

Significance
Plant leaves are colonized by a complex community of microbes that is shaped by host genetics. Although secreted metabolites are thought to mediate this effect, we investigated whether plants might also secrete RNA that could potentially structure microbial communities via cross-kingdom RNA interference. Here, we report that Arabidopsis leaves are covered with diverse RNAs of plant origin, including abundant tRNAs and tRNA-derived fragments. This leaf surface RNA is not associated with extracellular vesicles or protein complexes; however, it is less degraded than RNA found inside the extracellular spaces of leaves, suggesting that leaf surface RNA is secreted directly rather than exuded through stomata or hydathodes. We propose that this RNA plays a direct role in shaping the leaf microbiome.
Keywords: extracellular RNA, tRNA fragments, plant-microbe interactions, extracellular vesicles, Arabidopsis
Abstract
Transgenic expression of a double-stranded RNA in plants can induce silencing of homologous mRNAs in fungal pathogens. Although such host-induced gene silencing is well documented, the molecular mechanisms by which RNAs can move from the cytoplasm of plant cells across the plasma membrane of both the host cell and fungal cell are poorly understood. Indirect evidence suggests that this RNA transfer may occur at a very early stage of the infection process, prior to breach of the host cell wall, suggesting that silencing RNAs might be secreted onto leaf surfaces. To assess whether Arabidopsis plants possess a mechanism for secreting RNA onto leaf surfaces, we developed a protocol for isolating leaf surface RNA separately from intercellular (apoplastic) RNA. This protocol yielded abundant leaf surface RNA that displayed an RNA banding pattern distinct from apoplastic RNA, suggesting that it may be secreted directly onto the leaf surface rather than exuded through stomata or hydathodes. Notably, this RNA was not associated with either extracellular vesicles or protein complexes; however, RNA species longer than 100 nucleotides could be pelleted by ultracentrifugation. Furthermore, pelleting was inhibited by the divalent cation chelator EGTA, suggesting that these RNAs may form condensates on the leaf surface. These leaf surface RNAs are derived almost exclusively from Arabidopsis, but come from diverse genomic sources, including rRNA, tRNA, mRNA, intergenic RNA, microRNAs, and small interfering RNAs, with tRNAs especially enriched. We speculate that endogenous leaf surface RNA plays an important role in the assembly of distinct microbial communities on leaf surfaces.
Secretion of RNA into the extracellular environment is a well-conserved phenomenon as it is known to occur in all life forms (1–6). Recent studies in both mammalian and plant systems have revealed that the extracellular RNA (exRNA) pool is highly diverse, and a majority of exRNA is associated with RNA-binding proteins outside of extracellular vesicles (EVs) (7–9). However, there are only a few reports in plants wherein exRNAs other than those encapsulated inside EVs have been described (10, 11).
Most studies on plant exRNA have focused on small noncoding RNAs, including microRNAs (miRNAs) and small interfering RNAs (siRNAs), due to their roles in RNA interference (RNAi)-mediated gene silencing. The RNAi pathway is highly conserved and is triggered by dsRNA molecules that are recognized and processed into siRNAs by Dicer-like proteins (DCLs). The resulting siRNAs bind to Argonaute proteins (AGOs) to form RNA-induced silencing complexes (RISC), and subsequently, the siRNAs guide the RISC to target mRNA transcripts for degradation (12–15).
Two technologies that exploit RNAi to protect plants against invading phytopathogens have been successfully implemented. Host-induced gene silencing (HIGS) involves the incorporation of a transgene expressing a double-stranded RNA (dsRNA) in a plant that targets an essential pathogen gene (11). Spray-induced gene silencing (SIGS), in contrast, involves exogenous application of dsRNAs or siRNAs onto foliar surfaces. Both approaches have been shown to induce sequence-specific gene silencing in microbial pathogens, insects, and nematodes (16, 17). Notably, dsRNA sprayed onto barley leaves leads to the inhibition of fungal growth in nonsprayed distal tissues (16). Although the accumulation of unprocessed dsRNA was confirmed in distal tissues, the corresponding siRNAs were missing, suggesting that dsRNAs can be translocated within or on a plant leaf without being processed by DCLs, possibly in the apoplast.
Selective packaging inside plant EVs is the most widely studied trafficking system for silencing RNAs (10, 17–21). Two studies have reported the uptake of plant EVs carrying small RNAs by fungal cells, both in vitro and in vivo (18, 20); however, strong evidence supporting this mechanism of RNA uptake is still lacking. In particular, these studies did not assess whether RNA located outside of EVs contributes to HIGS, nor did they quantify the relative amounts of silencing RNAs located inside and outside of EVs in the apoplast. Additionally, if EVs are taken up via endocytosis, how silencing RNAs can then escape both the EV membrane and endosome membrane to engage the pathogen RNAi machinery is unknown.
In a recent report, Schlemmer et al. (22) isolated plant EVs from both dsRNA-expressing transgenic Arabidopsis plants and dsRNA-sprayed barley leaves to assess their effect on fungal growth. Surprisingly, both sources of plant EVs carried very small amounts of dsRNA-derived siRNAs and did not affect fungal growth, indicating a minor role of EVs in both HIGS and SIGS (22). There are no reports thus far confirming the presence of long dsRNA inside EVs. In another study, the exogenous application of sRNAs or dsRNAs targeting Botrytis DCL1 and DCL2 genes onto the surface of fruits, vegetables, and flowers was shown to significantly inhibit gray mold disease, suggesting that the pathogen is fully capable of taking up naked RNA (17).
Of particular note, HIGS-mediated resistance to a fungal pathogen can act prior to penetration of plant cell walls (23), which indicates silencing RNAs are being taken up from the leaf surface. For instance, transgenic rice lines expressing silencing RNAs targeting the MoAP1 gene of the fungal pathogen Magnaporthe oryzae displayed enhanced resistance to infection by M. oryzae. Although the fungal conidia germinated and formed appressoria, few of these formed infection hyphae, indicating that the fungus was blocked prior to its penetration into the plant cell wall. Similar observations were made for other fungal pathogens, including Puccinia triticina (24) and Puccinia striiformis f. sp. tritici (25, 26). These studies suggest that the silencing RNAs are taken up by the fungi from the leaf surface during the growth of the germ tube. Together, these observations led us to hypothesize that silencing RNAs produced by plants may be deposited onto the plant leaf surface, an extracellular fraction that has been overlooked in previous RNAi studies.
Here, we report that Arabidopsis leaf surfaces are coated with abundant RNA. Leaf surface RNA differs from apoplastic and cellular RNA both in composition and size. Furthermore, we found that the leaf surface RNA is not protected from endonuclease degradation either by EVs or RNA-binding proteins. In both the apoplast and leaf surface, we found tRNAs to be the most abundant, but leaf surface tRNAs were mostly intact, whereas apoplastic tRNAs were usually processed into tRNA halves (mainly produced by cleavage in the anticodon loop) and tRNA fragments (tRFs; produced by cleavage in the D loop). Such tRNA-derived molecules are now known to play gene regulatory roles in the context of plant–microbe interactions, which is independent of canonical tRNA function in translation (27, 28). We thus speculate that these extracellular tRNA-derived molecules may play a significant role in plant–microbe interactions. Additionally, we noted the enrichment of specific classes of miRNAs and siRNAs in the two extracellular fractions, which might also play a role in structuring the leaf microbiome and/or in mediating immune responses. Last, we found that leaf surface RNA forms cation-dependent condensates, which may contribute to the stability of these naked RNAs.
Results
Arabidopsis Secretes RNA Onto the Leaf Surface.
In our recent study, we demonstrated the presence of diverse species of RNA in the apoplast of Arabidopsis rosettes (11). To determine whether Arabidopsis plants also secrete RNA onto their leaf surface, we collected both leaf surface wash (LSW) and apoplastic wash fluid (AWF) from the same set of plants using the method developed in this study (SI Appendix, Fig. S1). To avoid microbial contamination, these two extracellular fractions were filtered through 0.2 µm filters before processing. Using trypan blue staining, we confirmed no ruptured cells (SI Appendix, Fig. S2), indicating that no cell damage occurred during LSW and AWF extraction. We then isolated RNA directly from the filtered LSW and AWF, without conducting any ultracentrifugation steps. Analysis of the purified RNA using denaturing RNA gel analysis revealed the presence of diverse species of long and small RNAs in both LSW and AWF (Fig. 1 A and B).
Fig. 1.
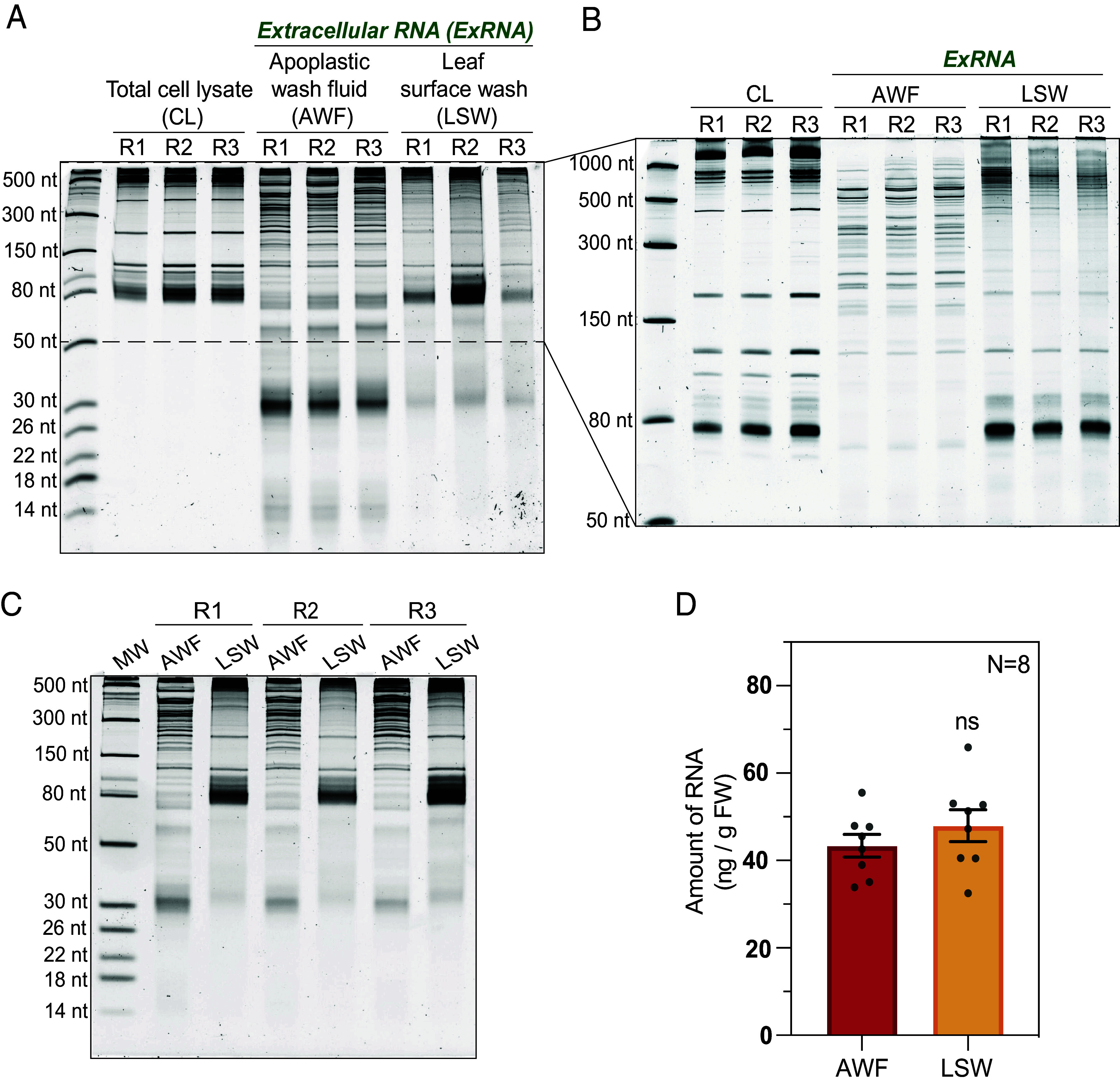
Plant leaves are coated with RNA. (A) Five-to-six-week-old Arabidopsis plants were used to collect leaf cell lysate (CL), AWF, and LSW from three biological replicates (R1, R2, and R3). 100 ng of RNA from each source was separated on a 15% denaturing polyacrylamide gel and stained with SYBR GOLD nucleic acid stain to visualize and compare the RNA-banding patterns. RNA size standards are shown in the leftmost lane. (B) CL, AWF, and LSW RNA (100 ng) was separated on a 10% denaturing polyacrylamide gel to better resolve the RNA between 50 to 1,000 nt. (C) To estimate the concentration of RNA in AWF and LSW isolated per gram fresh weight (ng/g FW) of leaf tissue, RNA separated on a 15% denaturing polyacrylamide gel was assessed through densitometry analysis using ImageJ software. (D) Bar graph represents the normalized amount of RNA isolated from eight independent replicates (N = 8; R4 to R8 shown in SI Appendix, Fig. S3) of AWF and LSW plotted with mean and SE. Student’s t test was performed to determine statistical differences in RNA concentration of AWF and LSW. ns: not significant.
Notably, equivalent RNA amounts were isolated from both leaf surface and apoplastic fluids [normalized per plant fresh weight (FW)] (Fig. 1 C and D and SI Appendix, Fig. S3). However, the RNA size profiles of the LSW and AWF fractions were substantially different from each other, and these two profiles differed from that observed for total cell lysate (CL) RNA, indicating that the two exRNA fractions were not contaminated with cellular RNA. Overall, apoplastic RNA was the most diverse fraction in terms of its size distribution. Although both extracellular fractions were enriched in RNAs smaller than 80 nt compared to total CL, species smaller than 60 nt were especially abundant in AWF. Of note, there was a strong accumulation of RNAs ranging from 30 to 35 nt in length in AWF, which were not as conspicuous in LSW. We observed a similar trend for tiny RNAs (<18 nt) (Fig. 1A). The differences in the banding pattern between LSW and AWF were also quite prominent in RNAs longer than 150 nt (Fig. 1B). The overall banding pattern of long RNAs (>1,000 nt) in LSW was more similar to that of CL than that of AWF.
Similar results were obtained when leaf surface RNA was isolated using an orthogonal technique of swabbing the adaxial and abaxial surfaces (SI Appendix, Fig. S4 A and B). Further analysis revealed that both leaf surfaces contained equivalent amounts of RNA despite the differences in the number of stomata and trichomes (SI Appendix, Fig. S4C). Together, these observations indicate that RNA on the leaf surface is unlikely to be derived from the apoplast, and thus is unlikely secreted out through stomata or hydathodes.
Unlike Apoplastic RNA, Leaf Surface RNAs Are Not Associated with Proteins and Can Be Fully Degraded by Endoribonucleases.
We have previously reported that RNA purified from P40 pellets isolated from AWF (obtained by centrifuging AWF at 40,000 g) is partially resistant to RNase A treatment, most likely due to its association with proteins, but not due to its packaging inside EVs (11). To analyze the role of proteins in protecting exRNAs (in both LSW and AWF) from ribonuclease digestion, we first treated both fractions with increasing concentrations of RNase A for 1 h at room temperature (RT) (Fig. 2A and SI Appendix, Fig. S5A).
Fig. 2.

LSW RNA is sensitive to RNase A and is depleted in proteins. (A) LSW and AWF samples were treated with increasing concentrations of RNase A as indicated. Treatments were performed at RT for 1 h, except for the indicated mock sample. RNA abundance in each gel lane was estimated by densitometry and expressed relative to Mock (4 °C). (B) Ribonuclease protection assay of AWF and LSW. AWF and LSW samples were treated with RNase A (0.1 µg/mL), or trypsin followed by RNase A, or TX-100 followed by trypsin followed by RNase A. RNA abundance in each gel lane was estimated by densitometry and expressed relative to the negative control kept on ice. The experiments in panels A and B were repeated twice on different days (additional replicate is shown in SI Appendix, Fig. S5). (C) Silver-stained SDS-PAGE showing the total protein profile of AWF and LSW. The amount of protein loaded in each lane was normalized by leaf fresh weight. MW: molecular weight markers (additional biological replicates (R5-R8) are provided in SI Appendix, Fig. S6A). (D) Amount of protein in AWF and LSW expressed as µg of protein per gram of leaf fresh weight. The amount of protein in each sample was estimated by densitometry using the silver-stained gels presented in panel C and SI Appendix, Fig. S6. The graph shows the mean ± SEM. Values from eight biological replicates were normally distributed and significant differences were calculated using Welch’s t test: ****P < 0.0001. (E) Immunodetection of EV-markers (PEN1, PATL1, ANNX1, and ANNX2) and RNA-binding proteins (GRP7, PR5, ANNX1, and ANNX2) in AWF and LSW fractions. The amount of protein loaded per fraction was normalized by leaf FW (additional replicates are shown in SI Appendix, Fig. S6B).
We observed a greater degradation of AWF RNA with an increase in the concentration of RNase A (from 0.1 to 9 µg/mL), but the overall banding pattern remained the same independent of the RNase A concentration, suggesting some degree of protection for RNAs in the AWF. Although AWF RNA was partly protected from digestion by RNase A, there was almost complete digestion of longer RNAs, and we noticed a change in the size distribution of RNAs smaller than 150 nt. Such alteration is indicative of partial digestion of RNAs, revealing an incomplete protection of RNA molecules. To further assess whether proteins were involved, we performed an RNase A protection assay using 0.1 µg/mL of RNase A followed by RNA isolation using TRIzol (Fig. 2B and SI Appendix, Fig. S5B). RNA gel analysis revealed that approximately 65 to 80% of the RNA in AWF was readily digested after treatment with only RNase A. Pretreatment with trypsin enabled an additional 10 to 15% of the RNA to be digested, but not all RNA. Notably, pretreatment with detergent prior to trypsin had no impact on digestion by RNase A, indicating that this RNA is not packaged inside EVs. These results are consistent with our previous reports using P40 pellets and indicate that proteins contribute to protecting apoplastic RNA from endoribonucleases (11).
We also assessed whether LSW RNA is also partially protected by proteins. We observed that treatment of LSW with just 0.1 µg/mL of RNase A alone was sufficient to degrade nearly all of the RNA in the LSW (Fig. 2 A and B and SI Appendix, Fig. S5), indicating that RNAs on the leaf surface are not protected by either proteins or EVs from exogenous RNases. Notably, a band corresponding in size to tRNA halves only disappeared with the highest concentration of RNase A used in this study, suggesting that these are partially resistant to degradation.
Since RNAs on the leaf surface are not protected from endoribonuclease digestion, we then tested the abundance of proteins associated with LSW RNA. Silver-stained SDS-PAGE analysis of AWF and LSW proteins, normalized by plant FW, revealed that there was substantially less protein in LSW compared to AWF (Fig. 2 C and D and SI Appendix, Fig. S6A). We performed immunoblot analysis to specifically assay the EV-marker proteins PENETRATION 1 (PEN1) and PATELLIN 1 (PATL1) (21) (Fig. 2E and SI Appendix, Fig. S6B) and RNA-binding proteins, including ANNEXIN 1 and 2 (ANN1 and ANN2) (19) in the two extracellular fractions. We also tested the extravesicular RNA binding proteins PATHOGENESIS-RELATED GENE 5 (PR5) and GLYCINE-RICH RNA-BINDING PROTEIN 7 (GRP7) (29–31). Surprisingly, LSW contained a very small amount of these proteins relative to AWF (Fig. 2E and SI Appendix, Fig. S6B). These results confirm a depletion of RNA-binding or EV-marker proteins in LSW, further suggesting that LSW may also lack RNases, which could account for the lower level of processing observed in leaf surface RNA compared to the apoplastic RNA.
AWF and LSW exRNAs Display Reduced Abundance of Many Transcripts Relative to Total CL RNA.
To investigate the long RNA content of AWF and LSW, we generated standard RNA sequencing libraries using random primers and without employing an rRNA depletion step to capture all exRNA, including degraded mRNAs. We used the standard Tuxedo pipeline (32) and the Arabidopsis TAIR10 annotation to identify differences in transcript abundance between the three fractions, CL, AWF, and LSW. In comparing AWF and LSW to CL, we found 300 and 297 genes, respectively, that were differentially expressed (DE; Fig. 3A, SI Appendix, Fig. S7, and Dataset S1). The majority of these DE genes were downregulated, demonstrating that both AWF and LSW fractions are gene-depleted relative to total CL RNA.
Fig. 3.

exRNA exhibits a distinct long and small RNA composition compared to cell lysate. (A) Number of differentially expressed genes found in the comparison of each fraction, down-regulated in pink and up-regulated in green. CLribo indicates an rRNA-depleted RNA sample, which showed no enrichment in mRNA-encoding genes. (B) Genomic origin of RNA reads based on the categories established in the TAIR 10 genome version. RNAs that mapped to the genome were categorized by origin and plotted by relative abundance in reads per million (RPM). The x axis represents the feature of interest. Note the differing y axis values between the three panels. MIR, microRNA precursors; snoRNA, small nucleolar RNA; TAS, trans-acting siRNA precursors; TE, Transposable elements; snRNA, small nuclear RNAs; tRNA, transfer RNA precursors; cDNA, complementary DNA; Intergenic regions; rRNA, ribosomal RNAs; The y axis indicates the relative cumulative abundance in RPM, calculated as indicated before. The three colored shades represent three independent biological replicates, with each replicate derived from 18 Arabidopsis plants. (C) sRNA size distribution of reads mapping to the Arabidopsis genome (TAIR version 10). The abundance of each size class was calculated for each sample independently, and normalized to the total number of library reads. The x axis indicates the sRNA size, from 10 to 50 nucleotides long, and the y axis indicates its abundance in RPM reads. (D) Genomic origin of small RNA reads based on the categories established in the TAIR 10 genome version. RNAs that mapped to the genome were categorized by origin and plotted by relative abundance in RPM. cDNA, complementary DNA; Pol4, Polymerase IV-dependent products; rRNA, ribosomal RNAs; TE, Transposable elements; tRNA, transfer RNAs; miR, microRNAs; snoRNA, small nucleolar RNA; snRNA, small nuclear RNAs; TAS, trans-acting siRNA. The three colored shades represent three independent biological replicates, with each replicate derived from 18 Arabidopsis plants.
We also analyzed the origin of the RNAs captured in each fraction (Fig. 3B). We observed that all fractions have similar levels of RNAs from rRNA, cDNA, and intergenic regions and that these are the most abundant ones. Interestingly, long reads originating from tRNAs were highly enriched in LSW compared to AWF. Notably, the exRNA fractions were depleted in reads originating from all the other minor features, including miRNA precursors, suggesting that these are particularly unstable in the extracellular environment or that they are not selectively secreted.
AWF and LSW Exhibit a Distinct Small RNA Composition Compared to Total CL.
To compare the content of leaf surface RNA to apoplastic and cellular RNA, we performed sRNA sequence analysis. We sequenced sRNA from three biological replicates of each fraction, including total CL, AWF, and LSW (a total of nine libraries). More than 70% of AWF and LSW RNA reads were derived from Arabidopsis (SI Appendix, Table S1), indicating that LSW RNA is not due to environmental contamination. To further support this conclusion, we also isolated AWF and LSW RNA from maize (Zea mays) seedlings and mapped these reads to both the Arabidopsis and maize genomes (SI Appendix, Table S1). 65 to 80% of these reads mapped to the maize genome, while only 17 to 26% mapped to the Arabidopsis genome. The latter likely represent conserved miRNA and tRNA sequences.
Total CL RNA displayed two predominant peaks at 21 and 24 nt, mostly corresponding to miRNAs and heterochromatic siRNAs (SI Appendix, Fig. S8). However, AWF and LSW RNA exhibited different size distribution patterns with peaks at 16 and 31 nt, and 16, 20, and 32 nt, respectively (Fig. 3C). To understand the nature of these AWF and LSW sRNAs, we analyzed their genomic origin. We observed that most of the sRNAs in the extracellular fractions originated from rRNAs, cDNA, tRNAs, and products that were dependent on RNA polymerase IV (Pol IV) (Fig. 3D and SI Appendix, Fig. S9). We also observed that the AWF and LSW RNAs exhibited statistically significant enrichment in sRNAs derived from tRNAs when compared to total CL and statistically significant depletion in miRNAs, tasiRNAs, and rRNA-derived fragments (SI Appendix, Fig. S9).
Specific miRNAs Differentially Accumulate in LSW and AWF.
We observed that the size distribution of reads mapping to miRNAs was different in each fraction, shifting from mainly 21 nt in CL to 18 nt in AWF, and 20 nt in LSW (SI Appendix, Fig. S8A), and a similar trend was seen for tasiRNAs (SI Appendix, Fig. S8B). These differences in size are due to the trimming of extracellular miRNAs at their 3′ ends (SI Appendix, Fig. S10), suggesting that they are processed by an extracellular 3′ exonuclease.
Since the size of the miRNA influences AGO loading and its subsequent activity, we performed a differential expression analysis considering only reads that mapped to known miRNAs with lengths 20, 21, or 22 nt long. Although miRNAs are typically underrepresented in the AWF and LSW fractions compared to total CL RNA, we observed that 19 miRNAs had a higher number of reads in AWF and/or LSW, compared to total CL (SI Appendix, Fig. S11). Out of these, eight miRNAs, belonging to three families, miR8167, miR5653, and miR5659, had a higher abundance in AWF than in LSW and CL (SI Appendix, Fig. S11A). The other eleven miRNAs had a higher abundance in LSW than in the other two fractions. These miRNAs belong to six distinct, but highly conserved miRNA families, including miR156, miR169, miR172, miR5014, miR773, and miR829 (SI Appendix, Fig. S11B). Plants overexpressing miR156 have been found to secrete this miRNA into the growth medium, which is then taken up by wild-type plants in coculture, resulting in downregulation of target genes (33).
We also observed that LSW miRNAs, but not AWF miRNAs, are globally enriched in miRNA* strands (SI Appendix, Fig. S12A). This enrichment suggested that some LSW miRNAs may be double-stranded. We thus assessed the miRNA to miRNA* ratio for the three most abundant LSW miRNAs (combined strand count). All three displayed a ratio close to 50% (SI Appendix, Fig. S12B), suggesting that these may, indeed, exist in a double-stranded form on the leaf surface.
tRNA Halves Are Enriched in Extracellular Fractions.
As noted in Fig. 3D, our sRNA sequence analysis revealed that LSW and AWF RNA are enriched in reads that mapped to tRNA genes. Since tRFs have recently been shown to possess biological activities independent of amino acid delivery to ribosomes (34–36), we analyzed these reads using the Unitas pipeline (37), which classifies tRNA-derived fragments into 5′ tRFs (5′ end to D-loop), 5′ tR-halves (5′ end to anticodon-loop), 3′ tRFs (TψC-loop to 3′ end, but without CCA addition), 3′ CCA-tRFs (TψC-loop to 3′CCA), 3′ tR-halves (anticodon-loop to 3′ end) and misc.-tRFs (miscellaneous tRFs; any reads that map to the mature tRNA but do not align to the very 5′ or the very 3′ ends). The majority of LSW reads corresponded to 3′ tR-halves, while most of AWF reads were classified in the misc-tRF category (Fig. 4A), consistent with a higher level of degradation in AWF samples resulting in the removal of 5′ ends and/or 3′ ends of tRNAs. We then plotted length distributions of misc.-tRFs (Fig. 4B and SI Appendix, Fig. S13 A and B). This analysis revealed that AWF misc-tRFs had peaks at 17 and 18, 28 to 30, and 32 to 35 nt. The first peak corresponds to 5′ and 3′ tRFs that are missing the first or last nucleotide. To test whether the other two peaks corresponded to tRNA halves lacking the 5′ and 3′ ends of the tRNAs, we reassigned misc.-tRFs as misc.-5′ tR-halves (if they started within the first four nucleotides of the tRNA and were longer than 28 nt), and to misc.-3′ tR-halves (if they started after position 29 and were longer than 27 nt) (SI Appendix, Fig. S13C). The majority of AWF misc.-tRFs were reassigned as misc.-3′ tR-halves, with only a few percent assigned to misc.-5′ tR-halves. Taken together, these results confirm that LSW and AWF are enriched in tRNA halves, especially 3′ tR-halves.
Fig. 4.

LSW and AWF are enriched in tRNA halves. (A) Abundance in RPM of tRNA-derived small RNAs as classified by unitas (5′-tR-halves, 3′-tR-halves, 5′-tRFs, 3′-tRFs, 3′-CCA-tRFs, tRNA leader, and tRF-1) across different samples. (B) Length distribution of reads classified as misc-tRF in AWF samples. The colors represent replicates.
To evaluate the accumulation of specific tRNA families, we combined all reads originating from tRNAs based on their anticodons. We identified a total of 94 different anticodon tRNAs in our samples, consisting of 50 nuclear, 29 chloroplastic, and 15 mitochondrial tRNAs. Of these 94 tRNAs, a small subset dominated the read count, with tRNAGluCUC/UUC being especially abundant, topping the list for AWF, LSW, and CL (Dataset S3). This abundance likely reflects both expression and resistance to extracellular RNases.
tRNAs Are Less Processed in LSW than in AWF.
The above sequence analyses indicated that AWF tRNAs are more degraded/processed than total CL tRNAs. Processing of tRNA by endoribonucleases in the extracellular environment has previously been reported in mammals. Although tRNA processing in plants has been well studied, whether this occurs primarily in intracellular or extracellular locations has not been assessed. Cleavage of tRNAs by endoribonucleases primarily occurs within tRNA loops (38, 39), and the primary enzymes implicated are endoribonucleases belonging to the RNase T2 family (34). The cleavage of tRNA by T2 RNases has been shown to result mostly in 3′ tRNA-derived fragments with 5′-OH ends and 5′ fragments with a terminal 2′,3′- cyclic phosphate (cP) or 3′ phosphate (P), both of which are recalcitrant to the sRNA sequence library preparation used in this work. Only a low percentage of tRNA-derived fragments possess the conventional 3′-OH and 5′-P ends, and therefore are likely sequenced (27, 40–42). As an alternative approach, we selected four tRNAs, tRNAGly, tRNAGlu, tRNAAla, and tRNALys, and analyzed them using RNA gel blot analysis (Fig. 5 and SI Appendix, Fig. S14).
Fig. 5.
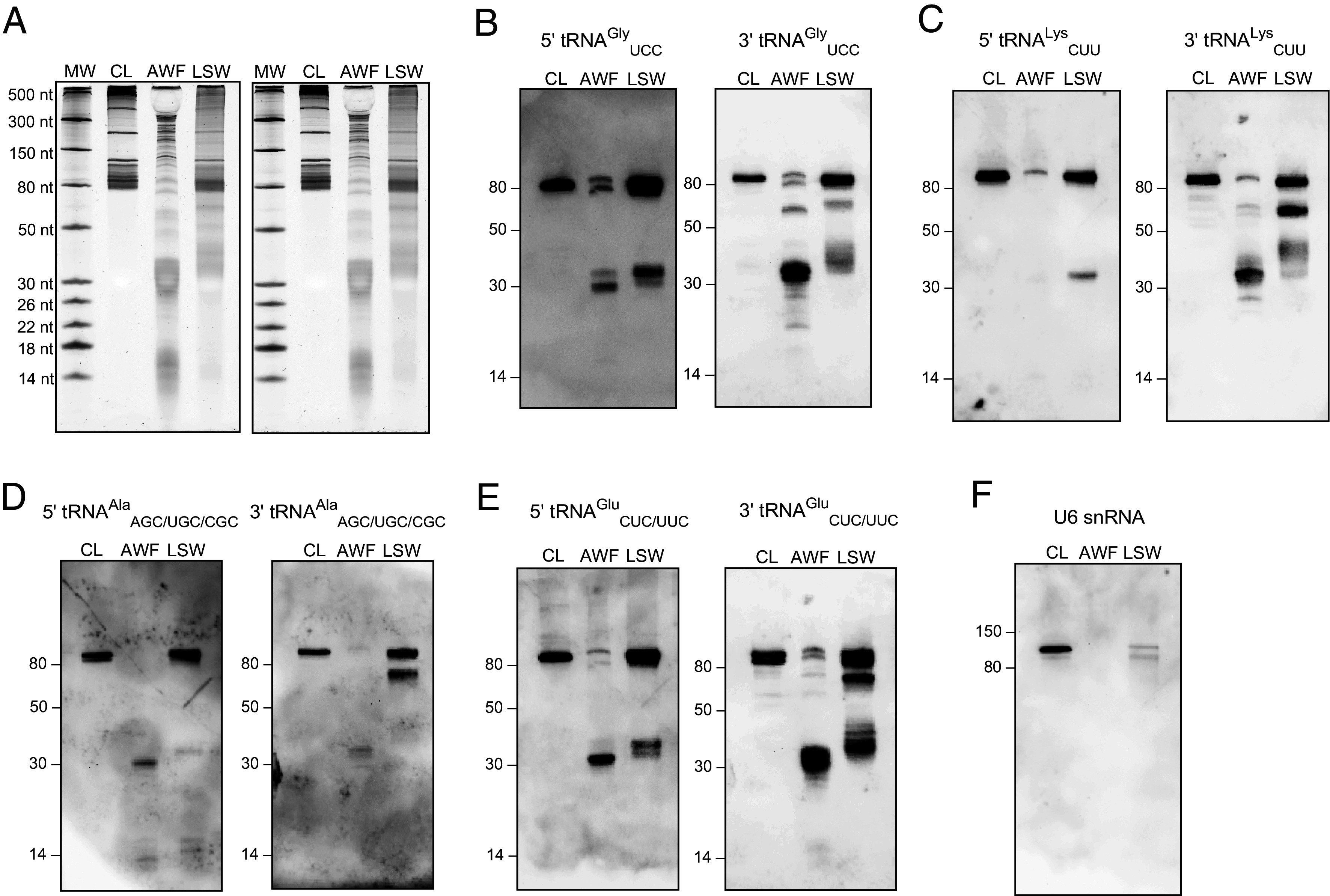
Extracellular fractions are enriched in tRFs compared to whole cell lysate. (A) 100 ng of RNA from CL, AWF, and LSW was separated on 15% denaturing polyacrylamide gels and stained with SYBR GOLD nucleic acid stain. (B–E) Upon blotting onto a positively charged nylon membrane, RNA was probed with DIG-labeled 5′ and 3′ probes against tRNAGly, tRNALys, tRNAAla, and tRNAGlu, respectively, to detect the tRNA halves (30 to 40 nt), and tiny tRFs (<18 nt) in the CL, AWF, and LSW samples. Full-length tRNAs (>80 nt) and three-quarter (3/4) tRFs (60 to 70 nt) were also detected in these samples. (F) RNA was probed with a DIG-labeled probe against U6 snRNA to assess its presence in AWF and LSW. Other replicate of this experiment is presented in SI Appendix, Fig. S14.
Three of these tRNAs, tRNAGlyUCC, tRNAGluCUC/UUC, and tRNAAlaAGC were selected based on their high abundance in our sRNA sequence data (Dataset S3). tRNALysCUU was selected because it did not occur in the top 10 list of tRNAs in our sRNA sequence but is known to accumulate stably in mammals without undergoing extensive cleavage (43). To enable the detection of both 5′ and 3′ tRNA-derived fragments, along with the corresponding full-length tRNAs, we designed two probes for each tRNA, one complementary to the 5′ tR-half (5′ probe) and the other to the 3′ tR-half (3′ probe) (SI Appendix, Table S2).
These RNA blot analyses confirmed that tRNA halves accumulate to higher levels in AWF than in LSW or leaf surface swab (LSS) RNA (Fig. 5 and SI Appendix, Figs. S14 and S15). Notably, we could not detect any tRNA halves in CL RNA, indicating that cleavage of tRNAs rarely occurs inside plant cells. Interestingly, we found that 3′ tR-halves accumulate even more compared to their 5′ counterparts in both AWF and LSW, suggesting that 3′ tR-halves are more stable than 5′ tR-halves in the extracellular milieu. This was especially noticeable for tRNALysCUU for which 5′ tR-halves seem to be highly unstable, while the 3′ tR-halves exhibited stable accumulation in AWF (Fig. 5C and SI Appendix, Figs. S14C and S15D). Strikingly, by using the 3′ probes, we were able to identify a class of tRNA-derived fragments ranging in size from ~60 to 70 nt in both AWF and LSW, which we termed tRNA three-quarters (hereafter referred to as tR-3/4). These are likely produced after cleavage only in the D-loop of the full-length tRNA (Fig. 5 B–E and SI Appendix, Figs. S14 B–E and S15 B and D). Moreover, we noted a high accumulation of full-length tRNAs in LSW compared to AWF, suggesting that the tRNAs are much less processed in LSW than in AWF. The occurrence of less tRNA processing in LSW compared to AWF could be attributed to the absence or very low abundance of RNA-processing enzymes on the leaf surface compared to the apoplast (Fig. 2 C and D and SI Appendix, Fig. S6).
We also observed a differential accumulation of fragments derived from the same parental tRNAs but with slightly different lengths. In mammals, tRNAGly has been shown to undergo sequential cleavage, first cleaved at the anticodon loop, generating 34 and 35 nt 5′ tR-halves that rapidly disappear, which are subsequently replaced by highly stable shorter fragments of approximately 30 and 31 nt (43). We observed a higher accumulation of 34 and 35 nt 5′ tR-halves of tRNAGly in LSW, and stable accumulation of the corresponding 30 and 31 nt 5′ tR-halves in AWF (Fig. 5B and SI Appendix, Fig. S15B). We also observed a similar trend upon specific assessment of tRNA halves derived from tRNAGly in our sequencing data. This further substantiates our hypothesis that there is less processing of RNA in LSW compared to AWF.
As a control, we used a probe specific to the U6 small nuclear RNA (snRNA) (Fig. 5F and SI Appendix, Figs. S14F and S15C). We detected little to no U6 RNA in LSW and AWF RNA, suggesting that U6 is either not secreted or secreted and is highly sensitive to extracellular RNases in AWF.
exRNA Is Not Enriched in m6A Modification.
Previously, we reported that exRNA isolated from AWF is enriched in the posttranscriptional modification N6-methyl adenine (m6A) (11). To test whether exRNA isolated from LSW is also enriched in m6A, we performed an ELISA-based m6A quantification method that has recently been commercialized (44). These analyses revealed that exRNA isolated from AWF and LSW is not enriched but depleted in m6A modification compared to total CL RNA (SI Appendix, Fig. S16A), which is contrary to what we observed in our previous work (11). This observation indicated that commercial anti-m6A antibodies might cross-react with a non-RNA molecule that copurifies with RNA extracted from leaf-derived samples using Trizol. We tested for potential contaminants using the anti-pectin antibody JIM7 as well as treating samples with DNase, but were unable to identify the source of the cross-reacting signal (SI Appendix, Figs. S16 B–D and S17).
exRNAs May Form Cation-Dependent Aggregates or Condensates In Vivo.
In our previous work, we found that apoplastic RNAs longer than 50 nt could be pelleted by ultracentrifugation at 100,000 g, indicating that this RNA was associated with some kind of particle. We thus assessed whether RNAs found on the leaf surface were also associated with a particle of some kind that could contribute to their stability. We ultracentrifuged AWF and LSW at 40,000 g (P40), and then the supernatant of this pellet at 100,000 g (P100-P40). These speeds are commonly used to isolate different subpopulations of plant EVs, but we did not expect single RNA molecules to pellet even at 100,000 g. We then analyzed the RNA content of P40 and P100-P40 pellets along with the supernatant of the P100-P40 pellet (S100). In concordance with our prior work (11), apoplastic RNAs longer than 35 to 40 nt pelleted at both speeds, although a higher amount was observed at 100,000 g (Fig. 6A).
Fig. 6.

Long RNAs in LSW form cation-dependent aggregates or condensates. (A) Profile of RNAs in P40, P100-P40 pellets, and supernatants (S100) obtained using AWF and LSW as the starting material. Volumes of starting AWF and LSW were normalized by plant fresh weight. This experiment was repeated at least three times with similar results. (B) Proteins do not play a significant role in the pelleting of LSW RNA. LSW was mock-treated (−) or treated with Trypsin (+) and incubated at 30 °C for 1 h, followed by ultracentrifugation at 40,000 g. This experiment was repeated twice with similar results (additional replicate is provided in SI Appendix, Fig. S18). (C) Cations contribute to RNA particle formation. LSW was treated with increasing concentrations of EGTA as indicated for 20 min on ice, followed by ultracentrifugation at 40,000 g. This experiment was repeated three times with similar results (additional replicate is provided in SI Appendix, Fig. S18). (D) Cations contribute to the copelleting of pectin with LSW RNA. LSW was treated with 0 and 10 mM EGTA for 20 min on ice, followed by ultracentrifugation at 40,000 g. RNAs were isolated from P40 pellets and aliquots of 100 ng of RNA were dot-blotted onto a positively charged nylon membrane and then probed with the JIM7 (anti-pectin) antibody. Two independent replicates are presented here.
Surprisingly, despite the lack of proteins in LSW, long LSW RNAs also pelleted at both speeds (Fig. 6A). To confirm that proteins were not contributing to the pelleting of LSW RNA, we treated LSW RNA with trypsin prior to centrifugation. This treatment had no impact on RNA pelleting (Fig. 6B and SI Appendix, Fig. S18C), indicating that the pellets are unlikely to be protein:RNA complexes.
Considering that RNAs are negatively charged molecules, we then tested whether cations were involved in RNA pelleting. Divalent ions can bind directly to more than one phosphate group and form a bridge between different parts of an RNA molecule or between different RNA molecules (45). Since we used a buffer containing 2 mM CaCl2 to isolate LSW, we repeated the pelleting at 40,000 g (P40) after adding increasing concentrations of EGTA, a divalent cation chelator with a strong affinity for Ca2+, to the LSW. We observed a significant reduction in the pelleting of RNA with 5 and 10 mM of EGTA (Fig. 6C and SI Appendix, Fig. S18A); additionally, we eliminated Ca2+ from VIB and repeated the LSW isolation and pelleting, but no effect was observed in the amount of RNA that pelleted compared to the buffer containing Ca2+ (SI Appendix, Fig. S18B), suggesting that leaf surfaces possess a substantial amount of endogenously secreted Ca2+ or other cations that facilitate RNA aggregation or condensation on the leaf surface.
The promotion of RNA pelleting from LSW by cations suggested that there could be other negatively charged molecules associated with the RNA. An abundant negatively charged molecule secreted by plant cells is pectin, a cell wall polysaccharide that can be noncovalently crosslinked by Ca2+ ions. To assess whether pectin was present in P40 pellets, we use an anti-pectin antibody (JIM7). Since pectin is highly methylated, we also used 21 nt long oligonucleotides with and without a single modified methylated adenosine as negative controls to assess the specificity of JIM7. We confirmed the presence of pectin in RNA isolated from the CL, AWF, and LSW fractions (SI Appendix, Fig. S16B). We next assessed whether pectin precipitation was also inhibited by EGTA, similar to RNA. We observed a significant reduction in pectin in the P40 pellet in the presence of 10 mM EGTA compared to no EGTA (Fig. 6D), suggesting that pectin associates with RNA and could potentially stabilize RNA on the leaf surface.
Discussion
In our previous work, we reported that the plant leaf apoplast contains abundant and diverse species of RNA outside EVs, which are protected from RNase degradation by a group of extravesicular RNA-binding proteins (10, 11). These apoplastic RNAs have not been ascribed a biological function, but potential roles have been discussed in a recent review (1). Here, we have reported the existence of abundant plant RNAs on the leaf surface. The RNA content on the leaf surface is distinct from that of the apoplast, as it appears much less degraded, which suggests an absence of RNA processing enzymes on the leaf surface. Additionally, our RNase protection analyses indicated that although leaf surface RNAs are less processed than apoplastic RNAs, they are not protected by proteins or vesicles. Consistent with an absence of RNases on the leaf surface, we found very low amounts of protein overall in LSW samples.
In our previous reports, we and others demonstrated that most siRNAs that copellet with EVs are located outside of EVs and can be found associated with proteins (10, 11, 22). Moreover, the vast majority of sRNAs in the apoplast did not pellet, even after spinning at 100,000 g (11), a speed that has been extensively used to isolate sRNA-containing EVs (18, 19). Those results encouraged us to analyze the entire sRNA pool in the extracellular space without conducting any ultracentrifugation steps. Surprisingly, our results revealed that Arabidopsis does not seem to accumulate siRNAs/miRNAs in extracellular fractions, with most miRNAs identified by sRNA sequencing far more abundant inside cells than outside cells when quantified in terms of RPM.
The most abundant class of exRNAs that we found in the apoplast was tRFs and tRNA halves [collectively referred to as tRNA-derived RNAs (tDRs)]. In mammals, full-length tRNAs are secreted into the extracellular space from diverse cell types, where they are rapidly processed into tDRs by extracellular RNases (8, 9). Although this mechanism has not been explored in plants, we believe that the extracellular processing of tRNA also occurs in plants. The generation of tDRs in plants relies mainly on endonucleases belonging to the RNase T2 family (reviewed in ref. 34). Of particular relevance to our findings, these RNases contain signal peptides and are known to be secreted to the apoplast. Consistent with this, the ratio of tRNA halves to full-length tRNAs in the apoplast was very high, while the opposite was observed in RNA isolated from total cell lysate, suggesting a lack of processing of tRNAs inside the cell. Although the pattern observed could also indicate selective and rapid secretion of tDRs by cells, we believe it is far more likely that tDRs are generated outside the cell due to the presence of extracellular RNases in the apoplast.
Given the presence of RNases in the apoplast, it is somewhat surprising that single-stranded sRNAs such as tDRs are so stable in the apoplast. In mammals, it has been shown that some tDRs can acquire high resistance to RNases by forming self-protecting homo- and heterodimers or oligomers (46, 47). Moreover, a recent mammalian paper shows that after cleavage at the anticodon loop, the 5′ and 3′ halves of the original tRNA can be held together, thus stabilizing these tDRs (43). Whether similar processes contribute to tDR stability in plants is still unknown. Interestingly, according to our RNA gel blot analyses, 3′ tDRs are more stable than 5′ tDRs in the apoplast. We speculate that posttranscriptional modifications may help stabilize specific tDRs (48, 49). Alternatively, 3′ tDRs may fold into more stable secondary structures than 5′ tDRs.
We have also shown the existence of 3/4 length 3′ tRFs. These tRFs are highly abundant in the apoplast and are likely produced after cleavage in the D-loop. Of note, we did not observe any 3/4 length 5′ tRFs (produced by cleavage in the TψC loop), and we detected very few 3′ tRFs. This suggests that cleavage in the TψC is less common than in the two other loops. In addition, our results support the idea that cleavage in the TψC takes place only after cleavage in the anticodon loop.
Diverse biological functions of tDRs have been recently proposed in plants, including a role in regulating gene expression (reviewed in refs. 34 and 35). Although direct evidence of a functional role for extracellular tDRs in cross-kingdom gene regulation is lacking, the high accumulation of tDRs in the apoplast suggests that they are biologically relevant. A key question to be addressed in the future is whether plant cells can take up tDRs from the apoplast, which would indicate that tDRs could function in intercellular gene regulation. Another key question is whether plant tDRs are taken up by leaf bacteria and can impact bacterial growth. Recent work on the human oral microbiome suggests that this is likely (50, 51). In that work, it was shown that human saliva contains abundant tDRs, several of which bear similarity to tRNA sequences in specific oral bacterial species. Application of these tDRs to cultures of these bacterial species inhibited the growth of some species and not others, and this inhibition was sequence specific. Thus, human tDRs help structure the human oral microbiome. Notably, this effect was observed with naked RNA, thus association with protein or packaging inside EVs was not required for uptake of tDRs by bacteria.
Our finding that leaf surfaces are coated with RNA raises the question of how this RNA gets to the surface and whether it is being secreted directly from epidermal cells, passing through the cuticle, or instead, is secreted from mesophyll cells and passes through stomata. If the latter were the case, however, we would expect LSW RNA to look more like apoplastic RNA. Instead, we observed that LSW RNA appears much less degraded than apoplastic RNA, indicating that a mesophyll origin is unlikely. How RNA molecules pass through the hydrophobic cuticle layer and reach the leaf surface is unknown, but the lack of RNA-binding proteins in LSW makes it unlikely that RBPs function as RNA-carriers across the cuticle. We speculate that regions of higher permeability across the cuticle layer can facilitate RNA diffusion. For instance, the cuticle covering trichomes is known to be more permeable than the cuticle of pavement cells in tomato (52).
It is also possible that our isolation protocol partially extracts RNA from the cuticle. Leaf surface RNAs may be embedded inside the cuticular layer along with other high-molecular-weight polysaccharides, such as pectins, which are soluble in aqueous and acidic conditions, and can be noncovalently crosslinked by Ca2+ ions (53). During LSW extraction, we use a slightly acidic VIB buffer that contains Ca2+ along with a wetting agent (Silwet). These properties could facilitate the release of RNAs and polysaccharides from the cuticular layer while promoting their association with each other.
In support of the latter suggestion, RNA pelleting was highly reduced when LSW was pretreated with EGTA before ultracentrifugation, suggesting that aggregation of RNAs in LSW is mediated by Ca2+ and other divalent cations. We speculate that the presence of polygalacturonic acid/pectins and related polysaccharides on the leaf surface might contribute to RNA aggregation. These polysaccharide molecules are heavily negatively charged like RNA and might be involved in the formation of a complex mesh with RNA and Ca2+ leading to the formation of a gel-like substance.
In the quest to identify factors associated with the selective secretion and stabilization of leaf surface RNA, we also tested whether the secreted LSW RNA exhibits a higher level of posttranscriptional modification compared to AWF and CL RNA. Using an ELISA-based assay, we found that neither AWF nor LSW RNA is enriched in m6A modification relative to the total CL RNA. Therefore, it will be necessary to test for the enrichment of other modifications, which might provide insights into the secretion and stability of exRNA.
Our finding of extravesicular RNA on leaf surfaces reinforces our previous finding that extravesicular RNAs are main constituents of the exRNA pool in plants. Our work highlights the need to identify alternate mechanisms through which RNA might be secreted and trafficked independent of EVs. This surprising finding of RNA on the leaf surface opens important questions about the mechanisms involved and its function. For instance: What are the cellular sources of RNA on the leaf surface and in the apoplast? How does this RNA pass through the plasma membrane and cell wall? Does leaf surface RNA play a role in shaping the leaf microbiome? Exploring these questions along with the potential roles of the secreted RNA in plant–microbe interaction will provide valuable information for improving crop plant protection strategies, especially in the context of improving the efficacy of HIGS and SIGS.
Materials and Methods
Plant Materials and Growth Conditions.
Arabidopsis thaliana Col-0 seeds were grown in a temperature-controlled room under a 9 h light/15 h dark photoperiod with 150 µmol m2 s2 photosynthetic photon flux density. Additional details are provided in SI Appendix, Methods.
Isolation of LSW, AWF, and LSS.
For each replicate, LSW, AWF, and LSS samples were isolated from 6- to 7-wk-old Arabidopsis plants as depicted in SI Appendix, Figs. S1 and S4 and described in detail in SI Appendix, Methods. For all experiments, a biological replicate was considered as the batch of a given number of plants growing in the same 36-cell insert that were sown at least 1 wk apart from the other biological replicates.
Isolation of Particles from AWF and LSW.
To obtain pellets containing EVs and other particles, freshly isolated AWF and LSW were transferred to ultracentrifuge (UC) tubes and centrifuged at 40,000 g for 1 h at 4 °C. Where indicated, EGTA [Ethylene glycol- bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid; Sigma-Aldrich, USA, Product ID E4378) was added to the LSW at the specified concentration, mixed, and incubated for 20 min on ice before the ultracentrifugation step. For experiments involving RNase protection by proteins, LSW was treated with 1 µg/mL trypsin (Promega, Madison, WI) and incubated at 30 °C for 1 h followed by the addition of 1.5 mg/mL trypsin inhibitor (Worthington Biochemical Corp, Lakewood, NJ). To obtain P100-P40 pellets, the supernatant after the 40,000 g spin was transferred to UC tubes and centrifuged at 100,000 g for 1 h at 4 °C. Details of centrifuges, rotors, and tubes can be found in SI Appendix, Methods.
Quantification of Cell Rupture Using Trypan Blue Staining.
Leaves were harvested from three individual 6- to 7-wk-old Arabidopsis plants before and after LSW and AWF isolation. Leaves were stained with trypan blue and then destained using chloral hydrate solution, as described in SI Appendix, Methods. The leaves were mounted on glass slides with 25% glycerol solution and imaged using a light microscope (Invitrogen EVOS XL Core #AMEX1200).
RNA Extraction.
Total leaf RNA (cell lysate) was extracted from 100 mg of leaf tissue using TRIzol Reagent (Thermo Fisher Scientific™, Waltham, MA) as described in SI Appendix, Methods. The aqueous phase was transferred to microfuge tubes containing 10 µg of RNase-free glycogen, mixed with one volume of cold isopropanol, and incubated at –20 °C for an hour. RNA was then pelleted at 13,000 g for 20 min at 4 °C, washed twice using ice-cold 70% EtOH, and resuspended in 20 to 30 µL ultrapure DNase/RNase-free water.
To isolate RNA either from supernatant, AWF, LSW, or LSS, the RNA was first precipitated by mixing the required volume of supernatant, AWF, or LSW with 0.1 volume of 3 M sodium acetate (pH 5.2) and 1.0 volume of cold isopropanol, incubated at –20 °C for a minimum of 1 h to overnight, and then centrifuged at 13,000 g for 30 min at 4 °C. The pellets were washed twice with ice-cold 70% EtOH, resuspended in 100 µL of ultrapure DNase/RNase-free water (Invitrogen™, Waltham, MA), and transferred to 1.5 mL centrifuge tubes. Thereafter, 1 mL of TRIzol was added to each tube and RNA extraction was performed following the same procedure as described for the total leaf RNA. Finally, RNA pellets were resuspended in 10 µL of ultrapure DNase/RNase-free water and stored at –80 °C.
Ribonuclease Protection Assays.
To assess whether RNA in AWF and LSW was protected from ribonuclease digestion by either encapsulation inside of EVs or association with proteins, we followed our previously described protocol (11) with minor modifications as described in SI Appendix, Methods.
Denaturing Polyacrylamide Gel Electrophoresis of RNAs.
Mini gels (7.2 cm × 8.6 cm × 0.75 mm) containing 10% or 15% polyacrylamide and 7 M urea in 1× Tris-Boric Acid EDTA (TBE, pH 8.4) were made using 40% Acrylamide/Bis Solution, 37.5:1 (Bio-Rad, catalog no. 1610148). RNA samples were mixed (1:1) with 2× denaturing loading buffer (95% formamide, 10 mM EDTA, 0.02% SDS, 0.02% bromophenol blue, and 0.01% xylene cyanol), denatured at 65 °C for 5 min and resolved in 0.5× TBE running buffer at RT. For size standards, we used a 1:1 mix of Low Range ssRNA Ladder (New England Biolabs™, catalog no. N0364S) and 14 to 30 nt ssRNA Ladder Marker (Takara™, catalog no. 3416). Gels were stained with 1× SYBR Gold Nucleic Acid Gel Stain (Invitrogen™, catalog no. S11494) in 0.5× TBE for 10 min, washed twice with distilled water, and imaged using a Bio-Rad ChemiDoc imaging system.
DIG-labeled Northern Blots.
RNA samples resolved in denaturing polyacrylamide gels were transferred to positively charged nylon membranes (Cytiva, Hybond-N+, catalog no. 45-000-850) using a semidry Trans-Blot Transfer System (Bio-Rad, catalog No. 1703940) in 0.5× TBE at constant 20 V for 45 min. Membranes were UV cross-linked twice at 120,000 µJ/cm2 for 30 s using a UVC-508 Ultraviolet Cross-linker (Ultra-Lum) and prehybridized for 40 min at 42 °C in DIG Easy Hyb solution (Roche) containing 0.1 mg/mL of Poly(A). Following prehybridization, membranes were hybridized overnight at 42 °C with digoxigenin-labeled DNA probes. Oligonucleotides were labeled with the DIG Oligonucleotide Tailing Kit (Roche, catalog no. 03-353-583-910), following the manufacturer’s instructions. After hybridization, membranes were washed as described in SI Appendix, Methods and probed for 30 min with an alkaline phosphatase-labeled anti-digoxigenin antibody (Roche, catalog no. 11093274910) and then washed twice for 15 min with washing buffer [0.1 M maleic acid, 0.15 M NaCl, pH 7.5, 0.3% (v/v) Tween 20] and then incubated for 5 min in detection buffer (0.1 M Tris-HCl, 0.1 M NaCl, pH 9.5). Signals were then visualized using CDP-Star ready-to-use (Roche) and detected using the Bio-Rad ChemiDoc imaging system. DNA oligonucleotides used for hybridization probes are listed in SI Appendix, Table S2.
RNA and Protein Quantification.
To estimate the concentration of RNA isolated from cell lysates and AWFs, a NanoDrop (Thermo Fisher Scientific™) instrument was used. For LSW RNA, we estimated RNA concentrations using densitometric quantification of SYBR Gold-stained polyacrylamide gels as described in SI Appendix, Methods. This was necessary because LSW samples contained a contaminant that caused an overestimation of RNA concentration when using absorbance-based quantifications.
AWF and LSW normalized by plant fresh weight were precipitated by mixing with four volumes of ice-cold acetone and incubating on ice for at least 1 h or overnight at –20 °C. Then, the samples were centrifuged at 14,000 g for 10 min at 42 °C. The pellet was allowed to dry for no longer than 1 h and resuspended in filtered VIB. To determine protein concentrations in AWF, we employed the Bradford method (54) using bovine serum albumin as a standard. Due to the extremely low concentration of proteins in LSW samples, we had to perform densitometric quantification of silver-stained polyacrylamide gels as described in SI Appendix, Methods. Gel images were acquired using the Bio-Rad ChemiDoc imaging system. The densitometric ratio between AWF and LSW was calculated using the ImageJ software, and this ratio and Bradford estimation of AWF proteins were used to estimate the protein concentration of LSW samples.
Immunoblots.
For immunoblots, 30 µL of resuspended pellets were combined with 10 µL of 4× SDS loading buffer and then denatured at 95 °C for 5 min. Then, the samples were separated on stain-free gradient gels (4 to 20% Precise Protein Gels, Thermo Scientific) at 150 V for 1 h in 1× TBS electrophoresis running buffer [24.8 mM Tris base, 0.1% (w/v) sodium dodecyl sulfate, 192 mM glycine, pH 8.3]. The resolved proteins were visualized using the Bio-Rad ChemiDoc imaging system. The proteins were transferred to a nitrocellulose membrane (Amersham™ Protran® Premium Western blotting membrane, nitrocellulose) using the semidry Trans-Blot Transfer System in Transfer buffer, and the membrane was stained with Ponceau S stain (0.1% dye in 5% acetic acid solution) for about 10 min and cleared with water to visualize the protein transfer. Membranes were then washed and incubated with antibodies as described in SI Appendix, Methods. Protein bands were visualized using ProtoGlow ECL Substrate (National Diagnostics) and the Bio-Rad ChemiDoc Imaging System. The densitometric ratio between AWF and LSW was calculated using the ImageJ software.
Quantification of N6-Methyladenosine (m6A) in exRNA.
For all samples, equal amounts of RNA were prepared in equal volumes (6 µL) using UltraPure DNase/RNase-free distilled water (Invitrogen). m6A quantification using dot blots was performed using the protocol described by Zand Karimi et al. (11) and described in detail in SI Appendix, Methods. When indicated, RNA samples were treated with RNaseA or RNaseR as described previously. Following RNase treatments, RNA was purified by precipitation with ammonium acetate and ethanol to remove free nucleotides and other small degradation products. This precipitation step was repeated twice for optimal RNA purity.
Alternatively, EpiQuik™ m6A RNA Methylation Quantification Kit (Fluorometric) (catalog no. P-9008-48, EpigenTek) was used to determine the m6A percentage in exRNA following the manufacturer’s instructions. Both negative and positive RNA controls provided in this kit were used to quantity the percentage of m6A and the absolute amount of m6A in each sample.
Detection of Pectin in exRNA Fractions.
Dot blot analysis of all RNA samples was performed using the same protocol as for m6A quantification except using JIM7 as the primary antibody at a dilution of 1:10 (Kerafast, catalog no. ELD005) and horseradish peroxidase-labeled goat anti-rat as the secondary antibody at a dilution of 1:5,000 (Invitrogen, catalog no. 31470).
Statistical Analyses.
Statistical analyses and plotting of RNA and protein concentrations were performed using the GraphPad Prism 8.3.0 software (GraphPad Software, San Diego, CA). The specific statistical test used for each analysis is provided in the corresponding figure legend. The number of independent biological replicates (n) is indicated in each plot.
Preparation of sRNA Sequence and Standard RNA Sequence Libraries.
sRNA libraries were constructed using the RealSeq-AC kit version 2 (Realseq Biosciences, Santa Cruz, CA, catalog no. 500-00048;) as per the manufacturer’s instructions. We used 60 ng of DNase I-treated total RNA as starting material for constructing libraries (DNaseI: catalog no. EN0521; Thermoscientific). For the Cell lysate rRNA-depleted samples, we used the RiboMinus Plant Kit for RNA-seq (Invitrogen, catalog no. A10838-08) at 1/10th of the recommended volume of regents and sample. For RNAseq libraries, we used NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, catalog no. E7760L) protocol. We sent all sRNA and RNAseq libraries for sequencing to the University of Delaware Sequencing and Genotyping Center, where they were sequenced on a NovaSeq2000 instrument using 50-bp single-end reads for sRNA and 75-bp paired-end reads for RNAseq.
Sequence Data Analysis.
All sRNA libraries were analyzed as previously described (11), as summarized in SI Appendix, Methods. To analyze the tRNA-derived sRNAs from the sRNAseq data, we ran Unitas version 1.8.0 (37) using the Genomic tRNA database for Arabidopsis (data accessed on 5 March 2024) (55, 56) using default parameters. The absolute read counts from the Unitas pipeline were normalized against total number of input RPM. The plots were drawn using R. To analyze the miRNA/miRNA* ratio, we defined miRNA* as the member of the duplex with a lower abundance in CL for each miRNA duplex.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Acknowledgments
We thank Dorothee Staiger at the University of Bielefeld for providing seed of GRP7-GFP transgenic Arabidopsis and for supplying anti-GRP7 antisera, the Indiana University Physical Biochemistry Instrumentation Facility for access to ultracentrifuges and nanoparticle tracking equipment, and the University of Delaware Sequencing and Genotyping Center for assistance with the generation of sRNA-seq and RNA-seq data. We also thank David Daleke and Craig Pikaard for providing access to centrifuges, and members of the Innes and Meyers laboratories for valuable discussions. This work was supported by three grants from the United States NSF, IOS-2243531 and IOS-1842685 to R.W.I. and IOS-1842698 to B.C.M. and P.B., and IOS-2243534 to P.B.
Author contributions
M.L.B., M.S.-R., P.B., H.Z.K., B.C.M., and R.W.I. designed research; M.L.B., M.S.-R., P.B., H.Z.K., and M.M. performed research; M.L.B. and M.S.-R. contributed new reagents/analytic tools; M.L.B., M.S.-R., P.B., M.H.S.-R., and R.W.I. analyzed data; B.C.M. edited the paper; and M.L.B., M.S.-R., P.B., and R.W.I. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
RNA sequence data have been deposited in NCBI Gene Expression Omnibus, accession number: GSE283977 (57).
Supporting Information
References
- 1.Borniego M. L., Innes R. W., Extracellular RNA: Mechanisms of secretion and potential functions. J. Exp. Bot. 74, 2389–2404 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng A. P., et al. , Extracellular RNAs released by plant-associated fungi: From fundamental mechanisms to biotechnological applications. Appl. Microbiol. Biotechnol. 107, 5935–5945 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosal A., et al. , The extracellular RNA complement of Escherichia coli. Microbiol. Open 4, 252–266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nation G. K., Saffold C. E., Pua H. H., Secret messengers: Extracellular RNA communication in the immune system. Immunol. Rev. 304, 62–76 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang M., Dean R. A., Movement of small RNAs in and between plants and fungi. Mol. Plant Pathol. 21, 589–601 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiberg A., Jin H., Small RNAs—The secret agents in the plant–pathogen interactions. Curr. Opin. Plant Biol. 26, 87–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arroyo J. D., et al. , Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. U.S.A. 108, 5003–5008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tosar J. P., et al. , Fragmentation of extracellular ribosomes and tRNAs shapes the extracellular RNAome. Nucleic Acids Res. 48, 12874–12888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nechooshtan G., Yunusov D., Chang K., Gingeras T. R., Processing by RNase 1 forms tRNA halves and distinct Y RNA fragments in the extracellular environment. Nucleic Acids Res. 48, 8035–8049 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baldrich P., et al. , Plant extracellular vesicles contain diverse small RNA species and are enriched in 10- to 17-nucleotide “tiny” RNAs. Plant Cell 31, 315–324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zand Karimi H., et al. , Arabidopsis apoplastic fluid contains sRNA- and circular RNA-protein complexes that are located outside extracellular vesicles. Plant Cell 34, 1863–1881 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernstein E., Caudy A. A., Hammond S. M., Hannon G. J., Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Campo S., et al. , Identification of a novel microRNA (miRNA) from rice that targets an alternatively spliced transcript of the Nramp6 (Natural resistance-associated macrophage protein 6) gene involved in pathogen resistance. New Phytol. 199, 212–227 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Hammond S. M., Bernstein E., Beach D., Hannon G. J., An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Napoli C., Lemieux C., Jorgensen R., Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell 2, 279–289 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koch A., et al. , An RNAi-based control of Fusarium graminearum infections through spraying of long dsRNAs involves a plant passage and is controlled by the fungal silencing machinery. PLoS Pathog. 12, e1005901 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M., et al. , Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nat. Plants 2, 16151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai Q., et al. , Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 360, 1126–1129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He B., et al. , RNA-binding proteins contribute to small RNA loading in plant extracellular vesicles. Nat. Plants 7, 342–352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regente M., et al. , Plant extracellular vesicles are incorporated by a fungal pathogen and inhibit its growth. J. Exp. Bot. 68, 5485–5495 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Rutter B. D., Innes R. W., Extracellular vesicles isolated from the leaf apoplast carry stress-response proteins. Plant Physiol. 173, 728–741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlemmer T., et al. , Isolation and characterization of barley (Hordeum vulgare) extracellular vesicles to assess their role in RNA spray-based crop protection. Int. J. Mol. Sci. 22, 7212 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo X. Y., et al. , Host-induced gene silencing of MoAP1 confers broad-spectrum resistance to Magnaporthe oryzae. Front. Plant Sci. 10, 433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panwar V., McCallum B., Bakkeren G., Host-induced gene silencing of wheat leaf rust fungus Puccinia triticina pathogenicity genes mediated by the Barley stripe mosaic virus. Plant Mol. Biol. 81, 595–608 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Qi T., et al. , Host-induced gene silencing of an important pathogenicity factor PsCPK1 in Puccinia striiformis f. sp. tritici enhances resistance of wheat to stripe rust. Plant Biotechnol. J. 16, 797–807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu X., et al. , Silencing PsKPP4, a MAP kinase kinase kinase gene, reduces pathogenicity of the stripe rust fungus. Mol. Plant Pathol. 19, 2590–2602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu H., et al. , A 5′ tRNA-Ala-derived small RNA regulates anti-fungal defense in plants. Sci. China Life Sci. 65, 1–15 (2022). [DOI] [PubMed] [Google Scholar]
- 28.Ren B., Wang X., Duan J., Ma J., Rhizobial tRNA-derived small RNAs are signal molecules regulating plant nodulation. Science 365, 919–922 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Bach-Pages M., et al. , Discovering the RNA-binding proteome of plant leaves with an improved RNA interactome capture method. Biomolecules 10, 661 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Streitner C., et al. , An hnRNP-like RNA-binding protein affects alternative splicing by in vivo interaction with transcripts in Arabidopsis thaliana. Nucleic Acids Res. 40, 11240–11255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan Y., Ham B. K., Chong Y. H., Yeh S. D., Lucas W. J., A plant SMALL RNA-BINDING PROTEIN 1 family mediates cell-to-cell trafficking of RNAi signals. Mol. Plant 13, 321–335 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Pertea M., Kim D., Pertea G. M., Leek J. T., Salzberg S. L., Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Betti F., et al. , Exogenous miRNAs induce post-transcriptional gene silencing in plants. Nat. Plants 7, 1379–1388 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chery M., Drouard L., Plant tRNA functions beyond their major role in translation. J. Exp. Bot. 74, 2352–2363 (2023). [DOI] [PubMed] [Google Scholar]
- 35.Panstruga R., Spanu P., Transfer RNA and ribosomal RNA fragments—Emerging players in plant–microbe interactions. New Phytol. 241, 567–577 (2024). [DOI] [PubMed] [Google Scholar]
- 36.Alves C. S., Nogueira F. T. S., Plant small RNA world growing bigger: tRNA-derived fragments, longstanding players in regulatory processes. Front. Mol. Biosci. 8, 638911 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gebert D., Hewel C., Rosenkranz D., unitas: The universal tool for annotation of small RNAs. BMC Genomics 18, 644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alves C. S., et al. , Genome-wide identification and characterization of tRNA-derived RNA fragments in land plants. Plant Mol. Biol. 93, 35–48 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Megel C., et al. , Plant RNases T2, but not Dicer-like proteins, are major players of tRNA-derived fragments biogenesis. Nucleic Acids Res. 47, 941–952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shigematsu M., Kawamura T., Kirino Y., Generation of 2′,3′-cyclic phosphate-containing RNAs as a hidden layer of the transcriptome. Front. Genet. 9, 562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tosar J. P., Castellano M., Costa B., Cayota A., Small RNA structural biochemistry in a post-sequencing era. Nat. Protoc. 19, 595–602 (2024). [DOI] [PubMed] [Google Scholar]
- 42.Wang H., et al. , CPA-seq reveals small ncRNAs with methylated nucleosides and diverse termini. Cell Discov. 7, 25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costa B., et al. , Nicked tRNAs are stable reservoirs of tRNA halves in cells and biofluids. Proc. Natl. Acad. Sci. U.S.A. 120, e2216330120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ensinck I., et al. , m6A-ELISA, a simple method for quantifying N6-methyladenosine from mRNA populations. RNA 29, 705–712 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolev S. K., et al. , Interaction of Na(+), K(+), Mg(2+) and Ca(2+) counter cations with RNA. Metallomics 10, 659–678 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Lyons S. M., Gudanis D., Coyne S. M., Gdaniec Z., Ivanov P., Identification of functional tetramolecular RNA G-quadruplexes derived from transfer RNAs. Nat. Commun. 8, 1127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tosar J. P., et al. , Dimerization confers increased stability to nucleases in 5′ halves from glycine and glutamic acid tRNAs. Nucleic Acids Res. 46, 9081–9093 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Q., Zhang X., Shi J., Yan M., Zhou T., Origins and evolving functionalities of tRNA-derived small RNAs. Trends Biochem. Sci. 46, 790–804 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lorenz C., Lunse C. E., Morl M., tRNA modifications: Impact on structure and thermal adaptation. Biomolecules 7, 35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang M., et al. , Targeting Fusobacterium nucleatum through chemical modifications of host-derived transfer RNA fragments. ISME J. 17, 880–890 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He X., et al. , Human tRNA-derived small RNAs modulate host–oral microbial interactions. J. Dent. Res. 97, 1236–1243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fich E. A., Fisher J., Zamir D., Rose J. K. C., Transpiration from tomato fruit occurs primarily via trichome-associated transcuticular polar pores. Plant Physiol. 184, 1840–1852 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Braidwood L., Breuer C., Sugimoto K., My body is a cage: Mechanisms and modulation of plant cell growth. New Phytol. 201, 388–402 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Bradford M. M., A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976). [DOI] [PubMed] [Google Scholar]
- 55.Chan P. P., Lowe T. M., GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan P. P., Lowe T. M., GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baldrich P., et al. , Diverse plant RNAs coat Arabidopsis leaves and are distinct from apoplastic RNAs. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE283977. Deposited 10 December 2024. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Data Availability Statement
RNA sequence data have been deposited in NCBI Gene Expression Omnibus, accession number: GSE283977 (57).