

Abstract
Leber's hereditary optic neuropathy (LHON) is an ocular mitochondrial disease that involves the impairment of mitochondrial complex I, which is an important contributor to blindness among young adults across the globe. However, the disorder has no available cures, since the approved drug idebenone for LHON in Europe relies on bypassing complex I defects rather than fixing them. Herein, PARKIN mRNA-loaded nanoparticle (mNP)-engineered mitochondria (mNP-Mito) were designed to replace dysfunctional mitochondria with the delivery of exogenous mitochondria, normalizing the function of complex I for treating LHON. The mNP-Mito facilitated the supplementation of healthy mitochondria containing functional complex I via mitochondrial transfer, along with the elimination of dysfunctional mitochondria with impaired complex I via an enhanced PARKIN-mediated mitophagy process. In a mouse model induced with a complex I inhibitor (rotenone, Rot), mNP-Mito enhanced the presence of healthy mitochondria and exhibited a sharp increase in complex I activity (76.5%) compared to the group exposed to Rot damage (29.5%), which greatly promoted the restoration of ATP generation and mitigation of ocular mitochondrial disease-related phenotypes. This study highlights the significance of nanoengineered mitochondria as a promising and feasible tool for the replacement of dysfunctional mitochondria and the repair of mitochondrial function in mitochondrial disease therapies.
Key words: Engineered mitochondria, Mitochondrial transfer, Mitochondrial disease, Leber's hereditary optic neuropathy, Nanoparticle, Complex I defect, Mitochondrial function, Idebenone
Graphical abstract
PARKIN mRNA-loaded nanoparticle (mNP)-engineered mitochondria (mNP-Mito) were designed to replace dysfunctional mitochondria with the delivery of exogenous mitochondria, normalizing the function of complex I for Leber's hereditary optic neuropathy (LHON).
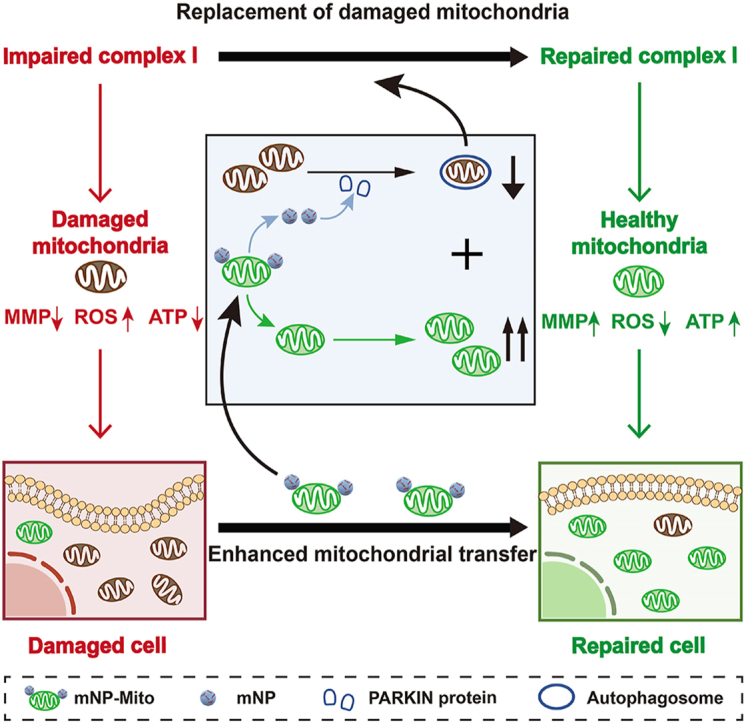
1. Introduction
Mitochondrial diseases are a group of threat disorders involving inherited or acquired impairments of mitochondrial function1,2. Leber's hereditary optic neuropathy (LHON) is a typical and aggressive ocular mitochondrial disease caused by dysfunction of complex I in the mitochondrial respiratory chain, leading to blindness, especially in young individuals3,4. Both blocked energy production and an increase in mitochondrial reactive oxygen species (mtROS) are the potential consequences of the impairment of complex I in LHON, contributing to mitochondrial dysfunction and central vision degeneration5. Although idebenone had been approved as adjuvant therapy for LHON in Europe, it relies on bypassing complex I defect rather than fixing the dysfunction of complex I6,7. Except for idebenone, there is no other efficient curative treatment for LHON in the clinic. Hence, current treatments are unmet clinical needs for LHON patients. There are some preclinical gene therapy studies for LHON based on allotopic expression, which import DNA into the nucleus and then guide the translated protein into the mitochondria to recover protein function (ClinicalTials.gov: NCT03293524, NCT02161380, and NCT03428178). In addition, given the intricate involvement of mitochondria in diverse metabolic networks, some mitochondria-targeted therapies, such as introducing a specific factor into mitochondria for the restoration of complex I, that alter mitochondrial inherent processes could potentially trigger cell death8, 9, 10, 11. Therefore, safe and efficient complex I restoration strategies are urgently needed for the treatment of LHON.
It is reported that the transfer of mitochondria between healthy and damaged cells through various mechanisms is a spontaneous way to replenish functional mitochondria in damaged cells and rescue damaged cells12,13. Inspired by this spontaneous process, mitochondrial transfer therapy emerges as a promising strategy to supply healthy mitochondria to damaged cells for enhancing mitochondrial function, which shows clinical and preclinical successes in the treatment of mitochondrial diseases14,15. Accumulating evidence shows that damaged and healthy mitochondria coexist in cells, and the dysfunctional mitochondrial contents tightly correlate with the disorder severity of mitochondrial diseases16, 17, 18. Transferring exogenous healthy mitochondria into damaged recipient cells lowers the ratio value of damage to healthy mitochondria19. It is an easier approach for replacing the dysfunctional complex I than other mitochondria-targeted techniques repairing complex I function, such as mitochondria-targeted nanoparticles and mitochondria-targeted gene editing20, 21, 22, 23, 24, 25. Replacing the dysfunctional complex I in damaged mitochondria with exogenous mitochondria may be a prospective strategy in LHON treatment. Exogenous mitochondria could be engulfed into damaged recipient cells via coculture26. Despite it being a simple process, the limited mitochondrial transfer efficiency due to the electrostatic repulsion-based barrier is still a challenge of mitochondrial transfer therapy in clinical application27, 28, 29. Moreover, within the same cells, damaged mitochondria carrying dysfunctional complex I remain exist. The accumulation of damaged mitochondria is not conducive to the restoration of mitochondrial functions and the remission of disease progression18,30, 31, 32, 33. Therefore, enhancing the efficiency of mitochondrial transfer and minimizing damaged mitochondria are pivotal endeavors to amplify the therapeutic effectiveness of mitochondrial transfer in LHON.
Herein, we designed PARKIN mRNA-loaded nanoparticle-engineered mitochondria (mNP-Mito) that promoted delivery efficiency of healthy exogenous mitochondria and mitophagy of damaged mitochondria, which rejuvenated the functionality of complex I for treating LHON. Briefly, normal cells and mouse heart tissues with a rich number of mitochondria were used as donors for the extraction of exogenous mitochondria. Considering the risk of damaged mitochondria carryover, mNP-Mito was constructed by adhesion PARKIN mRNA-loaded Lipofectamine 2000 nanoparticles (mNPs) to the surface of healthy mitochondria (Mito) through the electrostatic interaction (Fig. 1A). Because the dysfunction of complex I is an essential pathogenic factor for LHON, complex I inhibitor (rotenone, Rot)-induced cell and mouse model were introduced as proof-of-concept models to investigate the curative effect of mNP-Mito-mediated mitochondrial transfer. In Rot-induced cells, mNP-Mito was internalized into cells by macropinocytosis, and the exogenous mitochondria in mNP-Mito increased the proportion of healthy mitochondria with functional complex I. In addition, the modification of mNPs maintained exogenous mitochondrial activity in mNP-Mito and promoted mitophagy of damaged mitochondria. The synergistic regulation of healthy and damaged mitochondrial populations mediated by mNP-Mito restored the function of complex I and rescued cells from Rot damage (Fig. 1B). After intravitreal injection, mNP-Mito increased the activity of complex I and repaired the generation of ATP in the retina, and reverted disease-related molecular and biochemical phenotypes in Rot-induced experimental LHON-like mouse model. Our findings propose that employing nanoengineered mitochondria to restore mitochondrial function represents a viable and safe therapeutic strategy for addressing LHON and other mitochondrial diseases.
Figure 1.

The mNP-Mito restores mitochondrial functions for ocular mitochondrial disease treatment. (A) Design of mNP-Mito through attaching PARKIN mRNA-loaded nanoparticle to exogenous mitochondria; (B) The mNP-Mito restores complex I function by replacing dysfunctional mitochondria with healthy exogenous mitochondria.
2. Materials and methods
2.1. Materials
Lipofectamine™ 2000 (11668030), MitoTracker Green probe (M7514), MitoTracker Red probe (M7512), and MitoSOX (M36009) were purchased from Invitrogen (Carlsbad, CA, USA). Mitochondrial Isolation Kit (MP-007) was bought from Invent Biotechnologies (Plymouth, MN, USA). Tissue Mitochondria Isolation Kit (C3606), Mitochondrial Membrane Potential Assay Kit with JC-1 (C2006), and ATP Assay Kit (S0026) were gained from Beyotime (Shanghai, China). PARKIN-specific mRNA was synthesized by RiboBio (Guangzhou, China). The mRNA labeled with Cy3 or Cy5 fluorophore was custom-made by APExBIO (Houston, TX, USA). Rotenone (R105077) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Dulbecco's Modified Eagle Medium (KGL1206-500), and BCA Detection Kit (KGB2101-250) were obtained from KeyGEN BioTech (Nanjing, China). Janus green-b (G1570) and Mitochondrial Complex I/NADH-CoQ Reductase Activity Assay Kit (BC0510) were obtained from Solarbio Science & Technology Co., Ltd. (Beijing, China). DNA Extraction Kit and SYBR Green PCR Master Mix (DP304) were purchased from TIANGEN Biotech Co., Ltd. (Beijing, China). Calcein-AM/PI Double Stain Kit (40302ES50) was obtained from Yeasen Biotechnology Co., Ltd. (Shanghai, China). Chlorpromazine (CPZ), genistein (GEN), and amiloride (Ami) were obtained from J&K Chemical (Beijing, China). FAS eye fixation fluid was purchased from Servicebio (Wuhan, China).
NDUFB8 Rabbit Antibody (1:1000, ET7108-25) and UBIQUITI Rabbit Antibody (1:1000, ER31212) were gained from HuaBio (Hangzhou, China). β-Actin Mouse Monoclonal Antibody (1:1000, AF2811) was purchased from Beyotime (Shanghai, China). Anti-Complex I Antibody (1:1000, ab109798) and LC3B Mouse Monoclonal Antibody (1:1000, ab243506) were obtained from Abcam (Cambridge, England). FITC/Cy3 Goat Anti-Mouse IgG (H+L) (1:100, AS008/AS007), PARKIN Rabbit pAb (1:1000, A0968) and COX IV Rabbit pAb (1:1000, A6564) were purchased by ABclonal (Wuhan, China).
2.2. Cell culture
HeLa cell line was obtained by Cell Bank (Shanghai, China). HeLa cell lines with mitochondrial complex I damaged were generated using 0.1 μmol/L Rot as previously described34, 35, 36. The HeLa cells with or without Rot treatments were cultured in DMEM containing 10% fetal bovine serum, penicillin, and streptomycin.
2.3. Animal model
BALB/c mice were purchased from the East China Normal University Laboratory Animal Technology Co., Ltd. (Shanghai, China). All experimental procedures were executed according to the protocols approved by China Pharmaceutical University Animal Care and Use Committee (2021-11-009).
For the Rot-induced experimental LHON-like mouse model, BALB/c mice were anesthetized and treated with 3 μL 1, 2.5, or 5 mmol/L Rot (solvent: trilaurin) by intravitreal injection35, 36, 37, 38. The vehicle group was treated with 3 μL trilaurin.
2.4. Preparation and characterization of mNP-Mito
2.4.1. Preparation of mNP-Mito
The mNP-Mito was prepared by adhering to mNPs on the surface of extracted exogenous mitochondria. In brief, we isolated exogenous mitochondria with 40 μg mitochondrial proteins from 6.32 million HeLa cells according to the protocol of Mitochondrial Isolation Kits. The mNPs were constructed through adding 1 μg PARKIN mRNA into 2 μL Lipofectamine™ 2000. After incubation for 0.5 h at 4 °C, the prepared mNPs were mixed with exogenous mitochondria. The mNP-Mito were prepared after centrifugation (Dragon Laboratory Instruments Limited, D3024R, Beijing, China) at 16,000×g for 20 min to remove free mNPs. The average hydrodynamic sizes in the Mito and mNP-Mito groups were analyzed via dynamic light scattering (DLS, Brookhaven, ZataPlus, Upton, NY, USA), and the zeta potentials were also detected through Nano ZS Zetasizer DLS analyzer. The mNP-Mito were constructed by the binding of mNPs (0.05 μg PARKIN mRNA/eye) and exogenous mitochondria (10 μg/eye) extracted from 483.39 μg heart tissues of BALB/c mice in in vivo studies.
2.4.2. Determination of the mRNA contents in mNP-Mito
For determination of the mRNA contents in mNP-Mito, mRNA labeled with Cy3 fluorophore was loaded in mNPs to further construct Cy3-mNP-Mito. After 30 min of incubation and 20 min of centrifugation (16,000×g, Dragon Laboratory Instruments Limited), the sediment was collected and resuspended with PBS. The fluorescence intensity (FI) of Cy3-mRNA in the sediment and supernate was analyzed by a multi-mode microplate reader (Molecular Devices, SpectraMax ID5, San Jose, CA, USA). The amount of loaded Cy3-mRNA was calculated by FI of Cy3-mRNA.
2.4.3. Visualization of the adhesion of mNPs and isolated mitochondria
Cy3-mRNA (1 μg) was mixed with exogenous mitochondria (40 μg mitochondrial proteins) labeled with MitoTracker Green. The mNP-Mito and Mito were respectively seeded into the 35 mm glass bottom dishes, and the images were recorded by inverted fluorescence microscopy (Nikon, Tokyo, Japan) and grazing incidence structured illumination microscopy (GI-SIM, Carl Zeiss, Jena, Germany). Furthermore, the morphology of mNP-Mito was determined by scanning electron microscopy (SEM, Hitachi, Tokyo, Japan).
2.4.4. Detection of the exogenous mitochondrial activity
Janus green-b staining was carried out to examine the functionality of mitochondria after binding with mNPs. The images of extracted mitochondria with or without mNPs were collected by inverted fluorescence microscopy (Nikon). For the mitochondrial membrane potential (MMP) of mitochondria, the Mito group and mNP-Mito were stained with Mitochondrial Membrane Potential Assay Kit with JC-1 after Days 0, 1, and 3 of storage. The FI of the JC-1 probe was detected by multi-mode microplate reader (525 nm, Molecular Devices).
2.4.5. Stability of mNP-Mito
Cy3-mNPs were adhered to extracted mitochondria for Day 0 or 1 in PBS. Then, the solutions were centrifugated for 20 min at 16,000×g (Dragon Laboratory Instruments Limited). The supernates were detected by a multi-mode microplate reader (Molecular Devices), and the sediments were resuspended in Heparin solution for 30 min to replace the Cy3-mNPs on the surface of mitochondria. After 16,000×g centrifugation (Dragon Laboratory Instruments Limited) for 20 min, the supernate and sediment were analyzed by a multi-mode microplate reader (Molecular Devices) to calculate the contents of mRNA shedding from mNP-Mito and the contents of mRNA internalized into extracted mitochondria after different storage times.
2.5. Cellular internalization of exogenous mitochondria and mRNA
The normal donor HeLa cells were stained with MitoTracker Green/MitoTracker Red, and the MitoTracker Green/MitoTracker Red-labeled exogenous mitochondria were extracted after a series of rinses and centrifuges. And then, the Cy5-mRNA was loaded into Cy5-mNPs. Cy5-mNP-Mito was constructed by attaching Cy5-mNPs to MitoTracker Green/MitoTracker Red-stained isolated mitochondria.
HeLa cells were cultured into 24-well plate or 35 mm glass bottom dishes (1 × 105 cells/well) and then treated with 0.1 μmol/L Rot. After 24 h, the Rot-treated HeLa cells were respectively cultured with Cy5-mNP (0.4 μg/mL Cy5-mRNA), Cy5-mNP-1.5 (0.6 μg/mL Cy5-mRNA), Mito (80 μg/mL mitochondrial proteins) and Cy5-mNP-Mito (80 μg/mL mitochondrial proteins and 0.4 μg/mL Cy5-mRNA). After 1, 4, or 24 h, the cells were collected. The mean fluorescence intensity (MFI) of MitoTracker Green/Red and Cy5-mRNA were analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA) and the images were recorded by CLSM (LSM 700, Carl Zeiss, Jena, Germany).
The dose of mNP-Mito in vitro depended on the number of recipient cells. In the in vitro experiments of this study, unless otherwise specified, when the number of recipient cells was 1 × 105 cells/well, the administration dosage of mNP-Mito was 80 μg/mL mitochondrial proteins and 0.4 μg/mL Cy5-mRNA.
2.6. Cellular uptake mechanism study of mNP-Mito
For the mechanism of cellular uptake, Rot-treated cells in a 24-well plate (1 × 105) were covered with different cellular uptake inhibitors for 1 h, including 8.5 μg/mL CPZ, 56.75 μg/mL GEN, and 0.133 mg/mL Ami. Then, Cy5-mNP and Cy5-mNP-Mito were respectively added to a 24-well plate for 4 h. Flow cytometry (BD Biosciences) was used to evaluate the MFI of Cy5-mRNA.
2.7. Live cell imaging system
The normal donor HeLa cells were stained with MitoTracker Red, and the MitoTracker Red-labeled exogenous mitochondria were extracted after a series of rinses and centrifuges. The MitoTracker Red labeled mNP-Mito was prepared by attaching Cy5-mNPs to MitoTracker Red-stained isolated mitochondria.
The Rot-treated HeLa cells were cultured in the 35 mm glass dish (1 × 105 cells/well) and labeled using MitoTracker Green probe. Then, cells were treated with mNP-Mito for 4 h. The mode of live cell imaging in CLSM (Carl Zeiss) was performed to capture the cellular uptake of mNP-Mito.
2.8. Cellular internalization of exogenous mitochondria by CLSM (Carl Zeiss)
The normal donor HeLa cells were stained with MitoTracker Green, and the MitoTracker Green-labeled exogenous mitochondria were extracted after a series of rinses and centrifuges. The MitoTracker Green-labeled mNP-Mito was prepared through the binding of mNPs and MitoTracker Green-labeled exogenous mitochondria.
To explore the location of exogenous mitochondria and total cellular mitochondria in vitro, HeLa cell lines were incubated within a 35 mm glass dish (1 × 105 cells/well). After incubation for 14 h, HeLa cells were treated with Rot for 24 h and respectively cultured with mNP, mNP-1.5, MitoTracker Green-marked Mito, and MitoTracker Green-marked mNP-Mito for 24 h. Subsequently, total mitochondria within HeLa cell lines were stained with MitoTracker Red probe. CLSM (Carl Zeiss) was used to record the images in various treatment groups. The green fluorescence in CLSM images was exogenous mitochondria. The red fluorescence spots presented the total mitochondria within recipient cells, including endogenous mitochondria and internalized exogenous mitochondria.
2.9. Detection of mitophagy
The expression levels of PARKIN, LC3B, and mitochondrial PARKIN proteins were explored through Western blot assays. Rot-treated HeLa cells were cultured in the 6-plate or 10 cm culture dishes. The mNP, mNP-1.5, Mito, and mNP-Mito were incubated with Rot-treated HeLa cells for 24 h. The cellular or mitochondrial proteins were extracted through the whole cell lysis assay, and protein concentration was ascertained using the BCA Protein Assay Kit. The samples were respectively incubated with different antibodies, including PARKIN Rabbit pAb and LC3B Mouse Monoclonal Antibody. In addition, β-Actin Mouse Monoclonal Antibody and COX IV Rabbit pAb were used for cellular protein and mitochondrial protein reference antibodies. The band was collected using the chemiluminescence imaging system (5200, Tanon, Shanghai, China). The band intensity of PARKIN and β-Actin/COX IV proteins was quantified using integrated density value by ImageJ software (National Institutes of Health, NIH, Bethesda, MD, USA).
2.10. Function of mitochondrial complex I in vitro
2.10.1. Mitochondrial complex I activity
HeLa cells were cultured with 0.1 μmol/L Rot for 24 h, and different treatment groups were added to 6-well plate for 24 h. The cells were collected, and the complex I activity was detected by the Mitochondrial Complex I/NADH-CoQ Reductase Activity Assay Kit according to the protocols.
2.10.2. Mitochondrial complex I content
HeLa cells were collected after being treated with various groups, and immunofluorescence in fixed HeLa cells was carried out to detect the localization of complex I proteins. The antibodies were used as follows: anti-complex I antibody (1:1000) and FITC goat anti-mouse IgG (H+L) (1:100). The photos were recorded using CLSM (Carl Zeiss). FI of mitochondrial complex I proteins in immunofluorescence images was calculated using ImageJ software.
2.10.3. Expression of mitochondrial protein within complex I
Expression of mitochondrial protein (NDUFB8) was assessed via Western blot assay. Cell proteins in different Rot groups were extracted from HeLa cells by the whole cell lysis assay. The whole protein contents were tested by the BCA Detection Kit. Anti-NDUFB8 recombinant rabbit monoclonal antibody (1:1000) and β-Actin mouse monoclonal antibody (1:1000) were cultured with the bands. The band intensity of NDUFB8 and β-ACTIN proteins was quantified using integrated density value by ImageJ software.
2.11. MMP detection
To evaluate the change of MMP in Rot-treated HeLa cells, 0.1 μmol/L Rot-treated HeLa cells were cultured in the 35 mm glass bottom dishes (1 × 105 cells/well) or 24-well plate (1 × 105 cells/well). After 24 h, the cells from different treatment groups were stained by Mitochondrial Membrane Potential Assay Kit with JC-1 at 37 °C for 15 min. Then, the images were observed by CLSM (Carl Zeiss) at 525 nm and 490 nm excitation. In addition, a multi-mode microplate reader (Molecular Devices) was used to record the FI of JC-1 aggregates at 525 nm. Normalized FI was shown in Eq. (1):
| (1) |
2.12. ATP contents assay in vitro
HeLa cells were plated in a 6-well plate (2 × 105 cells/well) overnight and treated with 0.1 μmol/L Rot for 24 h. The Rot-treated HeLa cells were covered with mNP, mNP-1.5, Mito, and mNP-Mito for 24 h. The levels of the ATP generation were estimated by ATP Bioluminescent Assay Kit, and the chemiluminometer (Luminoskan Ascent, ThermoFisher Scientific, Waltham, MA, USA) was used to detect the luminescence in various groups.
2.13. Mitochondrial ROS level detection
35 mm glass dishes were used to culture HeLa cells (1 × 105 cells/well) for 14 h and treated with 0.1 μmol/L Rot for 24 h. The Rot-treated HeLa cells were cultured with mNP, mNP-1.5, Mito, and mNP-Mito. After incubation of 24 h, MitoSOX (0.5 μmol/L) as a mitochondrial ROS probe was carried out to assess the mitochondrial ROS level by CLSM (Carl Zeiss).
2.14. Live/dead cells double stain
After HeLa cells were treated with 0.1 μmol/L Rot for 12 h, various formulations were added into Rot-treated HeLa cells for 8 h. Then, the medium was removed, and cells were treated with a Calcein-AM/PI Double Stain Kit. The images were recorded using inverted fluorescence microscopy (Nikon) and shown by ImageJ software.
2.15. Internalization of exogenous mitochondria in vivo
The normal heart tissues of BALB/c mice were stained with MitoTracker Green, and the MitoTracker Green-labeled exogenous mitochondria were extracted after a series of rinses and centrifuges. The mNP-Mito was prepared through the binding of Cy3-mNPs and MitoTracker Green-stained mitochondria.
Given the distribution of MitoTracker Green-labeled Cy3-mNP-Mito in vivo, Cy3-mNP-Mito (10 μg mitochondrial proteins/eye and 0.05 μg Cy3-mRNA/eye) were injected into normal BALB/c mice. After 24 h, the eyes from BALB/c mice were analyzed using fluorescent photos.
2.16. Therapeutic effect of mNP-Mito in Rot-induced experimental LHON-like mouse model
2.16.1. Flowchart of mNP-Mito treatment
To establish a Rot-induced experimental LHON-like mouse model, BALB/c mice were anesthetized, and 3 μL 2.5 mmol/L Rot was injected into the vitreous chamber. After Days 3 and 10, the mice were assigned to different formulations, including vehicle, mNP (0.05 μg PARKIN mRNA/eye), mNP-1.5 (0.075 μg PARKIN mRNA/eye), Mito (10 μg mitochondrial proteins) and mNP-Mito (10 μg mitochondrial proteins and 0.05 μg PARKIN mRNA/eye). Subsequent pharmacological studies were conducted on Day 17 post-modeling.
2.16.2. H&E staining, ATP contents assay, and detection of complex I function in retina
After various treatments, eyes from various treatments were gathered in eye fixation fluid, and longitudinal retinal sections were prepared for H&E staining. The thickness of the retina in H&E sections was analyzed by the CaseViewer software (3DHISTECH, Budapest, Hungary). In addition, retinal tissues were rapidly ground in lysis buffer, and the levels of ATP generation in different groups were assessed using ATP Bioluminescent Assay Kit. Mitochondrial complex I in retinal layers was assessed according to “2.10.1. Mitochondrial complex I activity” and “2.10.3. Expression of mitochondrial protein within complex I”.
2.16.3. Mitophagy in retinal tissues detected by Western blot assay
After various treatments, eyes from various treatments were gathered, and retinal tissues were rapidly grinded in a lysis buffer. The levels of PARKIN proteins and mitochondrial PARKIN proteins within retinal tissues were referred to as “2.9. Detection of mitophagy”. The images of immunofluorescence staining of PARKIN proteins were shown using Pannoramic MIDI/250 (3DHISTECH, Budapest, Hungary) for localization of PARKIN protein expression in the eye tissues.
The ubiquitination of mitochondrial proteins was also detected by Western blot assay. In brief, mitochondrial proteins from retinal tissues were extracted by the Tissue Mitochondria Isolation Kit. Anti-UBIQUITI rabbit polyclonal antibody (1:1000) was employed as an antibody, with COX IV (1:1000) serving as a loading control.
2.16.4. Optomotor test
The optomotor test was performed as previously described39. In brief, each mouse was placed in a circular platform and encompassed in a black-and-white striped roller for 5 min. The roller was turned clockwise and counterclockwise for 2 min each, and the movement times of the head were recorded.
2.17. Safety assessment in vivo
According to the dose and frequency for intravitreal injection of mNP-Mito, the normal BALB/c mice were injected with 1-, 2-, and 4-fold the dose of mNP-Mito40. The body weights were recorded during dosing. After one week, the serum samples from BALB/c mice were prepared for the detection of ALT, AST, BUN, and inflammatory factors. The whole blood was collected, and the relative mtDNA copy number was assessed. In addition, main organs and eye tissues were collected, and the samples were investigated by H&E staining and organ index analysis. The safety of mNP-Mito in the Rot-induced mouse model was also detected.
2.18. Statistics
Quantitative data from the experiments were expressed as mean ± standard deviation (SD) with corresponding sample sizes (n). Statistical comparisons among multiple groups utilized the one-way analysis of variance (ANOVA) test. Post hoc tests, such as Tukey's honest significant difference (HSD) or Scheffe test, were employed for pairwise comparisons when homogeneity of variance was observed. In cases where homogeneity of variance was not met, the Games–Howell test was used for comparisons between multiple groups. All statistical tests were conducted as two-sided analyses. The software package SPSS 19.0 (IBM, Armonk, NY, USA) was employed for statistical analysis, while ImageJ software (NIH) was utilized for quantitative analysis of FI in confocal photos.
3. Results and discussion
3.1. Preparation and characterization of mNP-Mito
First, we extracted autologous mitochondria from HeLa cells and identified a linear relationship between mitochondrial proteins and the number of donor cells (Supporting Information Fig. S1A)41. In subsequent in vitro studies, mitochondria were isolated from the same number of cells in each group (6.32 million cells). Mitochondria within cells were stained with the MitoTracker Green probe, and these mitochondria were isolated from cells. We found that isolated mitochondria still exhibited green fluorescence (Fig. S1B)42. Hence, we used MitoTracker Green probe to trace exogenous mitochondria within mNP-Mito. In addition, we applied Lipofectamine 2000 to load functional PARKIN-specific mRNA (designated mNP) according to the protocol, and the effective diameter of mNP was 255.4 ± 6.9 nm (Fig. S1C). We constructed mNP-Mito through the electrostatic interaction between isolated healthy mitochondria (Mito group) and mNP (Fig. 2A). Isolated mitochondria were labeled with MitoTracker Green probe, and Cy3-labeled mRNA was loaded into mNP to form Cy3-mNP. The binding of Mito and Cy3-mNP was assessed by the colocalization of green-labeled Mito and Cy3-labeled mNP. The images of GI-SIM (Carl Zeiss) and inverted fluorescent microscope (Nikon) showed that green-labeled Mito and Cy3-labeled mNP were merged, indicating the binding of Mito and mNP (Fig. 2B and Fig. S1D). In addition, surface-engineering mitochondria with mNP was visualized by SEM (Hitachi) (Fig. 2C)43. DLS (Brookhaven) was used to detect the sizes and zeta potentials in the Mito group and the mNP-Mito group. As shown in Fig. 2D and E, the sizes in the mNP-Mito group were larger compared to the Mito group. The data of zeta potentials in the Mito and mNP-Mito groups showed that the binding of positive mNP in the surface of negative Mito led to the zeta potential of mNP-Mito was higher than that of isolated Mito (Fig. 2F). These results suggested that mNP-Mito was constructed via the modification of PARKIN mRNA-loaded mNP in the surface of extracted free mitochondria.
Figure 2.

Preparation and characterization of mNP-Mito. (A) Illustration of the preparation of mNP-Mito; (B) GI-SIM images of MitoTracker Green labeled Cy3-mNP-Mito; (C) SEM images of Mito and mNP-Mito. White: Mito, red: mNP; (D) The size of Mito detected by DLS (Brookhaven); (E) The size of mNP-Mito detected by DLS (Brookhaven); (F) Zeta potentials of mNP, Mito and mNP-Mito; (G) Encapsulation efficiency of Cy3-mNP-Mito calculated by the FI of Cy3-mRNA; (H) The MMP detection (JC-1 dye) before and after binding with mNP at different time points. In (F–H), data are mean ± SD (n = 3). Statistical significance was performed by one-way ANOVA with Games–Howell test (H).
To assess the mRNA encapsulation efficiency of mNP-Mito, Cy3-labeled mRNA was loaded into mNP-Mito to construct Cy3-mNP-Mito. After centrifugation (Dragon Laboratory Instruments Limited), the FI of Cy3-labeled mRNA in Cy3-mNP-Mito (sediment) and free Cy3-labeled mRNA (supernate) were investigated through multi-mode microplate reader (Molecular Devices), and mRNA encapsulation efficiency in Cy3-mNP-Mito was 15.59 ± 1.34% (Fig. 2G and Fig. S1E). In addition, we explored whether the modification of mNP affects mitochondrial activities. The mitochondrial activities were evaluated using Janus green-B dye. In operational mitochondria, cytochrome oxidase maintains the dye in the oxidized state, showing a blue-green color. In contrast, the dye undergoes a reduction in the absence of active cytochrome oxidase, transferring into colorless44. Janus green-B dye showed both blue-green in the Mito and mNP-Mito groups, suggesting that the activity of mitochondria remained with and without the modification of mNP (Fig. S1F). MMP of Mito and mNP-Mito was detected by JC-1 probe. We found that MMP of exogenous mitochondria in the Mito group had no significant difference from that in the mNP-Mito group on Day 0. After Day 1, MMP in the Mito group was obviously decreased, which was significantly lower than that in mNP-Mito (Fig. 2H). This indicated that MMP of isolated mitochondria was protected by the modification of mNP compared with free mitochondria and remained normal within 1 day. To explore the stability of mNP-Mito, we used heparin sodium to disturb the electrostatic interaction between mNP and Mito. After centrifugation for 20 min (16,000×g, Dragon Laboratory Instruments Limited), the contents of Cy3-labeled mRNA in sediments and supernates were analyzed via a multi-mode microplate reader (Molecular Devices). The Cy3-labeled mRNA contents in mitochondrial sediments were only 0.0009 μg, which was much lower than the loaded Cy3-labeled mRNA contents (0.1559 μg) in mNP-Mito, suggesting that the loaded mRNA was barely internalized into the exogenous mitochondria. 89.5% Cy3-mRNA was stably retained on the surface of mitochondria after Day 1 of culture in the medium at 4 °C (Fig. S1G). The mNP-Mito stained with fluorescence were named Cy3-mNP-Mito and Cy5-mNP-Mito. In addition, the PARKIN mRNA-loaded mNP-Mito was abbreviated as mNP-Mito.
3.2. mNP-Mito promotes the delivery of healthy mitochondria and induces mitophagy of dysfunctional mitochondria
To assess the relationship between mitochondrial dysfunction and the severity of LHON in vitro, Rot, as a typical small molecule, is carried out to disrupt the function of mitochondrial complex I and cause LHON-like phenotypes37,45,46. We identified the concentration of Rot in vitro via MTT assay. As shown in Supporting Information Fig. S2A, cellular mortality rose with the increasing Rot concentration, and cellular mortality rate was 51.8% after 24 h of 0.1 μmol/L Rot treatment. Additionally, we detected the impairment of mitochondrial complex I and mitochondrial functions under different concentrations of Rot, such as the expression of mitochondrial protein within complex I, mitochondrial complex I content, and MMP detection47. First, NADH ubiquinone dehydrogenase 1β subcomplex 8 (NDUFB8) is a vital protein within complex I. The level of NDUFB8 proteins was investigated via Western blot assay, and β-ACTIN was used as a loading control, showing the changes in mitochondrial complex I within the same cell number under different Rot concentrations. The result in Fig. S2B indicated that the level of NDUFB8 decreased under the application of 0.1 and 0.5 μmol/L Rot, confirming that Rot induced the damage of mitochondrial complex I. We further investigated complex I content via immunofluorescence staining under the treatments of different Rot concentrations. Higher Rot concentrations (0.1 and 0.5 μmol/L) caused a reduction in complex I content (Fig. S2C). Free mitochondria in different groups were extracted, and complex I content in free mitochondria was explored via immunofluorescence staining. MFI of complex I in extracted free mitochondria was detected by flow cytometer. As shown in Fig. S2D, we found that Rot significantly declined complex I content in extracted mitochondria. Additionally, JC-1 Kits were used to detect MMP in different treatment groups. FI of healthy mitochondria with normal MMP (J-aggregate) reduced, and FI of damaged mitochondria with declined MMP (J-monomer) rose with the increasing of Rot concentration, suggesting that Rot led to the damage of MMP (Fig. S2E and F).
We further explored the underlying mechanism related to the synergistic regulation of healthy and damaged mitochondria via mNP-Mito treatment in a Rot-induced cell model. The cells treated with PBS after 24 h of Rot-treatment, named the “Rot” group, served as negative control treatment. First, we determined the internalization of MitoTracker-labeled exogenous mitochondria in mNP-Mito. As shown in Fig. 3A, mNP-Mito facilitated significant cellular internalization of MitoTracker Green-labeled healthy mitochondria compared with the Mito group due to the increasing zeta potential in mNP-Mito (Fig. 2F). In the normal and Rot-treated HeLa cells, mNP-Mito had both good cellular uptake (Supporting Information Fig. S3A). Following a 24 h treatment with mNP-Mito, the fluorescent signals from the isolated mitochondria were found to be in close proximity to the Hoechst-labeled nucleus (Fig. 3B). Live cell imaging was further employed to observe the internalization of exogenous mitochondria after 4 h of mNP-Mito treatment (Fig. 3C and D). These results demonstrated that the red fluorescent signals representing exogenous mitochondria in mNP-Mito overlapped with the green fluorescent spots indicating mitochondria in the recipient cell (red: exogenous mitochondria in mNP-Mito, green: mitochondria in recipient cells). To explore the location of exogenous and endogenous mitochondria, we performed the MitoTracker Green probes to stain the extracted mitochondria within the Mito and mNP-Mito groups. The total mitochondria in cells were stained with a MitoTracker Red probe after different treatments. Therefore, green fluorescence was exogenous mitochondria, and the red fluorescence spots were the total mitochondria after the delivery of exogenous mitochondria. After various treatments, isolated mitochondria in the mNP-Mito group demonstrated greater internalization compared to the Mito group. The total mitochondria in mNP-Mito were comparable to the PBS group and obviously higher than those in the other groups, indicating that mNP had the potential to promote the delivery of healthy mitochondria (Fig. 3E).
Figure 3.

Delivery of healthy mitochondria and mitophagy of dysfunctional mitochondria mediated by mNP-Mito treatment. (A, B) Delivery of exogenous healthy mitochondria detected by flow cytometry (BD Biosciences) (A) and CLSM (Carl Zeiss) (B) (24 h); (C) Internalization of MitoTracker Red labeled exogenous mitochondria in mNP-Mito investigated by live cell imaging, red: mNP-Mito, green: mitochondria within recipient cell; (D) 3D images of mNP-Mito treatment after 4 h; (E) The total mitochondria and exogenous mitochondria were respectively stained by MitoTracker Red and Green probes; (F, G) Analysis of PARKIN protein; (H) Analysis of mitochondrial PARKIN protein; (I) The expression of LC3B-II and LC3B-I proteins without (−) or with (+) chloroquine; (J) Schematic illustration of mNP-Mito treatment in the Rot-induced cell model, (i) the supplement of healthy exogenous mitochondria, (ii) the elimination of damaged mitochondria. Data are mean ± SD (n = 3 biologically independent samples in (A), (F). Statistical significance was determined using one-way ANOVA with Games–Howell test (A) and Tukey's honest significant difference (HSD) post hoc test (F).
On the other hand, we investigated whether mNP-Mito induced mitophagy of damaged mitochondria in a Rot-treated cell model. The process of mitophagy, based on the Putative kinase 1 (PINK1)–PARKIN pathway, selectively eliminates damaged mitochondria within cells48,49. PINK1 is rapidly turned over by proteolysis in healthy mitochondria. When mitochondria become damaged, PINK1 proteolysis is inhibited, allowing the accumulation of PINK1 only in damaged mitochondria. The subsequent recruitment of PARKIN, specifically to organelles that are accumulating PINK1, ultimately leads to the degradation of damaged mitochondria. We first detected the internalization of genes in mNP-Mito. The cellular uptakes of mitochondria and genes in the MitoTracker Green-labeled Cy5-mNP-Mito group were investigated using CLSM (Carl Zeiss) and flow cytometry (BD Biosciences) (Fig. S3B‒D)50. After 1 h incubation with mNP-Mito, Cy5-labeled mRNA showed colocalization with green-stained exogenous mitochondria. However, a separation of two fluorescent signals occurred after 4 h treatment (Fig. S3B). The data confirmed that the mNP-Mito complex entered the cells in an intact form. With prolonged incubation, there was a gradual dissociation of mNP from Mito, enabling them to function independently. As shown in Fig. S3E, the cell uptake contents of Cy5-tagged mRNA in the Cy5-mNP-Mito group increased compared with Cy5-mNP but were similar to those in the 1.5-fold gene-loaded Cy5-mNP-1.5 group. This may be attributed to the addition of exogenous mitochondria, shifting the uptake pathway of Cy5-mRNA from clathrin-mediated endocytosis to macropinocytosis (Fig. S3F and G). This shift in endocytic mechanisms was beneficial for increasing the cellular uptake of Cy5-mRNA by reducing endosomal degradation processes51,52. We also detected PARKIN mRNA transfection and found that the cells treated with mNP-Mito expressed more PARKIN protein than the other groups (Fig. 3F and G). Mitophagy after mNP-Mito treatment was investigated. As shown in Fig. 3H, mNP-Mito treatment increased the expression of PARKIN in mitochondria, indicating the enhanced recruitment of PARKIN to the surface of damaged mitochondria. Moreover, we have used mitophagy specific pharmacologic inhibitor (chloroquine, 40 μmol/L) to determine the change in the expression of LC3B-II protein, which indicates the autophagy flux. The results in Fig. 3I showed that mNP-Mito increased the expression of LC3B-II/LC3B-I under chloroquine treatment, suggesting that mNP-Mito enhanced the autophagy flux and the activities of autophagy. The results indicated that PARKIN protein overexpression in the mNP-Mito group enhanced mitophagy of damaged mitochondria.
Together, mNP-Mito was internalized into Rot-treated cells by micropinocytosis, and mNPs were detached from exogenous mitochondria. On the one hand, exogenous mitochondria were used to replenish dysfunctional mitochondria, increasing the population of functional mitochondria. On the other hand, mNPs promoted the expression of PARKIN proteins, inducing mitophagy of dysfunctional mitochondria (Fig. 3J).
3.3. mNP-Mito repairs complex I function
As mentioned above, mNP-Mito promoted the delivery of healthy mitochondria and enhanced mitophagy of dysfunctional mitochondria, which had the potential for correcting the activity and content of complex I. We further investigated the restoration of mitochondrial complex I mediated by mNP-Mito. First, we determined the activity of mitochondrial complex I through the Mitochondrial Complex I/NADH-CoQ Reductase Activity Assay Kit. Rot declined the activity of mitochondrial complex I compared with the PBS group, while mNP-Mito obviously repaired the activity of mitochondrial complex I (Fig. 4A). In addition, mitochondrial complex I antibody was used to mark functional complex I within cells. Complex I content was explored through flow cytometry (BD Biosciences) and CLSM (Carl Zeiss). After isolating mitochondria from cells treated with different groups, these isolated mitochondria in different groups were subjected to immunofluorescence staining using complex I antibody. The MFI was further detected by flow cytometry (BD Biosciences). As shown in Fig. 4B, the content of mitochondrial complex I was restored under mNP-Mito treatment. CLSM images (Fig. 4C) and quantitative analysis (Fig. 4D) of immunofluorescence staining showed that the FI in mNP-Mito was higher than that in the other groups and similar to the PBS groups, indicating that mNP-Mito enabled the repairment of mitochondrial complex I. These results indicated that mitochondrial complex I function was compromised in Rot-damaged cells. Conversely, treatment with mNP-Mito led to an enhancement in mitochondrial complex I function.
Figure 4.

The restoration of mitochondrial complex I function. (A) Detection of mitochondrial complex I activity; (B) Detection of complex I content by flow cytometry (BD Biosciences) in extracted mitochondria from different groups; (C, D) Immunofluorescence staining of complex I observed by CLSM (Carl Zeiss) (C) and quantitative data calculated by ImageJ software (D); (E) MMP detection by CLSM (Carl Zeiss); (F) MMP detection in different groups by a multi-mode microplate reader (Molecular Devices); (G) Detection of ATP generation; (H) The level of mtROS in different groups determined by MitoSOX probe in vitro; (I) Cellular viability after various treatments tested by live/dead cell double staining. Data are mean ± SD (n = 3 in A, B, and F, n = 30 cells in D, n = 6 in G). Statistical significance was analyzed via one-way ANOVA with Tukey's honest significant difference (HSD) post hoc test (A, B) or Games–Howell test (F, G).
Mitochondrial bioenergetics is a central regulator of cellular metabolism and redox maintenance53. We explored the restoration of MMP, ATP generation, and mtROS level under mNP-Mito treatment. To detect the restoration of MMP in Rot-induced cells after different treatments, we determined different MMP statuses of mitochondria by JC-1 Detection Kits. Strong red fluorescence indicates normal mitochondria with higher MMP, while an increase in green fluorescence represents damaged mitochondria. As shown in Fig. 4E, the red fluorescence indicated healthy mitochondria obviously declined, and the green fluorescent spots were enhanced in the Rot group, suggesting that Rot led to a decrease in MMP. In contrast, mNP-Mito restored the MMP to a normal level. In addition, we detected the normalized FI of JC-1-stained cells. Cells treated with Rot showed MMP damage compared to the PBS group. Although the cells that were treated with mNP, 1.5-fold gene-loaded mNP (named mNP-1.5) or Mito achieved only moderate mitochondrial remediation, the cells treated with mNP-Mito showed an obvious increase in MMP (Fig. 4F). Mitochondrial complex I serves as the primary entry point for electrons into the respiratory chain and is proposed to be the rate-limiting step in overall respiration. It plays a pivotal role in energy metabolism and ATP generation. Being the largest and most intricate component of the respiratory chain, complex I holds a central position in cellular bioenergetics. Hence, we investigated whether mNP-Mito could enhance ATP generation by improving mitochondrial complex I function in Rot-damaged cells. As illustrated in Fig. 4G, the ATP content in the Mito group exhibited an increase compared to the Rot group. Notably, the mNP-Mito group demonstrated a superior capacity for restoring ATP generation compared to other treatment groups, displaying no significant difference from the PBS group. As the byproducts of the mitochondrial respiratory chain54, the level of mtROS was assessed by using MitoSOX probe55,56. The results demonstrated that mNP-Mito obviously decreased the cellular levels of mtROS (Fig. 4H). A live/dead cell double-stain assay was conducted to assess the cellular viability protective effect of mNP-Mito, allowing for a clear distinction between fluorescent staining of live and dead cells. As shown in Fig. 4I, the total cell counts in the Rot group declined compared with the other groups. Additionally, green fluorescence-labeled live cells exhibited an increase in the mNP-Mito group compared to the Rot group, following a decrease in red fluorescence-labeled dead cells. In summary, the provided data demonstrated that mNP-Mito facilitated the repair of complex I, which promoted the restoration of mitochondrial function and rescued cells from damage caused by Rot.
3.4. Modulation of healthy and damaged mitochondrial populations via mNP-Mito in vivo
A common feature observed in a mouse model is the dysfunction of mitochondrial complex I caused by the inhibitor Rot35,57. In order to determine the appropriate dosage for Rot modeling, we conducted an optomotor test to probe eye health and explore the progress of Rot-induced LHON in mice36. Supporting Information Fig. S4A showed that the number of head movements was obviously decreased in the groups treated with different concentrations of Rot compared to in the vehicle control group, indicating that there was a noticeable decline in vision under the application of Rot. The generation of ATP in the retina isolated from mice was detected to show the mitochondrial function. The data in Fig. S4B suggest a significant reduction in ATP generation following treatment with 2.5 and 5 mmol/L Rot (P < 0.001). These results suggested that there was a noticeable decline in ATP generation and vision after one week of treatment with 2.5 mmol/L Rot. Moreover, the impairment of complex I was investigated after Rot damage for one week. The expression of NDUFB8 in the retinal layer was detected by Western blot. After Day 7 of 2.5 mmol/L Rot treatment, the band in the 2.5 mmol/L group was weaker than that in the Vehicle and 1 mmol/L groups (Fig. S4C). The content of complex I was assessed by immunofluorescence staining using an anti-complex I antibody. With increasing Rot concentration, complex I content was decreased (Fig. S4D). These results indicated a close association between Rot-induced mitochondrial damage and the progression of ocular mitochondrial diseases. In the subsequent studies, we opted for a 2.5 mmol/L Rot concentration to establish a mouse model for ocular mitochondrial disease.
For the construction of mNP-Mito in vivo studies, the allogeneic mitochondria were extracted from BALB/c mouse heart tissues (Supporting Information Fig. S5), which are the classical mitochondrial donors with a rich number of mitochondria58. The in vivo efficacy of mNP-Mito in repairing mitochondrial complex I function hinged on its capacity to regulate both healthy and damaged mitochondria. Based on this, we first investigated the in vivo capability of mNP-Mito to enhance mitochondrial delivery and induce mitophagy. On the one hand, we determined that mNP-Mito promoted the internalization of MitoTracker Red-labeled exogenous mitochondria in the normal mouse's retina, including the retinal ganglion layer (RGL) (Fig. 5A). This indicated that mNP-Mito promoted the delivery of exogenous mitochondria in RGL, and even a small quantity of mNP-Mito was capable of reaching the inner nuclear layer (INL). After 24 h treatment of MitoTracker Green-labeled Cy3-mNP-Mito in normal mice, exogenous mitochondria labeled by the green fluorescent spots were colocalized with the red-labeled mRNA. This observation suggested that Cy3-mNPs remained attached to the surface of the exogenous mitochondria upon reaching the RGL (Fig. 5B).
Figure 5.

The synergistic regulation of healthy and damaged mitochondria via mNP-Mito in vivo. (A) Uptake of MitoTracker Red-marked mNP-Mito in retinal ganglionic cell layer of normal mice (RGL: retinal ganglion layer; IPL: inner plexiform layer; INL: inner nuclear layer; OPL: outer plexiform layer; ONL: outer nuclear layer) (24 h); (B) The distribution of MitoTracker Green-labeled Cy3-mNP-Mito in vivo (24 h), white arrowheads represent the merged spots of exogenous mitochondria and Cy3-mRNA in RGL; (C, D) PARKIN protein expression in the whole cells according to immunofluorescence staining (C) and Western blot (D) assays in Rot-induced mice, dashed line area: RGL; (E) Quantitative analysis of PARKIN expression using ImageJ software based on the data presented in (D); (F) Mitochondrial PARKIN protein expression and ubiquitination of mitochondrial protein in Rot-induced mice in various treatment groups. Data are shown as the mean ± SD, n = 3 independent samples per group in (E). Statistical significance was shown by one-way ANOVA with Tukey's honest significant difference (HSD) post hoc test (E).
On the other hand, we detected mNP-Mito-mediated mitophagy in vivo. First, the expression of functional gene (PARKIN mRNA) was determined in Rot-induced mice. The mNP-Mito treatment was administered on Days 3 and 10 following the induction of the model by Rot. Retinal layers in varying treatments were collected on Day 17 after Rot modeling, and PARKIN protein expressions were investigated by immunofluorescence staining and Western blot assays. In the mNP-Mito group, PARKIN protein expressions in the RGL were higher than in other treatment groups (Fig. 5C). Simultaneously, the total PARKIN expression in the retina was significantly higher compared to other treatment groups (Fig. 5D and E). PARKIN-mediated mitophagy was assessed by observing the recruitment of PARKIN to mitochondria and ubiquitination of mitochondrial proteins (Fig. 5F). The levels of UBIQUITI and mitochondrial PARKIN expressions were higher in the mNP-Mito group compared to the other treatment groups. These findings indicated that mNP-Mito enhanced the delivery of exogenous healthy mitochondria and boosted PARKIN-mediated mitophagy of damaged mitochondria by promoting PARKIN protein expression in vivo. Therefore, mNP-Mito demonstrated the potential for the restoration of complex I function in Rot-induced mice through the dual regulation of both healthy and damaged mitochondria.
3.5. mNP-Mito suppresses Rot-associated ocular mitochondrial disease pathologies in mice
We investigated the therapeutic gains of nanoengineered mitochondria to suppress the progress of the ocular mitochondrial disease by the repairment of mitochondrial complex I (Fig. 6A). According to these results in Fig. S4, 2.5 mmol/L Rot was used as an inhibitor to induce the damage in mitochondrial complex I. It has been reported that the binding sites of inhibitor Rot bind are within complex I, including decylubiquinone and ND4 subunit in complex I46.
Figure 6.

The mNP-Mito alleviates ocular mitochondrial disease-related pathologies in mice by restoring the function of complex I. (A) Flowchart of mNP-Mito treatment in Rot-induced experimental ocular mitochondrial disease-like mouse model; (B) Detection of complex I activity in retina after different groups; (C) Mitochondrial NDUFB8 protein expression in retina detected by Western blot; (D) Detection of ATP generation in retina; (E) H&E staining; (F) Change in the thickness of the retinal layer detected by H&E staining; (G) Optomotor tests. Data are shown as the mean ± SD, n = 3 independent samples per group in (B, C, F), n = 6 mice per group in (D) and (G). Statistical significance was shown by one-way ANOVA with Tukey's honest significant difference (HSD) post hoc test (B, F, G) or Games–Howell test (C, D).
In the Rot-induced mouse model, the repairment of complex I under mNP-Mito treatment was investigated by the activity and content of complex I. First, mitochondrial complex I activities in the retina were investigated through the Mitochondrial Complex I/NADH-CoQ Reductase Activity Assay Kit. Complex I activity was significantly inhibited by the application of 2.5 mmol/L Rot. The activities of complex I were restored after the treatment with mNP, mNP-1.5, and Mito, while the activity of complex I was higher than that in the other groups (Fig. 6B). In addition, the contents of complex I were detected by the expression of NDUFB8 protein. The content of complex I was obviously decreased in the Rot group compared to the vehicle group. The parameter was increased when the mice were treated with mNP, mNP-1.5, and Mito. The content of complex I was further increased in the mNP-Mito group (Fig. 6C). ATP generation in the retinal layer was investigated by the ATP Bioluminescent Assay Kit. As shown in Fig. 6D, the ATP contents in the retina were hindered in the Rot group, whereas the generation of ATP was a slight increase in the mNP, mNP-1.5, and Mito groups. The production of ATP in the mNP-Mito group was higher than that in the other treatment groups.
In the presence of Rot damage, the retinal layer experiences impairment and a subsequent decline in thickness. The assessment of the retinal layer was conducted through Hematoxylin and Eosin staining (H&E staining), and the thickness was measured using the scale bar in H&E staining images. Following various treatments, eye tissues were collected for H&E staining. The results depicted in Fig. 6E and F affirmed that the thickness of the retinal layer in the mNP-Mito group increased compared to other groups. These findings suggested that mNP-Mito facilitated the repair of the retinal structure.
An optomotor assay was employed to assess the vision of mice subjected to various treatments. Rot nearly eliminated head responses in the mice, whereas the mNP-Mito group exhibited a significantly higher number of head movements compared to the mNP, mNP-1.5, and Mito groups. Interestingly, the number of head movements in the mNP-Mito group did not significantly differ from that in the vehicle group, suggesting that mNP-Mito contributed to a partial restoration of eyesight in Rot-treated mice (Fig. 6G). In summary, these results indicated that mNP-Mito suppressed the progress of ocular mitochondrial disease by the repairment of complex I function.
3.6. Safety assessment in vivo
Under some mitochondrial disease statutes, mitochondrial abnormalities are systemic; hence, allogeneic mitochondria from healthy mice were used to prepare mNP-Mito in our study. The mtDNA encodes 13 proteins associated with mitochondrial function, and the mitochondrial proteins in allogeneic mitochondria from the same species are similar59, but the safety risks and immune response of exogenous mitochondria should be considered. A series of assays were carried out to explore the safety risks and immune response to serial intravitreal injections of mNP-Mito (Fig. 7A). We recorded the body weights (Fig. 7B) and organ index (Supporting Information Table S1) of BALB/c mice and found that these indicators were not obviously changed compared with the PBS group. Liver function (alanine transaminase, ALT; aspartate aminotransferase, AST) and kidney (blood urea nitrogen, BUN) function were normal after different treatments (Fig. 7C‒E). In addition, the mtDNA copy number in the bloodstream serves as an important indicator for detecting whether mitochondrial transfer triggers an inflammatory response. We also determined the mtDNA contents in the whole blood after 1-, 2- and 4-fold mNP-Mito treatments. The results suggested that mNP-Mito did not cause an increase in mtDNA in whole blood and induce a further inflammatory response (Fig. 7F). To evaluate whether mNP-Mito treatment could induce inflammatory cytokine release, we determined the contents of TNF-α, IL-1β, IL-6, and IFN-γ in eye tissues using ELISA assays. As shown in Fig. 7G‒J, these inflammatory cytokines in the eye were not significantly different at any dose compared with the PBS group. In addition, H&E staining of the retina indicated that 1-, 2- and 4-fold mNP-Mito treatments did not damage the retinal structures (Fig. 7K). We also investigated the safety of mNP-Mito in the rotenone-induced animal model. As shown in Supporting Information Fig. S6A, the mouse weight in the mNP-Mito group did not show significant changes compared to the vehicle group. The key indicators of kidney and liver functions (Fig. S6B‒D), as well as hematological parameters (Supporting Information Table S2), were within normal ranges after various group treatments. These results confirmed the safety of mNP-Mito in the rotenone-induced animal model. In summary, our results showed that the supplementation with a large number of mitochondria caused almost no significant damage to the organism, and some studies reported that the increased bioenergetics from the introduction of exogenous mitochondria returned to the normal level over time58. The safety risks, including proinflammatory responses, from the supplementation of allogeneic mitochondria and the introduction of lipid-based NPs were not obviously observed, which supports the broader potential application of mNP-Mito in mitochondrial diseases.
Figure 7.

The safety of mNP-Mito in normal BALB/c mice. (A) Schematic representation outlining the safety assessment; (B) The weight of BALB/c mouse; (C–E) The levels of ALT (C), AST (D), and BUN (E); (F) Relative mtDNA copy number in the whole blood after Low-, Mid- and High-Dose treatments; (G–J) The levels of TNF-α (G), IL-1β (H), IL-6 (I) and IFN-γ (J) in various treatments; (K) H&E staining of retina in normal BALB/c mice after different mNP-Mito treatments. Data are shown as the mean ± SD, n = 5 mice per group in (B) and (F), n = 3 mice per group in (C–E) and (G–J). Statistical significance was performed by one-way ANOVA with Tukey's honest significant difference (HSD) post hoc test (G, H, J) or Games–Howell test (F, I). Low-dose: 1-fold mNP-Mito, Mid-dose: 2-fold mNP-Mito, High-dose: 4-fold mNP-Mito. The dashed area: normal range in each indicator.
4. Conclusions
In conclusion, we showed that PARKIN mRNA-loaded nanoparticle (mNP)-engineered mitochondria (mNP-Mito) increased mitochondria with functional complex I via the delivery of healthy exogenous mitochondria and decreased mitochondria with impaired complex I via enhanced PARKIN-mediated mitophagy process, which normalized the function of complex I for LHON treatment. Using a set of testing indexes including mitochondrial complex I activity and content, we found that mNP-Mito restored complex I function and rescued cells from Rot damage. After intravitreal injection, mNP-Mito repaired the mitochondrial function in the retina and reverted disease-related molecular and biochemical phenotypes in the Rot-induced experimental LHON-like mouse model. In addition, several mitochondrial diseases experience dysfunction within the mitochondria, and the advancement of many such conditions is closely linked to mitochondrial dysfunction, including ocular disorders, pulmonary disorders, neurodegenerative disorders, and cardiomyopathy. Therefore, mNP-Mito-mediated enhanced mitochondrial transfer offers a feasible approach for the restoration of the mitochondrial function at the level of mitochondrial population, which is promising for the treatment of more mitochondrial diseases.
Author contributions
Yi Wang: Writing – original draft, Investigation, Funding acquisition, Data curation, Conceptualization. Nahui Liu: Writing – review & editing, Methodology, Data curation. Lifan Hu: Writing – original draft, Methodology, Data curation. Jingsong Yang: Writing – review & editing, Data curation. Mengmeng Han: Writing – review & editing, Methodology, Data curation. Tianjiao Zhou: Writing – review & editing, Methodology. Lei Xing: Writing – review & editing, Supervision, Project administration, Funding acquisition. Hulin Jiang: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (82020108029, 82304416, 82073398, 82302367, China). This work was also supported by the Natural Science Foundation of Jiangsu Province (BK20231016, BK20231019, China), the China Postdoctoral Science Foundation (grant Number: 2022M720173, China), Jiangsu Funding Program for Excellent Postdoctoral Talent (2023, China), the Fundamental Research Funds for the Central Universities (2632023GR20, China), State Key Laboratory of Natural Medicines China Pharmaceutical University (SKLNMZZ202021, China), Double First-class University Projects (CPU2018GY06, China) and Double First-Rate construction plan of China Pharmaceutical University (CPU2022QZ18, China). We would like to thank Xiaonan Ma of China Pharmaceutical University for providing technical assistance with Carl Zeiss LSM 800 on the Public Experimental Platform.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2024.08.007.
Contributor Information
Lei Xing, Email: xinglei6xl@163.com.
Hulin Jiang, Email: jianghulin3@163.com.
Appendix A. Supporting information
The following is the Supporting Information to this article:
References
- 1.Murphy M.P., Hartley R.C. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018;17:865–886. doi: 10.1038/nrd.2018.174. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y., Hu L.F., Zhou T.J., Qi L.Y., Xing L., Lee J., et al. Gene therapy strategies for rare monogenic disorders with nuclear or mitochondrial gene mutations. Biomaterials. 2021;277 doi: 10.1016/j.biomaterials.2021.121108. [DOI] [PubMed] [Google Scholar]
- 3.Stenton S.L., Sheremet N.L., Catarino C.B., Andreeva N.A., Assouline Z., Barboni P., et al. Impaired complex I repair causes recessive Leber's hereditary optic neuropathy. J Clin Investig. 2021;131 doi: 10.1172/JCI138267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyseng-Williamson K.A. Idebenone: a review in Leber's hereditary optic neuropathy. Drugs. 2016;76:805–813. doi: 10.1007/s40265-016-0574-3. [DOI] [PubMed] [Google Scholar]
- 5.Falabella M., Minczuk M., Hanna M.G., Viscomi C. Gene therapy for primary mitochondrial diseases: experimental advances and clinical challenges. Nat Rev Neurol. 2022;18:689–698. doi: 10.1038/s41582-022-00715-9. [DOI] [PubMed] [Google Scholar]
- 6.Newman N.J. Treatment of hereditary optic neuropathies. Nat Rev Neurol. 2012;8:545–556. doi: 10.1038/nrneurol.2012.167. [DOI] [PubMed] [Google Scholar]
- 7.Zuccarelli M., Vella-Szijj J., Serracino-Inglott A., Borg J.J. Treatment of Leber's hereditary optic neuropathy: an overview of recent developments. Eur J Ophthalmol. 2020;30:1220–1227. doi: 10.1177/1120672120936592. [DOI] [PubMed] [Google Scholar]
- 8.Chen P.F., Liu X., Gu C.H., Zhong P.Y., Song N., Li M.B., et al. A plant-derived natural photosynthetic system for improving cell anabolism. Nature. 2022;612:546–554. doi: 10.1038/s41586-022-05499-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada Y., Hibino M., Sasaki D., Abe J., Harashima H. Power of mitochondrial drug delivery systems to produce innovative nanomedicines. Adv Drug Deliv Rev. 2020;154‒155:187–209. doi: 10.1016/j.addr.2020.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Zeng Z., Fang C., Zhang Y., Chen C.X., Zhang Y.F., Zhang K. Mitochondria-targeted nanocarriers promote highly efficient cancer therapy: a review. Front Bioeng Biotechnol. 2021;9 doi: 10.3389/fbioe.2021.784602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiao R., Lin X., Zhang Q., Zhang Y., Qin W., Yang Q., et al. Anti-tumor immune potentiation targets-engineered nanobiotechnologies: design principles and applications. Prog Mater Sci. 2024;142 [Google Scholar]
- 12.Liu D., Gao Y., Liu J., Huang Y., Yin J., Feng Y., et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct Target Ther. 2021;6:65. doi: 10.1038/s41392-020-00440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brestoff J.R., Wilen C.B., Moley J.R., Li Y., Zou W., Malvin N.P., et al. Intercellular mitochondria transfer to macrophages regulates white adipose tissue homeostasis and is impaired in obesity. Cell Metab. 2021;33:270–282. doi: 10.1016/j.cmet.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chinnery P.F. Mitochondrial replacement in the clinic. N Engl J Med. 2020;382:1855–1857. doi: 10.1056/NEJMcibr2002015. [DOI] [PubMed] [Google Scholar]
- 15.Huang T., Zhang T., Jiang X., Li A., Su Y.Q., Bian Q., et al. Iron oxide nanoparticles augment the intercellular mitochondrial transfer-mediated therapy. Sci Adv. 2021;7 doi: 10.1126/sciadv.abj0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youssoufian H., Pyeritz R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 17.Gorman G.S., Chinnery P.F., DiMauro S., Hirano M., Koga Y., McFarland R., et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2 doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- 18.Stewart J.B., Chinnery P.F. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- 19.Borcherding N., Brestoff J.R. The power and potential of mitochondria transfer. Nature. 2023;623:283–291. doi: 10.1038/s41586-023-06537-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim K., Cho S.I., Kim J.S. Nuclear and mitochondrial DNA editing in human cells with zinc finger deaminases. Nat Commun. 2022;13:366. doi: 10.1038/s41467-022-27962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacman S.R., Kauppila J.H.K., Pereira C.V., Nissanka N., Miranda M. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med. 2018;24:1696–1700. doi: 10.1038/s41591-018-0166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mok B.Y., Moraes M.H., Zeng J., Bosch D.E. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–637. doi: 10.1038/s41586-020-2477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho S.I., Lee S., Mok Y.G., Lim K., Lee J., Lee J.M., et al. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell. 2022;185:1764–1776. doi: 10.1016/j.cell.2022.03.039. [DOI] [PubMed] [Google Scholar]
- 24.Sun J., Lo H.T.J. High-efficiency quantitative control of mitochondrial transfer based on droplet microfluidics and its application on muscle regeneration. Sci Adv. 2022;8 doi: 10.1126/sciadv.abp9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu C., Wan T., Wang H., Zhang S., Ping Y., Cheng Y.Y. A boronic acid-rich dendrimer with robust and unprecedented efficiency for cytosolic protein delivery and CRISPR-Cas9 gene editing. Sci Adv. 2019;5 doi: 10.1126/sciadv.aaw8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitani T., Kami D., Matoba S., Gojo S. Internalization of isolated functional mitochondria: involvement of macropinocytosis. J Cel Mol Med. 2014;18:1694–1703. doi: 10.1111/jcmm.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun X., Chen H., Gao R., Qu Y., Huang Y., Zhang N. Intravenous transplantation of an ischemic-specific peptide-TPP-mitochondrial compound alleviates myocardial ischemic reperfusion injury. ACS Nano. 2023;17:896–909. doi: 10.1021/acsnano.2c05286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu S., Zhang A., Li S., Chatterjee S., Qi R., Segura-Ibarra V., et al. Polymer functionalization of isolated mitochondria for cellular transplantation and metabolic phenotype alteration. Adv Sci. 2018;5 doi: 10.1002/advs.201700530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeda H., Kami D., Maeda R., Murata Y., Jo J.I., Kitani T., et al. TAT-dextran-mediated mitochondrial transfer enhances recovery from models of reperfusion injury in cultured cardiomyocytes. J Cel Mol Med. 2020;24:5007–5020. doi: 10.1111/jcmm.15120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danese A., Patergnani S., Maresca A., Peron C., Raimondi A., Caporali L., et al. Pathological mitophagy disrupts mitochondrial homeostasis in Leber's hereditary optic neuropathy. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma L.K., Tiwari M., Rai N.K., Bai Y. Mitophagy activation repairs Leber's hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum Mol Genet. 2019;28:422–433. doi: 10.1093/hmg/ddy354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan X.Y., Guo L., Chen L.N., Yin S., Wen J., Li S., et al. Reduction of mtDNA heteroplasmy in mitochondrial replacement therapy by inducing forced mitophagy. Nat Biomed Eng. 2022;6:339–350. doi: 10.1038/s41551-022-00881-7. [DOI] [PubMed] [Google Scholar]
- 33.Fang E.F., Hou Y., Palikaras K., Adriaanse B.A., Kerr J.S., Yang B., et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci. 2019;22:401–412. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou H., Chen J., Lu X., Shen C., Zeng J., Chen L., et al. Melatonin protects against rotenone-induced cell injury via inhibition of Omi and Bax-mediated autophagy in Hela cells. J Pineal Res. 2012;52:120–127. doi: 10.1111/j.1600-079X.2011.00926.x. [DOI] [PubMed] [Google Scholar]
- 35.Chadderton N., Palfi A., Millington-Ward S., Gobbo O., Overlack N., Carrigan M., et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur J Hum Genet. 2013;21:62–68. doi: 10.1038/ejhg.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heitz F.D., Erb M., Anklin C., Robay D., Pernet V., Gueven N. Idebenone protects against retinal damage and loss of vision in a mouse model of Leber's hereditary optic neuropathy. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai E.T., Peng S.Y. HLA-homozygous iPSC-derived mesenchymal stem cells rescue rotenone-induced experimental Leber's hereditary optic neuropathy-like models in vitro and in vivo. Cells. 2023;12:2617. doi: 10.3390/cells12222617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sauvage F., Nguyen V.P., Smedt S.D. Laser-induced nanobubbles safely ablate vitreous opacities in vivo. Nat Nanotechnol. 2022;17:552–559. doi: 10.1038/s41565-022-01086-4. [DOI] [PubMed] [Google Scholar]
- 39.Shi C., Yuan X., Chang K., Cho K.S., Xie X.S., Chen D.F., et al. Optimization of optomotor response-based visual function assessment in mice. Sci Rep. 2018;8:9708. doi: 10.1038/s41598-018-27329-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y., Wang T., Dong X., Zhu C., Peng Q., Liu C., et al. Salivary amylase-responsive buccal tablets wipe out chemotherapy-rooted refractory oral mucositis. Adv Sci. 2024;11 doi: 10.1002/advs.202308439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W., Shi K., Chu B., Wei X., Qian Z. Mitochondrial surface engineering for multidrug resistance reversal. Nano Lett. 2019;19:2905–2913. doi: 10.1021/acs.nanolett.8b05188. [DOI] [PubMed] [Google Scholar]
- 42.Doherty E., Perl A. Measurement of mitochondrial mass by flow cytometry during oxidative stress. React Oxyg Species. 2017;4:275–283. doi: 10.20455/ros.2017.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Q., Li H., Archibong E. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat Biomed Eng. 2021;5:1038–1047. doi: 10.1038/s41551-021-00712-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghazi-Khansari M., Mohammadi-Bardbori A., Hosseini M.J. Using Janus green B to study paraquat toxicity in rat liver mitochondria: role of ACE inhibitors (thiol and nonthiol ACEi) Ann N Y Acad Sci. 2006;1090:98–107. doi: 10.1196/annals.1378.010. [DOI] [PubMed] [Google Scholar]
- 45.Reeves M.B., Davies A.A., McSharry B.P., Wilkinson G.W., Sinclair J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science. 2007;316:1345–1348. doi: 10.1126/science.1142984. [DOI] [PubMed] [Google Scholar]
- 46.Kampjut D., Sazanov L.A. The coupling mechanism of mammalian respiratory complex I. Science. 2020;370 doi: 10.1126/science.abc4209. [DOI] [PubMed] [Google Scholar]
- 47.Trushina E., Trushin S., Hasan M.F. Mitochondrial complex I as a therapeutic target for Alzheimer's disease. Acta Pharm Sin B. 2022;12:483–495. doi: 10.1016/j.apsb.2021.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palikaras K., Lionaki E., Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cel Biol. 2018;20:1013–1022. doi: 10.1038/s41556-018-0176-2. [DOI] [PubMed] [Google Scholar]
- 49.Wang J., Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. 2020;10:1866–1879. doi: 10.1016/j.apsb.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang M., Kim Y.K., Cui P.F., Zhang J., Qiao J.B., He Y.J., et al. Folate-conjugated polyspermine for lung cancer-targeted gene therapy. Acta Pharm Sin B. 2016;6:336–343. doi: 10.1016/j.apsb.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qi L.Y., Wang Y., Hu L.F., Zhao P.S., Yu H.Y., Xing L., et al. Enhanced nuclear gene delivery via integrating and streamlining intracellular pathway. J Control Release. 2022;341:511–523. doi: 10.1016/j.jconrel.2021.11.046. [DOI] [PubMed] [Google Scholar]
- 52.Zhang W., Kang X., Yuan B., Wang H., Zhang T., Shi M., et al. Nano-structural effects on gene transfection: large, botryoid-shaped nanoparticles enhance DNA delivery via macropinocytosis and effective dissociation. Theranostics. 2019;9:1580–1598. doi: 10.7150/thno.30302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vercellino I., Sazanov L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cel Biol. 2022;23:141–161. doi: 10.1038/s41580-021-00415-0. [DOI] [PubMed] [Google Scholar]
- 54.Wang D., Zhu X., Wang X., Wang Q., Yan K., Zeng G., et al. Multichannel sonocatalysis amplifiers target IDH1-mutated tumor plasticity and attenuate Ros tolerance to repress malignant cholangiocarcinoma. Adv Funct Mater. 2023;33 [Google Scholar]
- 55.Wang Y., Hekimi S. Mitochondrial dysfunction and longevity in animals: untangling the knot. Science. 2015;350:1204–1207. doi: 10.1126/science.aac4357. [DOI] [PubMed] [Google Scholar]
- 56.Mao Z., Liu W., Huang Y., Sun T., Bao K., Feng J. Anti-aging effects of chlorpropamide depend on mitochondrial complex-II and the production of mitochondrial reactive oxygen species. Acta Pharm Sin B. 2022;12:665–677. doi: 10.1016/j.apsb.2021.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y., Hu L.F., Cui P.F., Qi L.Y., Xing L., Jiang H.L. Pathologically responsive mitochondrial gene therapy in an allotopic expression-independent manner cures Leber's hereditary optic neuropathy. Adv Mater. 2021;33 doi: 10.1002/adma.202103307. [DOI] [PubMed] [Google Scholar]
- 58.Ali Pour P., Kenney M.C., Kheradvar A. Bioenergetics consequences of mitochondrial transplantation in cardiomyocytes. J Am Heart Assoc. 2020;9 doi: 10.1161/JAHA.119.014501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Falkenberg M., Larsson N.G., Gustafsson C.M. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.