

Abstract
Enzymes that contain metal ions—that is, metalloenzymes—possess the reactivity of a transition metal centre and the potential of molecular evolution to modulate the reactivity and substrate-selectivity of the system1. By exploiting substrate promiscuity and protein engineering, the scope of reactions catalysed by native metalloenzymes has been expanded recently to include abiological transformations2,3. However, this strategy is limited by the inherent reactivity of metal centres in native metalloenzymes. To overcome this limitation, artificial metalloproteins have been created by incorporating complete, noble-metal complexes within proteins lacking native metal sites1,4,5. The interactions of the substrate with the protein in these systems are, however, distinct from those with the native protein because the metal complex occupies the substrate binding site. At the intersection of these approaches lies a third strategy, in which the native metal of a metalloenzyme is replaced with an abiological metal with reactivity different from that of the metal in a native protein6–8. This strategy could create artificial enzymes for abiological catalysis within the natural substrate binding site of an enzyme that can be subjected to directed evolution. Here we report the formal replacement of iron in Fe-porphyrin IX (Fe-PIX) proteins with abiological, noble metals to create enzymes that catalyse reactions not catalysed by native Fe-enzymes or other metalloenzymes9,10. In particular, we prepared modified myoglobins containing an Ir(Me) site that catalyse the functionalization of C–H bonds to form C–C bonds by carbene insertion and add carbenes to both β-substituted vinylarenes and unactivated aliphatic α-olefins. We conducted directed evolution of the Ir(Me)-myoglobin and generated mutants that form either enantiomer of the products of C–H insertion and catalyse the enantio- and diastereoselective cyclopropanation of unactivated olefins. The presented method of preparing artificial haem proteins containing abiological metal porphyrins sets the stage for the generation of artificial enzymes from innumerable combinations of PIX-protein scaffolds and unnatural metal cofactors to catalyse a wide range of abiological transformations.
To create artificial metalloenzymes formed by combining abiological metals and natural metalloprotein scaffolds, we focused on haem proteins, which contain Fe-porphyrin IX (Fe-PIX) as a metal cofactor. Native haem enzymes catalyse reactions that include C–H oxidation and halogenation11, and they have been successfully evolved to oxidize abiological substrates12,13. Fe-PIX proteins have also been shown to catalyse abiological reactions involving the addition and insertion of carbenes and nitrenes to olefins and X–H bonds2,3,9,14. However, the reactivity of the Fe-centre in haem proteins limits the scope of these transformations. For example, Fe-PIX proteins catalyse the cyclopropanation of activated terminal vinylarenes9,10, but they do not catalyse reactions with internal vinylarenes or unactivated alkenes. Likewise, they catalyse insertions of carbenes into reactive N–H and S–H bonds, but they do not catalyse the insertion into less reactive C–H bonds3,14.
Because the repertoire of reactions catalysed by free metal-porphyrin complexes of Ru (ref. 15), Rh (ref. 16) and Ir (ref. 17) is much greater than that of the free Fe-analogues, we hypothesized that their incorporation into PIX proteins could create new enzymes for abiological catalysis that is not possible with Fe-PIX enzymes. Artificial PIX proteins containing Mn, Cr, and Co cofactors have been prepared to mimic the intrinsic chemistry of the native haem proteins18–21, but the reactivities and selectivities of these processes are lower than those achieved in the same reactions catalysed by native Fe-PIX enzymes. Thus, artificially metallated PIX proteins that catalyse reactions that are not catalysed by native Fe-PIX proteins are unknown, and the current, inefficient methods to prepare PIX proteins containing non-native metals have hindered the potential for directed evolution of the resulting enzymes22–25.
To evaluate rapidly the potential of artificial [M]-PIX enzymes, we envisioned creating an array of catalysts formed by pairing numerous mutants of apo-PIX proteins and [M]-cofactors in a combinatorial fashion. Previously, apo-PIX proteins have been prepared from native Fe-PIX enzymes by acidic, denaturing extraction of the Fe-cofactor, followed by extensive dialysis to refold the protein22. This multistep process is too lengthy for directed evolution, and the harsh, acidic conditions are known to result in proteins that are heterogeneous in structure, which would be detrimental for selective catalysis26. Alternatively, Ru-, Mn- and Co-PIX proteins have been expressed directly23–25, but these methods are not general, require a gross excess of metal cofactor, and would require a time-consuming purification of each combination of metal and protein. To avoid the aforementioned liabilities of these reported methods in the creation of the proposed catalyst library, we sought to express directly and purify apo-PIX proteins lacking the entire haem unit and to reconstitute them with metal cofactors containing metals other than iron in a stoichiometric fashion (Fig. 1a).
Figure 1 |. Strategy for expedient preparation of [M]-PIX-proteins.
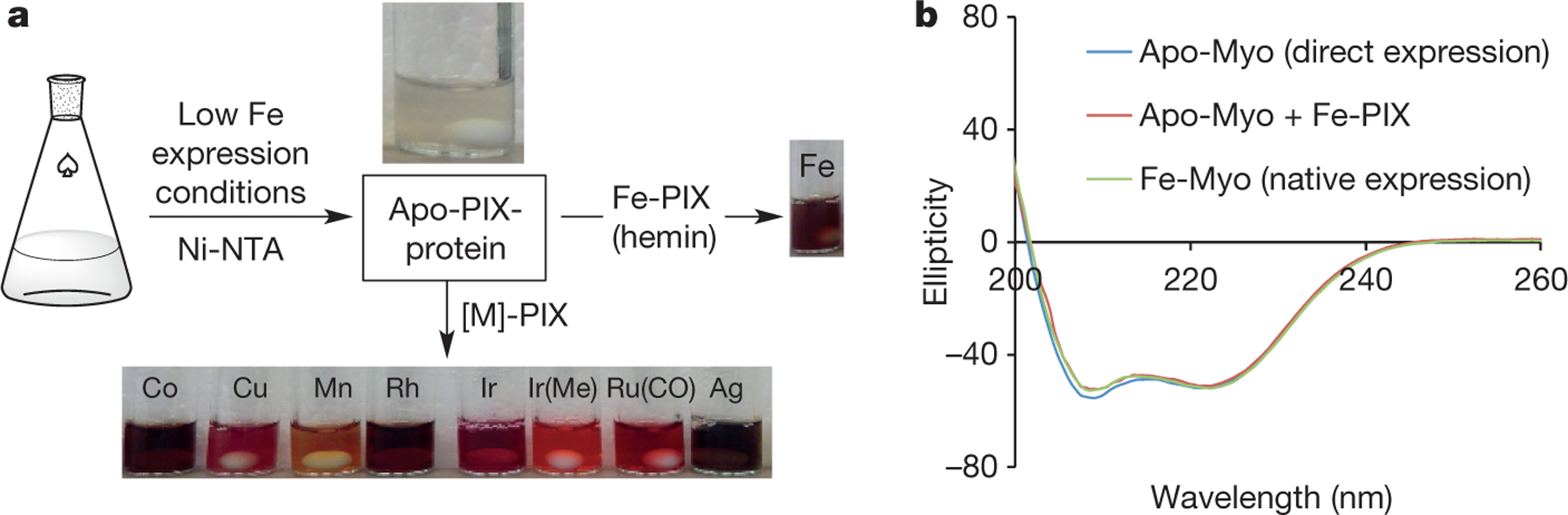
a, Direct expression, Ni-NTA purification, and diverse metallation of apo-PIX proteins to generate PIX proteins containing Co, Cu, Mn, Rh, Ir, Ru, and Ag sites or to regenerate the native Fe-containing protein. b, Comparison of the circular dichroism spectra obtained from directly expressed apo-Myo, the same protein reconstituted with Fe-PIX (hemin), and the same mutant expressed as a native Fe-PIX protein.
Evaluation of a series of expression conditions revealed those suitable for recombinant expression of the apo-form of haem proteins in Escherichia coli (Supplementary Tables 1 and 2). Under the optimized conditions (Supplementary Fig. 1 and Supplementary Table 1), involving minimal media lacking Fe to minimize the biosynthesis of hemin and low temperature to mitigate the instability of the apo-form, we expressed successfully the protein containing less than 5% of the Fe-PIX cofactor, as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). In particular, mutants of Physeter macrocephalus myoglobin (Myo) and Bacillus megaterium cytochrome P450 BM3h (P450) with and without an mOCR stability tag were overexpressed in high yields and purified (up to 70 mg l-1 of protein; Fig. 1a, Supplementary Tables 1 and 2)9,10,27. Circular dichroism spectroscopy revealed that these apo-proteins retain the fold of their native Fe-PIX analogues (Fig. 1b, Supplementary Fig. 2). The obtained apo-proteins were reconstituted quantitatively upon addition of stoichiometric amounts of various [M]-PIX cofactors, as determined by native nano electrospray ionization mass spectrometry (Supplementary Fig. 3). Moreover, reactions catalysed by reconstituted Fe-myoglobin and Fe-P450 occurred with the same enantioselectivities as those catalysed by native Fe-proteins (Supplementary Fig. 4)9,10, providing strong evidence that this method indeed generates [M]-PIX-proteins with the intact active site and with the cofactors bound at the native PIX-binding site. Further studies revealed that reconstituted mOCR-myoglobins are stable on storage; reactions catalysed by freshly prepared, frozen, and lyophilized enzymes proceeded with comparable enantioselectivity (see below, Supplementary Fig. 5).
Following this method, we directly expressed eight variants of apo-mOCR-Myo-H93X, each carrying a different mutation to the axial ligand position (H93X). Upon reconstitution of each variant with nine different porphyrin cofactors (containing Fe(Cl)-, Co(Cl)-, Cu-, Mn(Cl)-, Rh-, Ir(Cl)-, Ir(Me)-, Ru(CO)- and Ag-sites, Supplementary Table 2), we rapidly accessed 72 potential catalysts whose activity profiles are distinct from those of wild-type myoglobin, owing to the identity of the metal centre and the amino acid residue serving as the axial ligand (Fig. 2a, b)3.
Figure 2 |. Evaluation of artificial [M]-PIX mOCR-myoglobins as catalysts.
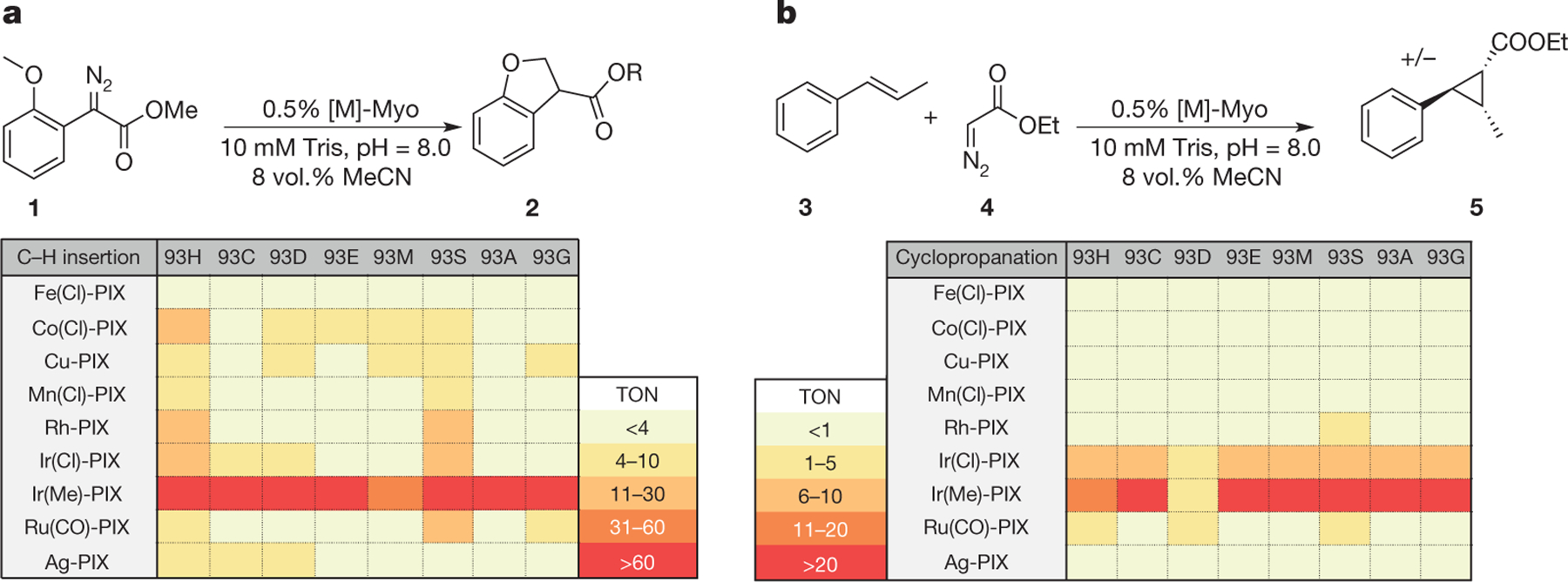
a, b, Catalysts for the insertion of carbenes into C–H bonds (a; 1 → 2) and for the addition of carbenes to an internal olefin (b; 3 + 4 → 5). TON, turnover number. Reaction conditions for the C–H insertion reaction: 10mM substrate and 0.5% catalyst in 250μl buffer (10mM Tris, pH 8.0 containing 8 vol.% MeCN). Reaction conditions for the cyclopropanation reaction: 10mM olefin, 30mM EDA, and 0.5% catalyst in 250μl buffer (10 mM Tris, pH 8.0 containing 8 vol.% MeCN).
Natural haem proteins functionalize C–H bonds to form C–O bonds11, but no haem protein is known to functionalize a C–H bond to form a C–C bond. To identify an enzyme for the insertion of a carbene into a C–H bond, the array of artificial [M]-mOCR-myoglobins containing various metals and axial ligands was evaluated for the reaction of diazoester 1 to form chiral dihydrobenzofuran 2 (Fig. 2a). All myoglobins formed from the native Fe-PIX cofactor were inactive, regardless of the axial ligand. In contrast, non-native metals formed active catalysts when paired with an appropriate axial ligand. The most active catalysts, those containing Ir(Me)-PIX, were formed by incorporating both an abiological metal (Ir) and an abiological axial ligand (-CH3) that cannot be incorporated though standard mutagenesis techniques. The eight myoglobins containing Ir(Me)-PIX formed enantioenriched dibenzohydrofuran 2 in up to 50% yield before any further mutagenesis (see below). Moreover, this artificial enzyme tolerated modifications to all portions of the substrate; diazoesters 6–11, containing varied ester, arene, and alkoxy functionalities (Fig. 3a, Supplementary Fig. 6), also underwent C–H insertion in the presence of Ir(Me)-PIX-Myo. Together, these results show that the multi-dimensional evaluation of reconstituted PIX-enzymes can identify new artificial metalloenzymes that catalyse reactions that biological Fe-PIX-proteins do not catalyse.
Figure 3 |. Summary of activities and selectivities for reactions catalysed by Ir(Me)-mOCR-Myo.
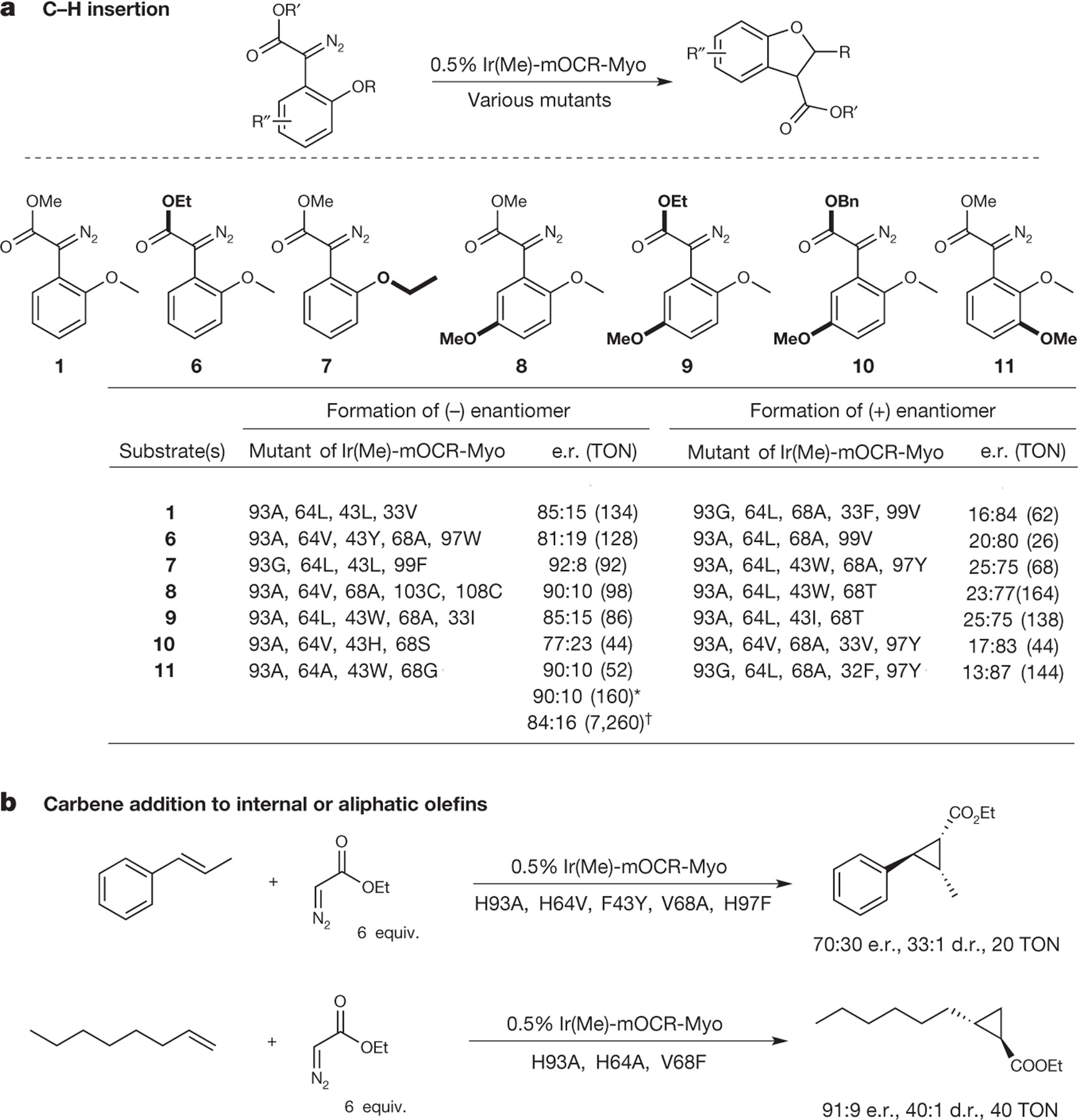
a, Substrates (1, 6–11) for C–H insertion reactions and the most selective mutants identified through the directed evolution. Notations (+)- and (−)-enantiomer distinguish the formation of opposite enantiomers of the product. *Reaction on a 0.12 mmol scale of substrate, TON based on isolated yield of product (80%). †Reaction on a 0.05 mmol scale of substrate with 0.0025% catalyst. Reaction conditions: 10 mM substrate and 0.5% catalyst in 250 μl buffer (10 mM Tris, pH 8.0 containing 8 vol.% MeCN). b, Carbene addition to internal and aliphatic olefins catalysed by mutants of Ir(Me)-mOCR Myo that were found to be the most selective. d.r., diastereomeric ratio. Reaction conditions: 10 mM olefin, 60 mM EDA, and 0.5% catalyst in 250 μl buffer (10 mM Tris, pH 8.0 containing 8 vol.% MeCN). EDA was added over 12 h via syringe pump.
A substantial benefit of using enzymes for synthetic applications is the potential to use directed evolution to obtain a catalyst with desired properties13,28. To develop an enantioselective catalyst for each of the seven substrates undergoing C–H insertion, we followed a hybrid strategy based on stepwise optimization of small sets of amino acids progressively more distal from the reaction site (Fig. 4). In the first phase, the axial ligand (H93) was modified to A or G and the residue directly above the metal centre (H64) was modified to A, V, L, or I (Fig. 4) to give an initial set of eight mutants. In the second phase, these initial eight mutants were modified at positions F43 and V68, which are located in the binding site (Fig. 4), to generate 225 prospective enzymes. To retain the hydrophobicity of the site that binds the porphyrin and substrate, only hydrophophic and uncharged residues (V, A, G, F, Y, S, T) were introduced at positions F43 and V68. Of these 225 mutants, 22 that were among the most selective for one or more of the substrates in Fig. 3 were subjected to a further round of evolution during which the residues at four additional positions (L32, F33, H97, and I99) were modified to generate 217 more mutants (Fig. 4).
Figure 4 |. Directed evolution strategy used to obtain Ir(Me)-mOCR-Myo mutants capable of producing either enantiomer of the products of C–H insertion reactions of varied substrates.
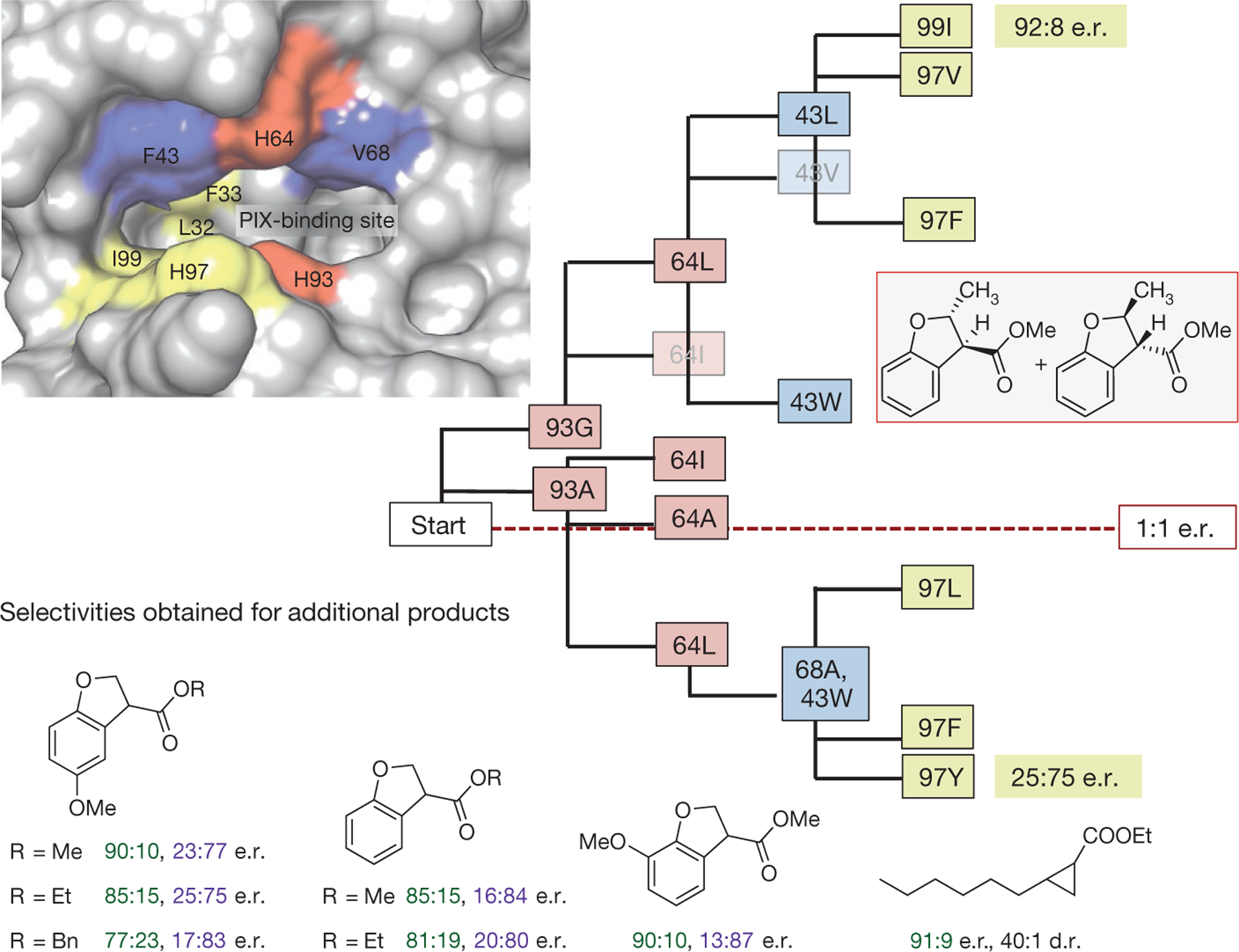
Inner sphere (red boxes), middle sphere (blue), and outer sphere (yellow) residues are highlighted in the depiction of the active site (top left) and in the evolutionary tree. (Image of active site and its surroundings produced in Chimera from PDB 1MBN30.) The strategy is exemplified by showing the enantioselectivies achieved in the formation of the product boxed on the right. Mutants positioned above the red dotted line formed predominantly the opposite enantiomer of those shown below the dotted red line. Some mutants are shown in boxes that are semitransparent for clarity of the figure. In the case of selectivities obtained for additional products, the e.r. values given in green and purple colour are those for reactions forming predominantly the opposite enantiomers. The reactions were run under the conditions described in Fig. 3.
The complete results of the carbene insertion reaction with these mutants are provided in Supplementary Tables 5–11 and are summarized in Fig. 3b and Supplementary Table 4. The directed evolution of Ir(Me)-myoglobins uncovered distinct enzymes catalysing the C–H functionalization to form either enantiomer of the products containing a new C–C bond formed from substrates 1 and 6–11. The reactions occurred with selectivities up to an enantiomeric ratio (e.r.) of 92:8 and with yields up to 97% (Fig. 4, Supplementary Fig. 7 and Supplementary Tables 4–11) with enzymes that were evolved from those giving nearly racemic product. The Ir-myoglobins are suitable catalysts for syntheticscale reactions; the carbene insertion of substrate 11 formed the product containing a new C–C bond in 80% isolated yield from a reaction of 28 mg of 11 with nearly the same enantioselectivity as observed on smaller scale (Fig. 3). A reaction conducted with a 40,000:1 ratio of substrate to Ir(Me)-mOCR-myo occurred with a turnover number (TON) of 7,200 (Fig. 3).
In contrast to the few directed evolutions of artificial enzymes reported previously28, our method of preparing variants of the Ir(Me)-PIX-enzyme enabled us to pursue an individual, eight-site evolutionary trajectory for the reaction of each substrate that identified catalysts selectively forming either enantiomer of all targeted products. These results demonstrate that Ir(Me)-PIX-myoglobins are highly evolvable for different substrates containing varied structural modifications. These results, along with the high isolated yield and the observation of high TONs, demonstrate that the direct expression of apo-Myo, the insertion of diverse [M]-PIX cofactors, and the subsequent directed evolution of the most active enzymes identified is a robust strategy that can be applied in a general way to create stereoselective enzymes for abiological catalysis that cannot be accomplished by any natural enzymes.
To assess the generality of this approach further, we sought catalysts for the cyclopropanation of internal alkenes and α-olefins that, like carbene insertion into C–H bonds, have not been accomplished with natural or artificial enzymes. As a starting point, we evaluated the two-dimensional array of [M]-PIX catalysts shown in Fig. 2b for the cyclopropanation of β-methylstyrene 3 with ethyl diazoacetate 4 (EDA). In agreement with literature reports10, Fe-PIX enzymes did not catalyse this reaction (Fig. 2b). In contrast, Rh-, Ru-, and Ir-PIX enzymes furnished the cyclopropane product 5. The enzyme containing Ir(Me)-PIX was the most active. Although further work is needed to obtain full conversion and high enantiomeric excess (e.e.), the reaction of EDA with β-methylstyrene catalysed by the Ir(Me)-mOCR-Myo mutant H93A, H64V, F43Y, V68A, H97F formed the cyclopropane 5 with a TON of 40 with 70:30 e.r., and with a high >33:1 ratio of diastereomers, favouring the trans isomer.
Having observed the expanded scope of enzyme-catalysed cyclopropanation, we assessed the ability of Ir(Me)-PIX enzymes to catalyse the cyclopropanation of 1-octene, an unactivated, aliphatic olefin. The series of Ir(Me)-PIX-Myo enzymes assessed for C–H insertion reactions were tested as catalysts for the reaction of EDA with 1-octene. Although the mutant H93A, H64A, V68F formed the products of C–H insertion from all substrates unselectively, the same mutant formed the product of cyclopropanation of 1-octene in an enantiomeric ratio of 91:9 and a trans:cis ratio of 40:1 (Figs 3b, 4; Supplementary Tables 12–14). Cyclopropanations of aliphatic alkenes catalysed by traditional metal complexes are typically conducted with an excess of the alkene29. In contrast, the Ir(Me)-PIX-Myo mutant catalyses the reaction with an excess of EDA (a TON of 42 with a 10:1 ratio of EDA:1-octene), suggesting that reactions can be developed with valuable alkenes as limiting reagent. The reactions with fewer equivalents of EDA occur with lower TON due to consumption of EDA by dimerization or O–H insertion of water. These cyclopropanations of unactivated alkenes show the broad potential to evolve artificial myoglobins containing abiological active sites for reactions that are not catalysed by enzymes containing native metals.
The work presented here demonstrates that unknown enzymatic reactivity can be achieved by incorporating just a metal ion with an accompanying small ligand into a well-known metalloprotein, while retaining the native structure of the active site. Selectivity for specific substrates, then, can be achieved readily by directed evolution. Considering the rich chemistry of free metalloporphyrins and the ease of preparation and evolution of haem proteins containing diverse metals by the methods just described, this methodology should seed the creation of many new artificial metalloenzymes with diverse, unnatural reactivity. Moreover, the facile, direct expression of apo-haem proteins could be used in tandem with strategies to incorporate highly active noble-metal complexes of ligands beyond porphyrins. Access to such a range of artificial haem proteins provides a nearly limitless opportunity to achieve catalytic reactions with selectivity derived from the interaction of the substrate with a natural, evolvable binding site.
Supplementary Material
Acknowledgements
This work was supported by the Director, Office of Science, of the US Department of Energy under contract no. DE-AC02–05CH11231, by the NSF (graduate research fellowship to H.M.K.), and the NWO Netherlands Organization for Scientific Research (Rubicon postdoctoral fellowship no. 680–50-1306 to P.D.). We thank the QB3 MacroLab facility (sub-cloning), the UC Berkeley DNA Sequencing Facility (plasmid sequencing), T. Iavarone and the QB3 Mass Spectrometry Facility (supported by NIH grant 1S10RR022393–01) for native NS-ESI-MS data and analysis, and H. Zhao (University of Illinois-Champaign Urbana) for the P411-CIS gene.
Footnotes
Supplementary Information is available in the online version of the paper.
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Lewis JC Artificial metalloenzymes and metallopeptide catalysts for organic synthesis. ACS Catal 3, 2954–2975 (2013). [Google Scholar]
- 2.Farwell CC, Zhang RK, McIntosh JA, Hyster TK & Arnold FH Enantioselective enzyme-catalyzed aziridination enabled by active-site evolution of a cytochrome P450. ACS Cent. Sci 1, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyster TK & Arnold FH P450BM3-axial mutations: a gateway to non-natural reactivity. Isr. J. Chem 55, 14–20 (2015). [Google Scholar]
- 4.Ringenberg MR & Ward TR Merging the best of two worlds: artificial metalloenzymes for enantioselective catalysis. Chem. Commun 47, 8470–8476 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Ward TR Artificial metalloenzymes based on the biotin−avidin technology: enantioselective catalysis and beyond. Acc. Chem. Res 44, 47–57 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Jing Q & Kazlauskas RJ Regioselective hydroformylation of styrene using rhodium-substituted carbonic anhydrase. ChemCatChem 2, 953–957 (2010). [Google Scholar]
- 7.Abe S et al. Polymerization of phenylacetylene by rhodium complexes within a discrete space of apo-ferritin. J. Am. Chem. Soc 131, 6958–6960 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Key HM, Clark DS & Hartwig JF Generation, characterization, and tunable reactivity of organometallic fragments bound to a protein ligand. J. Am. Chem. Soc 137, 8261–8268 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coelho PS, Brustad EM, Kannan A & Arnold FH Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–310 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Bordeaux M, Tyagi V & Fasan R Highly diastereoselective and enantioselective olefin cyclopropanation using engineered myoglobin-based catalysts. Angew. Chem. Int. Ed 54, 1744–1748 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ortiz de Montellano P Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev 110, 932–948 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters MW, Meinhold P, Glieder A & Arnold FH Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J. Am. Chem. Soc 125, 13442–13450 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Roiban G-D & Reetz MT Expanding the toolbox of organic chemists: directed evolution of P450 monooxygenases as catalysts in regio- and stereoselective oxidative hydroxylation. Chem. Commun 51, 2208–2224 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Tyagi V, Bonn RB & Fasan R Intermolecular carbene S-H insertion catalyzed by engineered myoglobin-based catalysts. Chem. Sci 6, 2488–2494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan KH, Guan X, Lo VKY & Che C-M Elevated catalytic activity of ruthenium(II)–porphyrin-catalyzed carbene/nitrene transfer and insertion reactions with N-heterocyclic carbene ligands. Angew. Chem. Int. Ed 53, 2982–2987 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Maxwell JL, O’Malley S, Brown KC & Kodadek T Shape-selective and asymmetric cyclopropanation of alkenes catalyzed by rhodium porphyrins. Organometallics 11, 645–652 (1992). [Google Scholar]
- 17.Anding BJ, Ellern A & Woo LK Olefin cyclopropanation catalyzed by iridium(III) porphyrin complexes. Organometallics 31, 3628–3635 (2012). [Google Scholar]
- 18.Carey JR et al. A site-selective dual anchoring strategy for artificial metalloprotein design. J. Am. Chem. Soc 126, 10812–10813 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Ohashi M et al. Preparation of artificial metalloenzymes by insertion of chromium(III) Schitf base complexes into apomyoglobin mutants. Angew. Chem. Int. Ed 42, 1005–1008 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Oohora K, Kihira Y, Mizohata E, Inoue T & Hayashi T C(sp3)–H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J. Am. Chem. Soc 135, 17282–17285 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Bordeaux M, Singh R & Fasan R Intramolecular C(sp3)–H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Bioorg. Med. Chem 22, 5697–5704 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teale FW Cleavage of the haem-protein link by acid methylethylketone. Biochim. Biophys. Acta 35, 543 (1959). [DOI] [PubMed] [Google Scholar]
- 23.Lelyveld VS, Brustad E, Arnold FH & Jasanotf A Metal-substituted protein MRI contrast agents engineered for enhanced relaxivity and ligand sensitivity. J. Am. Chem. Soc 133, 649–651 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawakami N, Shoji O & Watanabe Y Single-step reconstitution of apohemoproteins at the disruption stage of Escherichia coli cells. ChemBioChem 13, 2045–2047 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Woodward JJ, Martin NI & Marletta MA An Escherichia coli expression-based method for heme substitution. Nat. Methods 4, 43–45 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Paulson DR, Addison AW, Dolphin D & James BR Preparation of ruthenium(II) and ruthenium(III) myoglobin and the reaction of dioxygen, and carbon monoxide, with ruthenium(II) myoglobin. J. Biol. Chem 254, 7002–7006 (1979). [PubMed] [Google Scholar]
- 27.DelProposto J, Majmudar CY, Smith JL & Brown WC Mocr: a novel fusion tag for enhancing solubility that is compatible with structural biology applications. Protein Expr. Purif 63, 40–49 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ilie A & Reetz MT Directed evolution of artificial metalloenzymes. Isr. J. Chem 55, 51–60 (2015). [Google Scholar]
- 29.Suematsu H, Kanchiku S, Uchida T & Katsuki T Construction of aryliridiumsalen complexes: enantio- and cis-selective cyclopropanation of conjugated and nonconjugated olefins. J. Am. Chem. Soc 130, 10327–10337 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Watson HC The stereochemistry of the protein myoglobin. Protein Stereochem 4, 299 (1969). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.