

Abstract
We report here the first genome-wide functional genomic screen for longevity genes. We systematically surveyed Caenorhabditis elegans genes using large-scale RNA interference (RNAi), and found that RNAi inactivation of 89 genes extend C. elegans lifespan. Components of the daf-2/insulin-like signaling pathway are recovered, as well as genes that regulate metabolism, signal transduction, protein turnover, and gene expression. Many of these candidate longevity genes are conserved across animal phylogeny. Genetic interaction analyses with the new longevity genes indicate that some act upstream of the daf-16/FOXO transcription factor or the sir2.1 protein deacetylase, and others function independently of daf-16/FOXO and sir2.1, and might define new pathways to regulate lifespan.
Keywords: Longevity, aging, C. elegans, insulin signaling, RNAi screen, genomic
Diverse genetic and environmental factors regulate longevity in a wide range of organisms (Finch 1990), but features of lifespan control that might be universal to animals have begun to emerge (Hekimi and Guarente 2003; Tatar et al. 2003). In Caenorhabditis elegans, modulations of particular gene activities cause a profound increase in lifespan (Guarente and Kenyon 2000). Most notably, reduced activity of the C. elegans daf-2/insulin-like signaling pathway causes a more than two-fold lifespan increase (Kenyon et al. 1993; Morris et al. 1996; Kimura et al. 1997; Paradis and Ruvkun 1998; Paradis et al. 1999). Signaling from the DAF-2/insulin-like receptor antagonizes the forkhead (FOXO) transcription factor daf-16, the major effector of daf-2/insulin-like regulation of C. elegans lifespan (Lin et al. 1997; Ogg et al. 1997). Similar modulations of insulin-like signaling pathways in fruit fly and mouse also modify lifespan (Clancy et al. 2001; Tatar et al. 2001; Bluher et al. 2003; Holzenberger et al. 2003), indicating that this pathway is a universal longevity regulator. There are >30 insulin genes in C. elegans that might mediate input to the daf-2 pathway through environmental cues, such as nutritional status or growth conditions (Pierce et al. 2001). Consistent with this model, mutations abrogating sensory neurons also regulate C. elegans lifespan in a daf-16-dependent manner (Apfeld and Kenyon 1999).
The Sir2 histone deacetylase is another important longevity determinant that regulates lifespan in diverse species (Kaeberlein et al. 1999). In yeast, Sir2 facilitates heterochromatin formation, and by reducing recombination at the rDNA locus, Sir2 helps to maintain genomic stability and extends lifespan (Imai et al. 2000). Similar to yeast, overexpression of the C. elegans SIR2 homolog sir-2.1 also extends lifespan (Tissenbaum and Guarente 2001), although the molecular mechanism whereby sir-2.1 enhances longevity in nematode is less clear. Genetic studies indicate that sir-2.1 works upstream of daf-16 to regulate C. elegans lifespan (Tissenbaum and Guarente 2001). Consistent with this genetic placement, in mammals, the Sir2 homolog SIRT1 deacetylates FOXOs, the mammalian DAF-16 homologs, and directly modulates FOXOs activity (Brunet et al. 2004; Giannakou and Partridge 2004; Motta et al. 2004; van der Horst et al. 2004).
Interestingly, mutations inhibiting C. elegans germline proliferation also extend lifespan (Hsin and Kenyon 1999; Arantes-Oliveira et al. 2002). The germline produces a daf-16-dependent signal to regulate lifespan, but acts independently of the upstream daf-2 gene (Lin et al. 2001). Thus, daf-16 might act as a master regulator of lifespan, capable of integrating several different longevity signals.
A handful of genes have been found to regulate C. elegans lifespan through pathways independent of daf-16. The feeding-defective eat mutants live slightly longer in a daf-16-independent manner. The eat mutations are suggested to extend lifespan through a mechanism resembling caloric restriction (Lakowski and Hekimi 1998). Mutations in clk-1, a gene required for coenzyme Q synthesis, also extends lifespan independently of daf-16, and slow the rate of many physiological processes (Lakowski and Hekimi 1996). Similarly, reduced function of the mitochondrial oxidative phosphorylation machinery extends C. elegans lifespan independent of daf-16 (Feng et al. 2001; Dillin et al. 2002; Lee et al. 2003b). A small-scale RNA interference (RNAi) screen for longevity regulators revealed that inactivation of particular mitochondrial electron transport chain (ETC) components increases C. elegans lifespan (Lee et al. 2003b).
Despite the identification of many genes that can regulate lifespan, a comprehensive survey of all of the gene classes capable of limiting the longevity of an animal has not been reported. We report here a genome-wide RNAi lifespan screen and the identification of a large number of new longevity genes, many of which are highly conserved in other species. This screen reveals 89 gene inactivations that can extend C. elegans lifespan from 5% to 70%. Our screen represents the first genome-wide survey of gene inactivations that can prolong C. elegans lifespan and provides a global view of the gene classes and physiological processes that can regulate the longevity of an animal. The large number of new longevity regulators reported here provides an important entry point for further characterizations that will likely reveal important insights into the molecular control of organismal longevity.
Results and Discussion
Genome-wide RNAi screen
Using an RNAi bacterial library targeting >80% of the ∼19,000 C. elegans open reading frames (Fraser et al. 2000; Kamath et al. 2003) and a high-throughput lifespan assay, we screened for RNAi inactivations that extend the lifespan of wild-type N2 worms. The RNAi clones that were scored positive in the first round of primary screen were subjected to two more rounds of high-throughput lifespan assay. Of the 16,475 RNAi clones tested, ∼600 RNAi clones induced lifespan extension above a rather modest threshold in high-throughput screening. Of these ∼600 RNAi clones, we performed lifespan assays with many more timepoints using rrf-3(pk1426), a mutant with enhanced susceptibility to feeding RNAi (Simmer et al. 2002) and a normal lifespan (Lee et al. 2003a; Murphy et al. 2003). After three rounds of retesting, we identified 90 RNAi clones that significantly extended C. elegans lifespan (P-value < 0.05) in at least two of the lifespan experiments (Table 1; Supplementary Table 1). The plasmid construct for each of the final 90 RNAi clones was isolated and sequenced to verify its corresponding target gene (Table 1). The 90 RNAi constructs target 89 distinct genes, because RNAi constructs C01F6.7 and F23B2.1 both target the C01F6.6 gene. Significantly, most of the candidate longevity genes are conserved in evolutionarily diverse species. In fact, based on Ensembl annotations (http://www.ensembl.org), 32 of the candidate genes have a clear orthologous partner in Drosophila, mouse, and/or human. Even for the genes that are not annotated to have a clear ortholog, many of them display significant sequence homology to genes in diverse species. This finding suggests that characterization of the candidate longevity genes identified in our screen will likely reveal mechanisms that influence longevity in many different organisms.
Table 1.
A genome-wide screen identified 90 RNAi inactivations that induce lifespan extension
| Primer name | Gene name | Brief description | Functional group | Fly ortholog | Mouse ortholog | Human ortholog |
|---|---|---|---|---|---|---|
| F07 A5.1 | F07 A5.1 | Innexin, required for GAP junction | Cell structure | |||
| F35C8.6 | F35C8.6 | Profilin | Cell structure | |||
| K07H8.1 | K07H8.1 | Tubulin-specific chaperone | Cell structure | yes | yes | yes |
| W05B2.5 | W05B2.5 | Collagen | Cell structure | |||
| Y119D3_451.a | Y92C3A.1 | Cadherin domain | Cell structure | |||
| C35A11.3 | C35A11.3 | Mucin | Cell surface | |||
| F08H9.5 | F08H9.5 | C-type lectin | Cell surface | |||
| K11D12.1 | K11D12.1 | Mucin | Cell surface | |||
| ZK896.7 | ZK896.7 | C-type lectin | Cell surface | |||
| C13C4.2 | C13C4.2 | Nuclear hormone receptor | Gene expression | |||
| F15E6.1 | F15E6.1a | SET domain; PHD-finger | Gene expression | yes | ||
| F54C4.2 | F54C4.2 | Transcription elongation factor Spt4 | Gene expression | yes | yes | yes |
| R11E3.4 | R11E3.4 | SET domain | Gene expression | |||
| T01B10.4 | T01B10.4 | Nuclear hormone receptor | Gene expression | |||
| T09A5.8 | T09A5.8 | Chromo domain | Gene expression | |||
| ZC64.3 | ZC64.3 | Homeobox protein ceh-18 | Gene expression | |||
| T05A1.4 | T05A1.4 | Retrotransposon integrase | Integrase | |||
| D1054.8 | D1054.8 | Glucose/ribitol dehydrogenase | Metabolism | |||
| W09H1.5 | W09H1.5 | Zinc-containing alcohol dehydrogenase superfamily | Metabolism | yes | yes | yes |
| K08E3.5 | K08E3.5 | UTP-glucose-1-phosphate uridylyltransferase | Metabolism: galactose/starch/sucrose metabolism | yes | yes | yes |
| F57B10.3 | F57B10.3 | Phosphoglycerate mutase | Metabolism: glycolysis | |||
| F43G9.1 | F43G9.1 | Isocitrate dehydrogenase | Metabolism: TCA cycle | yes | yes | yes |
| F54H12.1 | F54H12.1 | Aconitase | Metabolism: TCA cycle | yes | yes | yes |
| F59B8.2 | F59B8.2 | Isocitrate dehydrogenase | Metabolism: TCA cycle | yes | yes | yes |
| B0261.4 | B0261.4 | Mitochondrial 39-S ribosomal protein L47 | Metabolism: ETC | yes | yes | yes |
| C33F10.12 | C33F10.12 | Mitochondrial carrier | Metabolism | |||
| C53B7.4 | C53B7.4 | Mitochondrial ATP synthase g subunit | Metabolism: ETC | yes | ||
| D2030.4 | D2030.4 | NADH:CoQ oxidoreductase subunit B18 | Metabolism: ETC | yes | yes | yes |
| F26E4.6 | F26E4.6 | Cytochrome C oxidase | Metabolism: ETC | |||
| F26E4.9 | F26E4.9 | Cytochrome C oxidase | Metabolism: ETC | yes | yes | yes |
| K04G7.4 | K04G7.4 | NADH dehydrogenase | Metabolism: ETC | yes | yes | yes |
| T20H4.5 | T20H4.5 | NADH-quinone oxidoreductase | Metabolism: ETC | yes | yes | yes |
| W09C5.8 | W09C5.8 | Cytochrome c oxidase subunit IV | Metabolism: ETC | yes | yes | yes |
| F55B11.1 | F55B11.1 | Xanthine Dehydrogenase (XDH) | Metabolism: purine metabolism | yes | yes | yes |
| Y71G12A_202.a | Y71G12B.4 | Peptidylglycine alpha-amidating monooxygenase | Neuropeptide synthesis | yes | ||
| W01A11.3 | W01A11.3 | Spectrin repeat | Nuclear migration | |||
| C08B11.7 | C08B11.7 | Ubiquitin C-terminal hydrolase | Protein turnover | yes | yes | yes |
| C16C10.7 | C16C10.7 | Ubiquitin E3 ligase | Protein turnover | yes | yes | yes |
| C51E3.7 | C51E3.7 | egl-3 prohormone convertase | Protein turnover | yes | yes | yes |
| F57C12.1 | F57C12.1 | Astacin Peptidase | Protein turnover | |||
| T05G5.10 | T05G5.10 | Eukaryotic initiation factor 5A iff-1 | Protein turnover | yes | ||
| Y71H2_380.a | Y71H2AR.2 | Papain family cysteine protease | Protein turnover | |||
| C01F6.7 | C01F6.6 | PDZ domain | Protein-protein interaction | |||
| F23B2.1 | C01F6.6 | PDZ domain | Protein-protein interaction | |||
| Y50D4A_25.a | C39F7.2 | Zinc (RING) finger, SPRY domain, Fibronectin type III domain | Protein-protein interaction | yes | yes | yes |
| F09F7.5 | F09F7.5 | P21-Rho-binding domain | Protein-protein interaction | |||
| F21H12.1 | F21H12.1 | WD domain | Protein-protein interaction | yes | yes | yes |
| Y39H10A_224.b | Y39H10A.6 | WD domain, C-terminal to LisH motif | Protein-protein interaction | yes | yes | yes |
| Y54E5A.7 | Y54E5A.7 | SPRY domain, C-terminal to LisH motif | Protein-protein interaction | yes | yes | yes |
| C36H8.1 | C36H8.1 | MSP (Major sperm protein) | Reproduction | |||
| C39E9.1 | C39E9.1 | Testis-specific protein TPX-1 like | Reproduction | |||
| Y39F10C.1 | Y39F10C.1 | Vitelline membrane outer layer protein I (VOMI) | Reproduction | |||
| B0334.8 | B0334.8 | age-1 Phosphatidylinositol 3- and 4-kinase | Signaling | yes | yes | yes |
| C01F6.1 | C01F6.1 | Copine | Signaling | |||
| C04H5.3 | C04H5.3 | Adenylate and Guanylate cyclase, natriuretic peptide receptor | Signaling | |||
| C12D8.10 | C12D8.10 | akt-1 serine/threonine kinase | Signaling | yes | yes | yes |
| C27B7.7 | C27B7.7 | Ig domain, Fibronectin type III domain | Signaling | |||
| C34B4.1 | C34B4.1 | PH domain | Signaling | yes | yes | yes |
| F35D2.3 | F35D2.3 | EGF-like | Signaling | |||
| K08D10.7 | K08D10.7 | Scramblase | Signaling | |||
| K10B4.3 | K10B4.3 | PH domain | Signaling | yes | yes | yes |
| T04A11.10 | T04A11.10 | 7TM receptor | Signaling | |||
| T05E12.4 | T05E12.4 | 7TM chemoreceptor, srw family | Signaling | |||
| T26H5.1 | T26H5.1 | 7Tm receptor | Signaling | |||
| Y46H3C_13.a | Y46H3C.1/.2b | 7TM chemoreceptor, srw family | Signaling | |||
| F40F8.5 | F40F8.5 | ABC transporter | Small molecule transport | |||
| C09B7.2 | C09B7.2 | Transposase | Transposase | |||
| C26B2.2 | C26B2.2 | unknown | ||||
| C32H11.1 | C32H11.1 | Domain of unknown function DUF141 | unknown | |||
| C32H11.5 | C32H11.5c | Domain of unknown function DUF148 | unknown | |||
| D2054.8 | E03H12.5 | unknown | ||||
| F45H10.4 | F45H10.4 | unknown | ||||
| F49F1.12 | F49F1.12 | unknown | ||||
| F55B11.3 | F55B11.5/.3d | unknown | ||||
| H06H21.8 | H06H21.8 | Domain of unknown function DUF227 | unknown | |||
| K10D2.2 | K10D2.2 | PAP/25A associated domain | unknown | |||
| R05A10.5 | R05A10.5 | unknown | ||||
| R08E3.3 | R08E3.3 | unknown | yes | yes | yes | |
| T06G6.4 | T06G6.4 | unknown | ||||
| T07A9.8 | T07A9.8 | unknown | yes | yes | yes | |
| Y43F8B.12 | Y43F8B.12 | unknown | ||||
| Y43H11_160.a | Y43h11AL.2 | unknown | yes | yes | ||
| Y46H3C_14.b | Y46H3C.6 | unknown | ||||
| Y53F4C.d | Y53F4B.23 | unknown | ||||
| Y56A3A.9 | Y56A3A.9 | unknown | ||||
| Y75B8A.13 | Y75B8A.13 | unknown | ||||
| Y75B8A.33 | Y75B8A.33 | unknown | ||||
| ZK520.2 | ZK520.2 | unknown | ||||
| C18E9.4 | C18E9.4/.10e | |||||
| Y37D8A.12 | Y37D8A.12f |
F15E6.1 RNAi clone may hybridize to both F15E6.1 and Y51H4A.12, two PHD domain-containing proteins.
Y46H3C_13.a RNAi clone spans both Y46H3C.1 and Y46H3C.2, two highly similar genes coding for 7TM chemoreceptor in the srw family.
C32H11.5 RNAi clone may hybridize to multiple genes (Kamath et al. 2003); C32H11.5; C32H11.6; C32H11.8; C32H11.11; all of which contain DUF148.
F55B11.3 RNAi clone may hybridize to both F55B11.5 and F55B11.3, two highly similar genes of unknown function.
C18E9.4 RNAi clone spans both C18E9.4, which encodes a NADH-ubiquinone oxidoreductase, and C18E9.10, which encodes an unknown protein.
Y37D8A.12 RNAi clone may hybridize to multiple genes (Kamath et al. 2003).
The efficacy of our RNAi screen is validated by the recovery of genes and gene classes previously characterized to regulate C. elegans lifespan. We found that RNAi of age-1 as well as akt-1, both of which are components of the daf-2/insulin-like signaling pathway, caused robust lifespan extension (Table 2; Supplementary Table 1). age-1 mutations are well-established to extend lifespan (Morris et al. 1996). Interestingly, although akt-1 is a well-characterized component of the daf-2 signaling pathway, an akt-1 deletion mutant was previously reported to exhibit normal lifespan (Hertweck et al. 2004). The lifespan extension phenotype caused by akt-1 RNAi (Table 2) might only be apparent when akt-1 activity is reduced by RNAi knockdown, and not when akt-1 function is completely lost, as in the case of a deletion mutant. An alternative hypothesis that nonspecific knockdown of additional gene(s) by the akt-1 RNAi construct is also possible, though Blastn analysis reveals additional genes with only 17 bp of sequence identity to akt-1.
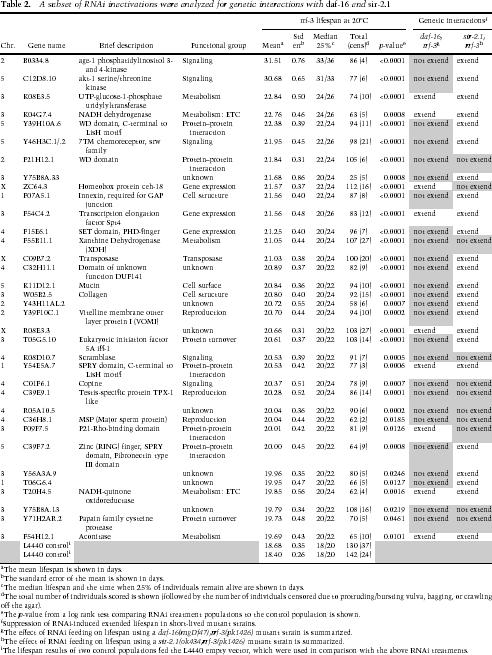
Table 2.
A subset of RNAi inactivations were analyzed for genetic interactions with daf-16 and sir-2.1
We also found that RNAi of several subunits of the mitochondrial ETC significantly extended C. elegans lifespan. This is consistent with previous genetics and RNAi studies indicating that reduced mitochondrial electron transport can lead to lifespan increase in C. elegans (Feng et al. 2001; Dillin et al. 2002; Lee et al. 2003b).
Longevity genes constitute diverse functional groups
Among the new candidate longevity genes that are annotated with a possible function (∼75%, 66/89) (Table 1; Supplementary Fig. 1), a large number (∼25%, 17/66) are involved in some aspect of metabolism, such as carbohydrate metabolism, alcohol metabolism, citric acid cycle, oxidative phosphorylation, and purine metabolism (Table 1). Although it is not clear how RNAi inactivation of each of the enzymes in these different metabolic processes regulates lifespan, an attractive hypothesis is that reduced function of many of these enzymes decreases the tempo or mode of energy generation, which might regulate longevity through mechanisms similar to that of caloric restriction.
Among the longevity genes predicted to participate in energy metabolism, >50% correspond to subunits of the ETC (complex I: D2030.4, K04G7.4, T20H4.5; complex IV: F26E4.6, F26E4.9, W09C5.8; complex V: C53B7.4.). Although complex III subunit was not found in our current screen, we previously showed that RNAi inactivation of the complex III subunit T02H6.11 could extend lifespan (Lee et al. 2003b). Complex II subunit has not been identified in any of our screens. Complex II is made up of a relatively small number of subunits and may represent a smaller target size for our screen. We hypothesize that reduced function of the different ETC components regulates lifespan through a common molecular mechanism, probably by lowering ATP generation and/or free radical production. Indeed, four of the RNAi inactivations identified here were characterized previously to induce lower ATP production and pleiotropic phenotypes, including small body size, slow pumping, slow growth rate, and sterility (Dillin et al. 2002; Lee et al. 2003b). We analyzed the three additional ETC subunits identified in the current screen and found that they also induced growth rate and reproduction defect (Supplementary Table 3). Interestingly, when worms are exposed to RNAi prior to embryogenesis, a much more severe growth phenotype was detected (Supplementary Table 3). Oxidative phosphorylation is required for C. elegans survival; lifespan extension is likely caused by partial reduction of mitochondrial electron transport activity, as severe reduction of ETC function causes larval arrest and lethality (Supplementary Table 3; Tsang and Lemire 2003).
Nuclear genes that are essential to mitochondrial function are not fully described in the C. elegans genome. To assess which of the 90 RNAi inactivations regulate mitochondrial function, we used the reporter hsp-6::gfp(zcIs13), which encodes a mitochondrial chaperone (Yoneda et al. 2004), to monitor a mitochondrial unfolded protein response. As expected, RNAi inactivation of the candidate genes predicted to function in the mitochondria, such as ETC subunits (D2030.4, K04G7.4, T20H4.5, F26E4.6, F26E4.9, W09C5.8, C53B7.4) and aconitase (F54H12.1), induced hsp-6::GFP expression (Fig. 1). ETC components work as large protein complexes; RNAi inactivation of one subunit will likely interfere with assembly of the entire protein complex and cause accumulation of unfolded proteins in the mitochondria (Yoneda et al. 2004). Interestingly, in addition to the candidate genes that are annotated to be mitochondrial, we found RNAi inactivation of four additional longevity genes [C39F7.2, Y53F4B.23, Y75B8A.33, Y92C3A.1(cdh-12)] also induced hsp-6::GFP, albeit to a lesser degree than RNAi of ETC subunits (Fig. 1). These results suggest that these four candidate genes might also function in the mitochondria or be necessary for mitochondrial biogenesis. Two of these genes encode proteins of unknown function (Y53F4B.23, Y75B8A.33), whereas one of them encodes a protein predicted to contain various protein–protein interaction domains (C39F7.2), and the remaining one encodes a cadherin homolog (cdh-12). The cadherin domain is usually present in proteins required for cell–cell junction formation. It is curious that cdh-12 RNAi inactivation should produce a mitochondrial unfolded protein response. Blastn analysis indicates that the RNAi construct Y119D3_451.a clearly targets gene cdh-12, but it also displays longer than 20-bp sequence homology to a number of other C. elegans genes, and may knockdown expression of additional gene(s). It is also important to note that for the genes that do not function as large protein complexes in the mitochondria, RNAi inactivations are not expected to induce a robust misfolded protein response, and will not be detected by a survey using the hsp-6::gfp reporter. Therefore, it is very possible that additional candidate longevity genes recovered from our screen function in mitochondria.
Figure 1.

RNAi inactivation of multiple candidate longevity genes induces hsp-6::GFP expression. (A) Basal levels of hsp-6::GFP expression in worms treated with control RNAi (L4440 empty vector). (B) Greatly enhanced hsp-6::GFP expression in worms treated with RNAi targeting F26E4.6 (cytochrome C oxidase subunit) (Yoneda et al. 2004). (C) A representative image of enhanced hsp-6::GFP expression in worms treated with a “group 1” RNAi. Group 1 RNAi include F54H12.1, C53B7.4, D2030.4, F26E4.6, F26E4.9, K04G7.4, T20H4.5, and W09C5.8. (D) A representative picture of somewhat enhanced hsp-6::GFP expression in worms treated with a “group 2” RNAi. Group 2 RNAi include B0261.4, C39F7.2, Y53F4B.23, Y75B8A.33, and Y92C3A.1.
In addition to oxidative phosphorylation components, the longevity RNAi screens identified several enzymes required for carbohydrate metabolism (e.g., UTP-glucose-1-phosphate uridylyltransferase K08E3.5), as well as the citric acid cycle (e.g., aconitate hydratase F54H12.1, isocitrate dehydrogenase F43G9.1 and F59B8.2), that regulate C. elegans lifespan. Carbohydrate metabolism and citric acid cycle are processes important to generating fuel necessary for oxidative phosphorylation. One interesting speculation is that RNAi of the carbohydrate metabolic enzymes and the citric acid cycle genes, extend lifespan via mechanisms similar to that of reduced oxidative phosphorylation.
On the other hand, it is also possible that RNAi inactivation of some of the specific metabolic enzymes block particular pathways, leading to elevated or reduced levels of certain metabolic intermediates that might be especially important for longevity.
A number of the longevity genes are annotated to participate in signal transduction. For instance, in addition to akt-1, two additional candidate longevity genes (C34B4.1, K10B4.3) encoding proteins with PH domains were identified. A well-established function of the PH domain is the binding of phosphatidylinositol lipids; an intriguing possibility is that these two candidate genes might respond to age-1 signaling to regulate C. elegans lifespan. Interestingly, RNAi of C34B4.1 or K10B4.3 required daf-16 to increase lifespan, suggesting that they act upstream of daf-16 (see below; Supplementary Table 1).
Four RNAi constructs identified in our screen inactivate annotated G-protein coupled receptors (GPCR) (T04A11.10, T05E12.4, T26H5.1, Y46H3C_13.a). Although RNAi construct Y46H3C_13.a might target genes Y46H3C.1 and Y46H3C.2, both of the probable target genes are annotated to be GPCRs. In C. elegans, GPCRs often act as chemosensory receptors, but many are predicted to be neuropeptide receptors as well (Bargmann 1998). Sensory neurons regulate C. elegans lifespan (Apfeld and Kenyon 1999); it is possible that some of the GPCRs we identified participate in sensory perception and transmit environmental cues to downstream signaling pathways. For example, production of various insulins in these sensory neurons might regulate longevity. However, because feeding RNAi is thought to work inefficiently in C. elegans neurons, neuronal-specific genes are unlikely to be recovered in our screen. On the other hand, GPCRs have a wide range of substrates. Recently, citric acid cycle intermediates have been identified to bind to GPCRs and trigger downstream signaling (He et al. 2004). This is especially intriguing, considering that our results indicate that RNAi inactivation of citric cycle enzymes promote C. elegans longevity.
Other signaling proteins recovered in our screen include a copine protein (C01F6.1), an evolutionarily conserved family of proteins that binds phospholipids in a Ca2+-dependent manner (Tomsig and Creutz 2002), and a guanylate cyclase (C04H5.3) that might act as a natriuretic receptor. Furthermore, a number of the longevity genes encode proteins that contain domains that might act as cell-surface receptors, such as EGF-like domains (F35D2.3) and Ig domains (C27B7.7). Lastly, a few longevity regulatory genes are predicted to encode scaffolding proteins, containing multiple protein–protein interaction motifs, such as WD domains (F21H12.1, Y39H10A.6), Zinc/RING fingers (C39F7.2), or PDZ domains (C01F6.6). These findings together suggest that these candidate longevity genes might participate in various signaling pathways. Given the importance of insulin-like signaling in lifespan in diverse species, it will be important to find new components of the insulin-like signaling pathway, as well as genes that might act in novel signaling pathways to regulate longevity.
Our screen revealed that RNAi of iff-1 (T05G5.10), a C. elegans homolog of the translation initiation factor eIF5A, extends lifespan. Interestingly, protein synthesis has been suggested to regulate longevity in other animals. In the long-lived Snell dwarf mouse, translation initiation is decreased compared with control (Hsieh and Papaconstantinou 2004). Reduced translation initiation is proposed to be a consequence of reduced insulin and mTOR signaling in these long-lived mice.
In addition, several genes likely involved in protein processing/degradation were also identified in our screen. These include several proteases (C51E3.7, F57C12.1, Y71H2AR.2), an ubiquitin ligase (C16C10.7), and an ubiquitin C-terminal hydrolase (C08B11.7), which functions to remove ubiquitin from modified proteins. Among the proteases, one particularly interesting candidate gene is egl-3 (C51E3.7), which encodes a prohormone convertase (Kass et al. 2001). Prohormone convertases mediate the proteolytic processing of various hormones and neural peptides, including insulin. It is possible that RNAi of egl-3 extends lifespan by inactivating the processing of certain insulin(s). It is interesting to note that egl-3 RNAi required daf-16 activity to prolong lifespan (see below; Supplementary Table 1), suggesting that egl-3 acts upstream of daf-16.
A number of the candidate longevity genes are predicted to encode proteins that might regulate gene expression. These include proteins with motifs that might modify chromatin structures (such as PHD domains, SET domains, or chromo domains) (F15E6.1, R11E3.4, T09A5.8), established transcription factors (such as SPT-4 F54C4.2, CEH-18 ZC64.3), and annotated nuclear hormone receptors (NHR) (C13C4.2, T01B10.4). Similar to that in yeast, SIR-2.1 may regulate C. elegans lifespan by modifying global or local chromatin structures. Some of the chromatin factors we identified might cooperate with sir-2.1 to modulate longevity. Furthermore, the NHR DAF-12 acts downstream of DAF-16 to modulate lifespan and dauer formation. It will be very interesting to test whether some of the NHRs identified here might functionally interact with DAF-12.
Additional longevity regulatory genes include several proteins likely involved in reproduction, such as a vitelline membrane outer layer protein (Y39F10C.1), a major sperm protein (C36H8.1), and a testis-specific TPX protein (C39E9.1). A prominent theory of aging postulates that the vast amount of energy an animal spends on reproduction will limit the resources remaining for life maintenance. RNAi inactivation of these genes may cause lifespan extension by reducing energy expenditure through reproduction. However, it is important to note that many examples counter to this theory exist (Finch 1990), and many sterile mutants in C. elegans show normal lifespan.
We also identified an annotated transposase (C09B7.2) and a possible retrotransposon integrase (T05A1.4) in our screen. In a recent genome-wide expression profiling of aging C. elegans (Lund et al. 2002), transposases are among the groups of genes whose expression is elevated during aging. Increased transposase activity might cause increased transposon hopping and genomic instability. It is possible that RNAi inactivation of one or more transposases, and/or retrotransposon integrases, might extend lifespan by increasing genomic stability.
To determine the comprehensiveness of our genome-wide screen, we compared the longevity genes we identified with the representative longevity genes that have been reported previously. In the daf-2/insulin signaling pathway, reduction in daf-2, age-1, pdk-1, akt-1, sgk-1, and ins-18 gene activities extend lifespan. Of these genes, age-1, akt-1, daf-2, and ins-18 are represented in the 80% coverage RNAi library. Three of these four genes, age-1, akt-1, and daf-2, emerged in the high-throughput screen without reference to their identity, and two of these, age-1 and akt-1, were intense enough to pass the secondary screens. The daf-2 RNAi clone did not pass our subsequent multiple rounds of retest, probably because of its weaker phenotype (∼20% lifespan extension induced by the particular daf-2 RNAi clone in the library vs. ∼50% lifespan extension induced by age-1 or akt-1 RNAi) and the variability of RNAi. We did not detect a lifespan phenotype with ins-18 RNAi. This is not surprising, as ins-18 RNAi was capable of extending lifespan only upon multiple generations of feeding inactivation (Kawano et al. 2000), a strategy that is very different from our high-throughput RNAi strategy.
Previous genetic screens identified mitochondrial genes that affect C. elegans lifespan were as follows: clk-1, isp-1, and lrs-2 (Lakowski and Hekimi 1996; Feng et al. 2001; Lee et al. 2003b). Only the clk-1 RNAi construct is present in the RNAi library, and we were not able to detect a lifespan phenotype associated with clk-1 RNAi. RNAi inactivation of the mitochondrial genes atp-3 (F27C1.7), nuo-2 (T10E9.7), cyc-1 (C54G4.8), cco-1 (F26E4.9), D2030.4, T02H6.11, F26E4.6, W09C5.8, B0261.4, T06D8.6, F13G3.7, and K01C8.7 (Dillin et al. 2002; Lee et al. 2003b), have previously been reported to extend lifespan. Eight of these 12 genes were recovered in our high-throughput screens, and five were among the final 89 candidate longevity genes: cco-1 (F26E4.9), D2030.4, F26E4.6, W09C5.8, and B0261.4 (Table 1). In addition, our genome-wide screen identified seven additional mitochondrial function genes that can affect lifespan (F43G9.1, F54H12.1, F59B8.2, C33F10.12, C53B7.4, K04G7.4, and T20H4.5) (Table 1).
Previous forward genetic screens also revealed that loss-of-function mutations of a number of genes important for sensory neuron development extend lifespan (Apfeld and Kenyon 1999). Our RNAi screen did not recover any gene that is implicated to affect sensory neuron development. This is not surprising, considering that feeding RNAi does not work well in neurons, and we have expected our screen to miss the group of longevity genes that specifically function in the nervous system.
The eating defective (eat) mutants were specifically examined for a lifespan phenotype to show that caloric restriction also extends lifespan in C. elegans (Lakowski and Hekimi 1998). Among the long-lived eat mutants, eat-1 (T11B7.4), eat-3 (D2013.5), and eat-6 (B0365.3) RNAi are represented in the library, but were not identified in our screen. However, we did recover eat-4 RNAi in our high-throughput screens (among the 600 candidates). Our subsequent analyses indicate that the eat-1, eat-3, or eat-6 RNAi constructs did not significantly prolong lifespan under our RNAi conditions.
Loss-of-function mutation of glp-1 results in loss of germline stem cells and extended lifespan (Arantes-Oliveira et al. 2002). Although glp-1 RNAi is represented in the RNAi library, our analyses showed that the glp-1 RNAi construct was not able to induce lifespan extension, probably because our feeding RNAi approach does not deplete glp-1 early enough to ablate the germline stem cells.
Finally, from a microarray experiment focusing on identifying possible DAF-16 downstream genes, Murphy et al. (2003) showed that RNAi inactivation of ∼20 putative DAF-16 downstream genes resulted in a lifespan increase that ranged from 3% to 55%. Because of the modest lifespan extension caused by some of these RNAi inactivations, we do not expect to recover many of these 20 genes in our screen. Interestingly, F55G11.5 and gcy-18 (ZK896.8) were among the 600 candidates we identified in our high-throughput screenings. Furthermore, we identified additional candidate genes that may participate in functions similar to some of the DAF-16 downstream genes, and may affect lifespan via similar mechanisms. For example, we identified gcy-23 in our screen, which may affect similar cellular processes as gcy-18 (Murphy et al. 2003). Although we did not recover the DAF-16 downstream genes vit-2 and vit-5 (vitellogenin) in our screen (Murphy et al. 2003), we did recover a vitellogenin membrane outer layer protein (Y39F10C.1). Lastly, although we did not recover the DAF-16 downstream genes C32H11.10 and C32H11.12 (Murphy et al. 2003), both encoding DUF141 proteins, we did recover five different genes predicted to encode DUF141 proteins from our high-throughput screenings, and the DUF141 encoding gene C32H11.1 exhibited a robust and consistent phenotype and was among the final 89 longevity candidate genes (Table 1).
Our comparison indicates that although we attempted to be comprehensive, our genome-wide screen is not saturating. This is likely due to the intrinsic limitations that feeding RNAi tends to be variable and often only gives a hypomorphic phenotype. Furthermore, our high-throughput feeding RNAi strategy exposed animals to RNAi starting as L1s, which might not be optimal depending on the temporal requirement of the longevity genes. It is important to note that our RNAi screen represents an unbiased screening strategy, and our screen is very successful in recovering many of the known longevity genes identified through previous unbiased screens (e.g., insulin-signaling components, mitochondrial genes). The hit rate for our screen regarding the other longevity genes identified through candidate gene approach is lower (e.g., eat genes). This is likely because the lifespan phenotype associated with these genes is modest and is easily missed in high-throughput screenings. Although our screen did not recover all of the previously reported longevity genes, very often, our screen revealed candidate longevity genes that are predicted to participate in functions similar to that of the longevity genes reported previously. This indicates that our screen appears to have targeted most of the gene classes that, when inactivated, can promote longevity, and suggests that our genome-wide RNAi screen is valuable in providing a global view of many of the gene classes that normally limit lifespan.
Genomic classification of longevity genes
Some of the longevity genes recovered from our screen were closely located on the chromosomes. For instance, four different cosmids were found to each contain at least two candidate longevity genes (C01F6.1 and C01F6.6, C32H11.1 and C32H11.11, F26E4.6 and F26E4.9, F55B11.1 and F55B11.11). We suspect that some of these candidate genes might be different genes of the same operon, and their RNAi might affect all of the genes on the same operon and cause a common phenotypic outcome. However, a close inspection of each of the genomic regions did not reveal that any of the genes listed above might constitute part of an operon (Blumenthal et al. 2002). Therefore, the fact that some of the candidate longevity genes are closely located might be due to their related function on longevity control or might simply be coincidence.
We attempted to classify the candidate longevity genes by examining whether they share any similar expression pattern. Using the TopoExpression Mountains characterized by Kim et al. (2001), we found that the candidate longevity genes fit into many different expression groups. Interestingly, five expression mountains are significantly overrepresented (P < 0.001) (Supplementary Table 2). These include the mountains enriched in genes involved in biosynthesis, carbohydrate metabolism, energy generation, mitochondrial, and protein expression/energy generation, respectively. Further inspection reveals that a similar subset of candidate genes, the ones annotated with a metabolic function (Table 1), fit into each of these five different mountains. Over-representation of these five expression mountains among our candidate longevity genes is consistent with our finding that energy metabolism represents a major gene class important for C. elegans lifespan.
We also used the genome-wide protein–protein interaction maps published by Li et al. (2004) to examine whether some of the candidate longevity genes might participate in common protein complexes. Two-hybrid protein–protein interaction data are available for only 25 of the 89 candidate genes. Among these 25 predicted proteins, no direct protein–protein interaction was detected in the two-hybrid screen (Li et al. 2004). However, several of the candidate genes identified in our screen might form a complex with each other through one bridging protein (Supplementary Fig. 2). We did not look for complex formation between the candidates that might be mediated by more than one bridging protein, as such a survey revealed complicated interaction maps that are difficult to interpret. Although each pair-wise interaction detected in the two-hybrid screen might occur individually under different circumstances, it is also possible that the linked interactions detected by the genomic two-hybrid screen suggest that some of the candidate longevity genes might function as protein complexes and cooperate to influence lifespan. It will be interesting to test whether RNAi inactivation of the two-hybrid interactors of the candidate longevity genes might also cause a lifespan phenotype in future investigations.
Dauer analysis
Because the prominent longevity pathway daf-2/insulin-like signaling is also critical for controlling dauer formation, we sought to determine whether any of the new longevity candidate genes might also affect dauer formation. Feeding RNAi in general is not effective in inducing dauer formation, probably because it does not efficiently knockdown neuronal genes. We utilized a sensitive assay in which we examined daf-2(e1370) worms for an ability to recover from dauer arrest at the semi-nonpermissive temperature 22°C (Lee et al. 2003a; Murphy et al. 2003). We surveyed the 90-candidate longevity RNAi. Although the survey was done blind, we successfully identified age-1 and akt-1 RNAi to enhance the dauer formation phenotype of daf-2(e1370) mutant (Supplementary Table 4). We additionally found that RNAi of K08E3.5 and F15E6.1 moderately enhanced the dauer formation phenotype, suggesting that these candidate genes might have roles in both longevity and dauer development (Supplementary Table 4). Interestingly, RNAi of most of the ETC genes also enhanced the dauer phenotype. This is likely due to the slower growth of the RNAi animals, which would delay dauer exit and act like an enhancer in our assay. Alternatively, these animals might be hypometabolic and might be dauer prone.
Genetic interaction of longevity genes
One method of organizing the long lists of longevity regulators into pathways is by genetic interaction analysis with other known longevity mutants. Therefore, we tested whether RNAi of a subset of the candidates might require daf-16 or sir-2.1 activity to regulate lifespan by performing RNAi lifespan experiments using either a daf-16 deletion or a sir-2.1 deletion mutant strain. To ensure that the different genetic backgrounds do not affect the efficiency of feeding RNAi, we used several test RNAi clones that produce visible phenotypes (e.g., dpy-13, hmr-1 [Kennedy et al. 2004]) and demonstrated that the rrf-3(pk1426);daf-16(mgDf47) and rrf-3(pk1426);sir-2.1(ok434) mutants exhibited similar dpy-13 and hmr-1 phenotypes compared with rrf-3(pk1426) (data not shown). We have thus far tested the longevity RNAi candidates that exhibited the most robust phenotypes, those with a P-value ≤ 0.001 in the lifespan experiments using rrf-3 mutant at 25°C (Supplementary Table 1). A total of 35 candidate longevity genes were chosen for our genetic interaction analyses, and they fall into functional groups that display a distribution similar to that of the entire set of candidate genes (Table 2).
It is interesting to note that the rrf-3(pk1426);sir-2.1 (ok434) mutant displayed a slightly shorter lifespan compared with rrf-3(pk1426), an observation that is consistent with what is known about Sir2 deletion in yeast. However, the lifespan shortening of rrf-3(pk1426);sir-2.1 (ok434) is not as dramatic as that of the rrf-3(pk1426); daf-16(mgDf47) mutant (Supplementary Table 1). Our results indicate that daf-16 and sir-2.1 show overlapping, yet distinct genetic interactions with the panel of candidate longevity genes (Table 2; Supplementary Table 1). Of the 35 candidate genes tested, RNAi inactivation of 26 of them requires daf-16 activity to extend lifespan, whereas RNAi of 11 candidates requires sir-2.1 activity to extend lifespan. Furthermore, whereas a number of candidate genes require both daf-16 and sir-2.1 activity to extend lifespan (nine candidates), a number of candidate genes require only daf-16 (17 candidates) or only sir-2.1 (two candidates) activity to regulate lifespan (Table 2). These results suggest that daf-16 and sir-2.1 cooperate to regulate lifespan in some instances. However, both daf-16 and sir-2.1 have significant independent activity that is important for lifespan regulation.
Mammalian SIRT1 affects gene expression through modification of multiple chromatin proteins; C. elegans SIR-2.1 may similarly modify multiple substrates. Loss of daf-16 suppresses the lifespan extension caused by overexpression of SIR-2.1 (Tissenbaum and Guarente 2001), indicating that daf-16 is a major output of sir-2.1. Our analyses suggest that sir-2.1 is likely to have additional downstream mediators that also regulate longevity. In addition, both age-1 and akt-1 RNAi robustly extend the lifespan of sir-2.1 deletion mutant animals. These results indicate that, even in the case that sir-2.1 acts through daf-16 to modulate lifespan, sir-2.1 is likely to act either upstream of age-1 or in a pathway that is parallel to the canonical daf-2/age-1/akt-1 signaling cascade, but converges onto daf-16.
Considering that SPT-4, CEH-18, and DAF-16 are all transcription factors, it is possible that they function as a protein complex that regulates gene expression and longevity. However, our genetic interaction results argue against this possibility. RNAi inactivation of the transcriptional elongation factor spt-4 or the POU-domain transcription factor ceh-18 extends daf-16 null mutant lifespan, suggesting that SPT-4 and CEH-18 likely regulate lifespan by functioning independent of DAF-16. RNAi of ceh-18 requires sir-2.1 activity to extend lifespan, suggesting that ceh-18 might function upstream of sir-2.1, or cooperate with sir-2.1 to regulate lifespan in a daf-16-independent manner.
Several longevity genes depend on both daf-16 and sir-2.1 activities to regulate lifespan. These include a WD domain protein (F21H12.1), a scramblase (K08D10.7), a copine protein (C01F6.1), a cysteine protease (Y71H2AR.2), a protein related to male fertility (C36H8.1), and two unknown proteins (R05A10.5 and Y75B8A.13) (Table 2). These genes might act upstream of both daf-16 and sir-2.1, or they might cooperate with both daf-16 and sir-2.1 to affect lifespan. A noticeable finding from our genetic interaction analyses is that a substantial number of the candidate longevity genes require daf-16 activity to regulate lifespan. This includes a number of proteins with unknown function, as well as proteins predicted to participate in signal transduction. Thus, in addition to age-1 and akt-1, a large number of factors might function through daf-16 to regulate lifespan. This finding is consistent with the emerging view that DAF-16 acts as a central integrator of multiple different longevity signals.
Of all the longevity regulatory genes identified by the RNAi screen, age-1 or akt-1 RNAi induces the most robust lifespan extension phenotype (Table 2; Supplementary Table 1). The daf-2/age-1 insulin-like signaling pathway was the first signaling pathway identified to dramatically regulate C. elegans lifespan through forward genetic screens. It is interesting that this pathway stands out in two different screening strategies. It is likely that insulin-like regulation of C. elegans lifespan is so potent because inactivation of insulin signaling mimics a physiological regulatory event; in the dauer animal, insulin signaling is physiologically decreased (Riddle and Albert 1997) and a cascade of gene regulatory responses, including protection from free radicals and a change in metabolic rate ensues (Vanfleteren and De Vreese 1995; Honda and Honda 1999; Van Voorhies and Ward 1999; Murphy et al. 2003). Because inactivation of insulin-signaling pathway components induces a change in lifespan that has probably been under genetic selection for long-lived and stress-resistant dauer stage animals, it is not surprising that it is a robust regulator of C. elegans longevity.
Our screen also identified a large number of new candidate longevity genes that are predicted to participate in diverse biological functions. Although RNAi inactivation of many of these candidate genes did not produce a phenotype as robust as that of age-1 or akt-1 RNAi, their weaker phenotype might be due to insufficient gene knockdown, redundant function, or the need for precise gene knockdown in specific cells and/or developmental times. Some of these candidate longevity genes belong to functional classes that have not been implicated in longevity control before. Characterization of these candidate longevity genes will provide novel insights into how lifespan might be regulated in C. elegans.
Materials and methods
Strains
We used the following C. elegans strains: wild-type N2 Bristol, rrf-3(pk1426), daf-16(mgDf47);rrf-3(pk1426), sir-2.1(ok434);rrf-3(pk1426), and hsp-6::gfp(zcIs13) (a kind gift from David Ron, Skirball Institute, New York).
Genome-wide RNAi lifespan screen
We carried out a large-scale RNAi screen using wild-type N2 strain as described previously (Lee et al. 2003b). Briefly, each RNAi colony was grown overnight in LB with 50 μg/mL of ampicillin and then seeded onto 24-well RNAi agar plates containing 5 mM isoprophylthiogalactoside (IPTG). The RNAi bacteria were induced overnight at room temperature for dsRNA expression. We then added ∼20–40 synchronized larval stage-one animals to each well, allowed worms to develop to adults, and then added 5-fluorodexoyuridine (FUDR) solution to a final concentration of 0.1 mg/mL. Worms were kept at 25°C and their lifespan was monitored. Worms feeding on bacteria carrying the empty vector (L4440) were used as control. Worms feeding on RNAi bacteria expressing dsRNA targeting the daf-2 gene was used as control (daf-2 RNAi construct kindly provided by M. Vidal, Harvard Medical School, Boston, MA). We routinely screened ∼500–1000 RNAi clones in one experiment. For each screening experiment, at least 48 wells of L4440 control were included. We monitored the control worm populations regularly. At the time when all of the control worms were dead, we scored each well containing the different RNAi bacteria for possible live worms. RNAi wells in which live worms were observed were scored as positives. These “positive” RNAi clones were retested at least two more times using similar high-throughput screening strategy described above.
RNAi clones that were scored as positive in rescreening were deemed “primary positives”. Primary positives were retested two or three times in conventional RNAi lifespan assays (see below) using the rrf-3(pk1426) strain. Because each of the final 90 RNAi clones were scored positive at least twice in our initial high-throughput screening using N2 worms, and were subsequently scored positive at least two more times using rrf-3(pk1426) worms in conventional lifespan assays, we are confident that reduced function of each of these genes contributes to lifespan extension. Because RNAi often only partially inactivates a gene function, it is very possible that a more severe attenuation of some of these genes would extend lifespan more. This is, in fact, observed with our positive control daf-2 RNAi. The control daf-2 RNAi construct we used (kindly provided by M. Vidal) induces ∼50% lifespan increase, and the daf-2 RNAi construct from the genome-wide library only induces ∼20% lifespan increase. In contrast, a temperature-sensitive genetic mutation of daf-2 extends lifespan up to threefold (Kenyon et al. 1993; Morris et al. 1996; Kimura et al. 1997; Paradis and Ruvkun 1998; Paradis et al. 1999). Some of the RNAi clones induced lifespan extension in only two of the three retests. Variability of RNAi efficacy among experiments has been observed previously (Simmer et al. 2003).
RNAi lifespan assay
RNAi bacteria were prepared as described above. Gravid rrf-3(pk1426) worms were allowed to lay eggs onto RNAi bacteria plates containing 2 mM IPTG at 15°C for ∼12 h. The progeny were allowed to grow to adults at 25°C. At 25°C, a large proportion of the rrf-3(pk1426) worms became sterile adults. Approximately 40–60 of the adult animals undergoing RNAi treatment were then transferred to RNAi agar plates containing 0.05–0.1 mg/mL of FUDR that were seeded with freshly induced RNAi bacteria of interest. The animals were then kept at either 25°C or 20°C, and scored every 2 d by gentle prodding with a platinum wire to test for live or dead. To ensure the continued efficacy of RNAi knockdown, animals were fed freshly induced RNAi bacteria every 5 d. Lifespan is defined as the time elapsed from when worms were put on FUDR plates (adult lifespan = 0) to when they were scored as dead. Worms that died of protruding/bursting vulva, bagging, or crawling off the agar were censored.
For epistasis analysis, RNAi lifespan assays were performed as described above, except either daf-16(mgDf47);rrf-3(pk1426) or sir-2.1(ok434);rrf-3(pk1426) worms were used.
Statistical analysis
Statistical analyses were performed using the software SPSS (SPSS Inc.). The survival experience of each RNAi-treated population is compared with that of the population treated with control RNAi using the log rank test. A P-value < 0.05 was considered as significantly different from control.
hsp-6::gfp assay
Transgenic animals carrying an integrated transgene of hsp-6::gfp (Yoneda et al. 2004) were allowed to lay eggs onto RNAi bacteria as described above. The fluorescence intensity of each population was monitored as soon as worms reached young adulthood. Images of each population of RNAi-treated transgenic animals were captured using the OpenLab software. All of the images were taken with the same amount of exposure time.
Informatics
Analysis of how the candidate longevity genes fit into the TOPO expression mountains was performed via http://workhorse.stanford.edu/cgi-bin/gl/gene_list.cgi?set=2 (see also Kim et al. 2001). Analysis of possible protein–protein interaction among the candidate longevity genes was performed via the Interactome query page (http://vidal.dfci.harvard.edu).
Acknowledgments
We are grateful to J. Ahringer and colleagues for providing the genome-wide RNAi library, D. Ron for providing the hsp-6::gfp strain, C. Benedetti for advice on the hsp-6::gfp reporter, M. Vidal for the daf-2 RNAi construct, J. Xu and P. Hurd for technical support, N. Amin for assistance in some of the lifespan assays, E. Zaydfudim for assistance in sequencing of RNAi constructs, K. Kemphues for critical reading of the manuscript, members of G.R.'s and S.S.L.'s laboratories for helpful discussions, and the CGC for providing strains. This work was supported in part by a Damon Runyon post-doctoral fellowship and a grant from the National Institute of Aging to S.S.L., and grants from the National Institutes of Health to G.R.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1308205.
Corresponding authors.
References
- Apfeld J. and Kenyon, C. 1999. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402: 804–809. [DOI] [PubMed] [Google Scholar]
- Arantes-Oliveira N., Apfeld, J., Dillin, A., and Kenyon, C. 2002. Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295: 502–505. [DOI] [PubMed] [Google Scholar]
- Bargmann C.I. 1998. Neurobiology of the Caenorhabditis elegans genome. Science 282: 2028–2033. [DOI] [PubMed] [Google Scholar]
- Bluher M., Kahn, B.B., and Kahn, C.R. 2003. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299: 572–574. [DOI] [PubMed] [Google Scholar]
- Blumenthal T., Evans, D., Link, C.D., Guffanti, A., Lawson, D., Thierry-Mieg, J., Thierry-Mieg, D., Chiu, W.L., Duke, K., Kiraly, M., et al. 2002. A global analysis of Caenorhabditis elegans operons. Nature 417: 851–854. [DOI] [PubMed] [Google Scholar]
- Brunet A., Sweeney, L.B., Sturgill, J.F., Chua, K.F., Greer, P.L., Lin, Y., Tran, H., Ross, S.E., Mostoslavsky, R., Cohen, H.Y., et al. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015. [DOI] [PubMed] [Google Scholar]
- Clancy D.J., Gems, D., Harshman, L.G., Oldham, S., Stocker, H., Hafen, E., Leevers, S.J., and Partridge, L. 2001. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292: 104–106. [DOI] [PubMed] [Google Scholar]
- Dillin A., Hsu, A.L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A.G., Kamath, R.S., Ahringer, J., and Kenyon, C. 2002. Rates of behavior and aging specified by mitochondrial function during development. Science 298: 2398–2401. [DOI] [PubMed] [Google Scholar]
- Feng J., Bussiere, F., and Hekimi, S. 2001. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1: 633–644. [DOI] [PubMed] [Google Scholar]
- Finch C.E. 1990. Longevity, senescence, and the genome. The University of Chicago, Chicago, IL.
- Fraser A.G., Kamath, R.S., Zipperlen, P., Martinez-Campos, M., Sohrmann, M., and Ahringer, J. 2000. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408: 325–330. [DOI] [PubMed] [Google Scholar]
- Giannakou M.E. and Partridge, L. 2004. The interaction between FOXO and SIRT1: Tipping the balance towards survival. Trends Cell. Biol. 14: 408–412. [DOI] [PubMed] [Google Scholar]
- Guarente L. and Kenyon, C. 2000. Genetic pathways that regulate ageing in model organisms. Nature 408: 255–262. [DOI] [PubMed] [Google Scholar]
- He W., Miao, F.J., Lin, D.C., Schwandner, R.T., Wang, Z., Gao, J., Chen, J.L., Tian, H., and Ling, L. 2004. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429: 188–193. [DOI] [PubMed] [Google Scholar]
- Hekimi S. and Guarente, L. 2003. Genetics and the specificity of the aging process. Science 299: 1351–1354. [DOI] [PubMed] [Google Scholar]
- Hertweck M., Gobel, C., and Baumeister, R. 2004. C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev. Cell 6: 577–588. [DOI] [PubMed] [Google Scholar]
- Holzenberger M., Dupont, J., Ducos, B., Leneuve, P., Geloen, A., Even, P.C., Cervera, P., and Le Bouc, Y. 2003. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421: 182–187. [DOI] [PubMed] [Google Scholar]
- Honda Y. and Honda, S. 1999. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 13: 1385–1393. [PubMed] [Google Scholar]
- Hsieh C.C. and Papaconstantinou, J. 2004. Akt/PKB and p38 MAPK signaling, translational initiation and longevity in Snell dwarf mouse livers. Mech. Ageing Dev. 125: 785–798. [DOI] [PubMed] [Google Scholar]
- Hsin H. and Kenyon, C. 1999. Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399: 362–366. [DOI] [PubMed] [Google Scholar]
- Imai S., Armstrong, C.M., Kaeberlein, M., and Guarente, L. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M., McVey, M., and Guarente, L. 1999. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes & Dev. 13: 2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath R.S., Fraser, A.G., Dong, Y., Poulin, G., Durbin, R., Gotta, M., Kanapin, A., Le Bot, N., Moreno, S., Sohrmann, M., et al. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237. [DOI] [PubMed] [Google Scholar]
- Kass J., Jacob, T.C., Kim, P., and Kaplan, J.M. 2001. The EGL-3 proprotein convertase regulates mechanosensory responses of Caenorhabditis elegans. J. Neurosci. 21: 9265–9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T., Ito, Y., Ishiguro, M., Takuwa, K., Nakajima, T., and Kimura, Y. 2000. Molecular cloning and characterization of a new insulin/IGF-like peptide of the nematode Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 273: 431–436. [DOI] [PubMed] [Google Scholar]
- Kennedy S., Wang, D., and Ruvkun, G. 2004. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature 427: 645–649. [DOI] [PubMed] [Google Scholar]
- Kenyon C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. 1993. A C. elegans mutant that lives twice as long as wild type. Nature 366: 461–464. [DOI] [PubMed] [Google Scholar]
- Kim S.K., Lund, J., Kiraly, M., Duke, K., Jiang, M., Stuart, J.M., Eizinger, A., Wylie, B.N., and Davidson, G.S. 2001. A gene expression map for Caenorhabditis elegans. Science 293: 2087–2092. [DOI] [PubMed] [Google Scholar]
- Kimura K.D., Tissenbaum, H.A., Liu, Y., and Ruvkun, G. 1997. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277: 942–946. [DOI] [PubMed] [Google Scholar]
- Lakowski B. and Hekimi, S. 1996. Determination of life-span in Caenorhabditis elegans by four clock genes. Science 272: 1010–1013. [DOI] [PubMed] [Google Scholar]
- ____. 1998. The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. 95: 13091–13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.S., Kennedy, S., Tolonen, A.C., and Ruvkun, G. 2003a. DAF-16 target genes that control C. elegans life-span and metabolism. Science 300: 644–647. [DOI] [PubMed] [Google Scholar]
- Lee S.S., Lee, R.Y., Fraser, A.G., Kamath, R.S., Ahringer, J., and Ruvkun, G. 2003b. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33: 40–48. [DOI] [PubMed] [Google Scholar]
- Li S., Armstrong, C.M., Bertin, N., Ge, H., Milstein, S., Boxem, M., Vidalain, P.O., Han, J.D., Chesneau, A., Hao, T., et al. 2004. A map of the interactome network of the metazoan C. elegans. Science 303: 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K., Dorman, J.B., Rodan, A., and Kenyon, C. 1997. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278: 1319–1322. [DOI] [PubMed] [Google Scholar]
- Lin K., Hsin, H., Libina, N., and Kenyon, C. 2001. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28: 139–145. [DOI] [PubMed] [Google Scholar]
- Lund J., Tedesco, P., Duke, K., Wang, J., Kim, S., and Johnson, T. 2002. Transcriptional profile of aging in C. elegans. Curr. Biol. 12: 1566. [DOI] [PubMed] [Google Scholar]
- Morris J.Z., Tissenbaum, H.A., and Ruvkun, G. 1996. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382: 536–539. [DOI] [PubMed] [Google Scholar]
- Motta M.C., Divecha, N., Lemieux, M., Kamel, C., Chen, D., Gu, W., Bultsma, Y., McBurney, M., and Guarente, L. 2004. Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563. [DOI] [PubMed] [Google Scholar]
- Murphy C.T., McCarroll, S.A., Bargmann, C.I., Fraser, A., Kamath, R.S., Ahringer, J., Li, H., and Kenyon, C. 2003. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424: 277–283. [DOI] [PubMed] [Google Scholar]
- Ogg S., Paradis, S., Gottlieb, S., Patterson, G.I., Lee, L., Tissenbaum, H.A., and Ruvkun, G. 1997. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389: 994–999. [DOI] [PubMed] [Google Scholar]
- Paradis S. and Ruvkun, G. 1998. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes & Dev. 12: 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S., Ailion, M., Toker, A., Thomas, J.H., and Ruvkun, G. 1999. A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes & Dev. 13: 1438–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce S.B., Costa, M., Wisotzkey, R., Devadhar, S., Homburger, S.A., Buchman, A.R., Ferguson, K.C., Heller, J., Platt, D.M., Pasquinelli, A.A., et al. 2001. Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes & Dev. 15: 672–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle D.L. and Albert, P.S. 1997. Genetic and environmental regulation of dauer larva development. In C. elegans II (eds. D. Riddle et al.), pp. 739–768. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- Simmer F., Tijsterman, M., Parrish, S., Koushika, S.P., Nonet, M.L., Fire, A., Ahringer, J., and Plasterk, R.H. 2002. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr. Biol. 12: 1317–1319. [DOI] [PubMed] [Google Scholar]
- Simmer F., Moorman, C., van der Linden, A.M., Kuijk, E., van den Berghe, P.V., Kamath, R.S., Fraser, A.G., Ahringer, J., and Plasterk, R.H. 2003. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 1: E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M., Kopelman, A., Epstein, D., Tu, M.P., Yin, C.M., and Garofalo, R.S. 2001. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292: 107–110. [DOI] [PubMed] [Google Scholar]
- Tatar M., Bartke, A., and Antebi, A. 2003. The endocrine regulation of aging by insulin-like signals. Science 299: 1346–1351. [DOI] [PubMed] [Google Scholar]
- Tissenbaum H.A. and Guarente, L. 2001. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410: 227–230. [DOI] [PubMed] [Google Scholar]
- Tomsig J.L. and Creutz, C.E. 2002. Copines: A ubiquitous family of Ca(2+)-dependent phospholipid-binding proteins. Cell. Mol. Life Sci. 59: 1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.Y. and Lemire, B.D. 2003. Mitochondrial ATP synthase controls larval development cell nonautonomously in Caenorhabditis elegans. Dev. Dyn. 226: 719–726. [DOI] [PubMed] [Google Scholar]
- van der Horst A., Tertoolen, L.G., de Vries-Smits, L.M., Frye, R.A., Medema, R.H., and Burgering, B.M. 2004. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 279: 28873–28879. [DOI] [PubMed] [Google Scholar]
- Van Voorhies W.A. and Ward, S. 1999. Genetic and environmental conditions that increase longevity in Caenorhabditis elegans decrease metabolic rate. Proc. Natl. Acad. Sci. 96: 11399–11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanfleteren J.R. and De Vreese, A. 1995. The gerontogenes age-1 and daf-2 determine metabolic rate potential in aging Caenorhabditis elegans. FASEB J. 9: 1355–1361. [DOI] [PubMed] [Google Scholar]
- Yoneda T., Benedetti, C., Urano, F., Clark, S.G., Harding, H.P., and Ron, D. 2004. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell. Sci. 117: 4055–4066. [DOI] [PubMed] [Google Scholar]