MDH (14 nM) at 25°C in the presence of 50S particle (filled circle) and 23S rRNA (open circle) of E.coli ribosome after 40 min denaturation by 6 M Gdn–HCl at 20°C. Inset, the first order plot of these time course data. The rate constants determined for the 50S assisted (filled circle) and 23S rRNA assisted (open circle) reactivation of s-MDH from these plots are K1 = 1.6 × 10–3 s–1 and K1 = 2.6 × 10–3 s–1, respectively.
Fluorescence energy transfer to study dimerisation
Two identical aliquots of the enzyme (4 mg/ml) were labelled with FITC and RITC, respectively. The labelled proteins were purified on a Sephadex G-50 column. The incubation conditions and duration of the labelling reaction were controlled in such a way that a 1:1 ‘enzyme:probe’ labelling ratio was obtained. Labelling caused no loss of biochemical activity of s-MDH and did not interfere with the denaturation and refolding of the enzyme. After denaturation and reconstitution the labels were well-retained.
FITC labelled and RITC labelled s-MDH were mixed in equal proportions, and with this mixture subunit reassociation was studied after denaturation with 6 M Gdn–HCl (enzyme concentration 30 nM during refolding). Figure 3 shows the energy transfer profile between FITC and RITC during renaturation. The whole range of emission of FITC and RITC was scanned to obtain the total profile of fluorescence emission of both the donor (FITC) and acceptor (RITC) when excited at 495 nm (excitation λmax of FITC). The emission spectrum of the mixture is the control (Fig. 3, spectrum ‘a’). It shows a single peak at 520 nm, which is due to FITC. No fluorescence peak is obtained at 595 nm where the RITC fluorescence emission peak is expected. So the mixture effectively shows no energy transfer. But while refolding (both unassisted and 50S/23S assisted) immediately after dilution of the denaturant, the FITC fluorescence is partially quenched and RITC fluorescence is enhanced, showing energy transfer between them (Fig. 3, spectra ‘b’ and ‘c’). This is due to the formation of dimers having both the labels (FITC–RITC dimers). During self-folding there is small increase in energy transfer in the first 10–12 min but after that no further significant change is observed in the 40 min of study (Fig. 3, b1, b2 and b3).
Figure 3.

Subunit association of s-MDH studied by energy transfer. FITC labelled s-MDH and RITC labelled s-MDH were mixed in equal proportions. Excitation wavelength was set at 495 nm (excitation λmax of FITC) and the whole range of FITC and RITC emission was monitored. (a) The control spectrum, which shows the fluorescence emission of the mixture (30 nM). Only FITC fluorescence was observed (λmax at 520 nm). Absence of RITC fluorescence indicated that no energy transfer had occurred. (b) The fluorescence energy transfer during self-reconstitution of the labelled s-MDH mixture (30 nM); (b1) immediately after the manual mixing time, (b2) at 5 min and (b3) at 12, 20, 30 and 60 min. (c) The fluorescence energy transfer shown during 23S rRNA (30 nM) assisted refolding of the labelled s-MDH mixture (30 nM). The first spectrum was taken immediately after the manual mixing time and then at 5, 10, 20, 30 and 60 min. Within this period no increase in energy transfer was seen. Inset, the fluorescence energy transfer in the presence of 30 nM 50S particle.
In the presence of 23S rRNA the extent of energy transfer is 3-fold enhanced (as estimated from the efficiency of energy transfer) (Fig. 3, spectra ‘c’). But in this case no further increase in energy transfer is found after the first reading, taken immediately after the manual mixing time. This result suggests that the net dimer content, including the incompletely folded and properly folded ones (D* and D), does not change much in this period, though this phase involves significant recovery of activity. Using the transfer efficiency (70%) and the R0 value (40 Å) the distance between FITC and RITC labels was calculated to be 34.7 Å. Similar results are obtained while refolding with 50S subunit, which is summarised in the inset of Figure 3.
Reassociation studied by chemical cross-linking
The kinetics of reassociation of oligomeric proteins can be analysed by fast chemical cross-linking with gluteraldehyde during refolding. The cross-linked products are then subjected to SDS–PAGE, and from the gel the proportion of different folding species can be estimated at any time point using a gel-documentation system. Figure 4A and B reflects the time course of self-folding and 23S rRNA assisted folding of s-MDH, respectively, studied in this way. In SDS–PAGE the cross-linked native s-MDH dimers produce a single band of 70 kDa (active dimer or D) and uncross-linked s-MDH produces a band of 35 kDa (monomer or M). During refolding, besides these two bands another band, which migrates a little faster than the D band, is also observed. This band is probably due to an incompletely folded dimer species (D*), as suggested by Rudolph et al. (1). As evident from Figure 4A (i) and (ii), during self-folding of s-MDH both the ‘M’ and ‘D*’ species contribute to the formation of the active dimer ‘D’. Hence the folding involves two parallel pathways. At the first time point the amplitude of the ‘M’ and ‘D*’ species was ∼70 and 30%, respectively. This result agrees well with the previous observation of Rudolph et al. (1). Figure 4B (i) and (ii) are the results of the similar experiment where refolding of s-MDH is done in the presence of 23S rRNA. The profile is distinctly different from that of the self-folding. In this case, even at the first time point (1 min) no monomer species are found. Instead the dimeric intermediate (‘D*’) is found as the major species. The formation of the native dimer is traced against time, which showed first order kinetics with a rate constant K1 = 2.7 × 10–3 s–1 that is almost same as that determined previously in the case of the 23S rRNA assisted reactivation of s-MDH.
Figure 4.

The kinetics of refolding and reassociation of s-MDH at 25°C as determined from chemical cross-linking with gluteraldehyde. (A) Unassisted folding. (i) The band pattern obtained in the SDS–PAGE in 1 min (lane 1), 3 min (lane 2), 5 min (lane 3), 7 min (lane 4), 10 min (lane 5), 15 min (lane 6), 25 min (lane 7) and 45 min (lane 8). Cross-linked native s-MDH is run in lane 9 and molecular weight markers in lane 10. The monomer (M), inactive dimer (D*) and active dimer (D) populations are labelled. s-MDH concentration during refolding was 40 µg/ml. (ii) The relative proportions of M, D* and D with time as determined using Bio-Rad gel-documentation system. (B) 23S rRNA assisted folding. (i) The band pattern obtained in the SDS–PAGE representing cross-linking in 1 min (lane 2), 3 min (lane 3), 5 min (lane 4), 7 min (lane 5), 10 min (lane 6), 15 min (lane 7), 25 min (lane 8) and 30 min (lane 9) during refolding. Lane 1 shows molecular weight markers and native cross-linked s-MDH is run in lane 10. (ii) The relative proportion of D and D* with time. The formation of D with time was fitted in a first order plot with rate constant K1 = 2.7 × 10–3 s–1 (inset).
Effect of RNA1 and RNA2 in folding and subunit reassociation of s-MDH
We have reported before that the 660 nt in vitro transcript of domain V of 23S rRNA from B.subtilis and the 595 nt in vitro transcript of domain V of 23S rRNA from E.coli could refold denatured proteins including s-MDH, but none of the smaller transcripts containing different fragments of domain V showed folding activity (10). Later we observed that a mixture of two in vitro transcribed major fragments of domain V of B.subtilis 23S rRNA (RNA1 and RNA2), which can essentially form the whole domain V when put together (Fig. 1), can also refold proteins (14). This study was further extended using carbonic anhydrase as the study system and it was found that sequential interaction of RNA1 and RNA2 with the protein was needed for a successful folding. To test whether this observation was of any general significance, we performed similar experiments with dimeric s-MDH. Increasing concentration of RNA1 inhibits reactivation of denatured s-MDH (Fig. 5). To check how RNA1 is interfering with its folding, gel filtration experiments are performed with the refolding mixture (s-MDH:RNA1 = 1:4). The RNA is labelled with [α-32P]UTP and the protein is a 1:1 (M/M) mixture of FITC labelled s-MDH and RITC labelled s-MDH. This mixture of FITC labelled and RITC labelled s-MDH is used to identify the oligomeric state of the protein during RNA-assisted refolding. From our previous observation it is known that no fluorescence energy transfer takes place in this mixture when FITC is excited at 495 nm (Fig. 3, spectrum ‘a’). However, while refolding after denaturation, dimers are formed at random, and this can be detected by quenched FITC fluorescence at 520 nm and enhanced RITC fluorescence at 595 nm due to energy transfer. Figure 6 shows the elution profile of the refolding mixture in a Sephadex G-100 column. The profile showed that RNA1 and s-MDH are co-eluted in a single peak (Fig. 6C). When excited at 495 nm it shows only FITC fluorescence, and no RITC fluorescence is found. This absence of energy transfer proves that the RNA1 bound species of s-MDH are monomers. These fractions containing s-MDH have no enzyme activity. This is probably due to the fact that the monomers of s-MDH do not possess biochemical activity (18).
Figure 5.

Extent of reactivation of s-MDH (10 nM) with or without stoichiometric amount of ribosomal folding modulators after denaturation by 6 M Gdn–HCl at 20°C for 40 min. The reactivation is inhibited in the presence of increasing concentrations of RNA1.
Figure 6.

Sephadex G-100 gel filtration profile of (A) 32P labelled RNA1, (B) s-MDH (1:1 FITC and RITC labelled s-MDH mixture, 30 nM), (C) s-MDH (30 nM), while refolding in the presence of 150 nM RNA1 and (D) s-MDH (30 nM), while refolding in the presence of 100 nM RNA2 added in the 100 nM RNA1 containing refolding mixture. The 32P counts were used to trace RNA1 (filled triangle). The enzyme was traced by FITC labels (open circle). Excitation was done at 495 nm and FITC emission at 520 nm was monitored. Keeping the excitation wavelength the same RITC fluorescence emission at 595 nm was also monitored (filled circle) to see whether there was any energy transfer.
When stoichiometric amounts of RNA2 are added to the refolding mixture containing RNA1, regaining of activity starts immediately. The kinetics of reactivation involves a first order reaction with rate constant 2.7 × 10–3 s–1 (data not shown). When gel filtration is performed with this refolding mixture, RNAs (traced by 32P) and s-MDH (traced by FITC and RITC fluorescence at 520 and 595 nm respectively, exciting at 495 nm) are eluted in separate fractions (Fig. 6D). The fraction containing s-MDH shows both FITC and RITC fluorescence when excited at 495 nm. The FITC fluorescence is quenched to some extent when compared with the previous set with only RNA1. This quenched FITC fluorescence and enhanced RITC fluorescence clearly indicates that energy transfer has occurred due to the formation of FITC–RITC labelled dimers after the release of the trapped intermediates from RNA1 by RNA2.
DISCUSSION
The protein folding problem has been one of the major research topics of the last few decades. It has been attacked using different approaches but there still remain many missing links to obtain the whole scenario. For oligomeric proteins the folding problem is even more difficult because here subunit association plays a major role, in addition to chain folding. It is obvious that prior to the association step some preliminary structure has to be formed, but the extent of this prerequisite structure varies in different oligomeric systems and depends on the complexity of the structure of the subunit interfaces. In in vitro refolding experiments, the formation of this basic structure is often very fast and is not included in the folding pathway since it does not influence the rate limiting step of folding. Nevertheless to study subunit association designing in vitro refolding experiments with the oligomeric proteins are quite realistic because in the cell the subunit association most probably takes place after release of the full length chain from the ribosome. Whether the monomers are fully folded by that time or not is still a point of debate. Many arguments have come up in recent years in favour of co-translational folding, suggesting the involvement of the ribosome in the process (19,20). There are reports that with increasing length the nascent peptide gets progressively cross-linked to the nucleotides in different domains of 23S rRNA. Nascent peptides of the same length but of different amino acid sequence produce different cross-linking patterns, leading to the conclusion that they assume different conformations on the ribosome (21). However the ∼100 Å long peptide exit tunnel (width between 10 and 28 Å at the maximum) seen in the 2.4 Å crystal structure of the 50S subunit of Haloarcula marismortui (22) excludes the possibility of complex structure formation inside the ribosome during chain synthesis.
In the last couple of years our laboratory has been engaged in studying the role of the ribosome and its components in the refolding of many proteins in vitro. The gradual outcome of our investigation from both the in vitro and in vivo experiments is that the 50S subunit functions to facilitate protein folding and that the domain V of 23S rRNA is the part mostly involved in it. This domain is also responsible for the peptidyl transferase activity. In this study we have chosen s-MDH to study how chain folding and subunit reassociation take place in this dimeric protein in the presence of E.coli 50S and/or its active folding module 23S rRNA, with the hope that this will mimic the situation inside the cell at least to some extent. The study chiefly employs two well-known processes, namely fluorescence energy transfer between two external fluorescent probes (donor FITC and acceptor RITC) and chemical cross-linking with gluteraldehyde along with reactivation and gel filtration chromatographic experiments.
Self-folding of s-MDH has been studied in considerable detail by Rudolph et al. (1). The kinetics was determined by two parallel first order folding reactions. Initially ‘structured monomers’ (M) with some secondary structure are formed very rapidly by hydrophobic collapse. Since their formation is not rate limiting this step is not included in the self-folding pathway. The majority of these structured monomers (∼70%) fold to yield the ‘folded monomers’ in a first order reaction (K1 = 1.3 × 10–3 s–1); these then associate to form the active dimers (D). The rest of the molecules assemble very fast, similarly to a diffusion controlled process, to form ‘incompletely folded dimers’ (D*) which further fold to form the active dimer (D) with a much slower rate (K2 = 7.0 × 10–5 s–1). Our observation from the time course of self-folding studied by gluteraldehyde cross-linking followed by further analysis of the cross-linked products in SDS–PAGE agrees well with this. In this context it is relevant to mention that Staniforth et al. have carried out extensive study on the role of GroEL/GroES in the folding of s-MDH and have reported that the chaperone could neither influence the yield nor the rate of folding (2). This report prompted us more to study the role of ribosome and its active folding modules in this dimeric system.
The reactivation of s-MDH in the presence of E.coli 50S and the 23S rRNA revealed kinetics which are quite different from its self-folding kinetics. In both cases only one first order reaction was observed with rate constant K1 = 1.6 × 10–3 s–1 in the case of 50S assisted folding, and K1 = 2.6 × 10–3 s–1 in the case of 23S rRNA assisted folding. However, whether this is due to isomerisation at the monomer level (M*→M) or dimer level (D*→D) could not be predicted from this result. To understand this it was important to study the subunit association of s-MDH in the presence of 50S and/or 23S rRNA.
The fluorescence energy transfer technique has been used in many systems to study association. For studying association in s-MDH the FITC–RITC donor−acceptor pair was chosen (R0 = 40 Å). Different aliquots of s-MDH were labelled with either FITC or RITC at a labelling ratio of 1:1. The labelling did not interfere with the enzyme activity and did not affect the reactivation with or without 50S and/or 23S rRNA. The FITC labelled and RITC labelled s-MDH were mixed in 1:1 proportion. When excited at 495 nm (excitation λmax of FITC), this mixture showed no RITC fluorescence; hence no energy transfer had occurred. However, energy transfer was seen when subunit reassociation was studied with the mix of FITC and RITC labelled s-MDH, after denaturation with 6 M Gdn–HCl. FITC fluorescence was quenched (λmax at 520 nm), and RITC fluorescence was enhanced (λmax at 595 nm). Actually, upon random association three types of dimers were expected to be formed: FITC–FITC, RITC–RITC and FITC–RITC labelled dimers. When FITC was excited (at 495 nm), the first one would show only FITC fluorescence, the second one would not fluoresce but the third one would show FITC as well as RITC fluorescence as a result of FITC→RITC energy transfer. Thus, this energy transfer process was successfully utilised to monitor the subunit association.
In the case of unassisted folding the energy transfer increased for ∼10–12 min and then saturated. In the presence of 50S subunit or 23S rRNA the extent of energy transfer was ∼3-fold greater than the self-folding in the first time point and no further increase in energy transfer was observed. The findings from the report of Rudolph et al. (1) and from our observation from the chemical cross-linking experiments suggest that during self-folding ∼30% of the molecules undergo instant dimerisation (D*). Thus most probably interaction with the 50S and/or 23S rRNA instantly induces formation of (30 × 3)% i.e. ∼90% dimers. That is why no further increase of energy transfer was obtained after the first reading. But the 50S and 23S assisted reactivation experiments performed in parallel showed a slow increase in activity in the first 20 min with the rate constants mentioned before. This must be reflecting the folding of the rapidly formed inactive dimers to the active ones. The efficiency of energy transfer was 70% and the distance calculated between the probes was 34.7 Å.
This result was further confirmed by chemical cross-linking experiments with gluteraldehyde followed by its analysis in SDS–PAGE. Chemical cross-linking during self-folding produced similar results, as was previously reported by Rudolph et al. (1) (stated earlier). But in the case of 23S assisted folding, even at the first time point no monomeric species was found. Instead only the slightly faster migrating dimeric intermediates were present. The reason why these dimeric intermediates migrated faster than the native dimers is probably that as they are less compact than the native dimers they have fewer cross-links and hence they can bind more SDS than the native dimers. But the compact native dimers having greater cross-links can bind less SDS and thus migrate a little slower than the less compact intermediates. With time these intermediates fold to form the active dimers. The rate constant of formation of these active dimers as determined from the chemical cross-linking experiments was almost the same as that found from the 23S rRNA assisted reactivation. This explains why 23S rRNA induced reactivation showed only a single first order kinetics instead of two parallel first order kinetics as seen in the case of its self-folding. Similar experiments with the 50S subunit could not be successfully performed since the ribosomal proteins interfered with the detection of the s-MDH molecules in the SDS–PAGE. However, the combined observation from the experiments described above clearly suggests that 50S and/or 23S rRNA assisted folding of s-MDH is distinctly different from its self-folding or chaperone assisted folding.
Furthermore, studying energy transfer coupled with the gel filtration chromatography experiments using the smaller transcripts of domain V of B.subtilis 23S RNA as the folding modulator throws some light on how 23S rRNA acts on its substrates. The 23S rRNA from many bacterial species including E.coli and B.subtilis have identical secondary structures where the nucleotides in the single-stranded regions, especially the central loop of domain V, are invariant. The nucleotides vary sometimes in the double-stranded regions from one bacterial species to another, but the conformations remain the same. The cloned domain V of B.subtilis 23S rDNA has convenient restriction sites that are lacking in the corresponding region of the E.coli 23S rDNA. Therefore the smaller transcripts were generated using the B.subtilis construct. These are named RNA1 (337 nt RNA fragment containing the central loop of domain V) and RNA2 (425 nt fragment from the 5′ end of domain V containing mainly the upper stem–loop regions) (Fig. 1).
During refolding RNA1 binds the structured monomers of s-MDH immediately after the hydrophobic collapse before they can slip into the misfolded state. In this case, the enzyme molecules co-eluted with RNA1. These molecules were confirmed as monomers as they showed no energy transfer (unquenched FITC emission and no RITC emission). Due to the binding of RNA1, subunit association was prevented and, hence, reactivation was also blocked. As soon as RNA2 was added in the refolding mixture it released the trapped monomeric intermediates from RNA1, and by this time the subunit interacting sites were modified in such a way that they could immediately dimerise, which was reflected by instant energy transfer. These are the faster migrating inactive dimeric intermediates seen by chemical cross-linking experiments which fold to form the active ones. In SDS–PAGE this inactive dimeric species forms bands identical to those seen in the case of the self-folding, yet from the folding rates (D*→D: K2 = 7 × 10–5 s–1 in the case of self-folding and K1 = 2.6 × 10–3 s–1 in the case of 23S rRNA assiated folding) it appears that they are not the same. The dimeric intermediate formed during self-folding is rather incorrectly folded and this is why the rate of its folding is so slow. On the other hand the dimeric intermediate formed during 23S assisted folding is probably more structured. Recently another group has published that ribosome bound nascent chain of tailspike protein from phage P22 displays epitopes not detected in the early in vitro folding intermediates (23). These observations are too preliminary to extrapolate to ‘in vivo’ conditions, but putting these findings together the role of ribosome in protein folding becomes more clear. Probably during synthesis the RNA1 region of 23S RNA protects the nascent chain from misfolding by holding it and when the trap is released by the interaction of RNA2 it is already directed to the proper folding channel.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Derek Logan for helping with the manuscript preparation. This work was supported by grants from Government of India agencies, CSIR (Grant No. 37/0935/97 EMR-II) and DBT (Grant No. BT/TF/15/15/91). S.C.S. and S.C. were University Grant Commission (India) funded senior research fellows, S.P. was a CSIR funded senior research fellow.
REFERENCES
- 1.Rudolph R., Fuchs,I. and Jaenicke,R. (1986) Reassociation of dimeric cytoplasmic malate dehydrogenase is determined by slow and very slow folding reactions. Biochemistry, 25, 1662–1669. [DOI] [PubMed] [Google Scholar]
- 2.Staniforth R.A., Cortes,A., Burston,S.G., Atkinson,T., Holbrook,J.J. and Clarke,A.R. (1994) The stability and hydrophobicity of cytosolic and mitochondrial malate dehydrogenases and their relation to chaperonin-assisted folding. FEBS Lett., 344, 129–135. [DOI] [PubMed] [Google Scholar]
- 3.Das B., Chattopadhyay,S. and DasGupta,C. (1992) Reactivation of denatured fungal glucose-6-phosphate dehydrogenase and E. coli alkaline phosphatase with E.coli ribosome. Biochem. Biophys. Res. Commun., 183, 774–780. [DOI] [PubMed] [Google Scholar]
- 4.Chattopadhyay S., Das,B., Bera,A.K. and DasGupta,C. (1994) Refolding of denatured lactate dehydrogenase by E. coli ribosomes. Biochem. J., 300, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bera A.K., Das,B., Chattopadhyay,S. and DasGupta,C. (1994) Refolding of denatured restriction endonucleases with ribosomal preparations from Methanosarcina barkeri. Biochem. Mol. Biol. Int., 32, 315–323. [PubMed] [Google Scholar]
- 6.Das B., Chattopadhyay,S., Bera,A.K. and DasGupta,C. (1996) In vitro protein folding by ribosomes from Escherichia coli, wheat germ and rat liver: the role of the 50S particle and its 23S rRNA. Eur. J. Biochem., 235, 613–621. [DOI] [PubMed] [Google Scholar]
- 7.Kudlicki W., Coffman,A., Kramer,G. and Hardesty,B. (1997) Ribosomes and ribosomal RNA as chaperone for folding of proteins. Folding Des., 2, 101–108. [DOI] [PubMed]
- 8.Chattopadhyay S., Das,B. and DasGupta,C. (1996) Reactivation of denatured proteins by 23S ribosomal RNA: role of domain V. Proc. Natl Acad. Sci. USA, 93, 8284–8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chattopadhyay S., Pal,S., Pal,D., Sarkar,D., Chandra,S. and DasGupta,C. (1999) Protein folding in Escherichia coli: role of 23S ribosomal RNA. Biochim. Biophys. Acta, 1429, 293–298. [DOI] [PubMed] [Google Scholar]
- 10.Pal D., Chattopadhyay,S., Chandra,S., Sarkar,D., Chakraborty,A. and DasGupta,C. (1997) Reactivation of denatured proteins by domain V of bacterial 23S rRNA. Nucleic Acids Res., 25, 5047–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fairclough R.H. and Cantor,C.R. (1978) The use of singlet-singlet energy transfer to study macromolecular assemblies. Methods Enzymol., 48, 347–379. [DOI] [PubMed] [Google Scholar]
- 12.Hermann R., Jaenicke,R. and Rudolph,R. (1981) Analysis of the reconstitution of oligomeric enzymes by cross-linking with glutaraldehyde: kinetics of reassociation of lactic dehydrogenase. Biochemistry, 20, 5195–5201. [DOI] [PubMed] [Google Scholar]
- 13.Zettlmeissl G., Rudolph,R. and Jaenicke,R. (1982) Rate-determining folding and association reactions on the reconstitution pathway of porcine skeletal muscle lactic dehydrogenase after denaturation by guanidine hydrochloride. Biochemistry, 21, 3946–3950. [DOI] [PubMed] [Google Scholar]
- 14.Pal S., Chandra,S., Chowdhury,S., Sarkar,D., Ghosh,A.N. and DasGupta,C. (1999) Complementary role of two fragments of domain V of 23 S ribosomal RNA in protein folding. J. Biol. Chem., 274, 32771–32777. [DOI] [PubMed] [Google Scholar]
- 15.Frieden C., Honegger,J. and Gilbert,H.R. (1978) Malate dehydrogenases. The lack of evidence for dissociation of the dimeric enzyme in kinetic analyses. J. Biol. Chem., 253, 816–820. [PubMed] [Google Scholar]
- 16.Lakowicz J.R. (1983) Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- 17.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 18.Birktoft J.J., Rhodes,G. and Banaszak,L.J. (1989) Refined crystal structure of cytoplasmic matate dehydrogenase at 2.5-Å resolution. Biochemistry, 28, 6065–6081. [DOI] [PubMed] [Google Scholar]
- 19.Baldwin T.O. (1999) Protein folding in vivo: the importance of ribosomes. Nature Cell Biol., 1, 154–155. [DOI] [PubMed] [Google Scholar]
- 20.Kramer G., Ramachandiran,V. and Hardesty,B. (2001) Cotranslational folding—omnia mea mecum porto? Int. J. Biochem. Cell Biol., 33, 541–553. [DOI] [PubMed] [Google Scholar]
- 21.Choi K.M. and Brimacombe,R. (1998) The path of the growing peptide chain through the 23S rRNA in the 50S ribosomal subunit: a comparative cross-linking study with three different peptide families. Nucleic Acids Res., 26, 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nissen P., Hansen,J., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The structural basis of ribosome activity in peptide bond synthesis. Science, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 23.Clark P.L. and King,J. (2001) A newly synthesized, ribosome-bound polypeptide chain adopts conformations dissimilar from early in vitro refolding intermediates. J. Biol. Chem., 276, 25411–25420. [DOI] [PubMed] [Google Scholar]