

Abstract
The neurovascular unit (NVU) is highly responsible for cerebral homeostasis and its dysfunction emerges as a critical contributor to Alzheimer's disease (AD) pathology. Hence, rescuing NVU dysfunction might be a viable approach to AD treatments. Here, we fabricated a self-regulated muti-functional nano-modulator (siR/PIO@RP) that can intelligently navigate to damaged blood-brain barrier and release therapeutical cargoes for synergetic AD therapy. The resulting siR/PIO@RP enables self-regulation of its distribution in accordance with the physio/pathological state (low/high RAGE expression) of the target site via a feedback loop. siR/PIO@RP is capable of performing intricate tasks and goes beyond the capabilities of single-target therapeutic agents utilized in AD therapy, such as reducing cerebral Aβ load, relieving neuroinflammation, and alleviating the dysfunction of NVU. Overall, the current study provides proof of concept that normalizing NVU holds promise as a means of alleviating AD symptoms.
Key words: Alzheimer's disease, Targeted delivery, Pathological blood–brain barrier, Neurovascular unit, Receptor for advanced glycation end products, Gene delivery, Anti-inflammation, Synergetic therapy
Graphical abstract
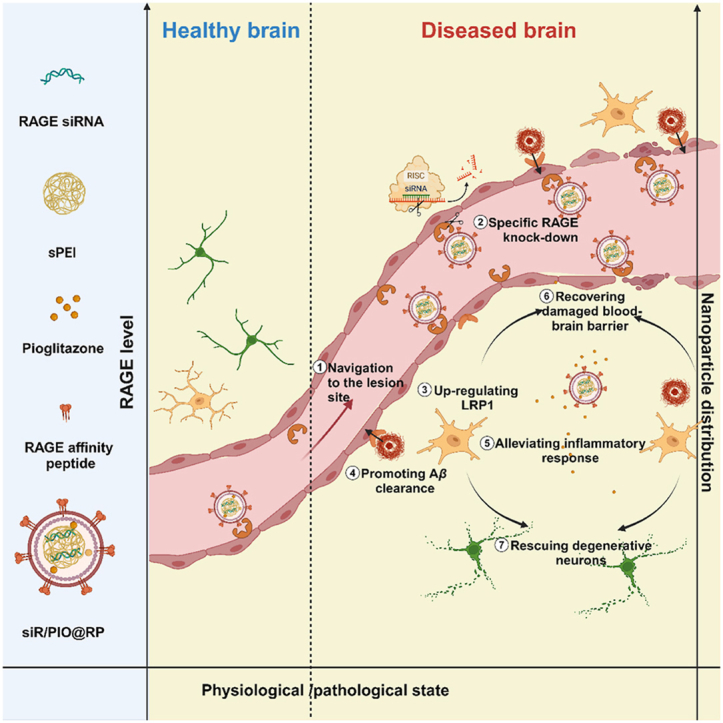
The multi-functional nano-modulator (siR/PIO@RP) preferably accumulated at lesioned brain area and rescued the impaired neurovascular unit for Alzheimer's disease therapy.
1. Introduction
Alzheimer's disease (AD), a progressive neurodegenerative disorder, is officially listed as the top ten leading causes of death over the years1,2. Despite a large monetary investment in research on AD treatment, the current therapeutic effect is still far from satisfactory. A study revealed that 99% of Alzheimer's drug trials failed, among the highest rate for all diseases3. Given the complex and ambiguous pathology of AD, the road to a cure is paved with obstacles4,5. Therefore, developing new recipes grasping the crucial links and restoring normal brain function during AD progression are urgently needed.
Recently, the neurovascular unit (NVU) dysfunction has gradually come under spotlight as a hypothesis of AD, suggesting that faulty clearance of amyloid β peptide (Aβ) through blood–brain barrier (BBB) and senescence of brain vascular system initiates permanent loss of neuronal function and cognitive impairment6, 7, 8. Multiple studies revealed that trans-BBB transport serves as a primary pathway for the removal of Aβ, accounting for 70%–85% of Aβ clearance9. In this process, Aβ binds to LRP1 (Low-density lipoprotein receptor-related protein 1), which is mainly expressed at the abluminal side of the BBB, and triggers phosphatidylinositol binding clathrin Assembly Protein (PICALM)/clathrin-dependent endocytosis of Aβ-LRP1 complexes and then Aβ-containing endocytic vesicles are guided towards exocytosis, ultimately leading to Aβ transcytosis10. However, a significant reduction in LRP1 expression is found during aging and AD progression and correlates with an increase of Aβ in the brain11. Furthermore, emerging evidence also demonstrates that there is a concomitant increased expression of receptor for advanced glycation end-products (RAGE) in suffered micro-vessels during the development of AD12,13. Besides functioning as the major Aβ influx receptor of BBB that accelerates brain Aβ accumulation, RAGE interacts with Aβ and amplifies a cascade of oxidative stress and inflammatory response, which leads to cerebrovascular damage and ultimately neurodegeneration14, 15, 16, 17, 18, 19. In addition to the pathological alterations of Aβ-related transporters, BBB breakdown is also a major risk factor for AD. The progressive BBB disruption increases vascular permeability and allows exogenous toxic substance to enter the brain, which triggers inflammation and oxidative stress, worsening the process of neurodegeneration20. Overall, the aforementioned evidence supported that cerebrovascular insults, including alterations in transport properties, a loss of tight junction integrity, and inflammatory reactions, could lead to the deterioration of AD. Considering all the pathological cerebrovascular alterations, it is plausible to conclude that the diseased BBB-targeted modulation strategy can be a promising remedy for AD treatment. However, the long-existing dilemma faced by traditional brain targeting systems, namely the potential risk caused by undifferentiated widespread distribution and extensive interaction with brain tissue, remains a tough nut to crack. Herein, we speculate that nanomedicine can provide a way out of such a dilemma by specifically targeting the damaged BBB and intelligently self-regulating its accumulation in targeted brain areas according to their physio/pathological state21.
To verify our hypothesis, we herein developed a multifunctional self-regulated nano-modulator by loading RAGE siRNA (siRAGE) and pioglitazone into a RAP (RAGE affinity peptide)-decorated GSH-responsive lipopolyplex (siR/PIO@RP) (Scheme 1). Considering the overexpression of RAGE on cerebro-vasculature during AD development, RAP, a peptide generated from a phage display library, that can bind to RAGE22, was selected as a motif to facilitate intelligent navigation of the damaged cerebral vasculature. RAGE-specific gene downregulation therapy based on small interfering RNA technology (siRNA) offered promising therapeutics for repairing cerebrovascular injuries by directly blocking causative gene expression with high targeting specificity. Meanwhile, given the dynamic expression of RAGE in different states (low expression in the healthy brain while high expression in the diseased brain), the built-in feedback loop with RAGE as a cornerstone enables self-regulatory distribution based on the physiological and pathological status of the target site via the interplay between RAP and RAGE. Besides, to combat challenges facing gene delivery, a GSH (glutathione)-sensitive lipopolyplex was fabricated. The contained disulfide-bonded cationic complexes could condense siRNA with high efficacy, promote lysosome escape, and facilitate siRNA release in intracellular redox environments, thus benefiting the efficacy of siRNA. What's more, a lipid layer that could mask positive charge and prolong circulating time21 was also implemented for further in-vivo application. Pioglitazone, a peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist, was also utilized in our system for its ability to promote Aβ clearance by upregulating LRP123. It was also proved that pioglitazone could rescue cerebrovascular function and selected markers of AD neuropathology (oxidative stress, inflammatory reaction, etc.)24. Overall, we assumed that our multifunctional nano-modulator (siR/PIO@RP) bearing self-regulation ability might specifically modulate the impaired cerebrovascular system during the occurrence and progression of AD. As we excepted, siR/PIO@RP sufficiently accumulated along diseased cerebral microvasculature, rescued the structure and function of NVU, reversed AD-related transcriptomic alterations, and demonstrated cognitive improvement in AD model mice, providing proof of concept that attenuating NVU dysfunction is conducive to AD treatment.
Scheme 1.

The scheme of siR/PIO@RP establishment and therapeutical mechanism. The multi-functional nano-modulator (siR/PIO@RP) preferably accumulated at lesioned brain area through the bind affinity between RAGE affinity peptide (RAP) and high expressed RAGE and rescued the impaired neurovascular unit for AD therapy.
2. Materials and methods
2.1. Materials
1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[succinimidyl succinate ester(polyethylene glycol)], was purchased from Ponsure Biological Co., Ltd. (Shanghai, China). Sphingomyelin was purchased from Jiuding Chemical Co., Ltd. (Shanghai, China). Polyethylenimine was purchased from RHAWN, and cholesterol and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased from AVT (shanghai) Pharmaceutical Tech Co., Ltd. (Shanghai, China). 1,2-Dioleoyl-sn-glycero-3-phospho-l-serine (DOPS), was purchased from Bide Pharmatech Co., Ltd. (Shanghai, China). Lys-Ala-Pro-Asp-Thr-Lys (Dde)-Thr-Gln was synthesized by Sangong Biotech (Shanghai, China). The RAGE siRNA1 (sense:GGAAGGAGGUCAAGUCCAATT, antisense: UUGGACUUGACCUCCUUCCTT), RAGE siRNA2 (sense: GCACUUAGAUGGGAAACUUTT, antisense: AAGUUUCCCAUCUAAGUGCTT) RAGE siRNA3 (sense: CCUCAGGUCCACUGGAUAATT, antisense: UUAUCCAGUGGACCUGAGGTT), RAGE siRNA4 (sense: GCCAGAAAUUGUGGAUCCUTT, antisense: AGGAUCCACAAUUUCUGGCTT) and negative control siRNA (sense: UUCUCCGAACGUGUCACGUTT, antisense: ACGUGACACGUUCGGAGAATT) were designed and purchased from Sinotech Genomics company (Shanghai, China). Amyloid-β (1–42) peptide (Aβ1‒42) and FITC-amyloid-β (1–42) peptide (FITC-Aβ1‒42) were purchased from GL Biochem Ltd. (Shanghai, China). The anti-amyloid oligomers antibody and anti-MAP2 antibody were purchased from Abcam (Cambridge, UK). The anti-RAGE antibody was purchased from Abclonal and the anti-ZO-1 antibody was obtained from Proteintech (Wuhan, China).
2.2. Synthesis of sPEI and DSPE-PEG-RAP
The synthesis was performed following the reported method with some modifications25. The pH of PEI800 (500 mg) aqueous solution was adjusted to 7.2 by dropwise adding 0.5 mol/L HCl. After the removal of water by evaporation, the yellow solid thus obtained dissolved in 30 ml of methanol in the bottleneck flask and the sample was purged with nitrogen 3 times. A calculated amount (5 times molar excess to PEI800) of propylene sulfide was added and the solution was reacted at 60 °C for 24 h. The content was evaporated to dryness under reduced pressure and taken up in methanol, followed by precipitation in cold diethyl ether twice. The obtained precipitate was dissolved in DMSO and stirred at room temperature for 48 h. After that, the product was purified by dialysis against water (MWCO 3500), lyophilized, and the chemical structure was confirmed by 1H NMR (in D2O, 400 MHz): 1H NMR (300 MHz, D2O): δ 3.14–2.66 (m, NCH2CH2N, NCH2CHMeS), 1.25–1.05 (m, CH3).
For the synthesis of DSPE-PEG-RAP, DSPE-PEG2000-NHS was dissolved in DMF at 10 mg/mL and then added with 1.3× molar RAP peptide and 2× molar excess trimethylamine (TEA). The solution was stirred at 40 °C under nitrogen for 24 h. After the reaction, the mixture was dialyzed against DMSO for 12 h and then against deionized water for another 24 h. The final product was obtained by freeze-drying and turned to time-of-flight mass spectrometry for successful structure verification.
2.3. Preparation and characterization of polyplexes with different weight ratios
The siRNA and sPEI were respectively dissolved in DEPC water to prepare stock solutions with a concentration of 1 mg/mL and 20 μmol/L. According to the preset weight ratios (w/w = 0, 0.3, 0.5, 1, 2, 3, 4), sPEI solution with various volumes was added to 2 nmol siRNA and then the mixture was incubated at room temperature for 20 min.
To confirm the siRNA encapsulation efficiency and GSH-responsiveness of polyplexes, a gel electrophoresis assay was carried out. Two percent of agarose in TAE buffer was subjected to microwave heating for dissolution and then GelRed was added as a nucleic acid indicator. Samples with different weight ratios were loaded and transferred for 20 min electrophoresis at 100 V. For the GSH-responsiveness test, the complexes were treated with 10 mmol/L GSH at 37 °C for 2 h and electrophoresis was performed as above-mentioned.
2.4. RAGE knockdown efficiency assessments of polyplexes
For RAGE knockdown efficiency assessment, 200,000 bEnd.3 cells in 6-well plates were treated with polyplexes containing 50 nmol RAGE siRNA for 48 h, with branched polyethyleneimine (PEI, 25 kDa) as a positive control and then harvested. PCR was performed as described previously26,27. Bands were incubated with primary antibody dilution overnight, followed by incubation with secondary antibody dilution for 2 h. Finally, bands were visualized with ECL detection reagents. All antibodies were diluted according to manufacturers’ recommendations.
2.5. Preparation and characterization of siR/PIO@RP
According to a previous report, RAP-modified pioglitazone-loaded lipopolyplex was prepared by reverse evaporation method26,28. Various lipids and pioglitazone were dissolved either in chloroform, ethanol, or methanol-ethanol (v/v = 1:1) with a concentration of 4 mg/mL. The organic phase was obtained by mixing cholesterol, sphingomyelin, DOPC, DOPS, DOPE, DSPE-PEG2000, DSPE-PEG2000-RAP, and pioglitazone were added drop by drop to the water phase. The mixture was transferred to ultrasonication for 5 min in an ice bath and the organic phase was removed using a rotary evaporator to get lipopolyplex solution. The siRAGE was mixed with sPEI for 30 min. Then the mixture was added to the lipopolyplex solution and incubated for 4 h at room temperature. The Dynamic light scattering (DLS) was performed to assess the size and zeta potential of siR/PIO@RP and the TEM was utilized to observe the morphology of siR/PIO@RP. To further determine DLC (%) and EE (%) of pioglitazone, HPLC analysis was carried out. The release process of pioglitazone was investigated via the dialysis method. Briefly, PIO@RP solution (1 mL, containing 1 mg pioglitazone) was sealed in a dialysis bag (MWCO = 2000 Da) and dialyzed against 10 mL of PBS containing 1% tween 80 at different pH (pH = 7.4 or 5.0). At predetermined time intervals, the dialysate was withdrawn for HPLC analysis.
2.6. Experimental cells and animals
The bEnd.3, BV2 and TC1 cell lines were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Mouse primary hippocampal neurons were purchased from Wuhan Warner Biotechnology Co., Ltd. (Wuhan, China). APP/PS1 mice were purchased from the Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). The animals were maintained in the specific pathogen-free animal facility at 22 ± 2 °C on a 12 h light–dark cycle with free access to food and water. All experimental procedures were executed according to the protocols approved by the Ethics Committee of Sichuan University (license No. K2022012).
2.7. Aβ1–42 oligomer preparation
Lyophilized human Aβ1–42 monomer peptide (GL Biochem, Shanghai, China) purified by HPLC (>95%) was used to prepare Aβ1‒42 oligomer as earlier described with slight modifications. Aβ1–42 was initially dissolved to Aβ1‒42 monomer dissolved in dry DMSO to a concentration of 2 mmol/L and was further diluted into ice-cold Opti-MEM to bring the peptide to a final concentration of 100 μmol/L. After vortexing for 30 s, the stock solution was incubated at 4 °C for 24 h before utilization.
2.8. Cell uptake and lysosomal escape assay
For cell uptake assay, bEnd.3 cells were seeded onto glass slides and pre-cultured by 10 μmol/L Aβ oligomer for 24 h. After that, the culture medium was removed and washed with PBS (pH = 7.2) twice. FITC-labeled nanoparticles were prepared and diluted in FBS-free medium to a concentration of 100 ng/mL. Then FITC-RP and FITC-NP were added to the plate respectively. After 1 h incubation, the cells were washed and stained with DAPI for 5 min to label the nuclei. Cells were further washed with PBS and fixed with 4% PFA before CLSM observation. As for lysosomal escape assay, bEnd.3 cells were plated onto glass slides. siRNA and lysosome were respectively labeled with FITC and lysotracker red DND99 for easy trace.
2.9. Cytotoxicity assay
To measure cell viability, MTT assay was performed. Specifically, bEnd.3 cells were seeded in 96-well plates and were administrated with different treatments. After that, 10 μL MTT (10 mg/mL, diluted in PBS; Sigma) was added to each well and fully mixed. After 4 h incubation at 37 °C, the medium was gently removed and replaced by 100 μL DMSO. After gently shaking the plate for 1 h at 37 °C. Absorbance was measured at 570 nm by a microplate reader. The relative cell viability was expressed as the absorbance relative to the control group.
2.10. Hemolytic test
First, the fresh blood of mice was collected into anticoagulant tubes and diluted with PBS solution followed by centrifugation at 2000 rpm for 5 min (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Then the supernatant was removed and the centrifugation process was repeated 3–5 times until the supernatant was colorless. The red blood cells in the bottom were suspended in PBS (pH 7.4) at a final concentration of 2%. The red cell suspension was respectively incubated with purified water (positive control), PBS buffer solution (negative control), and nanoparticles of different concentrations at 37 °C for 24 h. After centrifugation at 2000 rpm for 5 min, the photo was captured and supernatant was taken and its absorbance at 490 nm was measured. The hemolysis ratio was calculated as follows Eq. (1):
| Hemolysis rate (%) = [(ODsample‒ODnegative)/(ODpositive‒ODnegative)] × 100 | (1) |
2.11. Alleviating intracellular ROS
BV2 cells were planted in 12-well plates (80,000/well) and incubated with different formulations for 48 h and then Aβ for 24 h. After that, the medium was removed and treated with FBS-free medium containing 10 μmol/L DCFH-DA for 30 min at 37 °C. Then, cells were harvested and rinsed with PBS for three times. The fluorescent intensity was obtained by flow cytometer (FCM, Agilent NovoCyte, Palo Alto, USA).
2.12. Western blot
Cells or brain tissues were collected and lysed in RIPA containing 1 mmol/L PMSF buffer for protein expression analysis. Equivalent amounts of protein were loaded to the wells of sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by film transfer operation. Then the nitrocellulose membrane (Millipore) was blocked with 5% fat-free milk for 2 h at 37 °C and was incubated with primary antibodies overnight at 4 °C. The membranes were washed 5 times with TBST and then incubated with a secondary antibody (LI-COR, Lincoln, Nebraska, USA) for 2 h at room temperature. The images of Western blot bands were captured using the Odyssey infrared fluorescence imaging system (LI-COR, Lincoln, Nebraska, USA).
2.13. Quantitative real-time PCR
Total RNA was isolated from cultured bEnd.3 or brain tissues using a total cell/animal RNA kit (Foregene, Chengdu, China). RNA concentrations were evaluated by micro nucleic acid and protein quantifier. The Hifair® Ⅲ 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus) and Hieff® qPCR SYBR Green Master Mix (Low Rox Plus) (Yeasen, Shanghai, China) were employed to perform RT-qPCR according to the manufacturer's protocol.
2.14. BBB permeability measurements
The in vitro BBB barrier function was investigated by measuring the leakage of Na-F across endothelial cell monolayer, following a previously described protocol14. Briefly, bEnd.3 cells were cultured on 3 μm polycarbonate membrane transwell permeable inserts. Cells were subjected to different treatments for 2 days after achieving the desired confluence and further incubated with 10 μmol/L Aβ oligomer for 24 h to induce invitro BBB permeability. Then Hank's balanced salt solution (HBSS) was added into the basolateral compartments and the culture medium in the inserts was replaced by 0.3 mL HBSS assay buffer containing 10 μg/mL Na-F. 30 min later, HHBS buffer from the lower compartments were diluted measured by a microplate reader at wavelengths of 473 nm (excitation) and 535 nm (emission).
2.15. In vivo imaging in APP/PS1 model mice
Cy5-labeled siRNA was loaded into nanoparticles with or without RAP modification for further biodistribution track. Both Cy5-siR@NP and Cy5-siR@RP were injected intravenously into 8-month-old APP/PS1 mice. According to a previous report29, mice were sacrificed at 4 h post-injection to collect major organs and imaged ex vivo, and then fluorescence intensity was quantified by Lumina III Imaging System (PerkinElmer, Waltham, USA). The brains were fixed in 4% paraformaldehyde for 24 h and then followed by programmed dehydration before the frozen section was performed. The sections were stained with DAPI to show nuclei and were immediately observed under a fluorescence microscope (Leica, DMi8, Weztlar, Germany). To further determine the co-localization of different nanoparticles with RAGE, immunofluorescence staining was performed on RAGE, and images were captured by an inverted fluorescence microscope (Leica).
2.16. Drug treatment and memory evaluation
APP/PS1 transgenic mice were randomly divided into five groups and intravenously administrated with PBS, siR@RP, PIO@RP siR/PIO@NP and siR/PIO@RP. Wilde-type C57 mice were used as healthy controls. The mice were treated every 5 days at a dose of 0.5 mg/kg siRNA and 1 mg/kg pioglitazone. Mice were weighed every 5 days for toxicity assessment. After seven sessions of treatment, the Morris water maze test was utilized to evaluate the memory function. The round pool was set in a quiet test room with constant light during the experiment. The pool was evenly divided into four quadrants and each quadrant was marked with a different geometric Figure.
In the 3rd quadrant, a circular platform was positioned at the center, and the water level was kept at 1 cm higher than the platform. To prevent the mice from detecting the underwater platform, the water was stained with titanium dioxide. Training trials were conducted for four consecutive days before the spatial exploration test, where the platform was removed. Each mouse was released into the water from the opposite side of the original platform quadrant. The time spent in the target quadrant (the quadrant where the platform was originally placed), the number of times the animal entered the platform at the quadrant, and the escape latency (time from water entry to platform arrival for mice) were recorded as indicators of spatial memory.
2.17. Mechanism experiments after treatment
After the behavioral experiment, mice were sacrificed and brains were harvested for histological analysis, and gene and protein expression analysis. For histological analysis, the tissues were fixed in a 4% PFA fix solution and embedded in paraffin. Then paraffin-embedded tissues were sectioned into slices and subjected to HE and Nissl staining. Immunofluorescent staining of Aβ, ZO-1, and MAP2 was executed following the manufacturer's protocols. For protein expression analysis, brains were homogenized by a high-speed tissue grinder (Servicebio, Wuhan, China) to extract protein for further Western blot and ELISA assay. For gene analysis, total brain RNA was isolated and used for further RT-qPCR and transcriptome assay (PANOMIX Biomedical Tech Co., Ltd., Suzhou, China).
2.18. Evaluation of systemic toxicity
During the treatment, all mice were weighed once every three days. The weight changes of each group were counted after administration. Blood samples were taken from all mice after the MWM test, and then blood biochemical indexes were detected. The cumulative toxicity of the nanoparticles was observed by H&E staining on the visceral slices of mice after treatment.
2.19. Statistical analyses
The results are expressed as the mean ± SD, indicated by error bars in each graph. For statistical comparisons between two groups, Student's t-test analysis was applied, while for statistical comparisons between samples of three groups and above, one-way ANOVA Dunnett's test was used. For statistical analyses, P < 0.05 was considered statistically significant.
3. Results
3.1. Characterization of siR@sPEI and siR/PIO@RP
To synthesize a redox-sensitive polymer, low molecular weight (LMW) polyethyleneimine PEI600 was linked using disulfide-crosslinking (PEI-SS-PEI, named sPEI) by a reported method with some modifications25. Detailed information is provided in the Supporting information Fig. S1. The chemical structure of polymers was confirmed by 1H NMR (Supporting information Fig. S2). The DSPE-PEG-RAP was obtained and verified by time-of-flight mass spectrometry (Supporting information Fig. S3). Then, the ideal weight ratio was determined by both gel electrophoresis and gene transfection efficiency assessments. As demonstrated via gel retardation assays (Fig. 1A), sPEI showed comparable gene loading capacity with that of traditional gene transfection vector polyethyleneimine 25K (PEI25K) but showed lower toxicity (Supporting information Fig. S4). sPEI could completely constrain siRNA when the weight ratio was fixed at 1:1 or greater, while unmodified LMW PEI600 couldn't form complete complexation at a higher weight ratio of 4. To exert the optimal pharmacological effects of siRNA, the sequence of RAGE siRNA was screened by real-time PCR. The inhibitory efficiency of siRAGE-3 was approximately 100% in BV2 and 50% in TC1 cells (Supporting information Figs. S5A and S5B), ranked the best among all candidates. Therefore, siRAGE-3 was selected as the siRNA drug for the subsequent experiments. For further investigation of the most appropriate weight ratio, the silencing efficacy of sPEI and siRNA polyplexes at different weight ratios was explored, and the weight ratio of one demonstrated the best silencing effect with ∼40% down-regulation efficiency of RAGE in bEnd.3 (Fig. 1B), which was significantly higher than that of conventional vehicles PEI25K. Therefore, sPEI and siRNA polyplexes with a weight ratio of one were used for further experiments.
Figure 1.

Characterization of nanoparticles. (A) Gel retardation assay of various polyplexes at different weight ratios. (B) Representative PCR analysis of RAGE downregulation in bEnd.3 cells were treated with siR@sPEI at different weight ratios. NC, scrambled siRNA was loaded and used as a negative control. Data are presented as mean ± SD (n = 3). ∗P < 0.05, ∗∗P < 0.01, One way ANOVA Dunnett's test. Gel retardation assay of siR@sPEI and siR@PEI25K with or without GSH incubated when weight ratio was fixed at 1. (C) TEM images showing the morphic changes of siR@sPEI incubated with or without GSH, scattered fragments are shown by red arrows. (D) Hydration particle size and TEM image of siR/PIO@RP. Scale bar = 100 nm. (E) Zeta potential of different formulations. (F) Cellular uptake of FITC-RP inhibited by RAGE inhibitor FPS-ZM1 (5 × 10−6 mol/L) for 1 h in BV2, Data are presented as mean ± SD, n = 3, ∗∗P < 0.01 by two-tailed t-test. (G) Confocal imaging of cellular uptake of FITC-NP, 10:1 FITC-siR@RP 10:2 FITC-siR@RP 10:4 FITC-siR@RP by bEnd.3 cells. Scale bar = 50 μm. (H) Confocal Semi-quantification of the cellular uptake of FITC-NP, 10:1 FITC-siR@RP 10:2 FITC-siR@RP 10:4 FITC-siR@RP by bEnd.3 cells, n = 4, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001 by One way ANOVA Dunnett's test. (I) Lysosome escape test, scale bar = 25 μm. The X axis represents distance and the Y-axis represents fluorescence intensity.
In our design, the disulfide bond in sPEI would be cleaved at high glutathione (GSH) levels within the intracellular redox environment. Then, the siR@sPEI nanostructure is destroyed, resulting in siRNA release, which could be beneficial in terms of improving gene silencing efficacy. Therefore, the reductive responsiveness of the sPEI was investigated. As shown in Fig. 1C, the spherical structure was mostly broken and showed a disintegrating form after GSH incubation, resulting in the formation of many scattered fragments in the background. Also, obvious change was observed in siR@sPEI group after incubation with 10 mmol/L GSH (Supporting information Fig. S6A), while no such change was found in its GSH non-responsive control. Similarly, as shown in the agarose gel experiment, after 2 h incubation with 10 mmol/L GSH, the siRNA presented a tendency to release from the polyplexes at a weight ratio of 1, while under non-reducing conditions, the siRNA could not be released (Supporting information Fig. S6B), furthermore, there was no sign of the responsive release of siRNA in the control group. These results confirmed our hypothesis that our polyplexes could condense siRNA with high efficacy and exhibit the ability to release siRNA in response to GSH.
To meet the requirement for systematic administration, the positive charge of siR@sPEI polyplexes should be shielded to endow the delivery system with stability and longer circulation characteristics. Therefore, a pioglitazone-loaded lipid layer was utilized and the detailed preparation process is shown in Supporting information After decoration, siR/PIO@RP with a size of 148.4 ± 7.8 nm was obtained (Fig. 1D), and transmission electron microscopy (TEM) images demonstrated a typical lipid-vesicle structure (Fig. 1D). In addition, the zeta potential of siR/PIO@RP decreased from +18.4 ± 2.1 to −17.9 ± 1.3 mV, further confirming the successful coating of lipid layer (Fig. 1E). The size of siR/PIO@RP remained almost unchanged within 24 h in PBS and in 10% serum (Supporting information Fig. S7), and the PDI remained below 0.3, suggesting its good stability. The hemolysis ratio was lower than 2% within a wide range of concentrations, which means the lipid layer coating increases biosafety, thus facilitating in vivo administration (Supporting information Fig. S8). The DLC and EE rate of pioglitazone in siR/PIO@RP were 3.65% and 76.61%, respectively (Supporting information Fig. S9A). What's more, the amount of pioglitazone released from siR/PIO@RP was increased in acid pH compared with that in neutral pH, indicating that pioglitazone could be released from the carrier in lysosome after uptake by BBB endothelial cells (Supporting information Fig. S9B).
It was reported that the expression of RAGE is upregulated on the diseased BBB in AD patients, which might exacerbate the disease progression30,31. We thus investigated whether the RAP modification can facilitate the delivery of therapeutical components to diseased sites. According to previous reports31,32, we constructed a diseased BBB model by pre-incubating the bEnd.3 cells with 10 μmol/L Aβ oligomer, where RAGE expression was upregulated by 1.5 times (Supporting information Fig. S10A‒S10C). Considering the targeting efficiency was affected by the density of RAP peptide, the cellular uptake of FITC-RP with various densities of RAP peptide was evaluated by confocal imaging (Fig. 1G and H). The cellular uptake of RAP-LP increased as the RAP weight ratio increased (Fig. 1G and H), proving that RAP modification facilitated the internalization of nanoparticles. These results were consistent with those obtained by flow cytometry (Supporting information Fig. S11). Furthermore, RAGE inhibitor FPS-ZM1 inhibited the cellular uptake of RP but not NP (Fig. 1F), indicating that RAGE played a crucial role in promoting the cellular uptake of RP. These results indicated that modification of RAP might increase the diseased BBB targeting efficacy through its high affinity with RAGE.
To exert a pharmacological effect, it is crucial for siRNA to escape from the endo/lysosome. Lysotracker red was then used to label endosomes/lysosomes27 and siRNA was labeled with FITC fluorophore for convenient tracking. The results showed that after incubation of FITC-siR@RP with bEnd.3 cells for 0.5 h, a considerable overlap was observed between red and green fluorescence, suggesting that FITC-siR@RP presented in endosome/lysosome. However, with incubating time prolonging, the red fluorescence separated from the green fluorescence (Fig. 1I). This indicated our system could help siRNA successfully escape from endosome/lysosome, hence avoiding the degradation of siRAGE by “endosome/lysosome” pathway.
3.2. In vivo targeting efficacy of RAP
As previously mentioned, the upregulated RAGE was found in diseased cerebral endothelium in AD model, where a vicious cascade was present. Therefore, we attempted to investigate whether tethering RAP to lipoplexes was more effective in increasing its accumulation in damaged brains using AD model. We first explored the biodistribution with an IVIS Lumina III Imaging System. As demonstrated in Fig. 2A and B, Cy5-siRNA loaded RP showed stronger fluorescent signals in the brain compared with unmodified NP, while such phenomenon was not found in other main organs (Supporting information Fig. S12), suggesting that RAP modification was beneficial for specific pathological brain targeting. To further confirm that RAP can promote preferable pathological brain accumulation, we examined the brain distribution using confocal imagining. Confocal microscopy analysis found that RP accumulated at a level much higher than unmodified NP both in the cortex and hippocampus (Fig. 2C). Moreover, RAGE, a marker of damaged cerebral endothelial cells was immune-stained to study the enrichment of nanoparticles around pathological brain micro-vessels. As demonstrated in Fig. 2D, the fluorescence signal of accumulated RP around RAGE was higher than NP and there was a greater degree of overlap between RP and RAGE. These results proved that RAP modification could realize a preferential target of damaged brain microvasculature.
Figure 2.

In vivo targeting efficacy of nanoparticles in AD model mice. (A) Ex vivo imaging of AD mice brains excised at 4 h post-injection. (B) The semiquantitative data of fluorescence intensity, Data are presented as mean ± SD, n = 3, P = 0.056 by Student's t-test. (C) Distribution of Cy5-siR@NP and Cy5-siR@RP in the cortex and hippocampus. Scale bar = 100 μm. (D) Colocalization of Cy5-siR@RP and Cy5-siR@NP with RAGE. Scale bar = 20 μm.
3.3. siR/PIO@RP alleviates Aβ-induced neurovascular system impairment and facilitates LRP1 mediated Aβ efflux in vitro
A previous report33 showed that Aβ oligomer could induce BBB impairment and thus increasing its permeability, which might be the cause of Alzheimer's disease. We therefore employed an Aβ oligomer co-incubated bEnd.3 monolayer-based in vitro BBB model (Fig. 3B) to investigate whether our siR/PIO@RP could reserve the increased BBB permeability by measuring the permeability of Na-F. As shown in Fig. 3C, the BBB permeability increased after incubation with 10 μmol/L Aβ1–42 for 24 h, and the treatment with siR/PIO@RP could significantly rescue BBB breakdown, rendered as a ∼30% decreased Na-F fluorescence intensity. Studies demonstrated that tight junction (TJ) scaffold proteins of endothelial cells were involved in the integrity of the BBB34, the loss of which led to BBB impairment. Therefore, we evaluated the expression of TJ protein ZO-1. The results in Fig. 3A, E vividly showed that ZO-1 expression was largely reduced by Aβ oligomer incubation. After treatment with different formulations, the expression of ZO-1 was upregulated to varying degrees, with the most obvious upregulation observed in the siR/PIO@RP group. What's more, we also evaluated the cell viability of bEnd.3 by MTT. Similarly, siR/PIO@RP could restore the decrease in cell viability caused by Aβ (Fig. 3D), suggesting its neurovascular protection effect. Except the above-mentioned gains in recovering BBB morphology and structure, multiple literatures24,35 have illustrated the critical role of peroxisome proliferator-activated receptor agonist in recovering neurovascular dysfunction through its synergistic anti-inflammatory and ROS scavenging effects with siRAGE. We continued to investigate whether our siR/PIO@RP, a typical proliferator-activated receptor agonist pioglitazone-loaded nanocomposite, could exert such an effect. The neurovascular system in AD model featured wide inflammation, which results from Aβ overload and in turn promotes Aβ deposit36, 37, 38, 39. In this regard, we measured the expression of proinflammatory factors at mRNA level in BV2. As displayed in Fig. 3G and H, the mRNA level of IL-1β and IL-6 in Aβ oligomer-treated BV2 cells increased by 3.62 times and 1.32 times compared with Aβ untreated BV2. siR/PIO@RP noticeably reduced IL-6 level and IL-1β production by 48.99% and 82.20% respectively, indicating its anti-inflammation potential. The oxidative stress alleviation capacity of siR/PIO@RP was also determined by flow cytometry and demonstrated in Supporting information Fig. S13, siR/PIO@RP could decrease fluorescence intensity of DCFH-DA in BV2 by 14% compared with Aβ-induced positive control cells, suggesting its ROS elimination property.
Figure 3.

siR/PIO@RP reduced Aβ1‒42-induced BBB impairment in vitro. (A) ZO-1 immunofluorescence (red) in bEnd.3 cells following the different treatments. DAPI (blue) was used to stain nuclei. Scale bar = 50 μm. (B) The Schematic plot of in-vitro BBB model (C) BBB permeability was determined by the sodium fluorescein (Na-F) leakage test. Data are presented as mean ± SD (n = 3). (D) Cell viability of bEnd.3 cells were analyzed by MTT assay. Data are presented as mean ± SD (n = 7). (E) Western blot result of ZO-1. (F) Western blot result of LRP1. (G, H) Levels of IL-1β and IL-6 in BV2 cells following the treatments (n = 3). Data represent the mean ± SD. One way ANOVA Dunnett's test, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
What's more, it has been unveiled that pioglitazone could upregulate LRP1, whose expression is largely reduced on damaged brain microvascular cells, thus facilitating the efflux of Aβ through the BBB40, 41, 42. The results in Fig. 3F demonstrated that all pioglitazone-loaded preparations, PIO@RP, siR/PIO@NP, and siR/PIO@RP, could upregulate LRP1 protein expression in bEnd.3 cells, and the LRP1 upregulation effect could be maintained by PIO@RP ranging from 100 nmol/L to 5 μmol/L (PIO) (Supporting information Fig. S14). To verify that pioglitazone-loaded nanoparticles could promote BBB mediated Aβ transport, Aβ uptake was measured in bEnd.3 cells within 30 min by flow cytometry. The results (Supporting information Fig. S15) demonstrated that Aβ uptake by bEnd.3 cells significantly decreased after treatment with CoCl6, which significantly reduced the expression of LRP1, and PIO@RP could increase Aβ uptake by 1.82 times, confirming that PIO-loaded nanoparticles were beneficial for Aβ efflux through LRP1-mediated pathway. All results together proved that siR/PIO@RP could alleviate Aβ-induced neurovascular system impairment and facilitate LRP1-mediated Aβ efflux.
3.4. siR/PIO@RP improves learning and memory and reduces Aβ deposit in APP/PS1 mice
In order to investigate the effect on learning and memory function, APP/PS1 mice with different administrations and WT mice as a healthy control were subjected to Morris water maze (MWM) (Fig. 4A). After four days of training, the trajectories, number of platform crossings, time spent in target quadrant, and the latency to reach the platform of mice in different groups were recorded and analyzed. As demonstrated in Fig. 4C, AD mice exhibited aimless behavior, rendered as a circular searching track, whereas the routes of other mice after treatment were more targeted. According to heatmap, the strongest signal was detected in the vicinity of the target platform for mice of siR/PIO@RP group, suggesting its learning and memory improvement potential (Fig. 4C). Beyond that, the results in Fig. 4D showed that there was a significant decrease in target platform crossing times of AD mice compared with WT mice, while siR/PIO@RP administration largely increased the target platform crossing times by 1.5 times compared to AD group. It was also shown in Fig. 4E and F that the target platform retention time of siR/PIO@RP group increased by 14.7% relative to AD group and there was a 21.7% decrease in terms of escape latency time between the two groups. In general, all results present above suggested an improvement in cognition capacity by siR/PIO@RP treatment. In addition, the body weights during treatment were monitored (Fig. 4B) and major organs of mice were harvested for hematoxylin eosin (HE) stain to assess toxicity (Supporting information Fig. S16). As indicated by results, the weight of mice remained stable after siR/PIO@RP treatment and the HE stain did not reveal substantial organ damage, suggesting the safety of our system.
Figure 4.

Behavior evaluation examined by Morris Water Maze (MWM) and Aβ clearance effect of siR/PIO@RP. (A) Schematic diagram of the experimental timeline. APP/PS1 transgenic and WT mice were administrated with different formulations via tail intravenous injection every 5 days for seven cycles. Mice were then subjected to MWM for memory evaluation. (B) The body weight changes during treatment. (C) The representative heat maps of searching paths in Morris water maze. (D) Number of platform crossings. (E) Cumulative time in the quadrant of the platform of mice. (F) Escape latency of mice in different groups after treatment, Data are presented as mean ± SD (n = 10). (G) Aβ42 level in brain homogenates after different treatments determined by ELISA Data are presented as mean ± SD (n = 7), One way ANOVA Dunnett's test, ∗∗∗∗P < 0.0001. (H) Immunofluorescence staining of Aβ plaques in the cortex and hippocampus of mice in different groups. Scale bar = 500 μm.
Substantial research demonstrated that RAGE and LRP1-associated modulation could restore Aβ in/out brain balance, effectively reducing the deposition of Aβ in the brain43,44. We investigated if siR/PIO@RP could effectively regulate the expression of RAGE and LRP1 receptors as expected. As displayed by Western blot and semiquantitative analysis (Supporting information Figs. S17A, S17B, and S17D), RAGE protein was reduced while LRP1 protein was elevated after siR/PIO@RP treatment. What's more, PCR results in Supporting information Fig. S17C showed the mRNA expression level of RAGE was also decreased by siR/PIO@RP. To further test whether siR/PIO@RP could improve Aβ clearance like previous RAGE/LRP1 modulating strategies, anti-Aβ immunofluorescence analysis and immunohistochemical staining were applied. Results in Fig. 4H, and Supporting information Fig. S18 showed that Aβ accumulation was dramatically decreased both in the hippocampus and in the cortex after administrated with siR/PIO@RP. Similar results were also observed in Fig. 4G, where the quantification data of Aβ in brain homogenates were obtained by ELISA kit, showing siR/PIO@RP could reduce Aβ level in the brain of APP/PS1 mice. Together, these results demonstrated that siR/PIO@RP could efficiently clear Aβ deposition in the brain through regulating the expression of RAGE and LRP1.
3.5. siR/PIO@RP alleviates Aβ-induced neurovascular system impairment in vivo
Previous studies found that perivascular Aβ deposits could damage the structure and function of cerebral neurovascular units, aggravating AD progression45, 46, 47. Firstly, to test whether siR/PIO@RP could rescue the structural injury of brain micro-vessels, the expression of BBB tight-junction proteins ZO-1 was obtained by immune fluorescence staining. As shown in Fig. 5A, the ZO-1 expression in the AD group was tremendously decreased compared with WT mice, which could be reversed to varying degrees after different treatments, with the siR/PIO@RP group showing the highest ZO-1 expression. Given that inflammatory factors are also responsible for neurovascular injury, we further measured the levels of proinflammatory cytokines in the brain. The level of IL-6 in siR@RP, PIO@RP, siR/PIO@NP and siR/PIO@RP treated group decreased by 39.97%, 45.17%, 48.34%, and 53.59% respectively compared to the AD group (Fig. 5B). Likewise, the TNF-α level also showed a significant decrease in the different treated group (Fig. 5C), suggesting obvious anti-inflammatory effects of our preparations. Neurons are key and active players in the neurovascular system, the normal state of which closely relates to the learning and memory function. Therefore, we labeled neurons with microtubule-associated protein-2 (MAP2) to observe the difference in the neural structure after administration. It was vividly displayed in Fig. 5D, that there was a great loss of MAP2 in the AD group, while siR/PIO@RP treatment could dramatically reverse such loss. Furthermore, we also employed Nissl staining to investigate the functional state of neurons, as Nissl bodies are accepted as functional units for the synthesis of structural proteins, enzymes, and neuromodulators31,48. Deep blue stained Nissl bodies were detected in the cortical neuron cytoplasm of the WT group, while in AD mice, dissolution of Nissl bodies occurred and translucent voids were observed in the cytoplasm of nerve cells (Fig. 5E), indicating damaged neurons. After treatment, the pathological alterations were variously restored, especially in the siR/PIO@RP group, where the structure of Nissl bodies was restored to a normal WT-like state. Similar conclusions could be drawn from the results of HE, which was demonstrated in Supporting information Fig. S19. Compared with the WT group, a massive number of neurons with deep-stained nuclei and chromatin fixation were found in the CA3 hippocampus of mice in the APP/PS1 group, and such pathological alterations were recovered after siR/PIO@RP treatment. All the above results demonstrated that siR/PIO@RP could ameliorate neuronal pathology and antagonize Aβ induced damage in mouse hippocampal neurons.
Figure 5.

siR/PIO@RP reduced neurovascular impairment in vivo. (A) Immunofluorescence of ZO-1 in the cortex. Scale bar = 20 μm. (B, C) Expression of pro-inflammatory cytokines TNF-α and IL-6 in the brain after treatments (n = 4–5). Data represent the mean ± SD. One-way ANOVA Dunnett's test, ∗P < 0.05, ∗∗P < 0.01. (D) Immunofluorescence MAP2 (dendrite) in the cortex. Scale bar = 50 μm. (E) The Nissl staining of the brain after different administrations. The red arrow shows cell where the dissolution of Nissl bodies occurs: scale bar = 50 μm.
3.6. Transcriptomic effects of siR/PIO@RP on brain transcriptome
To investigate the effects of siR/PIO@RP on gene expression in the mouse brain, we performed RNA-sequencing on mice brains from wildtype (WT), AD, and siR/PIO@RP administrated AD mice. Compared with the AD group, a total of 1338 genes were significantly altered in expression after administrated with siR/PIO@RP, where 721 transcripts were upregulated (the red dots) and 617 genes were downregulated (the blue dots) (Fig. 6D). It was also demonstrated in the heatmap of differential gene clustering (Fig. 6A) that samples in WT and siR/PIO@RP group were clustered together and shared similar expression patterns, while samples in AD group were not and showed rather different expression patterns. What's more, there was an unexpectedly large overlap between WT vs. AD and siR/PIO@RP vs. AD, with 901 shared differentially expressed genes (DEG) between the comparisons (Fig. 6E). This might indicate that administration of siR/PIO@RP to AD mice reversed the original AD-like brain transcriptomic profiles in favor of WT-like transcriptomic characteristics. To identify the main biological functions performed by the DEG after being administrated with siR/PIO@RP, we conducted the Gene Ontology (GO) analysis. GO terms of our interests, such as the nervous system development, synapse and synapse signaling, cognition and memory, cell junction, etc., were found to be enriched in the DEG entries (Fig. 6C). Since we have verified that administration of siR/PIO@RP could alter several AD-associated pathological features, we thus further explored whether these AD-related pathologic changes could be manifested from the transcriptomic level. As seen in the KEGG pathway enrichment analysis (Fig. 6B), those well-documented AD-related signaling pathways were found in the list of DEG. For example, several synapse-related pathways, mTOR signaling pathway, PI3K/AKT signaling pathway, Wnt signaling pathway, and JAK-STAT signaling pathway were enriched, all of which were previously reported to be involved in the onset and progression of AD. Consistent with results in Fig. 6A and E, and Fig. 3A, the tight junction was found on the list. What's more, the enrichment of the AGE-RAGE signaling pathway and PPAR signaling pathway were also observed, which might be attributed to the pharmacologic effects of RAGE siRNA and pioglitazone. Overall, the aforementioned results supply transcriptomic proof that supports the effectiveness of siR/PIO@RP as a treatment for AD.
Figure 6.

In vivo transcriptomics analysis of APP/PS1 mice brains after therapy. (A) Heat map of differentially expressed genes (DEGs) in WT, APP/PS1, and siR/PIO@RP treated group. (B) KEGG pathway enrichment analysis of differentially expressed genes between APP/PS1 and siR/PIO@RP treated group. (C) Bar plot of GO enrichment analysis of genes between APP/PS1 and siR/PIO@RP treated group. Biological process, BP; Molecular function, MF; Cell component, CC. (D) Volcano map of DEGs between APP/PS1 and siR/PIO@RP treated group. ∗P < 0.05. |Fold change| ≥ 2. (E) Venn diagram of the overlap of DEGs in WT versus APP/PS1, APP/PS1 versus siR/PIO@RP, and WT versus siR/PIO@RP.
4. Discussion
AD is a troublesome neurodegenerative disorder with barely any effective therapeutics to halt its development due to complex pathogenesis49. The extracellular accumulation of amyloid-β plaques and aggregation of hyperphosphorylated tau protein are widely accepted as key causative factors for AD10. Despite all the efforts, strategies focused on interrupting the accumulation of Aβ and tau fail to achieve satisfying results. Poor BBB penetration has long been cited as the reason for poor drug responsiveness, and multiple existing approaches have attempted to improve brain penetration rate. For example, a glycosylated, siRNA-loaded nanomedicine (Gal-NP@siRNA) was developed to efficiently penetrate the blood-brain barrier (BBB) via glycemia-controlled glucose transporter-1 (Glut1) for AD treatment50. In the works of Guo group51, a BBB-penetrating peptide TGN was utilized to improve brain drug delivery. However, growing evidence suggests that BBB should not be viewed simply as an obstacle to intracerebral drug delivery, but rather as a potential target for AD treatment. Multiple investigations have illuminated that BBB breakdown usually precedes the occurrence of initial symptoms in AD and will aggravate as the disease progresses20,49. Disrupted transport and barrier properties of BBB, as well as deteriorating inflammatory microenvironment, are culpable for AD development.
Here, a self-regulated muti-functional nano-modulator (siR/PIO@RP) was fabricated. Our nanomedicine formulations enable the co-loading of pioglitazone and siRAGE, which could also be applied to the co-delivery of other chemical drugs and short nucleic acids. We demonstrated that the engineered siR/PIO@RP exhibited a vesicle-like structure and maintained high stability in various media, which is critical for in vivo application. In order to exert gene silencing effect, siRNA must escape from lysosomes and be released into the cytosol52. To this end, the disulfide-linked PEI (sPEI) with proton sponge effect was employed in our system. Results confirmed the admirable lysosome escape ability and GSH-sensitivity of our system. Furthermore, to improve the specific distribution of drugs at the lesion site, we incorporated RAP, a RAGE-targeting peptide into the nano-system. With high RAGE binding affinity, RAP bestowed increased accumulation around damaged brain blood vessels and intelligent self-regulation of distribution according to the physio/pathological state of the micro-environment. From this aspect, it may reduce the potential safety risks caused by widespread, non-specific distribution. After being treated with siR/PIO@RP, the abnormal upregulated RAGE and downregulated LRP1 were reversed both in vitro and in vivo, as well as the inflammatory microenvironment. What's more, the administration of siR/PIO@RP also leads to added benefits of reduced Aβ burden and the restored structure and function of neurons in APP/PS1 mice. Noticeably, the mechanisms underlying the in vivo therapeutic effects were first investigated through transcriptomic analysis. This revealed the possibility of multiple signaling pathways being involved, including the AGE-RAGE signaling pathway, the NF-kappa B signaling pathway, the PPAR signaling pathway, the JAK-STAT signaling pathway, and the neurotrophy signaling pathway. These findings were consistent with earlier reports53, 54, 55. It has been shown previously that RAGE mediated Aβ influx and enhanced Aβ accumulation, which in turn promote Aβ/RAGE interaction, activate NADPH oxidase-2 (NOX-2), and set off a series of events that accelerate the progression of AD. For example, endothelial was activated by NF-κB, accompanied by the secretion of proinflammatory cytokines and loss of TJ protein (e.g.) expression53. Therefore, we assumed that our nanomedicine could rescue TJ loss and relieve neuroinflammation through downregulating RAGE expression, which was attained by siRNA. We reasoned that by using siRNA to downregulate RAGE expression, our nanomedicine could prevent TJ loss and alleviate neuroinflammation. In-depth studies on PPAR agonists have also revealed their function in controlling neuro-inflammation, indicating that PPARs are important modulators of gene expression and interact with multiple signaling pathways, including AMPK, JAK/STAT, NF-κB, and MAPK56, 57, 58. Furthermore, PPARγ agonists have neuroprotective effects on the central nervous system (CNS), such as insulin sensitization and brain metabolite homeostasis. Neurite outgrowth and neuronal differentiation can be induced by PPARγ agonist55. Based on these results, it is reasonable to hypothesize that pioglitazone, a selective PPARγ agonist, may perform its intended function of mending NVU through anti-neuro-inflammation and enhancing metabolism and neurotrophic effect. Considering the versatile function of siR/PIO@RP, this nano-system might provide a proof of concept that a damaged neurovascular system displays a desired target for efficient AD therapy.
5. Conclusions
In this work, we developed a self-regulated muti-functional nano-modulator (termed siR/PIO@RP) to intelligently regulate diseased BBB for AD therapy. The modified RAGE affinity peptide (RAP) allows navigation to damaged BBB, where RAGE presents a high level. Subsequently, siRNA is released in response to elevated intracellular GSH levels, thereby effectively silencing RAGE and interrupting the RAGE/Aβ vicious cycle. Moreover, the co-loaded pioglitazone acts with RAGE siRNA to achieve added benefits such as anti-inflammation, damaged BBB restoration, and neurovascular protective effects, which are confirmed by in-vitro and in vivo results. Interestingly, it makes sense to know that after therapy, RAGE returns to its low expression level, removing the nano-modulator's ability to target the brain and potentially reducing pharmacogenetic toxicity. Collectively, these obtained results indicated that a multi-dimensional therapeutic strategy targeting the diseased neuro-vasculature seems to be a promising way out of the current dilemma in AD therapy.
Acknowledgments
The work was supported by Research and Development Program of Science and Technology Department of Sichuan Province (2022JDJQ0050, China), 111 Project (B18035, China) and the National Natural Science Foundation of China (No. 82373801, China).
Author contributions
Xue Xia: Writing – review & editing, Writing – original draft, Investigation. Ya Wei: Investigation. Qianqian Huang: Investigation. Yang Zhou: Investigation. Xiaorong Wang: Investigation. Yulong Shi:Investigation. Xiaotong Yang: Investigation. Wenqin Yang: Investigation. Yiwei Zhang: Investigation. Ting Lei: Investigation. Yuan Huang: Investigation. Hanmei Li: Writing – review & editing. Meng Qin: Writing – review & editing. Huile Gao: Conceptualization, Writing – review & editing.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2024.05.017.
Contributor Information
Meng Qin, Email: qinmeng212@scu.edu.cn.
Huile Gao, Email: gaohuile@scu.edu.cn.
Appendix A. Supporting information
The following is the Supporting information to this article:
References
- 1.2022 Alzheimer's disease facts and figures. Alzheimers Dement. 2022;18:700–789. doi: 10.1002/alz.12638. [DOI] [PubMed] [Google Scholar]
- 2.Jiang Y.F., Zeng Z.Y., Yao H.J., Guan Y., Jia P.P., Zhao X.L., et al. Treatment of Alzheimer's disease with small-molecule photosensitizers. Chin Chem Lett. 2023;34 [Google Scholar]
- 3.Moutinho S. The long road to a cure for Alzheimer's disease is paved with failures. Nat Med. 2022;28:2228–2231. doi: 10.1038/s41591-022-02062-0. [DOI] [PubMed] [Google Scholar]
- 4.Cheng G.W., Xie A.H., Yan Z., Zhu X.Z., Song Y.F., Chen T.K. Nanomedicines for Alzheimer's disease: therapies based on pathological mechanisms. Brain-X. 2023;1:e27. [Google Scholar]
- 5.Zeng H., Qi Y., Zhang Z., Liu C., Peng W., Zhang Y. Nanomaterials toward the treatment of Alzheimer's disease: recent advances and future trends. Chin Chem Lett. 2021;32:1857–1868. [Google Scholar]
- 6.Zlokovic B.V. Vascular disorder in Alzheimer's disease: role in pathogenesis of dementia and therapeutic targets. Adv Drug Deliv Rev. 2002;54:1553–1559. doi: 10.1016/s0169-409x(02)00150-3. [DOI] [PubMed] [Google Scholar]
- 7.Zlokovic B.V. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Wei Y., Xia X., Li H.M., Gao H.L. Influence factors on and potential strategies to amplify receptor-mediated nanodrug delivery across the blood–brain barrier. Expet Opin Drug Deliv. 2023;20:1713–1730. doi: 10.1080/17425247.2023.2245332. [DOI] [PubMed] [Google Scholar]
- 9.Nelson A.R., Sweeney M.D., Sagare A.P., Zlokovic B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta. 2016;1862:887–900. doi: 10.1016/j.bbadis.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Z., Sagare A.P., Ma Q., Halliday M.R., Kong P., Kisler K., et al. Central role for PICALM in amyloid-β blood‒brain barrier transcytosis and clearance. Nat Neurosci. 2015;18:978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donahue J.E., Flaherty S.L., Johanson C.E., Duncan J.A., Silverberg G.D., Miller M.C., et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 12.Deane R., Singh I., Sagare A.P., Bell R.D., Ross N.T., LaRue B., et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silverberg G.D., Miller M.C., Messier A.A., Majmudar S., Machan J.T., Donahue J.E., et al. Amyloid deposition and influx transporter expression at the blood‒brain barrier increase in normal aging. J Neuropathol Exp Neurol. 2010;69:98–108. doi: 10.1097/NEN.0b013e3181c8ad2f. [DOI] [PubMed] [Google Scholar]
- 14.Chen W.J., Chan Y.J., Wan W.B., Li Y.M., Zhang C.Y. Aβ1‒42 induces cell damage via RAGE-dependent endoplasmic reticulum stress in bEnd.3 cells. Exp Cell Res. 2018;362:83–89. doi: 10.1016/j.yexcr.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Harja E., Bu D.X., Hudson B.I., Chang J.S., Shen X., Hallam K., et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE–/– mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semchyshyn H. Is carbonyl/AGE/RAGE stress a hallmark of the brain aging? Pflügers Archiv. 2021;473:723–734. doi: 10.1007/s00424-021-02529-y. [DOI] [PubMed] [Google Scholar]
- 17.Body-Malapel M., Djouina M., Waxin C., Langlois A., Gower-Rousseau C., Zerbib P., et al. The RAGE signaling pathway is involved in intestinal inflammation and represents a promising therapeutic target for Inflammatory Bowel Diseases. Mucosal Immunol. 2019;12:468–478. doi: 10.1038/s41385-018-0119-z. [DOI] [PubMed] [Google Scholar]
- 18.Lee H.W., Gu M.J., Kim Y., Lee J.Y., Lee S., Choi I.W., et al. Glyoxal–lysine dimer, an advanced glycation end product, induces oxidative damage and inflammatory response by interacting with RAGE. Antioxidants (Basel) 2021;10:1486. doi: 10.3390/antiox10091486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Q.Q., Jiang C.Q., Xia X., Wang Y.F., Yan C.X., Wang X.R., et al. Pathological BBB crossing melanin-like nanoparticles as metal-ion chelators and neuroinflammation regulators against Alzheimer's disease. Research (Wash D C) 2023;6 doi: 10.34133/research.0180. 0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sweeney M.D., Sagare A.P., Zlokovic B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–150. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu R., Luo C., Pang Z.Q., Zhang J.M., Ruan S.B., Wu M.Y., et al. Advances of nanoparticles as drug delivery systems for disease diagnosis and treatment. Chin Chem Lett. 2023;34 [Google Scholar]
- 22.Cai C.Z., Dai X.Y., Zhu Y.J., Lian M.Y., Xiao F., Dong F., et al. A specific RAGE–binding peptide biopanning from phage display random peptide library that ameliorates symptoms in amyloid β peptide-mediated neuronal disorder. Appl Microbiol Biotechnol. 2016;100:825–835. doi: 10.1007/s00253-015-7001-7. [DOI] [PubMed] [Google Scholar]
- 23.Seok H., Lee M., Shin E., Yun M.R., Lee Y.H., Moon J.H., et al. Low-dose pioglitazone can ameliorate learning and memory impairment in a mouse model of dementia by increasing LRP1 expression in the hippocampus. Sci Rep. 2019;9:4414. doi: 10.1038/s41598-019-40736-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicolakakis N., Aboulkassim T., Ongali B., Lecrux C., Fernandes P., Rosa-Neto P., et al. Complete Rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor γ agonist. J Neurosci. 2008;28:9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Son S.J., Singha K., Kim W.J. Bioreducible BPEI–SS-PEG–cNGR polymer as a tumor-targeted nonviral gene carrier. Biomaterials. 2010;31:6344–6354. doi: 10.1016/j.biomaterials.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 26.Yang X.T., Yang W.Q., Xia X., Lei T., Yang Z.H., Jia W.F., et al. Intranasal delivery of BACE1 siRNA and rapamycin by dual targets modified nanoparticles for Alzheimer's disease therapy. Small. 2022;18 doi: 10.1002/smll.202203182. [DOI] [PubMed] [Google Scholar]
- 27.Cai L.L., Yang C.Y., Jia W.F., Liu Y.W., Xie R., Lei T., et al. Endo/lysosome-escapable delivery depot for improving BBB transcytosis and neuron targeted therapy of Alzheimer's disease. Adv Funct Mater. 2020;30 [Google Scholar]
- 28.Xu S.J., Ling S.B., Shan Q.N., Ye Q.W., Zhan Q.F., Jiang G.J., et al. Self–activated cascade–responsive sorafenib and USP22 shRNA co-delivery system for synergetic hepatocellular carcinoma therapy. Adv Sci (Weinh) 2021;8 doi: 10.1002/advs.202003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu P.X., Zhang T.Y., Chen Q.J., Li C., Chu Y.C., Guo Q., et al. Biomimetic dendrimer–peptide conjugates for early multi-target therapy of Alzheimer's disease by inflammatory microenvironment Modulation. Adv Mater. 2021;33 doi: 10.1002/adma.202100746. [DOI] [PubMed] [Google Scholar]
- 30.Yan S.D., Chen X., Fu J., Chen M., Zhu H., Roher A., et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 31.Lei T., Yang Z.H., Xia X., Chen Y.X., Yang X.T., Xie R., et al. A nanocleaner specifically penetrates the blood‒brain barrier at lesions to clean toxic proteins and regulate inflammation in Alzheimer's disease. Acta Pharm Sin B. 2021;11:4032–4044. doi: 10.1016/j.apsb.2021.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He X.Q., Wang X.R., Yang L.Y., Yang Z.H., Yu W.Q., Wang Y.Z., et al. Intelligent lesion blood–brain barrier targeting nano-missiles for Alzheimer's disease treatment by anti-neuroinflammation and neuroprotection. Acta Pharm Sin B. 2022;12:1987–1999. doi: 10.1016/j.apsb.2022.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan W.B., Cao L., Liu L.M., Zhang C.Y., Kalionis B., Tai X.T., et al. Aβ1–42 oligomer-induced leakage in an in vitro blood–brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J Neurochem. 2015;134:382–393. doi: 10.1111/jnc.13122. [DOI] [PubMed] [Google Scholar]
- 34.Zlokovic B.V. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Moon J.H., Kim H.J., Yang A.H., Kim H.M., Lee B.W., Kang E.S., et al. The effect of rosiglitazone on LRP1 expression and amyloid β uptake in human brain microvascular endothelial cells: a possible role of a low-dose thiazolidinedione for dementia treatment. Int J Neuropsychopharmacol. 2012;15:135–142. doi: 10.1017/S1461145711001611. [DOI] [PubMed] [Google Scholar]
- 36.Jones R.W. Inflammation and Alzheimer's disease. Lancet. 2001;358:436–437. doi: 10.1016/S0140-6736(01)05667-7. [DOI] [PubMed] [Google Scholar]
- 37.Guerrero A., De Strooper B., Arancibia-Cárcamo I.L. Cellular senescence at the crossroads of inflammation and Alzheimer's disease. Trends Neurosci. 2021;44:714–727. doi: 10.1016/j.tins.2021.06.007. [DOI] [PubMed] [Google Scholar]
- 38.Rubinsztein D.C. RIPK1 promotes inflammation and β-amyloid accumulation in Alzheimer's disease. Proc Natl Acad Sci U S A. 2017;114:10813–10814. doi: 10.1073/pnas.1715241114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spangenberg E.E., Green K.N. Inflammation in Alzheimer's disease: lessons learned from microglia-depletion models. Brain Behav Immun. 2017;61:1–11. doi: 10.1016/j.bbi.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikolakopoulou A.M., Wang Y., Ma Q., Sagare A.P., Montagne A., Huuskonen M.T., et al. Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. A. J Exp Med. 2021;218 doi: 10.1084/jem.20202207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X.P., Luo D.Z., Zheng M., Hao Y.W., Hou L., Zhang S.M. Effect of pioglitazone on insulin resistance in fructose-drinking rats correlates with AGEs/RAGE inhibition and block of NAPDH oxidase and NF kappa B activation. Eur J Pharmacol. 2010;629:153–158. doi: 10.1016/j.ejphar.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 42.Deane R., Wu Z., Sagare A., Davis J., Du Yan S., Hamm K., et al. LRP/Amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 43.Wang R., Xu Z.G., Li Y.F., Li W.J., Gao X.R., Liu C., et al. Lycopene can modulate the LRP1 and RAGE transporters expression at the choroid plexus in Alzheimer's disease rat. J Funct Foods. 2021;85 [Google Scholar]
- 44.Guo Y.X., He L.Y., Zhang M., Wang F., Liu F., Peng W.X. 1,25-Dihydroxyvitamin D3 regulates expression of LRP1 and RAGE in vitro and in vivo, enhancing Aβ1–40 brain-to-blood efflux and peripheral uptake transport. Neuroscience. 2016;322:28–38. doi: 10.1016/j.neuroscience.2016.01.041. [DOI] [PubMed] [Google Scholar]
- 45.Nortley R., Korte N., Izquierdo P., Hirunpattarasilp C., Mishra A., Jaunmuktane Z., et al. Amyloid β oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science (New York, NY) 2019;365 doi: 10.1126/science.aav9518.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andjelkovic A.V., Situ M., Citalan-Madrid A.F., Stamatovic S.M., Xiang J., Keep R.F. Blood–brain barrier dysfunction in normal aging and neurodegeneration: mechanisms, impact, and treatments. Stroke. 2023;54:661–672. doi: 10.1161/STROKEAHA.122.040578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang Z., Wong L.W., Su Y., Huang X., Wang N., Chen H., et al. Blood‒brain barrier integrity in the pathogenesis of Alzheimer's disease. Front Neuroendocrinol. 2020;59 doi: 10.1016/j.yfrne.2020.100857. [DOI] [PubMed] [Google Scholar]
- 48.Paul C.A., Beltz B., Berger-Sweeney J. The nissl stain: a stain for cell bodies in brain sections. CSH protocols. 2008;2008 doi: 10.1101/pdb.prot4805. pdb.prot4805. [DOI] [PubMed] [Google Scholar]
- 49.Petrovskaya A., Tverskoi A., Medvedeva A., Nazarova M. Is blood–brain barrier a probable mediator of non-invasive brain stimulation effects on Alzheimer's disease? Commun Biol. 2023;6:416. doi: 10.1038/s42003-023-04717-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y.T., Zhu F.Y., Liu Y., Zheng M., Wang Y.B., Zhang D.Y., et al. Blood–brain barrier-penetrating siRNA nanomedicine for Alzheimer's disease therapy. Sci Adv. 2020;6 doi: 10.1126/sciadv.abc7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo Q., Xu S.T., Yang P., Wang P.Z., Lu S., Sheng D.Y., et al. A dual-ligand fusion peptide improves the brain-neuron targeting of nanocarriers in Alzheimer's disease mice. J Control Release. 2020;320:347–362. doi: 10.1016/j.jconrel.2020.01.039. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y.Q., Guo R.R., Chen Y.H., Li T.C., Du W.Z., Xiang R.W., et al. Ionizable drug delivery systems for efficient and selective gene therapy. Mil Med Res. 2023;10:9. doi: 10.1186/s40779-023-00445-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carrano A., Hoozemans J.J., van der Vies S.M., Rozemuller A.J., van Horssen J., de Vries H.E. Amyloid beta induces oxidative stress-mediated blood–brain barrier changes in capillary amyloid angiopathy. Antioxidants Redox Signal. 2011;15:1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 54.Katsouri L., Lim Y.M., Blondrath K., Eleftheriadou I., Lombardero L., Birch A.M., et al. PPARγ-coactivator-1α gene transfer reduces neuronal loss and amyloid-β generation by reducing β-secretase in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2016;113:12292–12297. doi: 10.1073/pnas.1606171113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zolezzi J.M., Santos M.J., Bastías-Candia S., Pinto C., Godoy J.A., Inestrosa N.C. PPARs in the central nervous system: roles in neurodegeneration and neuroinflammation. Biol Rev Camb Phil Soc. 2017;92:2046–2069. doi: 10.1111/brv.12320. [DOI] [PubMed] [Google Scholar]
- 56.Titus C., Hoque M.T., Bendayan R. PPAR agonists for the treatment of neuroinflammatory diseases. Trends Pharmacol Sci. 2024;45:9–23. doi: 10.1016/j.tips.2023.11.004. [DOI] [PubMed] [Google Scholar]
- 57.Zhang N.X., Guan C., Liu Z.Y., Li C., Yang C.Y., Xu L.Y., et al. Calycosin attenuates renal ischemia/reperfusion injury by suppressing NF-κB mediated inflammation via PPARγ/EGR1 pathway. Front Pharmacol. 2022;13 doi: 10.3389/fphar.2022.970616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shang J.S., Mosure S.A., Zheng J., Brust R., Bass J., Nichols A., et al. A molecular switch regulating transcriptional repression and activation of PPARγ. Nat Commun. 2020;11:956. doi: 10.1038/s41467-020-14750-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.