

Abstract
The Keap1 (Kelch-like ECH-Associating Protein 1)–Nrf2 (Nuclear Factor Erythroid 2-Related Factor 2)-ARE (Antioxidant Response Element) signaling pathway plays a crucial role in the oxidative stress response and has been linked to the development and progression of various diseases. Its influence on cerebral ischemia/reperfusion (I/R) injury has garnered significant attention. In our study, we investigated the effect of compound 2, a non-covalent inhibitor of the Keap1–Nrf2 interaction, which was previously discovered by our research group. Specifically, we used 5-(3-(N-(carboxymethyl)naphthalene-2-sulfonamido)phenyl)-1-ethyl-1H-pyrrole-2-carboxylic acid (compound 2) to assess its therapeutic potential in a cerebral I/R injury model. The results demonstrated that compound 2 had a significant therapeutic effect, promoting the translocation of Nrf2 from the cytoplasm to the nucleus in diseased tissue. Additionally, it increased the production of key antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH).
The Keap1 (Kelch-like ECH-Associating Protein 1)–Nrf2 (Nuclear Factor Erythroid 2-Related Factor 2)-ARE (Antioxidant Response Element) signaling pathway plays a crucial role in the oxidative stress response and has been linked to the development and progression of various diseases.
1. Introduction
Cells are constantly exposed to reactive oxygen species (ROS) generated through various endogenous and exogenous processes. To maintain cellular homeostasis, healthy cells rely on both enzymatic and non-enzymatic antioxidants to neutralize these ROS. However, when the production of oxidants exceeds the capacity for clearance, the balance is disrupted, leading to a state of oxidative stress (OS).1 OS can damage cellular components, including membranes, lipids, proteins, lipoproteins, and DNA, contributing to the development of various diseases, disorders, and aging.
A key mechanism for regulating OS is the Keap1–Nrf2-ARE signaling pathway.2,3 Under normal conditions, Keap1 sequesters Nrf2, targeting it for ubiquitination and subsequent proteasomal degradation, thereby keeping Nrf2 levels low. However, under stress conditions, specific cysteine residues on Keap1 are modified, preventing Keap1 from functioning as an E3 ligase adaptor. This modification stabilizes Nrf2, allowing for its increased production through de novo protein synthesis. When Nrf2 levels surpass those of Keap1, Nrf2 escapes sequestration and translocates into the nucleus.4
In the nucleus, Nrf2 forms transcriptionally active heterodimers by binding to small musculoaponeurotic fibrosarcoma (sMaf) proteins.5 The Nrf2-sMaf heterodimer then recognizes antioxidant response elements (ARE) in the promoters of target genes, including those encoding antioxidant and phase II detoxification enzymes, thereby inducing their transcription6,7 (see Fig. 1 for illustration).
Fig. 1. Keap1–Nrf2-ARE signaling pathway.

Currently, Keap1–Nrf2 interaction inhibitors are classified into covalent and non-covalent types based on their mode of interaction with Keap1.8,9 Covalent inhibitors form covalent bonds with cysteine residues on Keap1. Compounds like dimethyl fumarate, bardoxolone methyl and CDDO-imidazolid are susceptible to off-target side effects.10,11 On the other hand, non-covalent inhibitors do not typically cause off-target effects but often contain multiple hydrophilic groups such as carboxyl groups, which can reduce the drug-likeness of the compounds, particularly for central nervous system diseases.11 Moreover, Keap1 has been leveraged as a novel E3 ubiquitin ligase tool, influencing the design of proteolysis targeting chimeras (PROTACs).12 Consequently, both covalent and non-covalent Keap1–Nrf2 inhibitors face significant challenges in achieving optimal drug-like properties.
Nrf2 is widely distributed in central nervous system tissues, such as the brain and spinal cord. Numerous studies have increasingly demonstrated that activation of Nrf2 through the Keap1–Nrf2-ARE signaling pathway effectively clears accumulated free radicals, inhibits oxidative stress, and mitigates inflammation. Consequently, this process plays a crucial role in preventing or alleviating cerebral ischemia/reperfusion (I/R) injury to the central nervous system.13,14 Tanshinone IIA (Fig. 2) has been found to enhance the proliferation of nerve cells while inhibiting the production of cellular reactive oxygen species. Additionally, it suppresses changes in mitochondrial membrane potential and activates the Keap1–Nrf2/ARE signaling pathway. Moreover, Tanshinone IIA induces and regulates the upregulation of downstream factors such as NQO1, HO-1, etc., providing cellular protection against cerebral infarction.15 Swertiamarin (Swe) (Fig. 2), an active secoiridoid glycoside compound isolated from Gentiana macrophylla Pall, emerges as a promising protective agent against cerebral I/R injury. It achieves this by suppressing oxidative stress through the activation of the Nrf2 protective pathway.16 Carnosic acid (Fig. 2) from Rosmarinus officinalis, a catechol-type electrophilic compound, provides neuronal protection both in vitro and in vivo by activating the Keap1/Nrf2 pathway through S-alkylation of targeted cysteines on Keap1.17
Fig. 2. Compounds affecting cerebral I/R injury via the Keap1/Nrf2 pathway.

In our previous study, we screened a compound library and identified compound 1 (Fig. 2), which demonstrated a dissociation constant (KD) of 5090 nM for Keap1.18 To improve its binding affinity, we employed scaffold hopping from compound 1 and developed a novel 5-phenyl-1H-pyrrole-2-carboxylic acid-based as a Keap1–Nrf2 inhibitor. Structure–activity relationship (SAR) studies identified compound 2 as the most potent non-covalent candidate (Fig. 2), with a KD2 of 42.2 nM against Keap1.
Compound 2 also exhibited significant protective effects against LPS-induced injury in BEAS-2B cells, where it promoted Nrf2 translocation to the nucleus. Subsequently, we evaluated its therapeutic potential in mouse models of acute lung injury (ALI). In the previous work, at a dose of 15 mg kg−1, compound 2 effectively alleviated ALI symptoms.18 Additionally, it enhanced Nrf2 nuclear translocation, increased Nrf2 levels, and upregulated the expression of antioxidant enzymes such as HO-1 and NQO1 in the affected tissues.18
Keap1/Nrf2 inhibitors for cerebral I/R injury are primarily derived from natural products, which generally exhibit weak affinity for the Keap1 protein, with some lacking reported affinity values altogether. This highlights the need to discover new, more potent inhibitors of the Keap1/Nrf2 interaction for the treatment of this condition. In response to this need, we plan to investigate the effects of compound 2—a novel non-covalent Keap1–Nrf2 interaction inhibitor discovered by our research group—on ischemic stroke.
2. Synthesis of compound 2
Compound 3 was brominated to produce compound 4, which was subsequently reacted with bromoethane. This intermediate then underwent a Suzuki coupling with 3-nitrophenylboronic acid to form compound 5. Compound 5 was reduced to yield the corresponding amine groups using iron powder. The amine was then sulfonamidated with an aryl sulfonyl chloride to generate key intermediates containing sulfonamide groups. These intermediates were then reacted with bromoacetic ester, followed by hydrolysis, leading to the synthesis of the target molecule 2 (Scheme 1).
Scheme 1. Reagents and conditions: (a): N-bromosuccinimide, THF : MeOH (2 : 1), 0 °C, 8 h; (b): (i): NaH, DMF, halogen-R1, r.t., 5 h; (ii): 3-nitrophenylboric acid, Pd(PPh3)4, Cs2CO3, dioxane : H2O (4 : 1), 85 °C, 3 h; (c): (i): Fe, NH4Cl, MeOH : H2O (2 : 1), r.t., 12 h; (ii): benzenesulfonyl chloride, pyridine, r.t., 3 h; (iii): ethyl 2-bromoacetate, K2CO3, KI, DMF, r.t., 5 h; (iv): NaOH, H2O : MeOH (1 : 1), HCl, r.t., 12 h.

3. The compound 2 inhibited cerebral I/R injury
In our previous research, ALI is regarded as a peripheral disease, and compound 2 has shown a significant protective effect in a mouse ALI model at a dose of 15 mg kg−1.18 Cerebral ischemia–reperfusion, however, is a central nervous system disease. Due to the structural and physicochemical properties of compound 2, its blood–brain barrier permeability is limited. Consequently, the dose of compound 2 was increased to 20 mg kg−1 in the mouse cerebral ischemia–reperfusion model to achieve effective therapeutic action in vivo. Whole cerebral sections were stained with 2,3,5-triphenyltetrazolium chloride (TTC). It is evident that both the sham group and the group pretreated with 20 mg kg−1 of compound 2 significantly reduced the percentage of brain infarction compared to the I/R group (Fig. 3).
Fig. 3. Compound 2 protected against cerebral I/R injury in MCAO mice. (A) Representative TTC staining of the cerebral infarct in brain. (B) The percentage of infarct volume was detected for each group, n = 6 per group. ***P < 0.001 versus the I/R group.

Superoxide dismutase (SOD) acts as a free radical scavenger, converting superoxide radicals to hydrogen peroxide and oxygen.19 Catalase (CAT) further metabolizes hydrogen peroxide, serving as a secondary defense against oxidative stress.20 Glutathione (GSH), an endogenous antioxidant, also plays a critical role in cellular defense.21 The activities of these enzymes, along with GSH levels, are commonly used as indicators of oxidative stress.22 To assess oxidative damage in our study, we measured SOD and CAT activities as well as GSH content (Fig. 4). In I/R group, the activities of GSH (Fig. 4A), SOD (Fig. 4B) and CAT (Fig. 4C) in brain tissue were significantly lower than those in the sham group. In the groups with 2 (20 mg kg−1), the content of GSH was markedly increased. Pre-treatment with 2 increased the SOD and CAT activities.
Fig. 4. (A) GSH-PX. (B) SOD. (C) CAT. n = 6 per group. ###P < 0.001, ##P < 0.01 versus the sham group. **P < 0.01, *P < 0.05 versus the I/R group.

In other words, compound 2 markedly mitigated oxidative damage in brain tissue. Western blotting results demonstrated that in the 2 + I/R group, the levels of nucleus protein Nrf2 were significantly elevated, hile the cytosolic protein levels of Nrf2 were degraded when compared to the I/R group. This indicated that Nrf2 could accumulate in the nucleus under the induction of compound 2 in the cerebral I/R injury mouse model (Fig. 5).
Fig. 5. Compound 2 could promote Nrf2 in the cytoplasm of brain tissue into the nucleus.

4. Parallel artificial membrane permeability assay of 2
We tested the permeability of compound 2 through parallel artificial membrane permeability assays (Table 1), and the results showed that the −log Pe of compound 2 was >9.2, indicating very low blood–brain barrier permeability. Despite this, compound 2 performed well in animal models of cerebral ischemia/reperfusion injury. A similar observation was made with the compound Swertiamain.16 To understand this, we analyzed the modeling process of the cerebral ischemia/reperfusion Injury animal model. Compound 2 was first injected intraperitoneally into mice before inducing the cerebral ischemia/reperfusion injury model. Research literature indicates that cerebral ischemia–reperfusion can increase the permeability of the blood–brain barrier, even leading to rupture,23–25 which leads to some cells and macromolecular proteins in peripheral blood entering the blood–brain barrier. And of course that includes our dicarboxylate compound 2. Both compound 2 and Swertiamain were intraperitoneally injected into mice before the induction of the injury model, leading to increased blood–brain barrier permeability during the modeling injury. Consequently, compound 2 was able to cross the blood–brain barrier.
Table 1. Permeability assay of 2.
| Compound | Test concentration (μM) | Pe × 10−6 (cm s−1) | −Log Pea | Recovery % |
|---|---|---|---|---|
| Methotrexateb | 10 | <0.0098 | >8.01 | <104.56 |
| Testosteroneb | 10 | 41.94 | 4.38 | 92.19 |
| 2 | 10 | <0.0006 | >9.20 | <106.81 |
−Log Pe < 6, hyperpermeability; −Log Pe > 6, low permeability.
The control group.
5. Conclusion
Keap1/Nrf2 inhibitors for cerebral I/R injury are primarily derived from natural products, many of which exhibit weak affinity for the Keap1 protein, and some lack reported affinity values altogether. Therefore, we aimed to investigate the effects of highly potent compound 2 on this condition. In the current study, compound 2 effectively mitigated cerebral I/R injury at a dose of 20 mg kg−1 in a cerebral ischemia model. Additionally, under oxidative stress conditions, compound 2 significantly increased the production of superoxide dismutase (SOD), catalase (CAT), and glutathione (GSH), while promoting the translocation of Nrf2 into the nucleus. These findings support the potential of novel Keap1–Nrf2 inhibitors in the treatment of both acute lung injury and cerebral ischemia.
Although compound 2 contains two hydrophilic carboxylic acid groups and has shown poor permeability across the blood–brain barrier in healthy conditions, as demonstrated in our parallel artificial membrane permeability assay, we found that ischemic stroke increases blood–brain barrier permeability. This increased permeability allows certain compounds, including compound 2, to penetrate the brain more easily. This explains why compound 2, despite its hydrophilic nature, is effective in a mouse model of ischemic stroke. Given the unique characteristics of ischemic stroke, non-covalent Keap1–Nrf2 inhibitors with high target affinity and hydrophilic properties hold great promise for the treatment of this condition.
6. Experimental section
6.1. Chemistry
All starting materials, solvents were obtained from commercial sources and were analytically pure. The 1H NMR and 13C NMR spectra were recorded on Bruker Avance 400 and 600 MHz spectrometers (Bruker Company, Germany) using tetramethylsilane as an internal standard and CDCl3 or methanol-d4 as solvents. Chemical shifts (d values) and coupling constants (J values) are given in ppm and hertz (Hz), respectively. High-resolution mass spectrometry data were acquired using a quadrupole time-of-flight micro mass spectrometer. Thin-layer chromatography (TLC) analyses were carried out on silica gel plates HGF254 (Qingdao Haiyang Chemical, China). Column chromatography separations were carried out on a silica gel 200–300 mesh. Computer aided drug design software is MOE2015. The purities of the compounds were analyzed by HPLC (Waters 2695) using a C18 column (QuikSep SP ODS-A, 4.6 × 250 mm, 5 μm) with 90% methanol/10% water as the mobile phase at a flow rate of 1 mL min−1, and all final compounds exhibited purities greater than 95%.
6.2. Ethyl 5-bromo-1H-pyrrole-2-carboxylate (4)
Compound 3 (3.087 g, 0.022 mol) were added to a three-necked round-bottomed flask under nitrogen atmosphere. Then to a solution of N-bromosuccinimide (3.937 g, 0.022 mol) in THF (40 mL) and MeOH (20 mL) were added into the system that protect from light with stirring at 0 °C. After stirring 6 h at 0 °C, the reaction was quenched with water (80 mL). The resulting mixture was extracted with EtOAc (3 × 90 mL), and the combined organic layers were washed with water (3 × 90 mL) and brine (80 mL), dried over Na2SO4, and concentrated under vacuum. The crude residue was purified by flash chromatography on silica gel, eluting with hexane/EtOAc (100 : 1) to provide the title compound 4 (2.269 g, 0.010 mol, yield: 47.30%) as white solid.1H NMR (400 MHz, chloroform-d) δ 10.00 (s, 1H), 7.05–6.83 (m, 1H), 6.37–6.20 (m, 1H), 4.41–4.32 (m, 2H), 1.38 (t, J = 7.0 Hz, 3H).
6.3. Ethyl 1-ethyl-5-(3-nitrophenyl)-1H-pyrrole-2-carboxylate (5)
NaH (15.0 mg, 0.616 mmol) was slowly added to a solution of compound 4 (112.0 mg, 0.513 mmol) in DMF (2 mL) with stirring at 0 °C. After stirring 0.5 h, bromoethane (73 mg, 0.667 mmol) were added into the mixture system at r.t. After stirring 2.5 h at r.t., the reaction was quenched with water (15 mL). The resulting mixture was extracted with EtOAc (3 × 20 mL), and the combined organic layers were washed with water (3 × 20 mL) and brine (15 mL), dried over Na2SO4, and concentrated under vacuum to provide the crude compound 3 (99.0 mg, 0.401 mmol, yield: 78.17%) as a faint yellow solid, which was directly used in the next step. Ethylated compound 4 (99.0 mg, 0.401 mmol), (3-nitrophenyl) boronic acid (100.0 mg, 0.602 mmol), Pd(PPh3)4 (46.0 mg, 0.040 mmol) and Cs2CO3 (261.0 mg, 0.802 mmol) were added to a three-necked round-bottomed flask under nitrogen atmosphere. Then dioxane (8 mL) and water (2 mL) were added into the mixture system with stirring. After stirring 2 h at 85 °C, the reaction was quenched with water (15 mL). The resulting mixture was extracted with EtOAc (3 × 20 mL), and the combined organic layers were washed with water (3 × 20 mL) and brine (15 mL), dried over Na2SO4, and concentrated under vacuum. The residue was purified by flash chromatography on silica gel, eluting with hexane/EtOAc (10 : 1) to provide the title compound 5 (83 mg, 0.287 mmol, yield: 71.57%) as white solid. 1H NMR (400 MHz, chloroform-d) δ 8.31–8.23 (m, 2H), 7.75 (d, J = 8.0 Hz, 1H), 7.65 (t, J = 8.0 Hz, 1H), 7.07 (d, J = 4.0 Hz, 1H), 6.26 (d, J = 4.0 Hz, 1H), 4.41–4.30 (m, 4H), 1.39 (t, J = 7.0 Hz, 3H), 1.32 (t, J = 7.0 Hz, 3H).
6.4. 5-(3-(N-(Carboxymethyl)naphthalene-2-sulfonamido)phenyl)-1-ethyl-1H-pyrrole-2-carboxylic acid (2)
6.4.1. Step 1
To a solution of compound 5 (83.0 mg, 0.287 mmol) in MeOH (4 mL) and H2O (2 mL), Fe (161.0 mg, 2.87 mmol), NH4Cl (154 mg, 2.87 mmol) were added to a one-necked flask. After stirring 12 h at r.t., the mixture was filtered with Celite, and the filtrate was dried over Na2SO4, and concentrated under vacuum to provide the crude compound ethyl 5-(3-aminophenyl)-1-ethyl-1H-pyrrole-2-carboxylate (58.0 mg, 0.225 mmol, yield: 78.23%) as a yellow solid, which was directly used in the next step.
6.4.2. Step 2
3-Methoxybenzenesulfonyl chloride (60.0 mg, 0.293 mmol) was added to a solution of compound ethyl 5-(3-aminophenyl)-1-ethyl-1H-pyrrole-2-carboxylate (58.0 mg, 0.225 mmol) in pyridine (2 mL) with stirring. After stirring 2 h at r.t., the reaction was quenched with water (15 mL). The resulting mixture was extracted with EtOAc (3 × 20 mL), and the combined organic layers were washed with water (3 × 20 mL) and brine (15 mL), dried over Na2SO4, and concentrated under vacuum to provide the crude compound ethyl 1-ethyl-5-(3-((3-methoxyphenyl) sulfonamido) phenyl)-1H-pyrrole-2-carboxylate (82 mg, 0.191 mmol, yield: 85.89%) as a faint yellow solid, which was directly used in the next step.
6.4.3. Step 3
To a solution of compound ethyl 1-ethyl-5-(3-((3-methoxyphenyl) sulfonamido) phenyl)-1H-pyrrole-2-carboxylate (82.0 mg, 0.191 mmol) in DMF (2 mL), K2CO3 (53.0 mg, 0.382 mmol), KI (16.0 mg, 0.096 mmol), ethyl 2-bromoacetate (42.0 mg, 0.25 mmol) was added were added to a one-necked flask. After stirring 3 h at r.t., the reaction was quenched with water (15 mL). The resulting mixture was extracted with EtOAc (3 × 20 mL), and the combined organic layers were washed with water (3 × 20 mL) and brine (15 mL), dried over Na2SO4, and concentrated under vacuum to provide the crude compound ethyl 5-(3-((N-(2-ethoxy-2-oxoethyl)-3-methoxyphenyl) sulfonamido) phenyl) -1-ethyl-1H-pyrrole-2-carboxylate (86 mg, 0.167 mmol, yield: 87.43%) as a faint yellow solid, which was directly used in the next step.
6.4.4. Step 4
Ethyl 5-(3-((N-(2-ethoxy-2-oxoethyl)-3-methoxyphenyl) sulfonamido) phenyl)-1-ethyl-1H-pyrrole-2-carboxylate (86 mg, 0.167 mmol) was added to a solution of NaOH (34.0 mg, 0.835 mmol) in H2O (10 mL) and MeOH (10 mL). After stirring 2 h at r.t., the reaction was quenched with 5% hydrochloric acid (15 mL). The resulting mixture was extracted with EtOAc (3 × 20 mL), and the combined organic layers were washed with water (3 × 20 mL) and brine (15 mL), dried over Na2SO4, and concentrated under vacuum. The crude residue was purified by flash chromatography on silica gel, eluting with DCM/MeOH (15 : 1) to provide the title compound 2. White solid (1690 mg, 3.528 mmol, yield: 66.92%). 1H NMR (400 MHz, methanol-d4) δ 8.26 (d, J = 2.0 Hz, 1H), 8.03–7.94 (m, 3H), 7.74–7.71 (m, 1H), 7.70–7.59 (m, 2H), 7.41–7.36 (m, 1H), 7.34–7.30 (m, 2H), 7.25 (t, J = 2.0 Hz, 1H), 6.95 (d, J = 4.0 Hz, 1H), 5.99 (d, J = 4.0 Hz, 1H), 4.08 (q, J = 7.0 Hz, 2H), 0.95 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, methanol-d4) δ 162.78, 140.28, 139.79, 135.50, 134.99, 133.26, 132.05, 128.93, 128.86, 128.82, 128.79, 128.75, 128.72, 128.44, 127.78, 127.60, 127.33, 122.65, 122.44, 118.30, 109.04, 62.89, 53.22, 40.04, 15.73. HRMS (ESI): calcd for C25H23N2O6S [M + H]+, 479.1277; found 479.1280. HPLC analysis: retention time = 2.5 min; peak area, >95% (210 254 nm).
7. Animal experiments
The protocol for animal procedures were approved by the animal care and use committee of Experimental Animal Center of Ningxia Medical University, Yinchuan, China (certificate no. SYXK Ningxia 2020-0001). All experimental animals were kept in an SPF-level laboratory with free access to standard dry diet and tap water, which were housed in an automated 12 h dark–light cycle at the temperature controlled at 22 ± 2 °C and the relative humidity of 50–60%. Besides, LPS was dissolved in PBS and 2 was dissolved in PBS (with 10% DMSO, 30% PEG-400 and 5% Tween-80).
8. IR injury induction and pretreatment
Thirty-six adult male Institute of Cancer Research (ICR) mice, weighing 20.0–25.0 g, were randomly divided into 3 groups: sham, control I/R, and 2 + I/R. Mice in the 2 + I/R group were administered compound 2 (20 mg kg−1) through intraperitoneal injection daily, while the other two groups received a blank solvent. One hour after administration, cerebral ischemia–reperfusion (IR) was induced on the seventh day. During the induction of cerebral IR injury, mice that had fasted for 2 hours were anesthetized with an intraperitoneal injection of 40 mg kg−1 sodium pentobarbital. Except for the sham group, mice in the other groups had their left middle cerebral artery (MCA) and external carotid artery (ECA) blocked, and a 4–0 monofilament nylon suture was inserted into their internal carotid artery (ICA) about 1 cm to induce left cerebral ischemia. After 1 hour of blockage, the suture was carefully removed to achieve reperfusion, and the wound was stitched. At 24 hours after reperfusion, all mice were sacrificed, and then the brains were quickly removed.
9. Brain-infarction volume
The brains were sliced into 2 mm thick serial coronal sections and exposed to 2% TTC staining fluid at 37 °C for 20 minutes. The resulting coronal sections, with unstained areas defined as infarcts, were analyzed for infarct volume using microscope image-analysis software (Image-Pro Plus, U.S.A.).
10. Western blotting
The ischemic hemispheres tissue were weighed and fully homogenized. The total proteins of ischemic hemispheres tissues were extracted with the whole-cell lysis assay kit (KeyGEN, China). In addition, the nuclear and cytoplasmic proteins were separated using a Cytoplasmic and Nuclear Extraction Kit (Invent Biotechnologies, China). Protein concentrations were determined by bicinchoninic acid (BCA) Protein Assay Kit (KeyGEN, China). The same amounts of protein samples proteins were resolved over 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, U.S.A.). The membranes were blocked for 1 h in PBS containing 5% nonfat milk and 0.1% Tween-20, and then they were probed with primary antibodies: anti-Nrf2 (Proteintech Cat# 16396-1-AP); anti-LaminB1 (Proteintech Cat# 12987-1-AP); anti-GAPDH (Proteintech Cat# 10494-1-AP) at 4 °C overnight. After washed with PBST, the membranes were probed with the secondary antibodies for 2 h at r.t. Protein signal was visualized using ECL reagents (Seven, China).
11. Determination of oxidative
The SOD, CAT and GSH were assessed by assay kits from Nanjing Jiancheng Bioengineering Institute. We disposed of the ischemic hemispheres tissue that was fully homogenized following the manufacturer's instruction. The protein concentrations were detected using the BCA Protein Assay Kit (KeyGEN, China). The absorbance was determined with a microplate reader.
12. Parallel artificial membrane permeability assay
12.1. Procedure
SOP No. SOP-DA-0002 V1.0-Parallel Artificial Membrane Permeability Assay (Lipid-PAMPA)
Prepare test compounds in DMSO at 10 mM and dilute with PBS by 1000-fold to 10 μM; sonicate porcine in dodecane at 20 mg mL−1; add 300 μL per well of 10 μM test compounds solution into donor compartment (bottom) in triplicate; add 5 μL per well porcine/dodecane into acceptor compartment (top), and then add 300 μL per well of PBS (pH = 7.4) into acceptor compartment within 10 minutes; transfer 50 μL of 10 μM test compounds to the sample plate containing 200 μL of cold methanol with IS* as C0; insert acceptor compartment into donor compartment and incubate at 25 °C for 16 hours; transfer 50 μL from the acceptor and donor compartment to the sample plate containing 200 μL of cold methanol with IS* after 16 hours' incubation; centrifuge the sample plate at 4 °C, 3220 g for 40 minutes; transfer 100 μL of the supernatant to analysis plate containing a proper volume of H2O for LC-MS/MS analysis.
12.2. Data analysis
Calculate effective permeability Pe (cm s−1) using Microsoft Excel.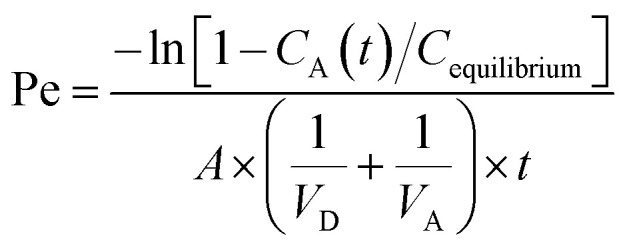
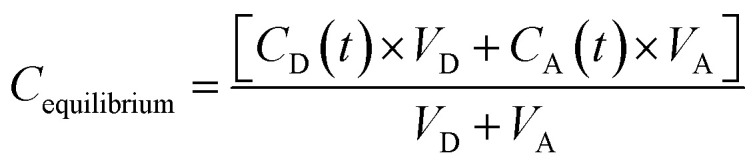
 VD = donor well volume = 0.3 mL; VA = acceptor well volume = 0.3 mL; A = effective filter area = f × 0.3 cm2; f = apparent porosity of the filter, 0.76 for MAIPN4550 (Merck Millipore); t = incubation time (s); CD(t) = compound concentration in donor compartment at time t; CA(t) = compound concentration in acceptor compartment at time t; C0 = initial compound concentration in donor compartment.
VD = donor well volume = 0.3 mL; VA = acceptor well volume = 0.3 mL; A = effective filter area = f × 0.3 cm2; f = apparent porosity of the filter, 0.76 for MAIPN4550 (Merck Millipore); t = incubation time (s); CD(t) = compound concentration in donor compartment at time t; CA(t) = compound concentration in acceptor compartment at time t; C0 = initial compound concentration in donor compartment.
Abbreviations
- HO-1
Heme oxygenase-1
- LPS
Lipopolysaccharide
- NQO-1, NADPH
Quinone oxidoreductase 1
- MCAO
Middle cerebral artery occlusion
- PEG
Polyethyleneglycol
- DMSO
Dimethyl sulfoxide
Data availability
The data that support the findings of this study are openly available.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgments
This project was supported by Doctoral Program Construction Project of University of Xizang Medicine in 2024-Tibetan Medicine Formula (BSDJS-XKJS-06); National Natural Science Foundation of China (Grant 82160658); Natural Science Foundation of Ningxia Province (No. 2024AAC03261); The Key Research and Development Program of Ningxia (2020BEB04020, 2023BEG02012, 2021BEG03104, 2021BEB04033).
References
- Borchers A. Pieler T. Genes. 2010;1:413–426. doi: 10.3390/genes1030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellezza I. Giambanco I. Minelli A. Donato R. Biochim. Biophys. Acta Mol. Cell Res. 2018;1865:721–733. doi: 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- Yu C. Xiao J. H. Oxid. Med. Cell. Longev. 2021;2021:6635460. doi: 10.1155/2021/6635460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L. Yamamoto M. Mol. Cell. Biol. 2020;40:e00099. doi: 10.1128/MCB.00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsu Y. Katsuoka F. Funayama R. Nagashima T. Nishida Y. Nakayama K. Engel J. D. Yamamoto M. Nucleic Acids Res. 2012;40:10228–10239. doi: 10.1093/nar/gks827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuoka F. Motohashi H. Engel J. D. Yamamoto M. J. Biol. Chem. 2005;280:4483–4490. doi: 10.1074/jbc.M411451200. [DOI] [PubMed] [Google Scholar]
- Katsuoka F. Motohashi H. Ishii T. Aburatani H. Engel J. D. Yamamoto M. Mol. Cell. Biol. 2005;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. E. Bayly A. R. Abell C. Skidmore J. Nat. Rev. Drug Discov. 2016;15:533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Wilson A. J. Kerns J. K. Callahan J. F. Moody C. J. J. Med. Chem. 2013;56:7463–7476. doi: 10.1021/jm400224q. [DOI] [PubMed] [Google Scholar]
- de Zeeuw D. Akizawa T. Audhya P. Bakris G. L. Chin M. Christ-Schmidt H. Goldsberry A. Houser M. Krauth M. Lambers Heerspink H. J. McMurray J. J. Meyer C. J. Parving H. H. Remuzzi G. Toto R. D. Vaziri N. D. Wanner C. Wittes J. Wrolstad D. Chertow G. M. N. Engl. J. Med. 2013;369:2492–2503. doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. Wang Y. You Q. Jiang Z. Expert Opin. Ther. Pat. 2020;30:209–225. doi: 10.1080/13543776.2020.1715365. [DOI] [PubMed] [Google Scholar]
- Lu M. Liu T. Jiao Q. Ji J. Tao M. Liu Y. You Q. Jiang Z. Eur. J. Med. Chem. 2018;146:251–259. doi: 10.1016/j.ejmech.2018.01.063. [DOI] [PubMed] [Google Scholar]
- Jiang S. Deng C. Lv J. Fan C. Hu W. Di S. Yan X. Ma Z. Liang Z. Yang Y. Mol. Neurobiol. 2017;54:1440–1455. doi: 10.1007/s12035-016-9707-7. [DOI] [PubMed] [Google Scholar]
- Liu L. Locascio L. M. Doré S. Front. Pharmacol. 2019;10:153. doi: 10.3389/fphar.2019.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X. He L. Gao W. Zhao Z. Cell Cycle. 2023;22:390–402. doi: 10.1080/15384101.2022.2119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Wei W. Lan X. Liu N. Li Y. Ma H. Sun T. Peng X. Zhuang C. Yu J. ACS Chem. Neurosci. 2019;10:2276–2286. doi: 10.1021/acschemneuro.8b00605. [DOI] [PubMed] [Google Scholar]
- Satoh T. Kosaka K. Itoh K. Kobayashi A. Yamamoto M. Kitajima Y. S. C. Cui J. Kamins J. Okamoto S. Izumi M. Shirasawa T. Lipton S. A. J. Neurochem. 2008;104:1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. Tie X. Zhi S. Wang Z. Zhao Q. Qu Z. Lu G. Li Q. Wu Y. Shi Y. Bioorg. Chem. 2024;153:107741. doi: 10.1016/j.bioorg.2024.107741. [DOI] [PubMed] [Google Scholar]
- Kelly P. J. Morrow J. D. Ning M. Koroshetz W. Lo E. H. Terry E. Milne G. L. Hubbard J. Lee H. Stevenson E. Lederer M. Furie K. L. Stroke. 2008;39:100–104. doi: 10.1161/STROKEAHA.107.488189. [DOI] [PubMed] [Google Scholar]
- Li Y. Bao Y. Jiang B. Wang Z. Liu Y. Zhang C. An L. Int. J. Dev. Neurosci. 2008;26:309–317. doi: 10.1016/j.ijdevneu.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Kho A. R. Choi B. Y. Lee S. H. Hong D. K. Lee S. H. Jeong J. H. Park K. H. Song H. K. Choi H. C. Suh S. W. Int. J. Mol. Sci. 2018;19:E1420. doi: 10.3390/ijms19051420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M. C. Lin M. C. Mong M. C. Lin C. Y. J. Agric. Food Chem. 2012;60:7697–7701. doi: 10.1021/jf302529x. [DOI] [PubMed] [Google Scholar]
- Simpkins A. N. Dias C. Leigh R. Stroke. 2016;47:2405–2408. doi: 10.1161/STROKEAHA.116.013805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo-Castro S. Sousa J. A. Martins E. Donato H. Nunes C. d'Almeida O. C. Castelo-Branco M. Abrunhosa A. Ferreira L. Sargento-Freitas J. Int. J. Stroke. 2023;18:783–794. doi: 10.1177/17474930231166306. [DOI] [PubMed] [Google Scholar]
- Mathias K. Machado R. S. Stork S. Dos Santos D. Joaquim L. Generoso J. Danielski L. G. Barichello T. Prophiro J. S. Petronilho F. Microvasc. Res. 2024;151:104621. doi: 10.1016/j.mvr.2023.104621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available.