

Abstract
An efficient stereoselective synthesis of 10-hydroxy-10-(1H-indol-3-yl)-9-(10H)-phenanthrene derivatives was realized through an organocatalyzed Friedel–Crafts reaction of phenanthrenequinones and indoles using a (S,S)-dimethylaminocyclohexyl-squaramide as the catalyst. Under the optimized conditions, the desired chiral products were obtained in good yields (73–90%) with moderate to high ee values (up to 97% ee). Two pairs of synthesized enantiomers were subjected to evaluation of their antiproliferative activities on four types of human cancer cell lines and one human umbilical vein endothelial cell line using the CCK-8 assay. The results indicated that stereoselectivity had obvious impacts on biological activity. (S)-4g was found to have optimal cytotoxicity against the A549 cell line and a good safety profile for human normal cells, which was better than the inhibitory activity of the positive control drug (doxorubicin).
Keywords: enantioselective, Friedel–Crafts reaction, organocatalysis, phenanthrenequinones, indoles, CCK-8 assay
1. Introduction
As well known, many chiral drugs exhibit different pharmacological activities and safety due to the difference of stereochemistry. For example, (S)-Ofloxacin, (S)-Finerenone, and (S)-Amlodipine show better bioactivity than do their R-enantiomers [1,2,3]. Thus, the development of enantioselective reactions to access optically active compounds would be of significance in drug discovery.
9,10-phenanthroquinone is an important skeleton that exists in many bioactive natural products and synthetic compounds [4,5,6,7,8,9,10], and they play an extremely important role in photochemistry, electrochemistry, and organic synthesis [11,12,13,14]. The C9 and C10 dicarbonyl groups of phenanthroquinone are prochiral nucleophilic addition sites, which could undergo asymmetric addition reactions with many nucleophiles to give chiral adducts. However, the enantioselective transformation with 9,10-phenanthroquinone as a substrate has been rarely reported [15,16].
Indole is an important heterocyclic structure found in various natural alkaloids and bioactive pharmaceuticals [17,18,19,20,21,22,23,24,25,26,27]. Due to their wide range of applications in the synthesis field, the direct functionalization of indoles has been extensively investigated, and many methods have been developed in great progress. Moreover, the asymmetric Friedel–Crafts alkylation of indoles with appropriate electrophilic reagents is one of the most important and efficient strategies to produce optically active indoles [28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44].
In 1998, the Yan group reported one example of photochemical synthesis of 10-hydroxy-10-(1H-indol-3-yl)-9-(10H)-phenanthrene via Friedel–Crafts reaction of 9,10-phenanthrene and indole. However, the racemic product was obtained because no asymmetric synthesis was involved (Figure 1a) [45].
Figure 1.

Friedel–Crafts reaction of 9,10-phenanthrene and indole.
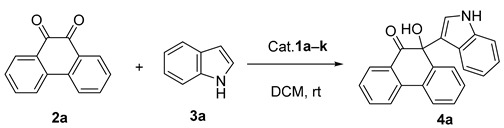
To the best of our knowledge, the asymmetric Friedel–Crafts reaction of phenanthrenequinones and indoles has never been reported to date. We herein first developed this reaction for the enantioselective construction of 10-hydroxy-10-(1H-indol-3-yl)-9-(10H)-phenanthrene (Figure 1b) by employing organocatalysts 1a–k (Figure 2). In addition, this work aimed to develop an efficient and enantioselective method for synthesizing phenanthrenequinone derivatives, evaluating their biological properties, and exploring the impact of stereochemistry on antitumor activity.
Figure 2.

The structure of screened organocatalysts (1a–1k).
2. Results and Discussion
We first applied the catalysts 1a–1k in the Friedel–Crafts reaction of phenanthrenequinone (2a) and indole (3a) to screen the optimal catalyst. The reaction was carried out with CH2Cl2 as a solvent in the presence of 10 mol% of catalysts at room temperature for 24 h (Table 1).
Table 1.
Asymmetric F–C reaction of phenanthroquinone and indole catalyzed by 1a–k a.
| ||||
|---|---|---|---|---|
| Entry | Catalyst | 2a:3a | Yield (%) b | %ee c |
| 1 | 1a | 1:1 | 43 | 20 |
| 2 | 1b | 1:1 | 45 | 5 |
| 3 | 1c | 1:1 | 40 | 10 |
| 4 | 1d | 1:1 | 42 | 22 |
| 5 | 1e | 1:1 | 40 | 21 |
| 6 | 1f | 1:1 | 45 | 30 |
| 7 | 1g | 1:1 | 42 | 26 |
| 8 | 1h | 1:1 | 40 | 15 |
| 9 | 1i | 1:1 | 41 | 24 |
| 10 | 1j | 1:1 | 45 | 40 |
| 11 | 1k | 1:1 | 40 | 20 |
| 12 | 1j | 1:3 | 53 | 42 |
| 13 | 1j | 1:5 | 78 | 48 |
| 14 | 1j | 1:6 | 85 | 65 |
| 15 | 1j | 1:7 | 90 | 70 |
| 16 | 1j | 1:8 | 88 | 66 |
| 17 | 1j | 1:10 | 90 | 60 |
a Reaction condition: phenanthrenequinone (0.10 mmol), indole (0.10–1.00 mmol) and catalysts (0.01 mmol) in CH2Cl2 (1.0 mL) at rt. b isolated yield. c Determined by HPLC analysis (Chiralcel OD-H).
All catalysts preceded the reaction smoothly to give the desired product 4a in low yields (40–45%) with 5–40% ees. Among them, (S,S)-dimethylaminocyclohexy-squaramide 1j was optimal in terms of the yield and enantioselectivity (entry 10). By monitoring the reaction (TLC), we found that indole was exhausted while the phenanthrenequinone was left obviously when the amount ratio of phenanthrenequinone (2a) to indole (3a) was 1:1. This might be the main reason for the low yields (entries 1–11). Therefore, we tried increasing the amount of indole by 3 to 10 equivalents in the reaction to improve the yield (entries 12–17). The results showed both yield and enantioselectivity were increased, and the optimal equivalent ratio was determined to be 1:7 of phenanthrenequinone to indole (entry 15), which would be further applied to the subsequent study.
To improve the enantioselectivity of the transformation, we investigated a variety of different reaction conditions (Table 2). The survey of solvents showed that CH2Cl2 was optimal in terms of the yield and enantioselectivity (entry 1), aligning with previous studies that CH2Cl2 was found to enhance reactivity and enantioselectivity in Friedel–Crafts reactions [39,40]. The screening of catalyst loading exhibited that 10 mol% of 1j was optimal. Reducing the catalyst loading to 5 mol% led to an obvious decrease in enantioselectivity and yield (entry 9 vs. entry 1), and 20 mol% loading was proven not beneficial for the catalysis performance (entry 10 vs. entry 1). When the reaction temperature was lowered from rt to 0 °C, both enantioselectivity and yield of product were decreased (entry 11 vs. entry 1). Furthermore, diluting the reaction concentration by half was detrimental for yield and enantiocontrol (entry 12 vs. entry 1). Additives such as 4 Å molecular sieves or benzoic acid offered no improvement in the asymmetric induction (entry 14). Based on these experiments, the optimized conditions were determined to be CH2Cl2 as the solvent with a 10 mol% loading of catalyst 1j at rt.
Table 2.
Screening of reaction conditions for the asymmetric F–C reaction catalyzed by 1j a.
| Entry | Solvent | Cat. Loading (% mmol) |
Yield (%) b | %ee c |
|---|---|---|---|---|
| 1 | CH2Cl2 | 10 | 90 | 70 |
| 2 | CHCl3 | 10 | 86 | 54 |
| 3 | (CH2)2Cl2 | 10 | 79 | 46 |
| 4 | Et2O | 10 | - | - |
| 5 | THF | 10 | - | - |
| 6 | PhMe | 10 | 76 | 14 |
| 7 | EA | 10 | 75 | 12 |
| 8 | ACN | 10 | 83 | 41 |
| 9 | CH2Cl2 | 5 | 77 | 50 |
| 10 | CH2Cl2 | 20 | 88 | 61 |
| 11 d | CH2Cl2 | 10 | 83 | 58 |
| 12 e | CH2Cl2 | 10 | 80 | 62 |
| 13 f | CH2Cl2 | 10 | 82 | 56 |
| 14 g | CH2Cl2 | 10 | 78 | 38 |
a Reaction condition: phenanthrenequinone (0.10 mmol), indole (0.70 mmol), and catalyst 1j in solvent. b isolated yield. c Determined by HPLC analysis (Chiralcel OD-H). d 0 °C. e 2 mL of solvent. f 4 Å MS (about 200 mg). g Benzoic acid (0.01 mmol).
Firstly, the reaction of different substituted indoles 3 with 9,10-phenanthrenequinone (2a) was investigated. As Table 3 shows, most of the screened indoles were tolerated to give the corresponding products in good yields with 48–97% ees. When various substituents (R2) on the phenyl ring (C4, C5, C6, and C7) of indole were investigated, it was found that screened substituents at the C4 position showed a detrimental impact on the performance of the catalyst, resulting in a failure of the reaction (entries 2, 3). Next, the electron-donating groups at C5 (Me and OMe) were found to be favorable for enantioselectivity (entries 6, 7); especially, 5-OMe indole as a substrate gave the best enantiomeric excess of 97%. However, the reaction using 5-NO2 indole as a nucleophile failed to proceed even by prolonging the reaction time to 48 h (entry 8). In addition, the electron-withdrawing or electron-donating group at the C6 position showed a similar effect on the stereoselectivity to give moderate ee values (entries 9–12). It is worth mentioning that substituents at the C7 position had a great influence on the action of the catalyst. The reaction of 7-Me and 7-OMe indole proceeded smoothly to afford products with moderate enantioselectivities (entries 15, 16), while halogen groups at the C7 position of indole caused the reaction not to proceed even by prolonging the reaction time (entries 13, 14). Furthermore, when 2,7-dibromo- or 3,6-dibromo-substituted phenanthrenequinones were used as substrates in the reaction, similar ee values were obtained compared to the results from the reaction of phenanthrenequinone 2a (entries 15–23), and the optimal enantioselectivities were observed in the reaction of 5-OMe indole (entries 19 and 23). Based on the above experimental results, the enantioselectivity was obviously affected by the substituted groups and their position. Among them, 5-OMe-substituted indole as a substrate gave the best enantiomeric excess (entries 7, 19, and 23).
Table 3.
Generality of the enantioselective F–C reaction of phenanthrenequinones and indoles a.

| |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yield (%) b | %ee c |
| 1 | H, H | H | 4a | 90 | 70 |
| 2 | H, H | 4-Cl | 4b | - | - |
| 3 | H, H | 4-Me | 4c | - | - |
| 4 | H, H | 5-F | 4d | 88 | 48 |
| 5 | H, H | 5-Cl | 4e | 85 | 48 |
| 6 | H, H | 5-Me | 4f | 87 | 80 |
| 7 | H, H | 5-OMe | 4g | 90 | 97 |
| 8 | H, H | 5-NO2 | 4h | - | - |
| 9 | H, H | 6-F | 4i | 84 | 54 |
| 10 | H, H | 6-Cl | 4j | 82 | 60 |
| 11 | H, H | 6-Me | 4k | 75 | 56 |
| 12 | H, H | 6-OMe | 4l | 80 | 54 |
| 13 | H, H | 7-F | 4m | - | - |
| 14 | H, H | 7-Cl | 4n | - | - |
| 15 | H, H | 7-Me | 4o | 78 | 52 |
| 16 | H, H | 7-OMe | 4p | 83 | 56 |
| 17 | 2-Br, 7-Br | H | 4q | 81 | 68 |
| 18 | 2-Br, 7-Br | 5-Me | 4r | 82 | 63 |
| 19 | 2-Br, 7-Br | 5-OMe | 4s | 85 | 94 |
| 20 | 2-Br, 7-Br | 6-Cl | 4t | 80 | 60 |
| 21 | 2-Br, 7-Br | 6-OMe | 4u | 78 | 52 |
| 22 | 3-Br, 6-Br | 5-Me | 4v | 83 | 60 |
| 23 | 3-Br, 6-Br | 5-OMe | 4w | 90 | 97 |
| 24 | 3-Br, 6-Br | 6-Cl | 4x | 75 | 58 |
| 25 | 3-Br, 6-Br | 6-OMe | 4y | 73 | 56 |
a Reaction condition: phenanthrenequinone (0.10 mmol), indole (0.70 mmol), and catalyst 1j (0.01 mmol) in CH2Cl2 (1 mL) at rt. b Isolated yield. c Determined by HPLC analysis (Chiralcel OD-H, Chiralpak AD-H, AS-H).
The absolute configuration of 10-hydroxy-10-(1H-indol-3-yl)-9-(10H)-phenanthrene products 4 were unambiguously assigned as R according to the X-ray crystal structure analysis of 4w (Figure 3) [46].
Figure 3.

X-ray crystal structure of 4w (CCDC 2375435).
A transition state model can be proposed according to the literature (Figure 4) [39,47]. As a bifunctional catalyst, the squaramide moiety of 1j activates and orients phenanthrenequinone through double H-bonding, while the tertiary amine of 1j activates indole through H-bonding. Then, Re face addition of indole to the activated phenanthrenequinone via favorable interaction leads to the formation of the (R)-product.
Figure 4.

Proposed transition state.
Next, we plan to study the antiproliferative activities of the obtained products. Moreover, to investigate the impact of the chiral center and electrical effect on activity, we chose products (R)-4g and (R)-4w with high ee values for the activity test. Their enantiomers of (S)-4g and (S)-4w were prepared by using the enantiomer of catalyst 1j. The structures of enantiomers and their ee values were shown in Figure 5, and all tested compounds were determined to have a purity of >95%.
Figure 5.

The structures of three pairs of enantiomers.
The antiproliferative activities of selected compounds and the positive control drug (doxorubicin) were evaluated on four types of human cancer cell lines and one human normal cell, human hepatocellular carcinoma cells (HepG-2), human non-small cell lung cancer cells (A549), human breast cancer cells (MCF-7), human gastric adenocarcinoma cells (SGC-7901), and human umbilical vein endothelial cells (HUVEC) using the CCK-8 assay (Table 4).
Table 4.
In vitro cytotoxicity of 10-hydroxy-10-(1H-indol-3-yl)-9-(10H)-phenanthrene derivatives against HepG-2, A549, MCF-7, SGC-7901, and HUVEC by CCK-8 assay (n = 3).
| Entry | Compounds | IC50 Values a (μM) | ||||
|---|---|---|---|---|---|---|
| HepG-2 | A549 | MCF-7 | SGC-7901 | HUVEC | ||
| 1 | (R)-4g | 81.8 ± 0.96 | 36.17 ± 0.69 | 59.95 ± 3.43 | 59.89 ± 0.68 | 61.3 ± 3.42 |
| 2 | (S)-4g | 36.65 ± 3.76 | 0.38 ± 0.26 | 34.15 ± 1.37 | 29.88 ± 0.45 | 28.97 ± 1.22 |
| 3 | (Racemic)-4g | - b | 2.5 ± 0.76 | - b | - b | - b |
| 4 | (R)-4w | 23.50 ± 0.41 | 16.58 ± 0.27 | 18.62 ± 0.95 | 17.07 ± 0.03 | 22.53 ± 0.32 |
| 5 | (S)-4w | 13.44 ± 0.51 | 12.79 ± 0.31 | 5.89 ± 0.57 | 16.69 ± 0.17 | 20.03 ± 0.20 |
| 6 | (Racemic)-4w | 14.15 ± 0.22 | 15.09 ± 0.28 | 15.18 ± 0.41 | 17.41 ± 0.08 | 16.06 ± 0.11 |
| 7 c | Doxorubicin | 0.12 ± 0.01 | 2.06 ± 0.14 | 1.05 ± 0.04 | 0.88 ± 0.09 | 0.44 ± 0.02 |
a IC50 is defined as the concentration, which results in a 50% decrease in cell number as compared with that of the control cultures in the absence of an inhibitor and were calculated using the respective regression analysis. b - = no biological activity. c Doxorubicin was employed as positive control.
The results showed that most of the tested compounds had cytotoxic activity against human cancer cell lines in a concentration-dependent manner (see Supporting Information). The stereoselectivity showed an obvious impact on activity. Generally, S-configuration compounds were found to be more effective than R-enantiomers or racemic products. Among them, (S)-4g exhibited superior cytotoxic activity against the A549 (IC50: 0.38 ± 0.26 μM), which was obviously better than its enantiomer (R)-4g and racemate (±)-4g. Moreover, it is worth mentioning that the IC50 value of (S)-4g against the A549 was significantly lower than the value of the positive control (doxorubicin, IC50: 2.06 ± 0.14 μM). Specifically, (S)-4g showed substantially less cytotoxicity to HUVEC (IC50: 28.97 ± 1.22 μM) than that of doxorubicin (IC50: 0.44 ± 0.02 μM).
3. Materials and Methods
3.1. Experimental
1H NMR and 13C NMR spectra were recorded on a Brucker spectrometer (Karlsurhe, Germany) at 500 or 400 and 125 or 100 MHz, respectively. using CDCl3 and DMSO-d6 as a solvent. The chemical shifts were reported in ppm, and the residual nondeuterated solvent was used as an internal standard (CDCl3, 7.26 and 77.0 ppm; DMSO, 2.5 and 39.5 ppm, respectively). The splitting patterns of the signals were reported as s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; and m, multiplet. High-resolution mass spectra (HRMS) were measured on a triple TOF 5600+ mass spectrometer (SCIEX, Concord, ON, Canada) equipped with an electrospray ionization (ESI) source in the negative-ion mode. Single-crystal structure was determined on Bruker D8 Venture (Karlsurhe, Germany). The enantiomeric excess (ee) values of the products were determined by chiral HPLC, using Daicel Chiralcel OD-H, Chiralpak AD-H, and Chiralpak AS-H columns (4.6 mm × 250 mm). The reactions were monitored by thin-layer chromatography (TLC). Purifications by column chromatography were conducted over silica gel (200–300 mesh). The organocatalysts 1a, 1b, and 1f–1k were purchased from Daicel Chiral Technologies (Shanghai, China), and the catalysts 1c–1e were synthesized according to the literature [48].
3.2. General Procedure for the Enantioselective Friedel–Crafts Reaction of Phenanthrenequinones and Indoles
To a tube, a mixture of phenanthrenequinones (0.1 mmol), indoles (0.7 mmol) and organocatalyst 1j (0.01 mmol), CH2Cl2 (1.0 mL) was added. The resulting mixture was stirred at 0 °C for 24 h. After the reaction was finished (monitored by TLC), the reaction was directly poured into a column chromatography on silica gel with hexane/EtOAc (4:1) as eluent to afford the new compounds 4a–y. The enantiomeric ratio was determined by HPLC analysis on a chiral Chiralcel OD-H, AD-H, AS-H column. Experimental data can be found in Supplementary Materials.
(R)-4a: yellow solid, mp: 95.2–96.1 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.98–7.93 (m, 1H), 7.92–7.84 (m, 3H), 7.79 (ddd, J = 7.5, 1.5, 0.5 Hz, 1H), 7.75–7.71 (m, 1H), 7.56 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.53–7.46 (m, 2H), 7.27–7.23 (m, 1H), 7.22–7.19 (m, 1H), 7.16–7.09 (m, 2H), 6.36 (dd, J = 3.0, 2.0 Hz, 1H), 4.80 (s, 1H); 13C NMR (125 MHz, Chloroform-d) δ 200.9, 139.7, 137.0, 136.6, 134.5, 130.0, 129.4, 128.8, 128.6, 128.4, 127.8, 127.7, 125.3, 124.2, 123.7, 122.7, 122.4, 120.4, 117.7, 111.3; HRMS (ESI) m/z: [M + Na]+ calcd for C22H15NO2Na: 348.1000; found 348.1005; [α]D25 = 25.7 (c 0.50, MeOH) (70% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 82:18, 1.0 mL/min, 254 nm), tR = 21.3 min (minor), 30.5 min (major).
(R)-4d: light yellow solid, mp: 174.1–175.0 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.99–7.95 (m, 1H), 7.95–7.87 (m, 3H), 7.84 (ddd, J = 7.5, 1.5, 0.5 Hz, 1H), 7.60 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.56–7.50 (m, 2H), 7.39 (ddt, J = 10.0, 2.5, 0.5 Hz, 1H), 7.31 (td, J = 7.5, 1.0 Hz, 1H), 7.16 (ddd, J =9.0, 4.5, 0.5 Hz, 1H), 6.91 (td, J = 9.0, 2.5 Hz, 1H), 6.46 (dd, J = 3.0, 1.0 Hz, 1H), 4.77 (s, 1H);13C NMR (125 MHz, Chloroform-d) δ 200.7, 158.0 (d, J = 247.5 Hz), 139.5, 137.0, 134.6, 133.1, 130.0, 129.4, 128.7, 128.6, 128.4, 127.8, 127.6, 125.8, 123.8, 122.8, 117.9, 111.9 (d, J = 15.0 Hz), 111.0 (d, J = 26.2 Hz), 105.6 (d, J = 25.0 Hz); HRMS (ESI) m/z: [M + Na]+ calcd for C22H14FNO2Na: 366.0906; found 366.0901; [α]D25 = +52.0 (c 0.51, MeOH) (48% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 13.4 min (minor), 22.3 min (major).
(R)-4e: light yellow solid, mp: 191.5–192.4 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.96–7.85 (m, 4H), 7.82 (ddd, J = 7.5, 1.5, 0.5 Hz, 1H), 7.71 (d, J = 2.0 Hz, 1H), 7.58 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.53–7.46 (m, 2H), 7.28 (td, J = 7.5, 1.0 Hz, 1H), 7.12 (dd, J = 8.5, 0.5 Hz, 1H), 7.08 (dd, J = 8.5, 2.0 Hz, 1H), 6.39 (t, J = 2.5 Hz, 1H), 4.76 (s, 1H); 13C NMR (125 MHz, Chloroform-d) δ 200.7, 139.4, 137.0, 135.0, 134.7, 129.9, 129.5, 128.8, 128.6, 128.5, 127.9, 127.6, 126.2, 125.4, 123.8, 122.9, 122.8, 120.1, 117.6, 112.3; HRMS (ESI) m/z: [M + Na]+ calcd for C22H14ClNO2Na: 382.0611; found 382.0617; [α]D25 = +42.3 (c 0.58, MeOH) (48% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 12.4 min (minor), 20.8 min (major).
(R)-4f: yellow solid, mp: 96.2–97.2 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.96–7.91 (m, 1H), 7.91–7.88 (m, 1H), 7.88–7.85 (m, 1H), 7.80 (dd, J = 7.5, 1.5 Hz, 2H), 7.59–7.52 (m, 2H), 7.52–7.46 (m, 2H), 7.25 (td, J = 7.5, 1.0 Hz, 1H), 7.08 (d, J = 8.5 Hz, 1H), 6.95 (dd, J = 8.5, 1.5 Hz, 1H), 6.28 (d, J = 2.5 Hz, 1H), 4.79 (s, 1H), 2.45 (s, 3H);13C NMR (125 MHz, Chloroform-d) δ 200.8, 139.8, 137.0, 135.0, 134.4, 130.0, 129.7, 129.4, 128.8, 128.5, 128.3, 127.9, 127.7, 125.5, 124.3, 124.1, 123.6, 122.7, 120.0, 117.0, 111.0, 21.6; HRMS (ESI) m/z: [M + Na]+ calcd for C23H17NO2Na: 362.1157; found: 362.1151; [α]D25 = 28.9 (c 0.57, MeOH) (80% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 11.2 min (minor), 16.8 min (major).
(R)-4g: yellow solid, mp: 200.8–201.8 °C; 1H NMR (500 MHz, Chloroform-d) δ 8.01–7.96 (m, 1H), 7.95–7.91 (m, 1H), 7.90 (ddt, J = 8.0, 1.0, 0.5 Hz, 1H), 7.84 (ddd, J = 7.5, 1.5, 0.5 Hz, 2H), 7.59 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.54–7.49 (m, 2H), 7.29 (td, J = 7.5, 1.0 Hz, 1H), 7.16–7.10 (m, 2H), 6.81 (ddd, J = 9.0, 2.5, 0.5 Hz, 1H), 6.40 (dd, J = 3.0, 0.5 Hz, 1H), 4.77 (s, 1H), 3.88 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 200.7, 154.5, 139.7, 137.0, 134.5, 131.7, 130.0, 129.4, 128.8, 128.6, 128.4, 127.8, 127.7, 125.7, 124.9, 123.6, 122.8, 117.1, 112.9, 112.0, 101.9, 55.8; HRMS (ESI) m/z: [M + Na]+ calcd for C23H17NO3Na: 378.1106; found: 378.1101; [α]D25= 35.6 (c 0.47, MeOH) (97% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR= 14.2 min (minor), 20.6 min (major).
(R)-4i: white solid, mp: 105.4–106.2 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.99–7.93 (m, 1H), 7.92–7.83 (m, 3H), 7.79 (ddd, J = 7.7, 1.5, 0.5 Hz, 1H), 7.65 (ddt, J = 8.7, 5.3, 0.7 Hz, 1H), 7.58 (ddd, J = 8.0, 7.4, 1.5 Hz, 1H), 7.52–7.46 (m, 2H), 7.31–7.26 (m, 1H), 6.93–6.85 (m, 2H), 6.35 (dd, J = 2.7, 1.5 Hz, 1H), 4.75 (s, 1H);13C NMR (125 MHz, Chloroform-d) δ 201.0, 160.0 (d, J = 237.5 Hz), 139.5, 137.1, 136.7, 134.6, 130.0, 129.4, 128.7, 128.6, 128.4, 127.8, 127.6, 124.5, 123.7, 122.8, 121.9, 121.5 (d, J = 10.0 Hz), 118.0, 109.2 (d, J = 25.0 Hz), 97.6 (d, J = 25.0 Hz); HRMS (ESI) m/z: [M + Na]+ calcd for C22H14FNO2Na: 366.0906; found 366.0910; [α]D25 = 18.6 (c 0.52, MeOH) (54% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 10.4 min (minor), 15.3 min (major).
(R)-4j: white solid, mp: 106.4–107.3 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.98–7.85 (m, 4H), 7.78 (ddd, J = 7.5, 1.5, 0.5 Hz, 1H), 7.65–7.60 (m, 1H), 7.58 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.53–7.46 (m, 2H), 7.30–7.26 (m, 1H), 7.20 (ddt, J = 2.0, 1.5, 0.5 Hz, 1H), 7.08 (dd, J = 8.5, 2.0 Hz, 1H), 6.58–6.13 (m, 1H), 4.76 (s, 1H);13C NMR (125 MHz, Chloroform-d) δ 200.9, 139.4, 137.0, 134.7, 130.0, 129.5, 128.8, 128.6, 128.5, 127.8, 127.6, 124.8, 123.9, 123.8, 122.8, 121.4, 121.2, 118.1, 111.2; HRMS (ESI) m/z: [M + Na]+ calcd for C22H14ClNO2Na: 382.0611; found 382.0615; [α]D25 = 36.4 (c 0.48, MeOH) (60% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 10.4 min (minor), 15.1 min (major).
(R)-4k: white solid, mp: 181.9– 182.7 °C;1H NMR (500 MHz, Chloroform-d) δ 8.03–7.95 (m, 1H), 7.94–7.90 (m, 1H), 7.90–7.86 (m, 1H), 7.85–7.75 (m, 2H), 7.60 (d, J = 8.0 Hz, 1H), 7.57 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.54–7.48 (m, 2H), 7.25 (td, J = 7.5, 1.0 Hz, 1H), 7.04–6.93 (m, 2H), 6.30 (d, J = 2.7 Hz, 1H), 4.80 (s, 1H), 2.41 (s, 3H);13C NMR (125 MHz, Chloroform-d) δ 200.9, 139.7, 137.1, 137.0, 134.4, 132.2, 130.0, 129.3, 128.8, 128.5, 128.3, 127.8, 127.6, 123.6, 123.1, 122.7, 122.2, 120.0, 117.5, 111.2, 21.5; HRMS (ESI) m/z: [M + Na]+ calcd for C23H17NO2Na: 362.1157; found: 362.1153; [α]D25 = 74.6 (c 0.60, MeOH) (56% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 13.6 min (minor), 16.6 min (major).
(R)-4l: yellow solid, mp: 101.0–102.0 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.98–7.91 (m, 1H), 7.91–7.87 (m, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.82–7.73 (m, 2H), 7.60–7.52 (m, 2H), 7.52–7.44 (m, 2H), 7.25 (td, J = 7.5, 1.0 Hz, 1H), 6.77 (dd, J = 9.0, 2.5 Hz, 1H), 6.65 (d, J = 2.5 Hz, 1H), 6.23 (d, J = 2.5 Hz, 1H), 4.76 (s, 1H), 3.75 (s, 3H); 13C NMR (126 MHz, Chloroform-d) δ 200.9, 156.5, 139.6, 137.5, 137.0, 134.5, 130.0, 129.3, 128.7, 128.6, 128.3, 127.8, 127.6, 123.6, 123.2, 122.7, 121.0, 119.6, 117.7, 110.3, 94.8, 55.5; HRMS (ESI) m/z: [M + Na]+ calcd for C23H17NO3Na: 378.1106; found: 378.1111; [α]D25 = 53.4 (c 0.50, MeOH) (54% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 17.2 min (minor), 24.6 min (major).
(R)-4o: yellow solid, mp: 140.6–141.7 °C; 1H NMR (500 MHz, Chloroform-d) δ 7.98–7.93 (m, 1H), 7.92–7.88 (m, 1H), 7.87 (dd, J = 8.0, 1.0 Hz, 1H), 7.81–7.72 (m, 2H), 7.59–7.53 (m, 2H), 7.53–7.46 (m, 2H), 7.25 (td, J = 7.5, 1.0 Hz, 1H), 7.03 (dd, J = 8.0, 7.0 Hz, 1H), 6.94 (dt, J = 7.0, 1.0 Hz, 1H), 6.37 (dd, J = 6.0, 2.5 Hz, 1H), 4.79 (s, 1H), 2.32 (s, 3H);13C NMR (125 MHz, Chloroform-d) δ 200.8, 139.7, 137.0, 136.2, 134.5, 130.0, 129.4, 128.8, 128.6, 128.4, 127.8, 127.7, 124.8, 123.9, 123.7, 123.0, 122.7, 120.6, 120.4, 118.3, 118.1, 16.4; HRMS (ESI) m/z: [M + Na]+ calcd for C23H17NO2Nal: 362.1157; found: 362.1152; [α]D25 = 49.1 (c 0.53, MeOH) (52% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 12.9 min (minor), 17.8 min (major).
(R)-4p: yellow solid, mp: 104.5–105.4 °C; 1H NMR (500 MHz, Chloroform-d) δ 8.12 (s, 1H), 8.03–7.97 (m, 1H), 7.95–7.91 (m, 1H), 7.91–7.86 (m, 1H), 7.82 (dd, J = 7.5, 1.5 Hz, 1H), 7.58 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.55–7.49 (m, 2H), 7.32 (dt, J = 8.0, 1.0 Hz, 1H), 7.27 (td, J = 7.5, 1.0 Hz,1H), 7.05 (t, J = 8.0 Hz, 1H), 6.61 (dd, J = 8.0, 0.5 Hz, 1H), 6.38 (d, J = 2.5 Hz, 1H), 4.81 (s, 1H), 3.88 (s, 3H);13C NMR (125 MHz, Chloroform-d) δ 200.8, 146.1, 139.7, 137.0, 134.46, 130.0, 129.4, 128.8, 128.6, 128.4, 127.8, 127.7, 127.3, 126.6, 123.7, 122.8, 120.9, 118.2, 113.0, 102.2, 55.2; HRMS (ESI) m/z: [M + Na]+ calcd for C23H17NO3Na: 378.1106; found: 378.1103; [α]D25 = 31.4 (c 0.55, MeOH) (56% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 24.9 min (minor), 28.1 min (major).
(R)-4q: orange solid, mp: 118.9–120.0 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.97 (d, J = 3.0 Hz, 1H), 8.07 (dd, J = 8.5, 4.5 Hz, 2H), 7.97 (d, J = 2.0 Hz, 1H), 7.81 (dd, J = 8.5, 2.5 Hz, 1H), 7.74 (dd, J = 8.5, 2.0 Hz, 1H), 7.70 (d, J = 2.0 Hz, 1H), 7.66–7.62 (m, 1H), 7.29 (dt, J = 8.0, 1.0 Hz, 1H), 7.05 (ddd, J = 8.0, 7.0, 1.5 Hz, 1H), 6.98 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H),6.53 (s, 1H), 6.39 (d, J = 2.5 Hz, 1H);13C NMR (125 MHz, DMSO-d6) δ 196.2, 143.9, 136.8, 136.6, 134.2, 131.4, 130.8, 130.2, 129.2, 128.4, 126.4, 126.0, 125.0, 124.6, 122.9, 121.8, 121.5, 120.6, 119.2, 114.7, 111.8, 77.5; HRMS (ESI) m/z: [M + Na]+ calcd for C22H13Br2NO2Na: 503.9211; found: 503.9215; [α]D25 = 58.0 (c 0.62, MeOH) (68% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 85:15, 1.0 mL/min, 254 nm), tR = 13.6 min (major), 16.2 min (minor).
(R)-4r: orange solid, mp: 103.2–104.2 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.83 (d, J = 3.0 Hz, 1H), 8.06 (dd, J = 8.5, 3.5 Hz, 2H), 7.95 (d, J = 2.0 Hz, 1H), 7.81 (dd, J = 8.5, 2.5 Hz, 1H), 7.73 (dd, J = 8.5, 2.0 Hz, 1H), 7.71 (d, J = 2.0 Hz, 1H), 7.47 (dd, J = 2.0, 1.0 Hz, 1H), 7.17 (dd, J = 8.5, 1.0 Hz, 1H), 6.88 (dd, J = 8.5, 1.5 Hz, 1H), 6.48 (s, 1H), 6.29 (d, J = 2.5 Hz, 1H), 2.35 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 196.0, 144.0, 136.8, 135.0, 134.2, 131.4, 131.0, 130.8, 130.3, 129.2, 128.4, 127.6, 126.9, 126.3, 126.0, 125.3, 124.6, 123.2, 122.9, 121.8, 120.2, 114.2, 111.5, 77.6, 21.4; HRMS (ESI) m/z: [M + Na]+ calcd for C23H15Br2NO2Na: 517.9367; found: 517.9361; [α]D25 = 80.4 (c 0.59, MeOH) (63% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 93:7, 1.0 mL/min, 254 nm), tR = 30.2 min (major), 33.1 min (minor).
(R)-4s: orange solid, mp: 101.5–102.7 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.85 (d, J = 3.0 Hz, 1H), 8.06 (dd, J = 8.5, 5.0 Hz, 2H), 7.98 (d, J = 2.0 Hz, 1H), 7.81 (dd, J = 8.5, 2.5 Hz, 1H), 7.76–7.71 (m, 2H), 7.18 (d, J = 9.0 Hz, 1H), 7.04 (d, J = 2.5 Hz, 1H), 6.70 (dd, J = 9.0, 2.5 Hz, 1H), 6.51 (s, 1H), 6.38 (d, J = 3.0 Hz, 1H), 3.72 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 195.9, 153.2, 143.8, 136.8, 134.2, 131.7, 131.4, 130.9, 130.3, 129.2, 128.4, 126.4, 126.0, 125.4, 125.3, 122.9, 121.9, 114.1, 112.5, 111.6, 102.3, 77.5, 55.2; HRMS (ESI) m/z: [M + Na]+ calcd for C23H15Br2NO3Na: 533.9316; found: 533.9319; [α]D25 = 23.4 (c 0.46, MeOH) (94% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 85:15, 1.0 mL/min, 254 nm), tR = 17.3 min (major), 33.7 min (minor).
(R)-4t: orange solid, mp: 194.1–195.2 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.07 (d, J = 2.5 Hz, 1H), 8.25 (d, J = 8.5 Hz, 1H), 8.13–8.06 (m, 3H), 7.96 (dd, J = 12.0, 2.5 Hz, 2H), 7.84 (dd, J = 8.5, 2.5 Hz, 1H), 7.75 (dd, J = 8.5, 2.0 Hz, 1H), 7.70 (d, J = 2.5 Hz, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.38–7.32 (m, 1H), 7.02 (dd, J = 8.5, 2.0 Hz, 1H), 6.58 (s, 1H), 6.42 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 196.1, 176.8, 137.4, 137.0, 136.9, 134.2, 133.5, 133.0, 131.5, 131.0, 130.5, 130.2, 129.2, 128.3, 126.8, 126.3, 126.0, 125.6, 123.7, 122.9, 122.7, 121.9, 121.8, 119.5, 115.1, 111.4, 77.3; HRMS (ESI) m/z: [M + Na]+ calcd for C22H12Br2ClNO2Na: 537.8821; found: 537.8826; [α]D25 = 73.1 (c 0.57, MeOH) (60% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 90:10, 1.0 mL/min, 254 nm), tR = 17.8 min (minor), 20.1 min (major).
(R)-4u: orange solid, mp: 71.9–72.8 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.74 (d, J = 2.5 Hz, 1H), 8.05 (dd, J = 8.5, 2.5 Hz, 2H), 7.96 (d, J = 2.0 Hz, 1H), 7.81 (dd, J = 8.5, 2.5 Hz, 1H), 7.72 (dd, J = 8.5, 2.0 Hz, 1H), 7.69 (d, J = 2.5 Hz, 1H), 7.49 (d, J = 9.0 Hz, 1H), 6.77 (d, J = 2.5 Hz, 1H), 6.65 (dd, J = 9.0, 2.5 Hz, 1H), 6.49 (s, 1H), 6.23 (d, J = 2.5 Hz, 1H), 3.71 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 196.3, 155.7, 143.9, 137.4, 136.8, 134.3, 131.4, 130.8, 130.2, 129.2, 128.4, 126.3, 126.0, 123.4, 122.9, 121.8, 121.2, 119.4, 114.8, 109.5, 94.8, 77.6, 55.1; HRMS (ESI) m/z: [M + Na]+ calcd for C23H15Br2NO3Na: 533.9316; found: 533.9312; [α]D25 = 23.7 (c 0.49, MeOH) (52% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 85:15, 1.0 mL/min, 254 nm), tR = 17.8 min (minor), 19.9 min (major).
(R)-4v: orange solid, mp: 213.9–214.5 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.79 (d, J = 3.0 Hz, 1H), 8.43 (dd, J = 6.0, 2.0 Hz, 2H), 7.79–7.71 (m, 2H), 7.61–7.51 (m, 2H), 7.51–7.44 (m, 1H), 7.21–7.09 (m, 1H), 6.87 (dd, J = 8.5, 1.5 Hz, 1H), 6.36 (s, 1H), 6.27 (d, J = 2.5 Hz, 1H), 2.39 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 196.9, 141.3, 136.7, 135.0, 132.4, 132.1, 131.0, 129.8, 129.1, 128.6, 128.4, 127.4, 127.0, 126.6, 125.4, 124.5, 123.0, 122.1, 120.3, 114.6, 111.4, 77.5, 21.4; HRMS (ESI) m/z: [M + Na]+ calcd for C23H15Br2NO2Na: 517.9367; found: 517.9363; [α]D25 = 84.18 (c 0.55, MeOH) (60% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 90:10, 1.0 mL/min, 254 nm), tR = 22.7 min (minor), 25.0 min (major).
(R)-4w: yellow solid, mp: 244.2–245.1 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.80 (d, J = 3.0 Hz, 1H), 8.44 (dd, J = 5.0, 1.5 Hz, 2H), 7.80 (d, J = 8.5 Hz, 1H), 7.76–7.72 (m, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.54 (dd, J = 8.0, 1.5 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 7.03 (d, J = 2.5 Hz, 1H), 6.69 (dd, J = 9.0, 2.5 Hz, 1H), 6.38 (dd, J = 11.0, 2.5 Hz, 2H), 3.72 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 196.8, 153.2, 141.1, 136.7, 132.4, 132.1, 131.7, 131.0, 129.9, 129.1, 128.7, 128.4, 127.0, 126.6, 125.5, 125.2, 122.1, 114.6, 112.4, 111.4, 102.4, 77.3, 55.2; HRMS (ESI) m/z: [M + Na]+ calcd for C23H15Br2NO3Na: 533.9316; found: 533.9311; [α]D25 = 581.8 (c 0.61, MeOH) (97% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 85:15, 1.0 mL/min, 254 nm), tR = 19.5 min (minor), 23.2 min (major).
(R)-4x: yellow solid, mp: 216.3–217.4 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.04 (d, J = 2.5 Hz, 1H), 8.64 (d, J = 2.0 Hz, 2H), 8.44 (dd, J = 6.0, 2.0 Hz, 2H), 7.93 (d, J = 8.5 Hz, 2H), 7.78–7.72 (m, 4H), 7.68 (d, J = 8.5 Hz, 1H), 7.61–7.51 (m, 2H), 7.32 (d, J = 2.0 Hz, 1H), 7.01 (dd, J = 8.5, 2.0 Hz, 1H), 6.48 (s, 1H), 6.41 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 197.0, 177.7, 140.9, 137.0, 136.7, 135.9, 132.7, 132.5, 132.2, 130.9, 130.8, 130.7, 130.0, 129.8, 129.1, 128.9, 128.1, 127.7, 127.1, 126.7, 126.3, 125.6, 123.8, 122.2, 122.1, 119.5, 115.6, 111.4, 77.2; HRMS (ESI) m/z: [M + Na]+ calcd for C22H12Br2ClNO2Na: 537.8821; found: 537.8825; [α]D25 =82.0 (c 0.49, MeOH) (58% ee); HPLC (Chiralpak AD-H, hexane:iPrOH = 70:30, 1.0 mL/min, 254 nm), tR = 15.9 min (major), 24.3 min (minor).
(R)-4y: yellow solid, mp: 122.7–123.3 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.71 (d, J = 2.5 Hz, 1H), 8.43 (dd, J = 8.0, 2.0 Hz, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.73 (dd, J = 8.5, 2.0 Hz, 1H), 7.58–7.53 (m, 2H), 7.52–7.48 (m, 1H), 6.76 (dd, J = 2.5, 0.5 Hz, 1H), 6.63 (dd, J = 9.0, 2.5 Hz, 1H), 6.37 (s, 1H), 6.21 (d, J = 2.5 Hz, 1H), 3.70 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 197.1, 155.6, 141.2, 137.4, 136.7, 132.4, 132.1, 130.9, 129.8, 129.0, 128.7, 128.4, 127.0, 126.6, 123.3, 122.0, 121.2, 119.4, 115.3, 109.3, 94.7, 77.4, 55.1; HRMS (ESI) m/z: [M + Na]+ calcd for C23H15Br2NO3Na: 533.9316; found: 533.9312; [α]D25 = 82.8 (c 0.52, MeOH) (56% ee); HPLC (Chiralpak AS-H, hexane:iPrOH = 80:20, 1.0 mL/min, 254 nm), tR = 17.4 min (minor), 26.4 min (major).
(S)-4g: yellow solid, mp: 200.5–201.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.83–10.69 (m, 1H), 8.11 (dd, J = 8.8, 4.0 Hz, 2H), 7.93–7.82 (m, 1H), 7.68 (d, J = 7.7 Hz, 1H), 7.63 (t, J = 7.6 Hz, 1H), 7.58–7.47 (m, 2H), 7.32 (t, J = 7.5 Hz, 1H), 7.15 (d, J = 8.8 Hz, 1H), 7.06 (d, J = 2.5 Hz, 1H), 6.67 (dd, J = 8.9, 2.5 Hz, 1H), 6.31 (d, J = 2.7 Hz, 1H), 6.24 (s, 1H), 3.70 (s, 3H); 13C NMR (101 MHz, DMSO) δ 198.6, 153.0, 141.5, 136.1, 134.2, 131.6, 129.8, 129.4, 129.0, 128.4, 128.4, 127.6, 127.0, 125.6, 125.0, 123.8, 123.2, 115.6, 112.2, 111.3, 102.5, 776, 55.2; HRMS (ESI) m/z: [M + Na]+ calcd for C23H17NO3Na: 378.1106; found: 378.1102; [α]D25 = 34.0 (c 0.50, MeOH) (91% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 75:25, 1.0 mL/min, 254 nm), tR = 13.9 min (major), 21.3 min (minor).
(S)-4w: yellow solid, mp: 243.8–244.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.86 (d, J = 2.9 Hz, 1H), 8.46 (dd, J = 3.4, 1.8 Hz, 2H), 7.82 (d, J = 8.4 Hz, 1H), 7.75 (dd, J = 8.4, 1.9 Hz, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.54 (dd, J = 8.2, 1.8 Hz, 1H), 7.18 (d, J = 8.8 Hz, 1H), 7.07 (d, J = 2.4 Hz, 1H), 6.70 (dd, J = 8.8, 2.5 Hz, 1H), 6.43 (s, 1H), 6.38 (d, J = 2.6 Hz, 1H); 13C NMR (101 MHz, DMF) δ 196.8, 153.2, 141.1, 136.7, 132.4, 132.1, 131.7, 131.0, 129.9, 129.1, 128.7, 128.4, 127.0, 126.6, 125.5, 125.2, 122.1, 114.6, 112.4, 111.5, 102.4, 77.4, 55.2; HRMS (ESI) m/z: [M + Na]+ calcd for C23H15Br2NO3Na: 533.9316; found: 533.9313; [α]D25 = 536.9 (c 0.51, MeOH) (91% ee); HPLC (Chiralcel OD-H, hexane:iPrOH = 85:15, 1.0 mL/min, 254 nm), tR = 19.0 min (major), 23.3 min (minor).
4. Conclusions
In summary, we have described the first enantioselective Friedel–Crafts reaction of phenanthrenequinones and indoles organocatalyzed by (S,S)-squaramide to synthesize 10-hydroxy-10-(1H-indol-3-yl)-9-(10H)-phenanthrene derivatives in good yield with up to 97% enantioselectivity. Moreover, we used our optimized conditions to expand upon the substrate scope of this reaction. In addition, two pairs of synthesized enantiomers were subjected to evaluation of their cytotoxic properties against different human cancer cell lines and one human umbilical vein endothelial cell. Compared to doxorubicin, (S)-4g was found to be better not only for activity but also for safety. Therefore, 10-hydroxy-10-(1H-indol-3-yl)-9-(10H)-phenanthrene derivatives might be developed as antitumor candidate compounds after further research.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules30010172/s1, Page S3–S24: 1H NMR and 13C NMR spectra; Page S25–S46: HPLC trace; Page S47–S48: X-Ray crystal data of compound 4w; Page S49–S50: In vitro cytotoxicity assay.
Author Contributions
Y.J. (Yan Jin) and Y.S. and are co-first authors; they contributed equally to this work. They performed the experiments and activity test, cultivated single crystals, acquired and analyzed the original data; Y.Y. performed the experiments; J.Z. and M.Z. conducted instrumental analysis; L.W. supervised the experiment, analyzed and checked all the data; Y.J. (Ying Jin) designed the research plan and provided funding supporting, wrote the draft and revised manuscript. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This research was funded by the Natural Science Foundation of Jilin province (No. 20230101226JC), the Department of Education of Jilin province (No. JJKH20240595KJ), Postgraduate Innovation Program Project of Jilin Medical University (2023zyc02) and Students’ Program for Innovation and Entrepreneurship Training (No. S202413706026).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Lee H.M., Kim H.D., Kim J.M., Kim J.K., Kim S.K. Molecular modeling study on the enantioselective binding of S-and R-ofloxacin to various dna sequences. J. Biomol. Struct. Dyn. 2007;25:231–241. doi: 10.1080/07391102.2007.10507172. [DOI] [PubMed] [Google Scholar]
- 2.Lerchen A., Gandhamsetty N., Farrar E.H.E., Winter N., Platzek J., Grayson M.N., Aggarwal V.K. Enantioselective total synthesis of (−)-Finerenone using asymmetric transfer hydrogenation. Angew. Chem. Int. Ed. 2020;132:23307–23311. doi: 10.1002/ange.202011256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalal J., Mohan J.C., Iyengar S.S., Hiremath J., Sathyamurthy I., Bansal S., Kahali D., Dasbiswas A. S-Amlodipine: An isomer with difference time to shift from racemic amlodipine. Int. J. Hypertens. 2018;2018:1–14. doi: 10.1155/2018/8681792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu D.L., Pan Y.C., Chen J.H. Chemical constituents, pharmacologic properties, and clinical applications of Bletilla striata. Front. Pharmacol. 2019;10:1168. doi: 10.3389/fphar.2019.01168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nano A., Dai J., Bailis J.M., Barton J.K. Rhodium complexes targeting DNA mismatches as a basis for new therapeutics in cancers deficient in mismatch repair. Biochemistry. 2021;60:2055–2063. doi: 10.1021/acs.biochem.1c00302. [DOI] [PubMed] [Google Scholar]
- 6.Wang R.B., Wei M., Wang X.R., Chen Y.S., Xiong Y.S., Chen J.X., Tan Y.H., Liao X.W., Wang J.T. Synthesis of ruthenium polypyridine complexes with benzyloxyl groups and their antibacterial activities against Staphylococcus aureus. J. Inorg. Biochem. 2022;236:111954. doi: 10.1016/j.jinorgbio.2022.111954. [DOI] [PubMed] [Google Scholar]
- 7.Guédouar H., Aloui F., Beltifa A., Mansour H.B., Hassine B.B. Synthesis and characterization of phenanthrene derivatives with anticancer property against human colon and epithelial cancer cell lines. Comptes Rendus Chim. 2017;20:841–849. doi: 10.1016/j.crci.2017.03.008. [DOI] [Google Scholar]
- 8.Csupor D., Kurtán T., Vollár M., Kúsz N., Kövér K.E., Mándi A., Szűcs P., Marschall M., Tahaei S.A.S., Zupkó I., et al. Pigments of the moss paraleucobryum longifolium: Isolation and structure elucidation of prenyl-substituted 8, 8′-linked 9, 10-phenanthrenequinone dimers. J. Nat. Prod. 2020;83:268–276. doi: 10.1021/acs.jnatprod.9b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo J.J., Dai J., Peng X.L., Wang Q., Wang S.X., Lou X.D., Xia F., Zhao Z.J., Tang B.Z. 9, 10-Phenanthrenequinone: A promising kernel to develop multifunctional antitumor systems for efficient type I photodynamic and photothermal synergistic therapy. ACS Nano. 2021;15:20042–20055. doi: 10.1021/acsnano.1c07730. [DOI] [PubMed] [Google Scholar]
- 10.Huang J.J., Chen Y.F., Guo Y.F., Bao M., Hong K.M., Zhang Y.Q., Hu W.H., Lei J.P., Liu Y.Q., Xu X.F. Synthesis of dihydrofuran-3-one and 9, 10-phenanthrenequinone hybrid molecules and biological evaluation against colon cancer cells as selective Akt kinase inhibitors. Mol. Divers. 2023;27:845–855. doi: 10.1007/s11030-022-10458-w. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y.X., Hu P., Huang Z.Y., Wang J.Y., Song H.Y., Chen X., Lin X., Wu T.M., Tan X.D. Significant enhancement of the polarization holographic performance of photopolymeric materials by introducing graphene oxide. ACS Appl. Mater. Inter. 2021;13:27500–27512. doi: 10.1021/acsami.1c07390. [DOI] [PubMed] [Google Scholar]
- 12.Karthika P., Ganesan S., Kamalakannan S., Prakash M. Design and synthesis of the D−π–A-structured coadsorbents with the phenanthraquinone core and its application in dye-sensitized solar cells. J. Phys. Chem. C. 2020;124:9886–9899. doi: 10.1021/acs.jpcc.9b12042. [DOI] [Google Scholar]
- 13.Li J.H., Hu P., Zeng Z.Y., Jin J.C., Wu J.H., Chen X., Liu J., Li Q.D., Chen M.Y., Zhang Z.Y., et al. Phenanthraquinone-doped polymethyl methacrylate photopolymer for holographic recording. Molecules. 2022;27:6283. doi: 10.3390/molecules27196283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X.H., Zhong H., Huang W., Sui Y., Sa R.J., Chen W.T., Zhou G.Y., Li X.D., Li D.F., Wen M.C., et al. The construction of conjugated organic polymers containing phenanthrenequinone redox centers for visible-light-driven H2O2 production from H2O and O2 without any additives. Chem. Eng. J. 2023;454:139929. doi: 10.1016/j.cej.2022.139929. [DOI] [Google Scholar]
- 15.Shao P.L., Chen X.Y., Sun L.H., Song Y. Enantioselective [4+2] cycloaddition of ketenes and 9,10-phenanthrenequinone catalyzed by N-heterocyclic carbenes. Tetrahedron. Lett. 2010;51:2316–2318. doi: 10.1016/j.tetlet.2010.02.122. [DOI] [Google Scholar]
- 16.Jiang Y., Fu J.H., Li T.Z., Sha F., Wu X.Y. Enantioselective direct vinylogous aldol-cyclization cascade reaction between β, γ-unsaturated amides and o-quinones. Org. Biomol. Chem. 2016;14:6435–6441. doi: 10.1039/C6OB00893C. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y., Cui Y., Lu L.Y., Gong Y.F., Han W., Piao G.S. Natural indole-containing alkaloids and their antibacterial activities. Arch. Pharm. 2020;353:2000120. doi: 10.1002/ardp.202000120. [DOI] [PubMed] [Google Scholar]
- 18.Song J.T., Zhang B., Li M., Zhang J.B. The current scenario of naturally occurring indole alkaloids with anticancer potential. Fitoterapia. 2023;165:105430. doi: 10.1016/j.fitote.2023.105430. [DOI] [PubMed] [Google Scholar]
- 19.Liu X.Y., Qin Y. Indole alkaloid synthesis facilitated by photoredox catalytic radical cascade reactions. Acc. Chem. Res. 2019;52:1877–1891. doi: 10.1021/acs.accounts.9b00246. [DOI] [PubMed] [Google Scholar]
- 20.El-Sharief A.M.S., Ammar Y.A., Belal A., El-Sharief M.A.M.S., Mohamed Y.A., Mehany A.B.M., Ali G.A.M.E., Ragab A. Design, synthesis, molecular docking and biological activity evaluation of some novel indole derivatives as potent anticancer active agents and apoptosis inducers. Bioorg. Chem. 2019;85:399–412. doi: 10.1016/j.bioorg.2019.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Reddy P., Guthridge K., Vassiliadis S., Hemsworth J., Hettiarachchige I., Spangenberg G., Rochfort S. Tremorgenic mycotoxins: Structure diversity and biological activity. Toxins. 2019;11:302. doi: 10.3390/toxins11050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gnanamani E., Yan X., Zare R.N. Chemoselective N-alkylation of indoles in aqueous microdroplets. Angew. Chem. Int. Ed. 2020;59:3069–3072. doi: 10.1002/anie.201913069. [DOI] [PubMed] [Google Scholar]
- 23.Sun P., Huang Y.Q., Chen S.H., Ma X.N., Yang Z.K., Wu J. Indole derivatives as agrochemicals: An overview. Chem. Soc. Rev. 2024;35:109005. doi: 10.1016/j.cclet.2023.109005. [DOI] [Google Scholar]
- 24.Cruz F.A., Zhu Y.M., Tercenio Q.D., Shen Z.M., Dong V.M. Alkyne hydroheteroarylation: Enantioselective coupling of indoles and alkynes via Rh-hydride catalysis. J. Am. Chem. Soc. 2017;139:10641–10644. doi: 10.1021/jacs.7b05893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang B.M., Ng X.Q., Zhao Y. Enantioselective synthesis of indoles through catalytic indolization. Chem. Catal. 2022;2:3048–3076. doi: 10.1016/j.checat.2022.10.004. [DOI] [Google Scholar]
- 26.Wang J.Y., Gao C.H., Ma C., Wu X.Y., Ni S.F., Tian W., Shi F. Design and catalytic asymmetric synthesis of furan-indole compounds bearing both axial and central chirality. Angew. Chem. Int. Ed. 2024;63:e202316454. doi: 10.1002/anie.202316454. [DOI] [PubMed] [Google Scholar]
- 27.Qi L.W., Mao J.H., Zhang J., Tan B. Organocatalytic asymmetric arylation of indoles enabled by azo groups. Nat. Chem. 2018;10:58–64. doi: 10.1038/nchem.2866. [DOI] [PubMed] [Google Scholar]
- 28.Ibáñez I., Kaneko M., Kamei Y., Tsutsumi R., Yamanaka M., Akiyama T. Enantioselective Friedel–Crafts alkylation reaction of indoles with α-trifluoromethylated β-nitrostyrenes catalyzed by chiral binol metal phosphate. ACS Catal. 2019;9:6903–6909. doi: 10.1021/acscatal.9b01811. [DOI] [Google Scholar]
- 29.Yu S.B., Cai Q.H., Wang C., Hou J.Q., Liang J.M., Jiao Z.J., Yao C., Li Y.M. Enantioselective Friedel–Crafts alkylation of indoles with β, γ-unsaturated α-ketoesters catalyzed by new copper (I) catalysts. J. Org. Chem. 2023;88:3046–3053. doi: 10.1021/acs.joc.2c02749. [DOI] [PubMed] [Google Scholar]
- 30.Fei Y.N., Hou J., Song H.Y., Yang M.L., Lei P., Ge X.Y., Wei H.B., Xu Y.Z., Xie W.Q. Catalytic asymmetric Friedel–Crafts alkylation of indole via in situ generated indol-2-one. Eur. J. Org. Chem. 2023;26:e202300122. doi: 10.1002/ejoc.202300122. [DOI] [Google Scholar]
- 31.Huang Z.L., Xu Y.Y., Lin W., Qian R., Zhang W., Li X. Asymmetric synthesis of acyclic N–N axially chiral indole compounds via catalytic N-acylation reaction. Org. Chem. Front. 2024;11:5437–5442. doi: 10.1039/D4QO01056F. [DOI] [Google Scholar]
- 32.You Y., Gan G.Y., Duan S.Y., Zhang Y.P., Li Q., Wang Z.H., Zhao J.Q., Liu X.L., Yuan W.C. Enantioselective synthesis of tryptanthrin derivatives enabled by an asymmetric aza-Friedel–Crafts reaction. Org. Chem. Front. 2023;10:5421–5427. doi: 10.1039/D3QO00961K. [DOI] [Google Scholar]
- 33.Cheng Y.S., Chan S.H., Rao G.A., Gurubrahamam R., Chen K. Enantioselective aza-Friedel-Crafts reaction of heteroarenes with in situ generated isoxazolium ions via chiral phosphoric acid catalysis. Adv. Synth. Catal. 2021;363:3502–3506. doi: 10.1002/adsc.202100408. [DOI] [Google Scholar]
- 34.Miyazaki Y., Zhou B., Tsuji H., Kawatsura M. Nickel-catalyzed asymmetric Friedel–Crafts propargylation of 3-substituted indoles with propargylic carbonates bearing an internal alkyne group. Org. Lett. 2020;22:2049–2053. doi: 10.1021/acs.orglett.0c00465. [DOI] [PubMed] [Google Scholar]
- 35.Shen M.L., Shen Y., Wang P.S. Merging visible-light photoredox and chiral phosphate catalysis for asymmetric Friedel–Crafts reaction with in situ generation of N-acyl imines. Org. Lett. 2019;21:2993–2997. doi: 10.1021/acs.orglett.9b00442. [DOI] [PubMed] [Google Scholar]
- 36.Kim Y., Lee J., Jung J.Y., Kim S.G. Chiral brønsted acid-catalyzed Friedel–Crafts reaction of 3-indolylsulfamidates with indoles: Synthesis of enantioenriched bisindolylmethane sulfamates. Tetrahedronl. Lett. 2019;60:1625–1630. doi: 10.1016/j.tetlet.2019.05.003. [DOI] [Google Scholar]
- 37.Yang Z.T., Yang W.L., Chen L., Sun H., Deng W.P. Organocatalytic enantioselective aza-Friedel-Crafts reactions of pyrazolinone ketimines with hydroxyindoles and electron-rich phenols. Adv. Synth. Catal. 2018;360:2049–2054. doi: 10.1002/adsc.201800181. [DOI] [Google Scholar]
- 38.Nakamura S., Furukawa T., Hatanaka T., Funahashi Y. Enantioselective aza-Friedel–Crafts reaction of cyclic ketimines with indoles using chiral imidazoline–phosphoric acid catalysts. Chem. Comm. 2018;54:3811–3814. doi: 10.1039/C8CC00594J. [DOI] [PubMed] [Google Scholar]
- 39.Nakashima K., Hanamura S., Imamura A., Matsushima Y., Hirashima S., Miura T. Asymmetric Friedel−Crafts alkylation of indoles with α, β-unsaturated trifluoromethyl ketones using a squaramide organocatalyst. Asian J. Org. Chem. 2022;11:e202200403. [Google Scholar]
- 40.Wang W., Xiong W.H., Wang J.P., Wang Q.A., Yang W. Brønsted acid-catalyzed asymmetric Friedel–Crafts alkylation of indoles with benzothiazole-bearing trifluoromethyl ketone hydrates. J. Org. Chem. 2020;85:4398–4407. doi: 10.1021/acs.joc.0c00116. [DOI] [PubMed] [Google Scholar]
- 41.Li J., Wei Z.L., Cao J.G., Liang D.P., Lin Y.J., Duan H.F. Aymmetric aza-Friedel–Crafts reaction of isatin-derived ketimines with indoles catalyzed by a chiral phase-transfer catalyst. J. Org. Chem. 2022;87:2532–2542. doi: 10.1021/acs.joc.1c02477. [DOI] [PubMed] [Google Scholar]
- 42.Ahmad T., Khan S., Ullah N. Recent advances in the catalytic asymmetric Friedel–Crafts reactions of indoles. ACS Omega. 2022;7:35446–35485. doi: 10.1021/acsomega.2c05022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu X., Zheng C., You S.L. Chiral brønsted acid-catalyzed intramolecular asymmetric dearomatization reaction of indoles with cyclobutanones via cascade Friedel–Crafts/Semipinacol rearrangement. J. Am. Chem. Soc. 2024;146:25878–25887. doi: 10.1021/jacs.4c09814. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J.W., Wu H., Zhang W.X., Wang L.M., Jin Y. Cinchona alkaloid silyl ether derivative catalyzed enantioselective Friedel-Crafts reaction of indoles with isatins. Chin. J. Org. Chem. 2021;41:1187–1192. doi: 10.6023/cjoc202009023. [DOI] [Google Scholar]
- 45.Wang Y.M., Wen Z., Chen X.M., Du D.M., Matsuura T., Meng J.B. Research on photochemical and thermochemical reactions between indole and quinones in the absence of solvent. J. Heterocyclic. Chem. 1998;35:313–316. doi: 10.1002/jhet.5570350209. [DOI] [Google Scholar]
- 46.CCDC 2375435 (4w) Contains the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge. [(accessed on 3 August 2024)]. Available online: https://www.ccdc.cam.ac.uk/structures/
- 47.Deng J., Zhang S.L., Ding P., Jiang H.L., Wang W., Li J. Facile creation of 3-indolyl-3-hydroxy-2-oxindoles by an organocatalytic enantioselective Friedel–Crafts reaction of indoles with isatins. Adv. Synth. Catal. 2010;352:833–838. doi: 10.1002/adsc.200900851. [DOI] [Google Scholar]
- 48.Zhang T.Y., He W., Zhao X.Y., Jin Y. Asymmetric oxaziridination catalyzed by cinchona alkaloid derivatives containing sulfide. Tetrahedron. 2013;69:7416–7422. doi: 10.1016/j.tet.2013.06.061. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are contained within the article and Supplementary Materials.