

Abstract
Anti-Markovnikov additions to alkenes have been a longstanding goal of catalysis, and anti-Markovnikov addition of arenes to alkenes would produce alkylarenes that are distinct from those formed by acid-catalysed processes. Existing hydroarylations are either directed or occur with low reactivity and low regioselectivity for the n-alkylarene. Herein, we report the first undirected hydroarylation of unactivated alkenes with unactivated arenes that occurs with high regioselectivity for the anti-Markovnikov product. The reaction occurs with a nickel catalyst ligated by a highly sterically hindered N-heterocyclic carbene. Catalytically relevant arene- and alkene-bound nickel complexes have been characterized, and the rate-limiting step was shown to be reductive elimination to form the C–C bond. Density functional theory calculations, combined with second-generation absolutely localized molecular orbital energy decomposition analysis, suggest that the difference in activity between catalysts containing large and small carbenes results more from stabilizing intramolecular non-covalent interactions in the secondary coordination sphere than from steric hindrance.
Linear alkylbenzenes are fundamental precursors to a variety of industrially relevant surfactants, detergents, plastics and fine chemicals1 with a global market value of over US$7.75 billion (ref.2). Although termed ‘linear alkylbenzenes’, the commercial process for their formation leads to mixtures of isomeric branched alkylbenzenes from the rearrangement of carbocationic intermediates in the alkylation step3. Because branched alkylarenes are more resistant to biodegradation4, their use as surfactants and detergents has led to the extensive pollution of rivers, lakes and oceans5,6.
Anti-Markovnikov transition-metal catalysed hydroarylation could lead to n-alkylbenzenes, but the reaction of unactivated alkenes with simple unactivated arenes lacking a directing group7–10 has not been reported with high selectivity for the linear product (Fig. 1a). The iridium, ruthenium and platinum systems reported by Periana11–15, Gunnoe16–18 and Goldberg19,20 and their co-workers all catalyse the reaction of benzene (1) with propylene (2) to provide alkylarenes 3 and 4 with moderate activity, but give nearly 1:1 ratios of the constitutional isomers (Fig. 1b). Gunnoe and co-workers recently reported a two-step synthesis of n-alkylarenes by rhodiumcatalysed oxidative alkenylation and hydrogenation, but the process involves two steps and a copper salt as the co-catalyst21,22.
Fig. 1 |. Transition metal-catalysed hydroarylation of unactivated alkenes with unactivated arenes.

a, Linear and branched isomeric products formed by undirected transition-metal-catalysed hydroarylation. b, State-of-the-art catalytic systems for the hydroarylation reaction of benzene with propylene with the turnover numbers (TONs) and linear/branched (l/b) ratios indicated. c, The linear-selective hydroarylation reaction described in this report.
The regioselectivity of published undirected hydroarylations is controlled by the regioselectivity of insertion of the alkene into the metal–aryl bond (1,2 vs 2,1 addition)13,17,23–25. By this mechanism, high selectivity for the linear product requires that insertion form a branched alkylmetal intermediate, but such an intermediate is typically less stable than the linear alkylmetal isomer. Moreover, the rates of these processes are likely limited by the counterbalancing electronic effects on the oxidative addition of an aryl C–H bond, which is often faster to more electron-rich metal centres than electron-poor metal centres24,26–31, and migratory insertion, which is often faster into metal–ligand bonds of more electron-poor metal centres than of more electron-rich metal centres32.
A catalytic cycle that operates by an alternative mechanism could enable higher levels of regioselectivity for the linear product and bypass the counterbalancing electronic effects on multiple steps. Previous research by our groups led to a nickel-catalysed, linear-selective hydroarylation with electron-deficient arenes33 and various electronically activated heteroarenes34, but the reactions of electron-neutral arenes, such as benzene or alkylarenes, occurred with low turnovers. Thus, an analysis of the factors controlling activity was needed to achieve the first addition of an unactivated arene to a terminal alkene in good yield with high linear selectivity. In the most favourable case, this reaction would occur with a catalyst based on an earth-abundant, non-precious metal.
We report here undirected hydroarylations of electron-neutral arenes with unconjugated terminal alkenes that occur in good yields with exceptionally high linear/branched selectivity (>50:1 in most cases, Fig. 1c) catalysed by a nickel complex containing an outsized N-heterocyclic carbene (NHC). The reaction occurs via the formation of an alkylnickel–aryl intermediate by an unusual ligand-to-ligand hydrogen transfer (LLHT) to the coordinated alkene33,35 and rate-determining reductive elimination. The results of computational studies imply that the attractive interactions of the large NHC ligand, rather than steric hindrance, lead to the high activity of the nickel–carbene complex.
Results and discussion
Reaction development.
Initial studies33 showed that the combination of [Ni(COD)2] (COD, cycloocta-1,5-diene) and the common NHC ligand IPr (L1; see Fig. 2a) led to the addition of benzene (1) to 1-decene (5) with a high linear/branched ratio of 19:1, but with a turnover of less than one. To increase the TONs, we evaluated the hydroarylation of 1-decene (5) with benzene (1) catalysed by [L–Ni(η6-C6H6)] complexes as catalyst precursors (Fig. 2a)36. The yields were more reproducible, particularly at low catalyst loadings (vide infra), when catalytic amounts of NaH and Na(acac) were added. Although the reaction catalysed by [L1–Ni(η6-C6H6)] provided only trace amounts of hydroarylation product 6, the reactions catalysed by the complex of the more sterically encumbered ligand IPr* (L2)37 and its more σ-donating analogue IPr*OMe (L3) occurred with several turnovers38. Further altering the substitution of the aromatic rings on the sidearms of the NHC with 3,5-dimethylphenyl groups (L4, m-XylIPr*OMe)39 or with 3,5-diethylphenyl groups (L5, DepIPr*OMe) led to catalysts that gave much higher yields of the hydroarylation product (56% for L4 and 54% for L5 after 24 h). Alkene isomerization during the reaction by an independent process33 decreased the concentration of the terminal alkene and reaction rates over time (Supplementary Figs. 4 and 5), but the catalysts were stable and 75 and 84% isolated yields of 6 were obtained, respectively, with L4 or L5 as ligand after 5 days (Table 1). The linear/branched selectivity when using L4 or L5 was unprecedently high at >50:1 (see above, Fig. 2a). To directly compare the relative rates of catalysts containing different NHC ligands, we measured the initial rates of the hydroarylation reaction catalysed by [L–Ni(η6-C6H6)] (Supplementary Fig. 17). The initial rate of hydroarylation with L4 as ligand was found to be 23 times greater than the initial rate of hydroarylation with L1 as ligand and two times greater than the initial rate of hydroarylation with L3 as ligand.
Fig. 2 |. Reaction development and characterization of [L4–Ni(η6-C6H6)].
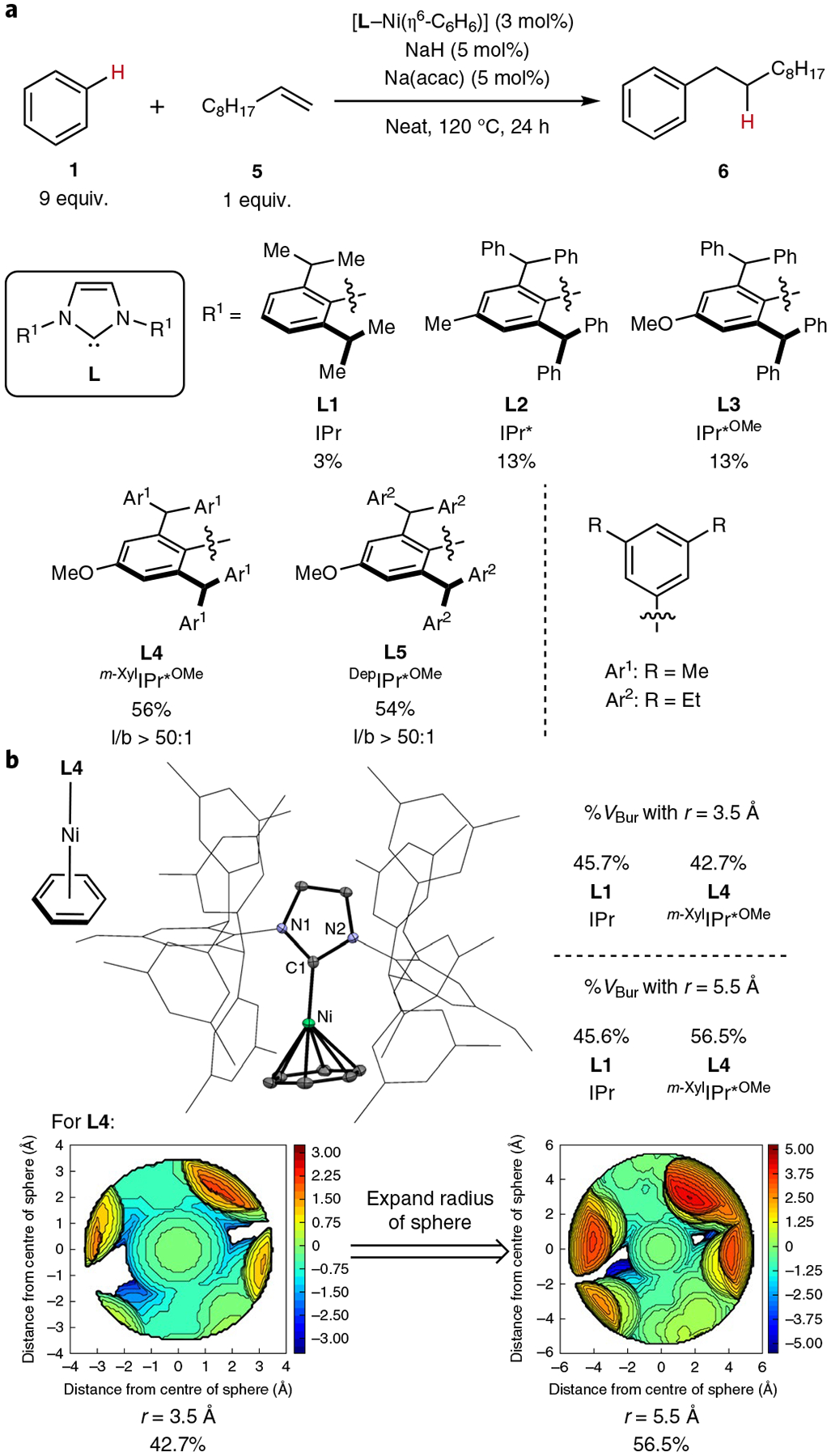
a, Identifying NhC ligands to achieve hydroarylation. Yields and linear/branched (l/b) ratios were determined by GC or NMr spectroscopic analysis of the crude reaction mixture. b, OrTEP diagram of [L4–Ni(η6-C6h6)] (thermal ellipsoids are shown at the 50% probability level), calculated %VBur values and steric map. An expanded radius of 5.5 Å for the %VBur calculation was necessary to account for the remote steric environment of L4. The origins of the steric maps depicted are centred 2.0 Å away from the carbene carbon atoms with the +z axis defined by the carbene carbons and the midpoints between the two backbone carbons, and the (x,z) planes are defined by the two backbone carbons and the carbene carbon atoms. Additional details of the calculations of the %VBur values can be found in the Supplementary Information. For clarity, all hydrogen atoms have been omitted from the OrTEP diagram, and NhC sidearms are represented in wire format.
Table 1 |.
Scope of the hydroarylation reaction

|
Determined by GC or NMr spectroscopic analysis of the crude reaction mixture.
18 equiv. of arene were added.
3 equiv. of arene were added.
Nah and Na(acac) were excluded.
Mesitylene was added as the reaction solvent.
Benzene underwent addition to a series of unactivated alkenes (Table 1) to give the product with a linear/branched selectivity >50:1 or with undetectable amounts of branched isomer in all cases. Because [L4–Ni(η6-C6H6)] and [L5–Ni(η6-C6H6)] catalyse in parallel the isomerization of alkenes, the reaction of benzene (1) with internal alkenes, such as trans-4-octene (7), also occurred to form the n-alkylarene products in good yields. Consistent with this observation, a mixture of cis- and trans-2-hexene (8) and cis- and trans-3-hexene (9) reacted to give the n-alkylarene product in 98% yield. Terminal alkenes bearing substituents at the α-position that inhibit or prevent isomerization reacted to full conversion within 24 h. For example, alkene 10 bearing a tertiary carbon at the α-position and alkene 11 bearing a quaternary carbon at the α-position reacted to give the resulting alkylarene products in >90% yield. Both protected primary alcohols (12) and vinyl siloxanes (13) were tolerated under the reaction conditions.
The reaction of tert-butylethylene (14) with benzene occurred in nearly quantitative yield after only 1 h under the standard conditions (Table 1, entry 1). The reaction with 3 mol% catalyst without added NaH and Na(acac) provided the product in nearly the same yield as that obtained with added NaH and Na(acac) (Table 1, entry 2). However, the basic additives increased the turnover numbers of reactions conducted with low catalyst loadings. With basic additives, the reaction with only 0.3 mol% catalyst formed the addition product in 85% yield (TON = 283, Table 1, entry 3), but without these additives, the reaction with 0.3 mol% catalyst formed the alkylarene in only 6% yield (Table 1, entry 4; for further details, see the Supplementary Information). The basic additives likely remove trace water, which affects reactions with low loadings.
The hydroarylation of unactivated alkenes also occurred with a variety of electron-neutral, electron-rich and electron-deficient arenes (Table 1). The reaction of alkene 14 with toluene provided n-alkylarene 15 in good yield as a mixture of m- and p-alkylarene isomers. Alkene 11 reacted with xylene isomers at the most sterically accessible position to give products 16 and 17 in moderate yields. Reactions with more electron-deficient fluoroarenes also proceeded in high yields (18–21). Finally, the intramolecular reaction of arene 22 containing a pendant alkene provided tetrahydronaphthalene 23 in 86% yield (by 1H NMR spectroscopy); no cyclized product from the potential intramolecular reaction to form a five-membered ring was observed.
Investigation of the reaction mechanism.
Having developed a method for the hydroarylation reaction between unactivated arenes and alkenes that occurs in good yields with high anti-Markovnikov regioselectivity, we sought to understand the origins of the high activity of the catalysts containing ligands L4 and L5. To do so, we determined experimentally the resting state of the catalyst with unhindered and hindered alkenes and the rate-determining step of the reaction, and we used a computational energy decomposition analysis40 to identify the intramolecular interactions leading to the high activity stemming from the large ligands.
Arene-bound NHC-ligated nickel complexes were prepared on the gram scale by the addition of the free carbene ligand to a Ni0 source in C6H6 as solvent under H2 pressure36,41. The solid-state structures of these complexes (Fig. 2b for [L4–Ni(η6-C6H6)]) illustrate the steric impact of the NHC ligand. In contrast to the symmetrical coordination of benzene in [L2–Ni(η6-C6H6)], the angle between the carbene carbon and the benzene ligand in [L4–Ni(η6-C6H6)] is significantly distorted from linearity (169.4°). This difference suggests that steric hindrance of the NHC ligand prevents a complete η6-interaction.
To quantify the steric properties of L4 further, the percent buried volume (%VBur)42,43 and steric map of L4 were calculated (Fig. 2b)44. With the standard parameters of Dorta et al.45 for the radius (r) of the sphere surrounding the metal centre of 3.5 Å, the proximal %VBur of L4 was smaller than that of L1 by 3%. However, with a radius of 5.5 Å to account for the remote steric environment of L4, the %VBur of L4 was greater than that of L1 by >10%. This difference is slightly greater than the difference between IPr and its N-(2,4,6-trimethylphenyl) analogue, known as IMes (8% difference in buried volume within 3.5 Å for l–AuCl)42, and correlates with the large difference in activity between the catalyst containing L1 and that containing L4 in the hydroarylation reaction. However, the large size of L4 cannot be solely responsible for the high reactivity of the catalyst containing L4 in alkene hydroarylation because the reactivities of complexes of L2 and L3 possessing similar steric properties (see the Supplementary Information for all %VBur values) are closer to that of the catalyst containing L1 than to that containing L4.
Monitoring the reaction catalysed by the complex containing L4 with a 13C-labelled carbene carbon (13CL4) by NMR spectroscopy revealed the resting states of the hydroarylation reactions with alkenes of varying sizes. The 13C NMR spectrum of the hydroarylation reaction between 1-d6 and the long-chain alkene 5 recorded at 100 °C contains a resonance at 204.7 ppm (Fig. 3a), matching that of bis-alkene complex 24 generated independently in solution by combining 2.2 equiv. of alkene 5 with [13CL4–Ni(η6-C6H6)] (Supplementary Figs. 7 and 8). The bis-propylene analogue 25 was isolated in 50% yield from the reaction of [L4–Ni(η6-C6H6)] and propylene (2; Fig. 3b) and fully characterized by NMR spectroscopy and single-crystal X-ray diffraction. The 13C NMR spectrum of the reaction of the more hindered tert-butylethylene (14) obtained at 25 °C contains a single resonance at 198.7 ppm (Supplementary Fig. 11) and the colour of the solution was yellow, matching the spectrum and colour of the alkene complex 26 generated in situ from [13CL4–Ni(η6-C6H6)] and 14 (vide infra). However, the 13C NMR spectrum recorded at 100 °C contains a resonance at 196.2 ppm, and the colour of the solution was red, matching those of [13CL4–Ni(η6-C6D6)]. Studies of the relative stabilities of the alkene and arene complexes by varying the amount of 14 in pentane under nitrogen (Fig. 3c,d) showed that both [L4–Ni(η6-C6H6)] and the alkene complex 26 were present at equilibrium at lower concentrations (0.007–0.2 M) of alkene, but the mono-alkene dinitrogen complex 26, which was identified by NMR and IR spectroscopy and single-crystal X-ray diffraction (Fig. 3e), was the only species observed at higher concentrations of alkene (0.3–0.5 M). Thus, the catalytically active species in the reactions of unhindered alkenes (such as 5) are bis-alkene complexes, whereas those in the reactions of hindered alkenes (such as 14) at elevated temperatures are an equilibrium mixture of mono-alkene and arene complexes.
Fig. 3 |. Observation and isolation of catalyst resting states.

a, Observation of the catalyst resting state in the hydroarylation reaction of 1-d6 with unhindered alkene 5 by 13C NMR spectroscopy. The resting state with unhindered alkene 5 was found to be complex 24. b, Preparation of bis-olefin complex 25 and the ORTEP diagram of the X-ray diffraction structure of 25 (thermal ellipsoids are shown at the 50% probability level). c, The equilibrium between [13CL4–Ni(η6-C6H6)] and the mono-olefin complex 26 was monitored by 13C NMR spectroscopy. The spectra resulting from the addition of 14 (0–60 equiv.) to a solution of [13CL4–Ni(η6-C6H6)] in C6D6 are shown. d, Expression for the equilibrium between arene- and alkene-bound 13CL4–Ni complexes and a plot of the ratio of nickel complexes with respect to [14]. We estimate the error in the integral ratios to be <5% based on the signal-to-noise ratio of the 13C NMR spectra. e, Preparation of mono-olefin complex 26 and ORTEP diagram of the the X-ray diffraction structure of 26 (thermal ellipsoids are shown at the 50% probability level). For clarity, all hydrogen atoms have been omitted from the ORTEP diagrams, and NHC sidearms are represented in wire format.
To identify the steps of the catalytic cycle, the initial rates of the hydroarylation of both unhindered alkene 5 and hindered alkene 11 with benzene (1) were measured at varying alkene concentrations (Fig. 4a). The initial rate of the hydroarylation reaction with terminal alkene 5 was inverse first order in the concentration of 5 (Fig. 4a, top), indicating that one equivalent of the unhindered alkene 5 dissociates from the bis-alkene resting state 24 prior to the binding of the arene and the rate-determining step of the reaction. The order in hindered alkene 11 depended on the concentration of 11 (Fig. 4a, bottom), which is consistent with the existence of an equilibrium between the catalytic resting states as a function of the nature and concentration of the alkene, as described above (see Fig. 3d)17,46. The first-order dependence of the reaction rate on [11] at low [11] indicates that replacement of the arene with one alkene precedes the rate-determining step of the catalytic process, whereas the zero-order dependence of the reaction rate on [11] at high [11] indicates that the resting state shifts to the mono-alkene complex 26 and that the highest-energy transition state contains an aryl or arene unit.
Fig. 4 |. Mechanistic experiments.

a, Dependence of the initial rate of the hydroarylation on alkene concentration for unhindered alkene 5 (top) or hindered alkene 11 (bottom). Error bars indicate a ±10% error in the initial rate. b, Deuterium incorporation was observed at the 2-alkenyl position of unreacted alkene 11 in the hydroarylation of 11 with 1-d6. c, A KIE experiment for the hydroarylation of alkene 11 was conducted in separate vessels, and the KIE was found to be 1.3 ± 0.1. This result indicates that hydrogen atom transfer is reversible.
Two sets of experiments revealed reversible steps within the catalytic cycle. First, the reaction of 1-d6 with alkene 11 led to 43% incorporation of deuterium at the 2-alkenyl position in unreacted 11 after 60% conversion (Fig. 4b). No deuterium incorporation into the terminal position of the alkene was observed. Second, the kinetic isotope effect (KIE) determined from separate reactions of alkene 11 with either 1 or 1-d6 was only 1.3 ± 0.1 (Fig. 4c)14,47. This KIE value contrasts with the measured KIE values of 2.1–2.5 for irreversible C–H activation during ruthenium-catalysed alkene hydroarylation17,27,28,31. The results of both experiments imply that the transfer of the arene hydrogen to the alkene by one or multiple steps is reversible. An overall catalytic cycle for the hydroarylation of unactivated alkenes consistent with our experimental results and previous reports33 is shown in Fig. 5a.
Fig. 5 |. Computational investigations.

a, Proposed mechanism for the nickel-catalysed hydroarylation of unactivated alkenes supported by mechanistic experiments and computations. b, Computed energetics for the hydroarylation reaction with catalysts containing L1 and L4. c, DFT-optimized geometries of TSRE. d, Energy decomposition analysis for the changes between the ground state and transition state for reductive elimination as a function of the carbene ligand. ΔEdist is the difference in energy between the most stable form of the free carbene ligand and free nickel fragment and the energy of the two components in their geometries of the complex, ΔΔEdist is the difference in distortion energies between the ground state and transition state and ΔΔΔEdist is the difference in ΔΔEdist for the pairs of ligands. ΔEint is the interaction energy of the two fragments in their distorted geometries and is decomposed as the sum of ΔEPauli, ΔEelstat, ΔEdisp, ΔEct and ΔEpol. In a similar manner, ΔΔE values are the difference in energies between the GS and the TS, and ΔΔΔE values are the difference in the component ΔΔE for pairs of ligands.
Computational studies using density functional theory (DFT) provided further insight into the mechanism of C−H bond cleavage (Fig. 5b, see the Supplementary Information for computational details). C−H activation could occur by oxidative addition of the C−H bond in benzene or by an alternative mechanism involving the direct transfer of the C–H bond of a coordinated arene to the bound alkene, known as ligand-to-ligand hydrogen transfer (LLHT)35. Previous computational studies on the nickel-catalysed hydroarylation of alkynes, hydroarylation with more acidic arenes, and hydroarylation with less hindered ligands indicated that the barrier to direct oxidative addition of the C−H bonds in benzene and other arenes to NHC-ligated Ni0 complexes is higher than that of a one-step transfer of the hydrogen from a coordinated arene to the coordinated π-system of the alkene or alkyne39,48. The barrier we computed for the LLHT process between benzene (1) and propene (2) ligands with the catalyst containing L4 (L4TSLLHT, 21.3 kcal mol−1), relative to the combination of benzene and the bis-alkene ground state (GS), was 5.2 kcal mol−1 lower in energy than that for the LLHT process with the catalyst containing the less hindered L1 (L1TSLLHT, 26.5 kcal mol−1) and 3 kcal mol−1 lower than the barrier for oxidative addition of the aryl C–H bond (see the Supplementary Information for further details).
However, the highest-energy transition state of the catalytic process deduced from experiment (vide supra) and computation is the reductive elimination to form the alkyl–aryl C−C bond from T-shaped 27, not the LLHT. The computed barrier (relative to the ground state) for reductive elimination from the alkyl aryl complex bound by L4 (L4TSRE) is 29.2 kcal mol−1, which is 3.9 kcal mol−1 lower than the computed barrier for the reductive elimination from the alkyl aryl complex bound by L1 (L1TSRE). This difference is consistent with the much higher activity of [L4–Ni(η6-C6H6)] as catalyst for the hydroarylation reaction compared with [L1–Ni(η6-C6H6)].
One might envision this difference in barriers to result from a steric effect on the rate of reductive elimination. However, computation of the geometry and non-covalent interactions provided a much different picture of the origin of the high activity of the complex of L4 compared with that of the smaller L1. Most striking, the geometrical parameters around the nickel atom in L4TSRE were found to be almost identical to those in L1TSRE (Fig. 5c), suggesting that an effect beyond simple steric effects within the transition state controls the rate.
To decipher the origin of the different barriers for reductive elimination from the complexes containing L1 and L4, we used the distortion/interaction or activation strain model pioneered by Houk and co-workers49 and the second-generation absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA) developed by Head-Gordon and co-workers40. Selected energy values from this analysis for the complexes of the four ligands L1−L4 are summarized in Fig. 5d. A comparison of the difference in distortion energies between pairs of complexes shows that the peripheral methyl groups in L4 (ΔΔΔEdist,L4-L3 = −1.5 kcal mol−1), in addition to the aromatic rings (ΔΔΔEdist,L2-L1 = −1.1 kcal mol−1), lead to distortion energies that favour reaction with L4 over reaction with the other ligands (ΔΔΔEdist,L4-L1 = −2.4 kcal mol−1). For all of the four NHC ligands, the NHC fragment is less distorted from the free carbene in the transition state for reductive elimination than it is in the ground state. However, this difference between the distortion energy of the carbene in the transition and ground states is greatest for L4 and contributes to a lower barrier for the reaction of the complex containing L4 than for the reactions of the complexes containing L1−L3 (Supplementary Table 6).
This energy decomposition analysis also shows that the aryl groups in ligands L2−L4, which are not present in L1, lead to interaction energies that cause the barriers for reactions catalysed by complexes of L4 (and L2 and L3) to be lower than those catalysed by the complex bearing the more common ligand L1 (ΔΔΔEint,L4-L1 = −4.3 kcal mol−1). An analysis of the major contributions to the difference between the interaction energies in the transition state for reductive elimination with one ligand and those in the transition state for reductive elimination with another ligand can be seen in Fig. 5d. The largest differences between these values for L4 compared with L1 are the Pauli repulsive (steric effects), electrostatic and London dispersion terms. Assessment of each pairwise difference in these contributions to the interaction energies for the reactions catalysed by the complexes of the series of ligands reveals the structural elements of the ligands that lead to these values. This analysis shows that the Pauli repulsion term strongly influences the barrier and results principally from the larger steric impact of L4. Particularly striking is that this Pauli term increases the barrier for the reaction with the large ligand relative to that for the reaction with the smaller ligand, rather than decreasing the barrier (ΔΔΔEPauli,L4-L1 = 5.2 kcal mol−1). Typically, one presumes that steric effects cause reductive elimination involving the coupling of two ligands at a single metal centre to be faster for complexes bearing more hindered ancillary ligands50.
In contrast, attractive electrostatic and London dispersion terms reduce the energy of the transition state containing L4 compared with the ground state more than they reduce this difference in energy for the complexes containing the other ligands. More specifically, the values in Fig. 5d show that that presence of the eight aryl groups in ligands L2−L4 lead to electrostatic interactions that are approximately 4 kcal mol−1 larger than the electrostatic interactions in the complexes of L1 (ΔΔΔEelstat,L2-L1 = −4.9 kcal mol−1 and ΔΔΔEelstat,L4-L1 = −3.7 kcal mol−1). A similar pairwise comparison of the London dispersion effects shows that the difference in values between the system containing L4 and the system containing L1 (ΔΔΔEdisp,L4-L1 = −4.3 kcal mol−1) results mainly from the presence of the 16 methyl groups on L4 that are not present in L1−L3.
Although the results above demonstrate that the peripheral methyl groups in L4 cause the difference between the stabilizing dispersive interactions in the ground and transition states containing L4 to be larger than this difference between those containing L3 (ΔΔΔEdisp,L4-L3 = −2.9 kcal mol−1), we further probed the origin of this difference in dispersive interactions with ligands L3 and L4. The methyl groups in L4 could participate in stabilizing dispersive interactions with each other and with other groups in the complex, or they could cause the positions of the aryl groups and the structure of the core of the complexes of L4 to be altered from those of L1−L3 in a way that leads to a larger difference in dispersion interactions between the ground and transition states bearing L4. To distinguish between these two possibilities, we performed the same energy decomposition analysis of the ground and transition states with L3 (lacking the peripheral methyl groups), but placing the atoms in the same positions as those in the lowest-energy geometry of L4. The results of this analysis are included in the Supplementary Information and show that the differences in dispersion interactions between the ground and transition states of the complexes containing L4 and L3 in the same geometry (the minimum-energy geometry of L4, ΔΔΔEdisp,L4-L3 in L4 geometry = −2.9 kcal mol−1; Supplementary Table 12) are similar to those of the ground and transition states containing L4 and L3 in their respective minimum-energy geometries. In other words, the stabilizing dispersion interactions in the complexes of L4 are due to direct interactions with the methyl groups, not to changes in the ligand geometry imparted by the methyl groups in L4. Finally, a graphical plot of the non-covalent interactions51,52 present in L4TSRE (Supplementary Fig. 18) corroborates the presence of significant stabilizing interactions involving the methyl groups.
Conclusion
In this work the first highly anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes has been accomplished, and many of the reactions occurred in good yields. Even internal alkenes reacted to give n-alkylarene products. The unparalleled selectivity and activity of this reaction was enabled by a nickel catalyst containing an extremely large N-heterocyclic carbene ligand that undergoes C–H activation by a mechanism that involves the formation of a linear alkyl–metal complex that undergoes reductive elimination to form the new carbon–carbon bond in the alkylarene product with rates that are enhanced by intramolecular non-covalent interactions. The results of our computational studies imply that the conventional view of how steric bulk favours reductive elimination does not apply to this system. Instead of accelerating the reductive elimination by steric repulsion, the multiple aryl groups in ligand L4 of the most active catalyst lead to favourable electrostatic interactions, and the large number of methyl groups in L4 leads to favourable London dispersion interactions and lower distortion energy. We anticipate that this reactivity and the analysis of its origins should aid the development of new methods for the functionalization of alkenes with additional strong, unactivated C–H or X–H bonds catalysed by complexes of nickel ligated by N-heterocyclic carbenes as well as by complexes of metals other than nickel with appropriate properties of the ancillary ligands.
Supplementary Material
Acknowledgements
We thank S. Arlow, C. Karmel and J. Wang for helpful discussions. We thank Y. Schramm for preliminary experiments. We acknowledge N. Settineri for X-ray crystallographic analysis. We thank M. Head-Gordon and M. Loipersberger for discussions on EDA calculations. This work was supported by the Director, Office of Science, of the U.S. Department of Energy under contract no. DE-AC02- 05CH11231, by the National Science Foundation (graduate research fellowship to N.I.S.) and by the Japan Society for the Promotion of Science (JSPS KAKENHI Grant Number JP15H05799). X-ray diffraction data were collected using an instrument funded by the NIH (S10-RR027172). Computations were performed on a computation cluster funded by the NIH (S10-OD023532). NMR spectroscopy was performed in the College of Chemistry’s NMR facility funded in part by the NIH (S10-OD024998).
Footnotes
Online content
Any Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-019-0409-4.
Competing interests
The authors declare no competing interests
Supplementary information is available for this paper at https://doi.org/10.1038/s41557-019-0409-4.
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers 1901576 ([L4–Ni(η6-C6H6)]), 1901577 (26) and 1901578 (25). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article and its Supplementary Information, or from the corresponding author upon reasonable request.
References
- 1.Kocal JA, Vora BV & Imai T Production of linear alkylbenzenes. Appl. Catal. A 221, 295–301 (2001). [Google Scholar]
- 2.Linear Alkyl Benzene Market Size, Share & Trends Analysis Report By Application (Heavy Duty Laundry, Laundry Powders, Washing Liquids, Industrial Cleaners, Household Cleaners), And Segment Forecasts, 2012–2020 (Grand View Research, 2017). [Google Scholar]
- 3.Röper M, Gehrer E, Narbeshuber T & Siegel W Acylation and alkylation in Ullmann’s Encyclopedia of Industrial Chemistry (Wiley-VCH, 2000). [Google Scholar]
- 4.de Almeida JLG, Dufaux M, Taarit YB & Naccache C Linear alkylbenzene. J. Am. Oil Chem. Soc 71, 675–694 (1994). [Google Scholar]
- 5.Ishiwatari R, Takada H, Yun S-J & Matsumoto E Alkylbenzene pollution of Tokyo Bay sediments. Nature 301, 599–600 (1983). [Google Scholar]
- 6.Macías-Zamora JV & Ramírez-Alvarez N Tracing sewage pollution using linear alkylbenzenes (LABs) in surface sediments at the south end of the Southern California Bight. Environ. Pollut 130, 229–238 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Murai S et al. Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature 366, 529–531 (1993). [Google Scholar]
- 8.Jun C-H, Hong J-B, Kim Y-H & Chung K-Y The catalytic alkylation of aromatic imines by Wilkinson’s complex: the domino reaction of hydroacylation and ortho-alkylation. Angew. Chem. Int. Ed 39, 3440–3442 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Gao K & Yoshikai N Cobalt–phenanthroline catalysts for the ortho alkylation of aromatic imines under mild reaction conditions. Angew. Chem. Int. Ed 50, 6888–6892 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Schinkel M, Marek I & Ackermann L Carboxylate-assisted ruthenium(II)-catalyzed hydroarylations of unactivated alkenes through C–H cleavage. Angew. Chem. Int. Ed 52, 3977–3980 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto T, Taube DJ, Periana RA, Taube H & Yoshida H Anti-Markovnikov olefin arylation catalyzed by an iridium complex. J. Am. Chem. Soc 122, 7414–7415 (2000). [Google Scholar]
- 12.Periana RA, Liu XY & Bhalla G Novel bis-acac-O,O–Ir(III) catalyst for anti-Markovnikov, hydroarylation of olefins operates by arene CH activation. Chem. Commun 24, 3000–3001 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Oxgaard J, Muller RP, Goddard WA & Periana RA Mechanism of homogeneous Ir(III) catalyzed regioselective arylation of olefins. J. Am. Chem. Soc 126, 352–363 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Bhalla G, Liu XY, Oxgaard J, Goddard WA & Periana RA Synthesis, structure, and reactivity of O-donor Ir(III) complexes: C–H activation studies with benzene. J. Am. Chem. Soc 127, 11372–11389 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Bhalla G, Oxgaard J, Goddard WA & Periana RA Anti-Markovnikov hydroarylation of unactivated olefins catalyzed by a bis-tropolonato iridium(III) organometallic complex. Organometallics 24, 3229–3232 (2005). [Google Scholar]
- 16.Lail M, Arrowood BN & Gunnoe TB Addition of arenes to ethylene and propene catalyzed by ruthenium. J. Am. Chem. Soc 125, 7506–7507 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Lail M et al. Experimental and computational studies of ruthenium(II)-catalyzed addition of arene C−H bonds to olefins. Organometallics 23, 5007–5020 (2004). [Google Scholar]
- 18.McKeown BA, Prince BM, Ramiro Z, Gunnoe TB & Cundari TR PtII-catalyzed hydrophenylation of α-olefins: variation of linear/branched products as a function of ligand donor ability. ACS Catal. 4, 1607–1615 (2014). [Google Scholar]
- 19.Luedtke AT & Goldberg KI Intermolecular hydroarylation of unactivated olefins catalyzed by homogeneous platinum complexes. Angew. Chem. Int. Ed 47, 7694–7696 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Clement ML, Grice KA, Luedtke AT, Kaminsky W & Goldberg KI Platinum(II) olefin hydroarylation catalysts: tuning selectivity for the anti-Markovnikov product. Chem. Eur. J 20, 17287–17291 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Webster-Gardiner MS et al. Catalytic synthesis of “super” linear alkenyl arenes using an easily prepared Rh(I) catalyst. J. Am. Chem. Soc 139, 5474–5480 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Chen J et al. Catalytic synthesis of superlinear alkenyl arenes using a Rh(I) catalyst supported by a “capping arene” ligand: access to aerobic catalysis. J. Am. Chem. Soc 140, 17007–17018 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Oxgaard J & Goddard WA Mechanism of Ru(II)-catalyzed olefin insertion and C−H activation from quantum chemical studies. J. Am. Chem. Soc 126, 442–443 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Oxgaard J, Periana RA & Goddard WA Mechanistic analysis of hydroarylation catalysts. J. Am. Chem. Soc 126, 11658–11665 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Suslick BA, Liberman-Martin AL, Wambach TC & Tilley TD Olefin hydroarylation catalyzed by (pyridyl-indolate)Pt(II) complexes: catalytic efficiencies and mechanistic aspects. ACS Catal. 7, 4313–4322 (2017). [Google Scholar]
- 26.Mann G, Shelby Q, Roy AH & Hartwig JF Electronic and steric effects on the reductive elimination of diaryl ethers from palladium(II). Organometallics 22, 2775–2789 (2003). [Google Scholar]
- 27.Foley NA et al. Comparative reactivity of TpRu(L)(NCMe)Ph (L = CO or PMe3): impact of ancillary ligand L on activation of carbon–hydrogen bonds including catalytic hydroarylation and hydrovinylation/oligomerization of ethylene. J. Am. Chem. Soc 129, 6765–6781 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Foley NA, Ke Z, Gunnoe TB, Cundari TR & Petersen JL Aromatic C−H activation and catalytic hydrophenylation of ethylene by TpRu{P(OCH2)3CEt}(NCMe)Ph. Organometallics 27, 3007–3017 (2008). [Google Scholar]
- 29.Foley NA, Lee JP, Ke Z, Gunnoe TB & Cundari TR Ru(II) catalysts supported by hydridotris(pyrazolyl)borate for the hydroarylation of olefins: reaction scope, mechanistic studies, and guides for the development of improved catalysts. Acc. Chem. Res 42, 585–597 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Joslin EE et al. Catalytic hydroarylation of ethylene using TpRu(L)(NCMe) Ph (L = 2,6,7-trioxa-1-phosphabicyclo[2,2,1]heptane): comparison to TpRu(L′)(NCMe)Ph systems (L′ = CO, PMe3, P(pyr)3, or P(OCH2)3CEt). Organometallics 31, 6851–6860 (2012). [Google Scholar]
- 31.Burgess SA et al. Hydrophenylation of ethylene using a cationic Ru(ii) catalyst: comparison to a neutral Ru(ii) catalyst. Chem. Sci 5, 4355–4366 (2014). [Google Scholar]
- 32.Malinoski JM & Brookhart M Polymerization and oligomerization of ethylene by cationic nickel(II) and palladium(II) complexes containing bidentate phenacyldiarylphosphine ligands. Organometallics 22, 5324–5335 (2003). [Google Scholar]
- 33.Bair JS et al. Linear-selective hydroarylation of unactivated terminal and internal olefins with trifluoromethyl-substituted arenes. J. Am. Chem. Soc 136, 13098–13101 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Schramm Y, Takeuchi M, Semba K, Nakao Y & Hartwig JF Anti-Markovnikov hydroheteroarylation of unactivated alkenes with indoles, pyrroles, benzofurans, and furans catalyzed by a nickel–N-heterocyclic carbene cystem. J. Am. Chem. Soc 137, 12215–12218 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Guihaumé J, Halbert S, Eisenstein O & Perutz RN Hydrofluoroarylation of alkynes with Ni catalysts. C−H activation via ligand-to-ligand hydrogen transfer, an alternative to oxidative addition. Organometallics 31, 1300–1314 (2012). [Google Scholar]
- 36.Hoshimoto Y, Hayashi Y, Suzuki H, Ohashi M & Ogoshi S One-pot, single-step, and gram-scale synthesis of nononuclear [(η6-arene)Ni(N-heterocyclic carbene)] complexes: useful precursors of the Ni0–NHC unit. Organometallics 33, 1276–1282 (2014). [Google Scholar]
- 37.Berthon-Gelloz G et al. IPr* an easily accessible highly hindered N-heterocyclic carbene. Dalton Trans. 39, 1444–1446 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Meiries S, Speck K, Cordes DB, Slawin AMZ & Nolan SP [Pd(IPr*OMe)(acac)Cl]: tuning the N-heterocyclic carbene in catalytic C−N bond formation. Organometallics 32, 330–339 (2013). [Google Scholar]
- 39.Okumura S et al. para-Selective alkylation of benzamides and aromatic ketones by cooperative nickel/aluminum catalysis. J. Am. Chem. Soc 138, 14699–14704 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Horn PR, Mao Y & Head-Gordon M Probing non-covalent interactions with a second generation energy decomposition analysis using absolutely localized molecular orbitals. Phys. Chem. Chem. Phys 18, 23067–23079 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Saper NI & Hartwig JF Mechanistic investigations of the hydrogenolysis of diaryl ethers catalyzed by nickel complexes of N-heterocyclic carbene ligands. J. Am. Chem. Soc 139, 17667–17676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clavier H & Nolan SP Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem. Commun 46, 841–861 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Hillier AC et al. A combined experimental and theoretical study examining the binding of N-heterocyclic carbenes (NHC) to the Cp*RuCl (Cp* = η5-C5Me5) moiety: insight into stereoelectronic differences between unsaturated and saturated NHC ligands. Organometallics 22, 4322–4326 (2003). [Google Scholar]
- 44.Falivene L et al. SambVca 2. A web tool for analyzing catalytic pockets with topographic steric maps. Organometallics 35, 2286–2293 (2016). [Google Scholar]
- 45.Dorta R et al. Steric and electronic properties of N-heterocyclic carbenes (NHC): a detailed study on their interaction with Ni(CO)4. J. Am. Chem. Soc 127, 2485–2496 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Matsumoto T, Periana RA, Taube DJ & Yoshida H Regioselective hydrophenylation of olefins catalyzed by an Ir(III) complex. J. Mol. Catal. A 180, 1–18 (2002). [Google Scholar]
- 47.Simmons EM & Hartwig JF On the interpretation of deuterium kinetic isotope effects in C−H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed 51, 3066–3072 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Tang S, Eisenstein O, Nakao Y & Sakaki S Aromatic C–H σ-bond activation by Ni0, Pd0, and Pt0 alkene complexes: concerted oxidative addition to metal vs ligand-to-ligand H transfer mechanism. Organometallics 36, 2761–2771 (2017). [Google Scholar]
- 49.Bickelhaupt FM & Houk KN Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem. Int. Ed 56, 10070–10086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartwig JF in Organotransition Metal Chemistry: From Bonding to Catalysis Ch. 8 (Univ. Science Books, 2010). [Google Scholar]
- 51.Johnson ER et al. Revealing noncovalent interactions. J. Am. Chem. Soc 132, 6498–6506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Contreras-García J et al. NCIPLOT: a program for plotting noncovalent interaction regions. J. Chem. Theory Comput 7, 625–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers 1901576 ([L4–Ni(η6-C6H6)]), 1901577 (26) and 1901578 (25). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article and its Supplementary Information, or from the corresponding author upon reasonable request.