


Abstract
We devised an indicator gene for retrotransposition based on an autocatalytic ribozyme element—the Tetrahymena thermophila 23S rRNA group I intron—which can self-splice in vitro and does not require—at variance with nuclear mRNA introns—any specific pathway and cellular component for the completion of the splicing process. Several constructs, with the Tetrahymena intron adequately modified so as to be inserted at various positions within a neomycin-containing cassette under conditions that restore the neomycin-coding sequence after splicing out of the intron, were assayed for splicing efficiency in mammalian cells in culture. We show, both by northern blot analysis and by the recovery of neomycin activity upon retroviral transduction of the cassettes, that splicing efficiency depends on both the local base pairing and the global position of the intron within the neomycin transcript, and that some constructs are functional. We further show that they allow the efficient sorting out of retrotransposition events when assayed, as a control, with a human LINE retrotransposon. These indicator genes should be of great help in elucidating the mechanisms of transposition of a series of retroelements associated with transcripts not prone to nuclear mRNA intron splicing and previously not opened to any retrotransposition assay.
INTRODUCTION
Mobile elements are universal components of living species, and constitute up to 40% of mammalian genomes. These elements can transpose into new genomic locations, sometimes resulting—depending on the target site—in phenotypically detectable mutations. However, a characterization of both the mechanisms involved in the mobility of transposons and the frequency of occurrence of transposition events has awaited, especially in the case of mammals which are not easily subject to large scale genetic assays, the construction of so-called ‘indicator genes for retrotransposition’ (1, reviewed in 2). These indicator genes take advantage, in the case of the widespread retrotransposons, of the nature of the expected molecular intermediate in the transposition event, i.e. the transposon RNA transcript, to monitor the mobility itself of the element and not simply phenotypic mutations possibly—and only possibly—associated with the de novo insertion of the element. Such indicator genes have allowed the monitoring of transposition both in cells in culture and in transgenic animals (1,3–7), for the two major classes of retrotransposons found in higher eucaryotes, namely the endogenous retroviruses and the LINE elements (reviewed in 2). These indicator genes further allowed the demonstration that mammalian cells possess an endogenous reverse transcription activity, which manifests itself by the generation of integrated cDNA copies of any cellular mRNAs (and therefore the formation of the so-called processed pseudogenes), and that the LINE retrotransposons are responsible for this activity (8).
A feature common to all devised indicator genes for retrotransposition is the presence of a ‘forward’ intronic sequence interrupting a selectable gene (e.g. the neomycin resistance gene) placed in ‘backward’ orientation relative to the retrotransposable element to be tagged. As a complete retrotransposition cycle includes transcription of the tagged retroelement followed by reverse transcription and integration of the intermediate RNA, elimination of the intron, by splicing, from the latter restores an active selectable gene in the retrotransposed copy. In all previous constructs, the introns used were nuclear mRNA introns which are non-autonomous sequences requiring specific pathways and cellular components for the completion of the splicing process. Actually, this feature might be a limitation in the study of retrotransposition, especially for the analysis of mobile elements which, for instance, possess a Pol III-driven promoter (e.g. the widespread Alu transposons, one million copies in the human genome), since it has been shown that in such transcripts nuclear mRNA introns are not spliced out (9). Some mobile structures present in organelles might similarly not be subject to nuclear mRNA intron splicing. Accordingly, we describe in this paper a newly devised indicator gene for retrotransposition, based on an autocatalytic ribozyme element—the Tetrahymena thermophila 23S rRNA group I intron—which can self-splice in vitro and therefore should function whatever the pathway used by the tagged retroelement.
The group I intron from T.thermophila catalyses its own excision from the precursor 23S rRNA without the aid of proteins, following a well characterized two-step pathway (10). In the context of the pre-rRNA transcript, the intron folds into a complex tertiary structure, forming an active site for the splicing of the adjacent exons (see Fig. 1A). This structure is achieved by base pairing interactions throughout the intron sequence forming RNA helices, which then fold into a specific conformation to create a catalytic core within the intron. The 5′ splice site is delineated by the P1 helix, which is formed by base pairing between the internal guide sequence (IGS) of the intron and the last six nucleotides of the 5′ exon. In the first step of splicing, P1 docks into the catalytic core, and the 5′ splice site is cleaved by an exogenous guanosine nucleophile. In the second step of splicing, the 5′ and 3′ exons are ligated together, and the intron is released (10). This group I intron can also self-splice in vivo from full-length and truncated versions of Tetrahymena rRNA. In addition, the Tetrahymena intron self-splices when inserted at the homologous position in the Escherichia coli rRNA gene, with a rate rivaling that of Tetrahymena rRNA processing. Efficient self-splicing from unnatural sequence contexts demonstrates that rRNA exon sequences are not a prerequisite for efficient in vivo catalysis (11–13). In addition, group I introns are not naturally found in E.coli, thus suggesting that species-specific cofactors are not required for efficient group I ribozyme activity in vivo. Finally, self-splicing of the Tetrahymena group I intron is supported by recent experiments with this intron introduced into nuclear or retroviral genes expressed in mammalian cells, where intron excision proceeds with high fidelity (14,15). However, these studies also showed that the sequences flanking the group I intron can significantly affect the catalytic efficiency of the ribozyme in vivo (14,15), most probably as a result of an inhibition by the exonic sequences of the adequate folding of the intronic domain.
Figure 1.

Structure of the wild-type T.thermophila rRNA group I intron and derivatives. (A) Predicted secondary structure of the Tetrahymena rRNA self-splicing intron with hairpins (Pi) schematized (according to 27). The 5′ and 3′ exonic sequences are in lower case and the intronic sequence in capital letters, with the splice sites indicated with arrows. The IGS (solid line) is essential for the splicing process and is involved in a complex association [see text and (B)]. (B) Schematic representation of the interactions involved in Tetrahymena intron self-splicing, with the pairing between the IGS and the 5′ and 3′ exonic sequences indicated with bars. The IGS sequence is underlined and the bulk of the intron sequence has been deleted. Mutations (indicated in light inverted) within the IGS and exonic sequences were generated (using appropriate primers and PCR amplification) to introduce the Tetrahymena intron into three distinct positions (the SphI, BanII and PstI sites) of the neomycin gene (see text and Fig. 2). The arrows delineate the 5′ and 3′ splice sites, and the exonic and intronic sequences are shown in lower and upper cases, respectively. The interactions of the IGS with the 5′ and 3′ exonic sequences (P1 and P10, respectively) are drawn simultaneously, for convenience.
Therefore, in this paper we have constructed and characterized a series of indicator genes (neoTET cassettes) which differ by the position of the Tetrahymena intron insertion within the neomycin-coding sequence and by the base pairing for the interactions between the intronic IGS and the imposed neomycin exonic sequences. We show, both by northern blot analysis and by the recovery of neomycin activity upon retroviral transduction of the neoTET cassettes, that some of the constructs are functional for splicing. We further show that they allow the efficient sorting out of retrotransposition events when assayed, as a control, with a human LINE retrotransposon. These newly devised neoTET retrotransposition cassettes should be of great help for the study of retroelements previously not opened to any assay.
MATERIALS AND METHODS
Plasmids
Construction of the neoTET and neoTNF cassettes. The modified T.thermophila 23S rRNA introns were introduced into three different sites of the neomycin gene, using PCR. Amplifications were performed with the proof-reading Pfu Taq polymerase (Promega) in 50 µl containing 5 µl buffer 10× [20 mM Tris–HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 100 µg/ml nuclease-free BSA and 0.1% Triton X-100], 0.2 mM each dNTP, 1.25 U of Pfu polymerase and 1 µg of DNA. The following primers were used (the IGS sequences are underlined and the 5′ and 3′ cleavage sites are indicated with a vertical bar): TET P 5′, 5′-T│AAATAGCAATATTGAACTTTGGGACGAAAAGTTATC-3′; TET P 3′, 5′-TTC│CGAGTACTCCAAACCTAATC-3′; TET P t/a 5′, 5′-TGT│AA(T/A)TAGCAATATAATGAATTGCAGGGAAAAGTTATC-3′; TET P t/a 3′, 5′-CGCT│ACTTAGCAATATGTATCAGTGGCGTGAAAAGTTATC-3′; TET B 5′, 5′-CGCT│ACTTAGCAATATGTATCAGTGGCGTGAAAAGTTATC-3′; TET B 3′, 5′-│CGAGTACTCCAAACCTAATCAAT-3′; TET S 5′, 5′-CAT│AAATAGCAATATAGGCGTTTGTGCCCAAAAGTTATCAG-3′; TET S 3′, 5′-A│CGAGTACTCCAAACCTAATCAATATACTTTCGCATACAAATTAGGTCCCAGCGG-3′. The TET S intron was amplified first, using p18R.TET(3) vector (gift from F. Michel) with the TET S primers (which allows removal of a 4U stretch in the intron P9.2 helix, see Fig. 1A); the PCR product was cloned into pSVneo* opened by SphI and blunt-ended. pSVneo* derives from the pMC1neo vector (Stratagene) by (i) introduction of a T-C transversion at the unique TTTT sequence in the neo-coding sequence, obtained by homologous replacement of the NcoI–NaeI neo fragment by a PCR fragment generated with the following primers: neo*, 5′-GTGACCCATGGCGATGCCTGCTTGCCGAATATCATGGTGGAGAATGGCCGCT-3′; neo4, 5′-TGGGCGAAGAACTCCAGCATGAGATC-3′; and (ii) replacing the tk promoter (XhoI-partial PstI blunt-ended fragment) of pMC1neo by the SV40 promoter (PvuII–HinDIII blunt-ended fragment) from pSVtkneo (3). The TET P, Pt, Pa and B introns were then amplified from pSVneoTET S using the corresponding primers and the PCR fragments were cloned into pSVneo* opened by PstI (P, Pt, Pa) or BanII (B) and blunt-ended. The structures of the 5′ and 3′ junctions of the inserted fragments were checked by sequencing (Applera). The pSVneoTNF reporter gene, marked with intron 2 of the murine TNFβ gene, was constructed similarly by PCR amplification using the following primers: TNF 5′, 5′-AGGTGAGGCAGCAAGAGATCTGGGGTGCTG-3′; TNF 3′, 5′-GCCTGGGACAGAAGAGAGTG-3′; and cloning of the PCR product into pSVneo* opened by BanII and blunt-ended.
The pCMVneoTET and pCMVneoTNF plasmids were constructed by introducing the neoTET cassettes and the neoTNF cassette (EagI–BamHI blunt-ended fragments) from the corresponding pSVneoTET and pSVneoTNF vectors into the pCMVβ vector (Clontech) opened by XhoI–NotI and blunt-ended.
The pCMVneoTET B/PstI and pCMVneoTET S/PstI plasmids were constructed by replacement of the EagI–RsrII fragment from pCMVneoTNF by the homologous fragments from pSVneoTET B/PstI and pSVneoTET S/PstI, respectively. The latter plasmids were constructed by introducing into the pSVneo* plasmid opened by PstI and blunt-ended, the modified TET B and TET S introns, obtained by PCR using pSVneoTET B and pSVneoTET S and the following primers: TET B/PstI 5′, 5′-CGCGCTACTTAGCAATATGTATCAGTG-3′; TET B/PstI 3′, 5′-AGCATCAGGCGAGTACTCCAAACCTAATC-3′; TET S/P 5′, 5′-GGGCATAAATAGCAATATAGGCGTTTG-3′; TET S/P 3′, 5′-AAGGCGCGACGAGTACTCCAAACCTAATC-3′. The pCMVneoTET Pt/BanII and pCMVneoTET Pt/SphI plasmids were constructed by replacement of the EagI–RsrII fragment from pCMVneoTNF by the homologous fragments from pMCIneoTET Pt/BanII and pMCIneoTET Pt/SphI, respectively. The latter plasmids were constructed by introducing into the pMCIneo plasmid (Stratagene) opened by BanII or SphI, respectively, and blunt-ended, the modified TET Pt intron obtained by PCR using pSVneoTET Pt and the following primers: TET Pt/BanII–SphI 5′, 5′-TCCTGTAATTAGCAA- TATAATGAATTGC-3′; TET Pt/BanII–SphI 3′, 5′-GAATGAACTCGAGTACTCCAAACCTAATC-3′.
The five pM2neoTET and the pM2neoTNF plasmids were constructed by inserting the PvuI–HinDIII blunt-ended fragments from the five pSVneoTET and the pSVneoTNF plasmids into the pMIGR.2 retroviral vector (gift from W. Wainchenker) opened by BglII–EcoRI and blunt-ended.
The p220.CMV-L1neoTET Pt and p220.CMV-L1neoTNF plasmids were constructed by replacement of the SfiI–BssHII fragment from p220.CMV-L1neoRT (8) by the homologous fragments from pSVneoTET Pt and pSVneoTNF, respectively.
Cells, transfections and retroviral infections
Human HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (Life Technologies), streptomycin (100 µg/ml) and penicillin (100 U/ml) at 37°C and 6% CO2. For selection of transformants, cells were seeded in 60-mm-diameter plates at a density of 106 cells per plate and allowed to settle for 24 h. Cells were transfected with 4 µg of DNA per plate using the Lipofectamine Plus kit (12 µl lipofectamine and 8 µl reagent; Life Technologies) following the manufacturer’s instructions. Transformants were selected with G418 (700 µg/ml; Life Technologies) or hygromycin (200 U/ml; Calbiochem).
Bosc23 ecotropic packaging cells and NIH3T3 murine cells were grown in DMEM supplemented with 10% fetal calf serum, streptomycin (100 µg/ml) and penicillin (100 U/ml) at 37°C and 6% CO2. Retroviral vectors containing the neo cassettes were first introduced into the Bosc23-packaging cells by transient transfection (4 µg DNA for 2 × 106 cells) with the Lipofectamine Plus kit (Life Technologies). The supernatants were collected 2 days post-transfection and used for infection of NIH3T3 cells (1 ml supernatant for 5 × 104 cells with 8 µg/ml polybrene). The cells were maintained in selective medium (500 µg/ml G418) for 2–3 weeks.
Nucleic acids extractions and analysis
Total RNA was extracted with the RNeasy kit (Qiagen). Ten micrograms of total RNA were electrophoresed on formaldehyde-agarose gels and transferred to Hybond-N membranes (Amersham Pharmacia Biotech). Membranes were hybridized with random-primed 32P-labeled DNA probes in Church solution at 65°C for 20 h, and washed once with 1× SSC, 0.1% SDS for 15 min at 65°C, once with 0.5× SSC, 0.1% SDS for 15 min at 65°C and once with 0.1× SSC, 0.1% SDS for 15 min at 65°C, as described previously (3).
Cellular DNA extractions were performed using a standard procedure (3). PCR amplifications were performed in 50 µl containing 15 µl of 3.3× Buffer II (tricine, potassium acetate, glycerol, DMSO), 1.1 mM Mg(OAc)2, 0.2 mM each dNTP, 1 µM each primer, 2 U rTth polymerase (Applera) and 1 µg of cellular DNA. After an initial step at 94°C (3 min), 40 cycles of amplification (40 s at 94°C, 60 s at 64°C and 2 min 30 s at 72°C) were carried out with following primers: neo12, 5′-GCCAAGCTCTTCAGCAATATCACG-3′; SV3, 5′-GCCTCTGAGCTATTCCAGAAGTAGTG-3′. PCR products were electrophoresed on agarose gels, eluted (Qiaquick, Qiagen), cloned into the pGEM-T easy vector II kit (Promega) and sequenced using the BigDye Terminator Cycle Sequencing v2.0 Ready Reaction kit (Applera).
RESULTS
Rationale and construction of modified Tetrahymena intron-marked neomycin reporter genes
The rationale of the construct was to introduce the Tetrahymena intron (Fig. 1A) within the neomycin gene, at different locations to circumvent the possible negative role of exonic sequences on intron folding, under conditions and after sequence alterations so as to (i) fulfill the required base pairing between the intronic and flanking sequences essential to maintain intron self-splicing activity (see below) and (ii) restore after splicing out of the intron the neomycin-coding sequence (for a functional indicator gene, see Fig. 4). The sequences of the 5′ and 3′ junctions between the Tetrahymena intron and the exonic sequences are very severely constrained, especially the sequence of the last few nucleotides of the 5′ and 3′ exons which both have to pair with the IGS of the Tetrahymena intron for efficient and precise splicing (Fig. 1B). Actually, the Tetrahymena intron inserted in the neomycin gene is expected to form a normal P1 helix (5′ exon-IGS pairing), including the required G·U wobble pair at the 5′ splice site (13,16,17). The P10 helix (IGS-3′ exon pairing) interaction is not as essential as P1, but it enhances precise exon ligation (18). Thus, we had to select insertion sites in the neomycin-coding sequence and acceptable mutations in nucleotides of the intronic IGS so as to generate complementary nucleotides between the IGS and neomycin RNA allowing formation of nine-nucleotide P1 and seven-nucleotide P10 interactions.
Figure 4.

Quantitative assay for the splicing of the modified Tetrahymena introns and for the neoTET cassettes. (A) Rationale of the assay: in the initial copy of the neoTET marked gene, the neomycin gene-coding sequence (neo) is interrupted by the Tetrahymena intron, and cells containing the marked gene should be G418-sensitive. Upon transcription of the marked gene, the Tetrahymena intron should be spliced out and, after reverse transcription and integration (i.e. retrotransposition), the neo gene in the transposed copy should be active and the cells be G418-resistant. (B) Experimental procedures for the assay of the neoTET cassette. The neoTET cassette was introduced into a Moloney murine leukemia virus-derived vector, as illustrated. The corresponding plasmids were then introduced by transfection into a viral packaging cell line (Bosc23); the supernatants of the transfected packaging cells were collected, and used to infect target NIH3T3 cells. The 3T3 cells were then submitted to G418 selection to quantify the number of G418-resistant cells. DNAs from the resulting clones were finally extracted and analyzed (D and E) for the precise splicing out of the Tetrahymena intron. (C) Splicing and reporter gene efficiency as measured by the number of G418-resistant clones after retroviral transduction of the neo-containing cassettes. G418 selections were performed on plates seeded with 5 × 104 cells (NIH3T3) per plate, and infected with 1 ml of transfected Bosc23 supernatant. The number of foci per plate is indicated for the five modified Tetrahymena intron-containing cassettes, the TNF intron-containing cassette, a positive control with an intronless neo cassette taken as a reference (100%) and a negative control without the neo gene; bars indicate standard deviations. (D) DNAs from individual clones were assayed by PCR for the presence of spliced copies, using primers bracketting the intronic domain. Positions of the primers used and sizes of the expected bands are indicated in the schema below the BET-labeled agarose gel. Bands of the expected size for the spliced-out intron (607 bp) are observed for all tested G418R clones, with a larger band (1020 bp) in the three control lanes (v, vector DNA). (E) Sequencing of the junctions of the spliced Tetrahymena introns. PCR products in (D) were sequenced, disclosing precise splicing out of the modified P, Pa and Pt Tetrahymena introns which restores the phase of the neo-coding sequence.
Accordingly, we constructed a series of intron-marked reporter genes in which the Tetrahymena intron was introduced into three different locations of the neomycin gene. Three of them were in a blunted PstI site (TET P, TET Pa, TET Pt), one in a blunted BanII site (TET B) and one in a blunted SphI site (TET S). All three sites were selected as they correspond to restriction enzymes with 3′ overhang allowing, after Klenow treatment, removal of at least four nucleotides within the neomycin sequence, thus facilitating introduction of mutations. The nucleotide sequences of the Tetrahymena intron-to-neomycin junctions were mutated using PCR and appropriate primers (see Materials and Methods) to optimize the P1 and P10 interactions (Fig. 1B) and conserve the neomycin-coding sequence after self-splicing. A control cassette was also constructed using intron 2 from the murine TNFβ nuclear gene, inserted into the BanII site (see Materials and Methods).
Self-splicing efficiency of the modified Tetrahymena introns: position and sequence effects
To test whether the modified Tetrahymena introns are functional for splicing in the context of the neomycin foreign exons, we introduced the intron-marked neomycin-coding sequence in an expression vector in which transcription is under the control of the CMV promoter and SV40 polyadenylation sequences. These constructs were then introduced into human HeLa cells by transfection. Three days post-transfection, total RNA was extracted and analyzed on northern blots (Fig. 2). As can be observed using a neomycin probe (Fig. 2, left), bands of 1.6 and 1.2 kb are detected. The former corresponds in size to unspliced transcripts, and the latter to spliced transcripts as assessed by the position of the band for the ‘none’ construct (intronless neomycin construct). Hybridization of the blot with an intronic sequence (Fig. 2, right) confirmed the nature of the bands, since only the 1.6 kb band was labeled. Accordingly, for three Tetrahymena constructs out of five (namely for the TET P, TET Pa, and TET Pt Tetrahymena intron insertions into the neomycin PstI site), a band corresponding to spliced transcripts can be observed, whereas no splicing is observed for the TET B and TET S constructs. The variable splicing level observed for the introns inserted at the single PstI site shows the importance of the IGS base pairing: quantitation of the northern blots (using a phosphoimager apparatus) actually yields an apparent splicing efficiency of 1.0, 2.8 and 5.1% for the closely related TET P, TET Pa and TET Pt constructs, respectively (the TET Pa and TET Pt constructs differ only by a single mispairing of the third A after the 5′ splice site). Similarly, the inefficient splicing for the TET B and TET S constructs might be accounted for by an inappropriate internal IGS base pairing, but also by a misfolding of the Tetrahymena intron due to its position within the neomycin transcript. To get further insight into this question, we devised additional constructs (i) with the ‘functional’ TET Pt intron together with its short exonic sequences involved in the IGS base pairing (namely the six and eight nucleotides from the 5′ and 3′ flanking sequences, respectively) inserted into the ‘non-functional’ BanII and SphI sites and, conversely, (ii) with the ‘non-functional’ TET B and TET S introns (also with their six/eight exonic nucleotides) inserted into the ‘functional’ PstI site. The splicing efficiency of these four constructs was assayed as described above, by northern blot analysis, and compared with the controls (Fig. 3). As illustrated in Figure 3, (i) positioning of the TET B and TET S introns at the PstI site resulted in some detectable splicing—although with much less efficiency than for the control TET Pt intron—and (ii) positioning of the TET Pt intron at the BanII and SphI sites resulted in a severely decreased splicing efficiency (associated, in the former case, with the generation of a band of abnormal size, most probably corresponding to an incomplete splicing event with only the 5′ exon-to-intron boundary processed). Altogether, these results demonstrate the importance for splicing efficiency of both the local IGS base pairing and the global position of the intron in the transcript.
Figure 2.

Assay for in vivo splicing of the modified Tetrahymena introns, as followed by northern blot analysis. Top, schematic representation of the neomycin gene, with the positions (and orientation, see arrows) of the inserted Tetrahymena introns as modified and depicted in Figure 1. The neomycin gene (in reverse orientation, see arrow) is under the transcriptional control of the CMV promoter (CMV) and the SV40 polyadenylation signal (pA). Bottom, in vivo splicing of the intron-containing neo constructs. The intron-containing neo constructs were introduced by transfection into HeLa cells, and RNAs extracted 3 days post-transfection. Northern blot analysis was performed using total RNA (10 µg per lane) and hybridization with a neo probe (left) and a TET probe (right). Bands of 1.6 and 1.2 kb should be observed for the unspliced and spliced transcripts, respectively (arrows). Lanes P, Pa, Pt, B and S, modified Tetrahymena introns (see Fig. 1); lane TNF, nuclear mRNA intron 2 from the TNF β gene inserted at the BanII site (see scheme above and Materials and Methods); lane none, intronless neo construct. r, ribosomal RNAs.
Figure 3.
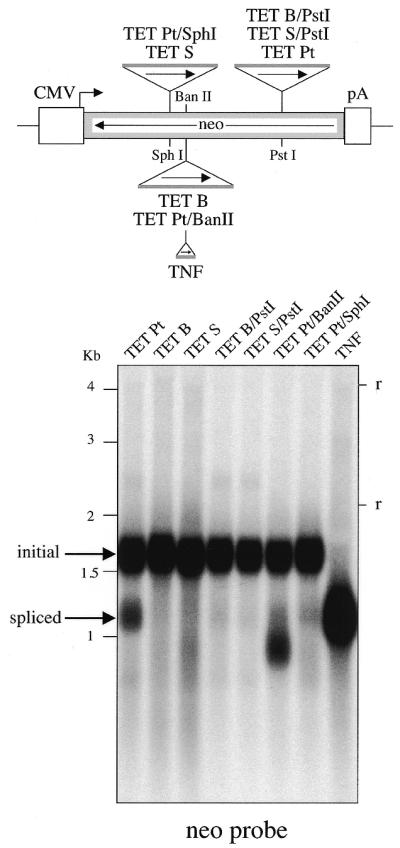
Position effects for the in vivo splicing of the modified Tetrahymena introns. Same experimental conditions as in Figure 2, with the TET Pt, TET B and TET S introns inserted at their initial positions (lanes TET Pt, TET B and TET S) and at alternate positions (lanes TET B/PstI, TET S/PstI, TET Pt/BanII and TET Pt/SphI).
Assay of the neoTET cassettes upon retroviral transduction
The intron-marked constructs above were then assayed for a complete retrotransposition cycle, using retroviral vectors. The rationale of the assay and the structure of the plasmids used are schematized in Figure 4A and B. The intron-marked neomycin-coding sequences together with a promoter for these sequences (i.e. the neoTET cassettes) were inserted in the indicated orientation into a retroviral vector whose LTRs are schematized. In the retroviral cycle (and similarly for retrotransposition) a LTR-driven transcript is generated, resulting in an intermediate RNA molecule in which the intron should be spliced out. Reverse transcription and integration by the retroviral machinery should then result in an integrated proviral copy where the intronless neomycin gene is active. The assay was performed as schematized in Figure 4B, for the five Tetrahymena intron-containing and control constructs. The intron-marked retroviral vectors were first introduced into Bosc23 packaging cells by transient transfection. The supernatants containing the viral particles were then collected and used to infect target NIH3T3 murine cells. The target cells were then submitted to G418 selection to isolate and quantify the expected neomycin-expressing cells (Fig. 4B). The results of three independent experiments are given in Figure 4C. Consistent with the data in Figure 2, no G418R clones were observed for the TET B and S cassettes, whereas the TET P, Pa and Pt cassettes yielded a significant number of clones, with the same rank order as found by northern blot analysis for intron splicing efficiency. The values are 12.4, 16.5 and 28.2% for the TET P, TET Pa and TET Pt cassettes, respectively, taking the intronless construct as a reference (100%, with 98.7% for the nuclear mRNA TNF intron). One important feature of the assay is that the DNA from the G418R clones can be isolated and the proviral copies characterized. An assay of the DNA of such clones for the presence of retrotransposed copies with a spliced out TET intron was carried out by both PCR (Fig. 4D) and sequencing after cloning (Fig. 4E). PCR amplification using primers bracketing the intronic domain of the neoTET cassettes yielded fragments of reduced size for all the G418R clones tested, as expected for the splicing out of the Tetrahymena intron (i.e. 607 versus 1020 bp, see Fig. 4D). The structure of the spliced junctions was further characterized after cloning and sequencing of the PCR products. As illustrated in Figure 4E, the sequences of the retrovirally transduced neoTET cassettes disclosed precise splicing out of the Tetrahymena intron, in agreement with the recovery of a fully coding neomycin gene.
Assay of the neoTET cassette for retrotransposition of human LINE elements
The neoTET Pt cassette which, among the neoTET cassettes, corresponds to the most efficient construct according to the above mentioned assays for Tetrahymena intron splicing, was selected for further analyses. It was inserted into a human LINE retrotransposon, downstream of the two ORFs of the element in order not to impair its transposition capacity, and the marked element was assayed together with the same element marked with the neoTNF construct and a previously devised neoRT indicator gene (1,3,8). As schematized in Figure 5A and B, the marked LINE elements were then introduced into episomal vectors containing the hygromycin resistance gene, and the retrotransposition assay was performed upon transfection of human HeLa cells with these constructs. An assay for retrotransposition was performed upon selection of the cells in G418-containing medium, either directly after transfection of the cells, or after an initial selection in an hygromycin-containing medium allowing the isolation of episomal vector-containing cell populations (Fig. 5A). The results of two independent transfection experiments are given in Figure 5C, and disclose G418R clones with the neoTET Pt marked LINEs, whatever the selection procedure used. Interestingly, the number of clones obtained with the neoTET cassette are closely related to those obtained with the neoRT cassette (3.9 × 10–4 and 4.7 × 10–4 events per cell per generation, respectively), and are only slightly lower than those obtained with the neoTNF cassette (2–4-fold, depending on the selection procedure). Finally, analysis of a series of positive clones that were recovered for further analysis provided evidence for retrotransposed copies for all constructs. This is illustrated in Figure 5D, where PCR amplifications of the DNA from G418R clones for the neoRT, neoTNF and neoTET cassettes were performed, using the same primers bracketing the intronic domains. Precise splicing out of the intron was ascertained by sequencing of some PCR products (data not shown).
Figure 5.

Quantitative assay for detection of retrotransposition of human LINEs marked with neo-cassettes. (A) Experimental procedures for detection of L1 retrotransposition events. The human L1.2 element (open box) was marked with a neo-cassette (neoTET, neoTNF or neoRT) and introduced into an hygromycin resistance gene-containing episomal vector. Marked L1-containing vectors were introduced into human cells by transfection, and then either cell transformants were isolated upon hygromycin selection and the resulting cell population (hygroR) was thereafter assayed for L1 retrotransposition upon G418 selection or, alternatively, transfected cells were directly submitted to G418 selection. (B) Structure of the three indicator genes (neoRT, neoTNF and neoTET) used for the L1 retrotransposition assay, with the RT and TNF introns being nuclear mRNA introns, and the Tetrahymena TET intron an autocatalytic group I intron. (C) Retrotransposition of the marked L1s. Numbers of foci per plates (5 × 105 and 5 × 104 cells per plate for direct and indirect G418 selections, respectively) are indicated as the mean of four independent transfections assays with the standard errors, together with the corresponding retrotransposition frequencies. (D) PCR analysis of the DNAs from initial and G418R cells assayed for the presence of retrotransposed copies with spliced-out introns, using the same primers bracketting the intronic domain for the three indicator genes. In the BET-labeled agarose gel, the first lane (hygR) for each construct corresponds to the DNA from the transfected hygroR cell population, before G418 selection, and the following lanes correspond to DNA from G418R cell populations (P) and clones (1–4). Bands of the expected size for the spliced-out intron are observed only for the G418R cells. Larger bands, corresponding for each construct to unspliced copies, are observed in the initial hygroR cell populations, before G418 selection.
DISCUSSION
We have constructed an indicator gene for retrotransposition which contains a self-splicing intron, the T.thermophila 23S rRNA group I intron. This indicator gene is functional as it allowed the monitoring of the mobility of the human LINE retrotransposon in HeLa cells, with an efficiency for detection of these events close to that reported for previously devised indicator genes. One essential improvement of the newly developed indicator gene is its versatility, as group I intron splicing is an auto-catalytic process not requiring any specific pathway or cellular component, at variance with the splicing of previously used nuclear mRNA introns (1–8). Accordingly, this indicator gene might be of great help in elucidating the retrotransposition mechanisms of, for instance, Pol III-driven retrotransposons, such as the human Alu sequences, which cannot afford splicing of nuclear mRNA introns (9), as well as of retroelements found in organelles.
One interesting outcome of the present analysis is the observation that splicing efficiency, as measured either by direct northern blot analysis or by the frequency of G418R clones in retrotransposition assays, is dramatically dependent both on the local base pairing of a few nucleotides from the exonic sequences with the IGS of the intron, as expected, and also on the global position of the inserted intron within the neo-containing transcripts. Indeed, the present data show that, for a given insertion site (e.g. the neomycin PstI site), splicing efficiency is dependent on refined modulations in the IGS-to-exons base pairing, with a 2-fold difference in splicing efficiency between the closely related TET Pa and TET Pt constructs which only differ by a mis-pairing (A-A versus A-T) at the P1 stem-to-loop junction. Conversely, splicing was found to be rather inefficient for the insertions within both the BanII and the SphI neomycin sites (which are close together), and we have shown, by displacing the various introns together with their flanking IGS-matching sequences, that it is essentially the positions of the intron which make the difference in splicing efficiency. Altogether, these results are consistent with previous observations that the exonic context might be deleterious for Tetrahymena intron splicing, by impeding the correct folding of the intronic RNA sequence (14,15). The present experiments show that splicing of the Tetrahymena intron can take place with significant efficiency within mammalian cells, as also observed for other constructs (14,15), provided that different sites are assayed.
Finally, a close comparison of the splicing efficiencies as measured by northern blot analysis and G418R clone frequencies discloses an interesting feature. Indeed, splicing efficiencies as measured by quantitative northern blot analysis are in the 1–5% range, whereas the values are in the 10–30% range when measuring the frequencies of G418R clones in either the retroviral transduction or the LINE retrotransposition assays. Although one might argue that such differences cannot be interpreted further, we do think that it is not the case, as the results were highly reproducible (over at least three independent experiments for each type of experiment) and as all measurements were quantified with appropriate controls. Accordingly, the observation that the steady state levels of spliced RNAs are at least 5-fold lower than expected from the retrotransposition rates suggests that the involvement of the marked RNAs in a retrotransposition process permits its more efficient splicing compared with that of the bulk of the marked transcripts. Actually, a simple molecular model can account for this result, if one considers that in both the retroviral transduction and the LINE retrotransposition assays the marked RNAs are expected to be sequestered for a non-negligible period of time in, respectively, a retroviral particle and a LINE ribonucleoparticle (2,19–21). Therefore, it is tempting to speculate that this sequestration favors intron splicing, either as a result of an increased stability of the RNA in the particle, thus allowing the kinetic process of intron splicing to proceed, or as a result of an improved folding of the intron RNA associated, for instance, with a spatial constraint or the presence of chaperone proteins within the corresponding particles. In fact, these two possibilities are non-exclusive, and are supported by a series of experimental data disclosing, for instance, that Tetrahymena intron splicing in mammalian cells is not as rapid as in its natural context (14), and that several retroviral proteins—and similarly the LINE ORF1 protein—have an RNA-binding activity (20,22–25) and could therefore act as chaperones (26). An alternative interpretation involving preferential ‘packaging’ of the spliced transcripts cannot be formally excluded, but is rendered unlikely by the fact that the transcript length, at least for the retroviral transduction experiment, is far from the maximum limiting value for packaging into retroviral particles (i.e. 5 ± 0.4 versus ∼10 kb).
Acknowledgments
ACKNOWLEDGEMENTS
We wish to especially acknowledge B. Sargueil and F. Michel for the generous gift of the p18R.TET(3) vector and for helpful discussions and advice. This work was supported by the CNRS and a grant from the Ligue Nationale contre le Cancer (Equipe Labellisée), and a fellowship from the ARC to C.E.
REFERENCES
- 1.Heidmann T., Heidmann,O. and Nicolas,J.-F. (1988) An indicator gene to demonstrate intracellular transposition of defective retroviruses. Proc. Natl Acad. Sci. USA, 85, 2219–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boeke J.D. and Stoye,J.P. (1997) Retrotransposons, endogenous retroviruses, and the evolution of retroelements. In Coffin,J.M., Hughes,S.H. and Varmus,H.E. (eds), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 343–435. [PubMed]
- 3.Heidmann O. and Heidmann,T. (1991) Retrotransposition of a mouse IAP sequence tagged with an indicator gene. Cell, 64, 159–170. [DOI] [PubMed] [Google Scholar]
- 4.Jensen S. and Heidmann,T. (1991) An indicator gene for detection of germline retrotransposition in transgenic Drosophila demonstrates RNA-mediated transposition of the LINE I element. EMBO J., 10, 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moran J.V., Holmes,S.E., Nass,T.P., DeBerardinis,R.J., Boeke,J.D. and Kazazian,H.H.J. (1996) High frequency retroposition in cultured mammalian cells. Cell, 87, 917–927. [DOI] [PubMed] [Google Scholar]
- 6.Tchénio T. and Heidmann,T. (1992) High-frequency intracellular transposition of a defective mammalian provirus detected by an in situ colorimetric assay. J. Virol., 66, 1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freeman J.D., Goodchild,N.L. and Mager,D.L. (1994) A modified indicator gene for selection of retrotransposition events in mammalian cells. Biotechniques, 17, 46, 48,–49, 52. [PubMed] [Google Scholar]
- 8.Esnault C., Maestre,J. and Heidmann,T. (2000) Human LINE retrotransposons generate processed pseudogenes. Nature Genet., 24, 363–367. [DOI] [PubMed] [Google Scholar]
- 9.Sisodia S., Sollner-Webb,B. and Cleveland,D. (1987) Specificity of RNA maturation pathways: RNAs transcribed by RNA polymerase III are not substrates for splicing or polyadenylation. Mol. Cell. Biol., 7, 3602–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cech T.R. (1990) Self-splicing of group I introns. Annu. Rev. Biochem., 59, 543–568. [DOI] [PubMed] [Google Scholar]
- 11.Price J.V. and Cech,T.R. (1985) Coupling of Tetrahymena ribosomal RNA splicing to β-galactosidase expression in Escherichia coli. Science, 228, 719–722. [DOI] [PubMed] [Google Scholar]
- 12.Roman J. and Woodson,S.A. (1998) Integration of the Tetrahymena group I intron into bacterial rRNA by reverse splicing in vivo. Proc. Natl Acad. Sci. USA, 95, 2134–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waring R.B., Ray,J.A., Edwards,S.W., Scazzocchio,C. and Davies,R.W. (1985) The Tetrahymena rRNA intron self-splices in E. coli: in vivo evidence for the importance of key base-paired regions of RNA for RNA enzyme function. Cell, 40, 371–380. [DOI] [PubMed] [Google Scholar]
- 14.Hagen M. and Cech,T.R. (1999) Self-splicing of the Tetrahymena intron from mRNA in mammalian cells. EMBO J., 18, 6491–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long M. and Sullenger,B. (1999) Evaluating group I intron catalytic efficiency in mammalian cells. Mol. Cell. Biol., 19, 6479–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strobel S.A. and Cech,T.R. (1995) Minor groove recognition of the conserved G.U pair at the Tetrahymena ribozyme reaction site. Science, 267, 675–679. [DOI] [PubMed] [Google Scholar]
- 17.Strobel S.A. and Cech,T.R. (1996) Exocyclic amine of the conserved G.U pair at the cleavage site of the Tetrahymena ribozyme contributes to 5′-splice site selection and transition state stabilization. Biochemistry, 35, 1201–1211. [DOI] [PubMed] [Google Scholar]
- 18.Suh E.R. and Waring,R.B. (1990) Base pairing between the 3′ exon and an internal guide sequence increases 3′ splice site specificity in the Tetrahymena self-splicing rRNA intron. Mol. Cell. Biol., 10, 2960–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deragon J.M., Sinnett,D. and Labuda,D. (1990) Reverse transcriptase activity from human embryonal carcinoma cells NTera2D1. EMBO J., 9, 3363–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hohjoh H. and Singer,M. (1996) Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J., 15, 630–639. [PMC free article] [PubMed] [Google Scholar]
- 21.Martin S.L. (1991) Ribonucleoprotein particles with LINE-1 RNA in mouse embryonal carcinoma cells. Mol. Cell. Biol., 11, 4804–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hohjoh H. and Singer,M.F. (1997) Sequence-specific single-strand RNA binding protein encoded by the human LINE-1 retrotransposon. EMBO J., 16, 6034–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin S.L., Li,J., Weisz,J.A., Wei,W., Gilbert,N., Ooi,S.L., Lawler,J.F., Ostertag,E.M., Kazazian,H.H., Boeke,J.D. and Moran,J.V. (2000) Deletion analysis defines distinct functional domains for protein-protein and nucleic acid interactions in the ORF1 protein of mouse LINE-1. J. Mol. Biol., 304, 11–20. [DOI] [PubMed] [Google Scholar]
- 24.Holmes S.E., Singer,M.F. and Swergild,G.D. (1992) Studies on p40, the leucine zipper motif containing protein encoded by the first open reading frame of an active human LINE-1 transposable element. J. Biol. Chem., 267, 19765–19768. [PubMed] [Google Scholar]
- 25.Kolosha V.O. and Martin,S.L. (1997) In vitro properties of the first ORF protein from mouse LINE-1 support its role in ribonucleoprotein particle formation during retrotransposition. Proc. Natl Acad. Sci. USA, 94, 10155–10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin S.L. and Bushman,F.D. (2001) Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Mol. Cell. Biol., 21, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cech T.R. (1988) Conserved sequences and structures of group I introns: building an active site for RNA catalysis—a review. Gene, 73, 259–271. [DOI] [PubMed] [Google Scholar]