

Abstract
Introduction and importance
In Morocco, diagnosing Gamma Sarcoglycanopathies mainly relies on histopathological analysis of muscle biopsies due to limited genetic and molecular research access. This study highlights the significance of muscle biopsies and explores potential predictive factors and possible correlation between histopathological abnormalities and clinical phenotypes.
Case presentation
Muscle biopsies of six patients diagnosed with γ-sarcoglycanopathy were collected over two years. Pathological analysis was initially performed on slides stained with Hematoxylin-Eosin and Gomori Trichrome. Additionally, cryosections marked for dystrophin, alpha, beta, and gamma sarcoglycans were reviewed. In the second phase of the analysis, formalin-fixed sections from each biopsy were immunostained for various markers: “anti-CD68” for macrophagic cells, “anti-CD56” for satellite cells, and “anti-CD31” for vascular capillary. These stained sections were then carefully examined.
Clinical discussion
The clinical presentation of the disease was uniform and consistent with Duchenne-like dystrophy. However, the histological abnormalities were heterogeneous and did not correlate with the severity of the clinical phenotype. The Loss of expression of a Sarcoglycan and earlier age of onset appear to be the most significant predictive markers of disease progression. Immuno-staining patterns for CD68, CD56, and CD31 indicated an impairment in the muscle regeneration process, probably, at an early stage of the disease.
Conclusion
This study's findings are crucial for understanding pathogenesis and identifying new therapeutic targets. However, because of the small sample size, further confirmation through a larger cohort is necessary.
Keywords: Muscle biopsy, Sarcoglycanopathy, Histology, Immunohistochemistry
Highlights
-
•
Diagnosing Gamma Sarcoglycanopathy can be challenging.
-
•
Potential correlation between histopathological abnormalities and clinical phenotype
-
•
Immunohistochemistry gives crucial for understanding the pathogenesis.
1. Introduction
Limb Girdle Muscular Dystrophies “LGMD” are heterogeneous hereditary muscle disorders characterized by progressive muscle weakness. The most common clinical form is autosomal recessive (AR). They show a degeneration/regeneration on muscle biopsy, usually associated with elevated serum creatine kinase levels, specific protein deficiency, and deleterious mutation [1,2]. LGMDR5, or γ-sarcoglycanopathy, is a common LGMD in North African populations due to the mutation c.525delT in the Sarcoglycan (SGC) gene. Its epidemiology is little known in Morocco and its prevalence within the Moroccan population has been little evaluated. The most recent study (based on a molecular epidemiological approach) estimated the frequency of carriers at 1/250, and the prevalence at approximately 1/20,492 considering the effect of consanguinity [3].
LGMDR5 is classified as severe autosomal recessive muscular dystrophy of the child and requires an early diagnostic strategy [4].
In Morocco, diagnosing muscular dystrophies can be challenging due to the low specificity of clinical characteristics and limited access to genetic and molecular research. Additionally, distinguishing between dystrophinopathy and Gamma Sarcoglycanopathy can be difficult. Therefore, histopathological and immunohistochemical analysis of muscle biopsies are essential for accurate diagnosis. A study by Ferreira AF et al. demonstrated that a thorough clinical evaluation, combined with immunohistochemical staining of muscle samples, effectively characterizes patients with limb-girdle muscular dystrophy (LGMD).
By applying this approach to a larger patient population, researchers may further enhance these diagnostic tools and better evaluate the cost-to-benefit ratio of molecular diagnostics [5].
This study evaluates several morphological parameters, including endomysial fibrosis, macrophage inflammatory infiltrates, satellite cells, and vascularization, to explore potential predictive factors and correlations between histopathological abnormalities and clinical phenotype.
2. Materials and methods
2.1. Patient population
We opted for a retrospective and descriptive study over two years, between January 2021 and December 2023. Our institution's Faculty of Medicine of Rabat's Ethics Committee does not require ethical approval for case series reporting. We maintained a high level of respect for both anonymity and confidentiality.
We identified 42 muscular dystrophies in our database including six cases of LGMDR5 that we retrieved from the archives of the Department of Pathology of Ibn Roched Hospital Center in Morocco. Clinical features including age, sex, family history, consanguinity, age of onset, motor deficit, assessment of serum creatine kinase (CK) levels, nerve conduction, and electromyographic (EMG) studies, presence or absence of orthopedic or cardiorespiratory complications, malformations, and cognitive disorders, were identified from the patient's medical files.
2.2. Histopathological analysis
The pathological findings from the hematoxylin-eosin and Gomori Trichrome slides were reviewed by two independent pathologists. The histological parameters assessed include uneven muscle fiber diameter, centralized nuclei, atrophic fibers, necrotic fibers, basophilic regeneration fibers, and endomysial fibrosis. Each of these parameters is rated based on its presence and distribution in the histological section as follows: (0) if the parameter is absent; (+ or mild) if it is rare and focal; (++or moderate) if it is moderate and intermediate between rare and numerous, and (+++or severe) if it is numerous and extensive.
Immunohistochemical stainings were reviewed on frozen sections on a DAKO Autostainer Link 48. The antibodies tested were anti-dystrophin 2 (cloneDy8/6C5, NovoCastra), anti-alpha Sarcoglycan (clone Ad1/20A6, NovoCastra), anti-beta Sarcoglycan (clone BetaSarc/5B1, NovoCastra), and anti-gamma Sarcoglycan (clone 35DAG/21B5, NovoCastra).
To assess potential alterations in the muscle microenvironment, we conducted immunostaining on formalin-fixed sections from each biopsy. This included staining for macrophagic cells using the anti-CD68 antibody (PMG1 clone), for satellite cells with the anti-CD56 antibody (NCAM clone), and for capillary vessels with the anti-CD31 antibody (clone JC70A).
No patient had genetic or molecular research.
The work has been reported in line with the PROCESS criteria [6].
3. Results
3.1. Clinical and demographic findings
Among the 158 muscle biopsies evaluated, 42 (26.5 %) corresponded to muscular dystrophy, of which 6 (14 %) were diagnosed as Gamma Sarcoglycanopathy (GSG). Furthermore, 59.5 % of patients with muscular dystrophy were children under the age of 19, with GSG accounting for 25 % of them.
The average age of patients at diagnosis was 10 years (ranging from 8 to 14 years). The average age of onset was 4 years (ranging from 3 to 5 years). The sex ratio was 1 (1 male = 1 female). Consanguinity of 1st and 2nd degree was observed in 3 out of 6 patients, with a family history of motor deficit (not investigated) in two cases.
Of the six patients, five were ambulatory and had pelvic girdle motor deficits. They experienced falls, difficulty climbing stairs, a waddling gait, and a positive Gowers sign. One of the 6 patients required a wheelchair due to a recurrent Botfoot, which worsened their motor deficit. Another ambulant child also had the same orthopedic malformation.
Only one of the six children was affected in the shoulder girdle. All patients had calf hypertrophy. Orthopedic complications were observed early in our patients, including scoliosis in 2/6 patients, Achilles tendon retraction in 4/6 patients, and hyperlordosis in 1/6 patients. There were no cognitive disorders or cardiorespiratory complications in any of the patients. The clinical characteristics at the time of diagnosis are outlined in Table 1.
Table 1.
Clinical features at the time of the diagnosis.
| Clinical parameters | Nombre of cases |
|---|---|
| Consanguinity | 3/6 |
| Familial history | 2/6 |
| Pelvic girdle motor deficit | 6/6 |
| Scapular motor deficit | 1/6 |
| Calf hypertrophy | 6/6 |
| Orthopedic malformation (clubfoot) | 2/6 |
| Orthopedic complications: Achilles tendon retraction Scoliosis Hyperlordosis |
4/6 2/6 1/6 |
| Cardio-respiratory complications | 0/6 |
| Cognitive disorders | 0/6 |
The CPK levels were elevated (1241–25,000 U/L) and the EMG showed myogenic activity in all patients.
3.2. Pathological findings
Histologically, all patients exhibited muscular dystrophy with uneven muscle fiber diameters, centralized nuclei, atrophic fibers, and necrotic fibers.
These characteristics were present in varying proportions, as shown in Table 2.
Table 2.
Distribution of histological parameters: 0 = absent, + = Mild, ++ = moderate, and +++ = severe.
| Patient | Uneven muscle fiber diameters | Centralized nuclei | Atrophic fibers | Necrotic fibers | Regenerative basophilic fibers | Endomysial fibrosis | Histological diagnosis |
|---|---|---|---|---|---|---|---|
| N°1 | +++ | ++ | +++ | +++ | + | +++ | Severe dystrophy |
| N°2 | + | + | + | + | − | + | Mild dystrophy |
| N°3 | + | + | + | + | − | + | Mild dystrophy |
| N°4 | + | + | + | + | + | + | Mild dystrophy |
| N°5 | ++ | ++ | ++ | ++ | + | ++ | Moderate dystrophy |
| N°6 | ++ | ++ | ++ | +++ | + | +++ | Severe Dystrophy |
Regenerative basophilic fibers were rare in 4 out of 6 patients and absent in 2 out of 6 patients. Endomysial fibrosis was extensive in 2/6 children, moderate in 1/6, and discreet in the other three.
Based on our morphological assessment, we categorized muscular dystrophy as mild in 3 out of 6 patients, moderate in 1 out of 6 patients, and severe in 2 out of 6 patients (Fig. 1).
Fig. 1.

(A, C, E): Histological appearance of varying severity of muscular dystrophy (Hematoxylin Eosin x 20). (B): microphotography of significant endomysial fibrosis, (D): Moderate endomysial fibrosis, and (F): absence of endomysial fibrosis (Trichrom of Gomori ×10).
Our immunohistochemical analysis showed that all patients had a complete loss of expression of anti-Gamma SG (GSG) and low expression of anti-Beta SG (BSG). Anti-Alpha SG (ASG) was normally expressed in 4 out of 6 patients and weakly expressed in 2 out of 6 patients. This leads us to two immunohistochemical patterns: The most frequently observed result in our population was “GSG negative/BSG low/ASG normal” which occurred in 4 out of 6 cases. The second observed pattern was “GSG negative/BSG low/ASG low”. Due to the small sample size, a proper statistical analysis was not feasible.
Dystrophin antibody was normally expressed in all cases (Fig. 2).
Fig. 2.

immunohistochemical profile of Sarcoglycan and Dystrophin2 expression (ASG = Alpha Sarcoglycan, BSG = Beta Sacrcoglycan, GSG = Gamma Sarcoglycan, Dys2 = Dystrophin2).
Immunostaining using anti-CD68, CD56, and anti-CD31 showed significant CD68-positive macrophagic inflammatory changes in 3 out of 6 patients (Fig. 3). A notable reduction in CD56-positive satellite cells with a clear decrease in CD31-positive capillaries was observed in all cases (Fig. 3).
Fig. 3.
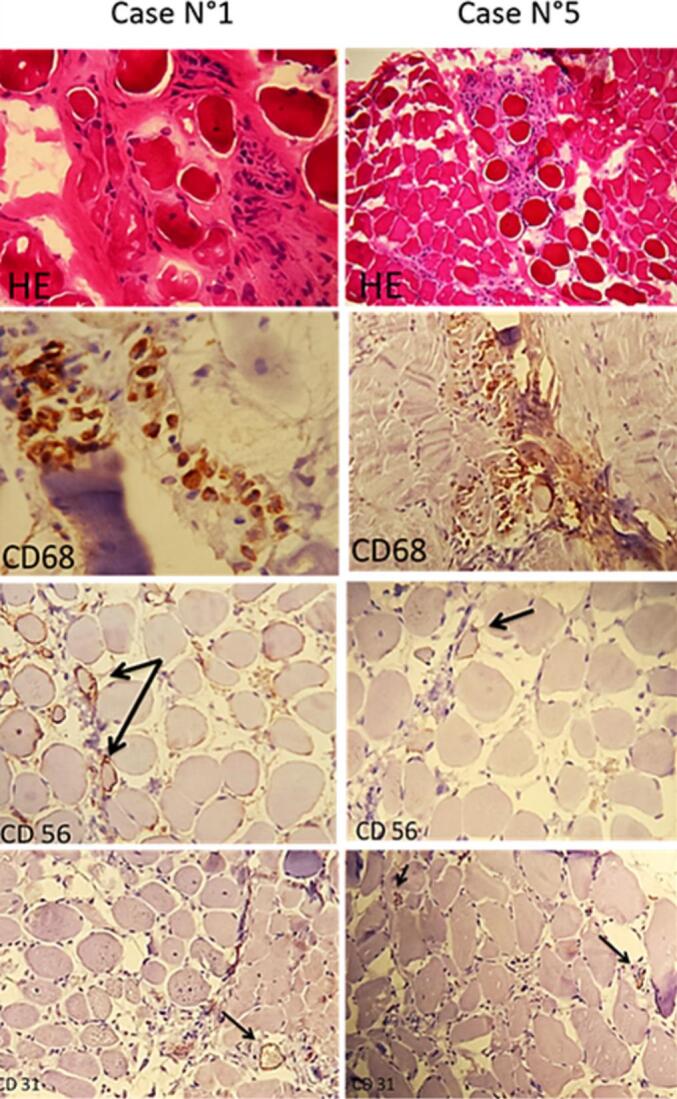
Histological appearance of the significant macrophagic infiltrate in cases N°1 and 5 and their respective CD68 immunostaining. We observed a clear decrease in the immunostaining of satellite cells for CD56 and capillaries for CD31 (arrow).
Table 3 summarizes the immunostaining results compared with our study's clinical and histological phenotypes.
Table 3.
Results of immunostaining by contributions in comparison with the observed clinical and histological phenotypes (A = Ambulant; WC = Wheelchair;Y = year; SG = Sarcoglycan; − = absent; + = mild; ++ = moderate; +++ = severe.
| Case | Age of onset | Motor status | Orthopedic complication | Histological diagnosis | Endomysial fibrosis | Gamma SG expression | CD68 staining | CD56 staining | CD31 staining |
|---|---|---|---|---|---|---|---|---|---|
| N°1 | 3 y | A at 8 y | Present | Severe dystrophy | +++ | − | +++ | + | + |
| N°2 | 4 y | WC at 14 y with clubfoot | Present | Mild dystrophy | + | − | + | + | + |
| N°3 | 5 y | A at 8 y | Absent | Mild dystrophy | + | − | − | + | + |
| N°4 | 3 y | A at 10 y | Present | Mild dystrophy | + | − | + | + | + |
| N°5 | 8 y | A at 11 y | Present | Moderate dystrophy | ++ | − | +++ | + | + |
| N°6 | 4 y | A at 10 y | Absent | Severe dystrophy | +++ | − | +++ | + | + |
4. Discussion
Diagnosing Limb Girdle Muscular Dystrophy (LGMD) in Morocco is quite challenging. This is mainly due to the non-specific clinical features and the lack of specialized laboratories that can identify the mutations causing the disease. In these cases, muscle biopsy remains essential for diagnosis. It helps confirm the presence of a dystrophic process and detects any underlying protein deficiencies, which can guide molecular diagnosis [[7], [8], [9]]. Furthermore, a detailed analysis of the muscle biopsy can provide prognostic markers, such as the degree of muscle fiber regeneration and the extent of endomysial fibrosis, which could be related to the severity of the clinical condition [7,10].
In this study, we investigated the role of muscle biopsy in diagnosing Gamma Sarcoglycanopathy (LGMDR5). This LGMD is associated with chromosome 13q12, encoding for the gamma sarcoglycan protein (GSG) [[11], [12], [13]], and is common in Morocco [3]. According to our lab data, LGMDR5 constituted 14 % of all muscular dystrophies diagnosed at any age and 24 % of those diagnosed in children. Our analysis revealed heterogeneous histological features within the same clinical phenotype (Duchenne-like). We found both mild and severe muscular dystrophy even for patients of the same age and similar motor status. As for the degree of endomysial fibrosis, it was superimposed on the degree of muscular dystrophy. Interestingly, we suggest that the severity of histological changes in muscular dystrophy may not consistently align with the clinical severity of the disease and might not be a dependable predictor of disease progression. Larger histopathological studies with significant sample sizes are needed to support this observation, as recent research increasingly focuses on immunohistochemical and molecular analyses to examine especially the correlation between phenotype and genotype [5,12].
On the other hand, all of our patients share two common features: they have completely lost their immunohistochemical expression of Gamma sarcoglycan, and they have experienced symptoms at an early age (on average, at 4 years old). These two factors could potentially explain the severity of their clinical phenotype [1]. A recent European study [14] showed that the mutations leading to the absence of expression of a Sarcoglycan, particularly a GSG, are associated with a more severe phenotype, with loss of walking occurring before age 18. According to this study, an earlier age of onset of the disease is associated with more significant progression [3,14].
The phenotypic heterogenicity and absence of significant predictive factors make patient monitoring and management challenging. Currently, there is no effective treatment available [15]. However, the results obtained in clinical trials using gene therapy over the last 20 years are encouraging despite their use in small groups of patients mainly because of the phenotypic heterogeneity of the patients [16].
In our research, we identified two distinct patterns of Sarcoglycan expression: “GSG negative/BSG low/ASG normal” and “GSG negative/BSG low/ASG low”. Interpreting these expression patterns could be challenging and may confuse. In practice, when dealing with a suspected sarcoglycanopathy case in a North African patient with normal Dystrophin expression, it's important to consider the prevalence patterns of this condition before conducting genetic analysis. In particular, the absence or reduction of Gamma SG expression may indicate the presence of the “c.525delT” mutation, which has a founder effect in the North African population [12,17].
Furthermore, our immunostaining showed a significant infiltrate of CD68+ macrophages in contact with necrotic fibers. This inflammatory process, initially stimulates satellite cell proliferation and promotes muscular regeneration [18,19], becomes chronic, and leads to muscle degeneration. Indeed, regardless of the severity of muscular dystrophy, we observed a clear reduction or even absence of CD56+ satellite cells, associated with scarcity of nuclear centralizations and basophilic regeneration fibers. The mechanisms of this transformation are still unclear. One possible explanation is that the inflammatory cells, particularly the macrophages that infiltrate the damaged muscle, produce an important amount of residual oxidative stress (ROS) [20]. Additionally, we observed a significant decrease in CD31+ capillaries in the endomysium of all types of muscular dystrophy, including the wild form. This indicates that capillary depletion is a crucial factor in muscular degeneration, and occurs early in the pathogenesis of Sarcoglycanopathy. Several studies have shown that the absence of dystrophin-associated proteins, such as sarcoglycans, affects angiogenesis. In the mdx mouse model for Duchenne muscular dystrophy [21], the lack of sarcoglycans in vascular smooth muscle disrupts vascular function, initiates cardiomyopathy, and worsens the clinical phenotype [22].
Our findings indicate an early deficiency in the muscular regeneration process involving the microenvironment of muscles (macrophagic inflammatory and capillary depletion), which could play a significant role in the progression of Gamma Sarcoglycanopathies and propose new targets for therapy [23].
5. Conclusions
This study revealed a diverse histological presentation of LGMDR5 associated with the same clinical phenotype. It suggested that there might be an early deficiency in the regeneration process involving the muscle microenvironment. We acknowledge that the small number of patients in our study means these findings require further investigation in the future with larger cohorts, as well as genetic and molecular analyses.
Patient consent
Written informed consent was obtained from the patients for publication and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Ethical approval
This report was prepared following the ethical standards of the institutional ethics committee and with the 1964 Helsinski Declaration. Our institution does not require ethical approval for reporting individual cases or case series, and we maintain high respect for anonymity and confidentiality.
Guarantor
Fouad Zouaidia, MD.
Sources of funding
The authors didn't use any funding sources, and this study has no sponsors.
CRediT authorship contribution statement
Hafsa E.L. Ouazzani: Writing – review & editing. Fouad Zouaidia: Supervision. Nadia Cherradi: Supervision. Mahdi Karkouri: Conceptualization, Methodology, Data curation.
Declaration of competing interest
There are no conflicts of interest for authors in this study.
Acknowledgement
Declared none.
Data availability
No new data were generated or analyzed in support of this research.
References
- 1.Vainzof M., Souza L.S., Gurgel-Giannetti J., Zatz M. Sarcoglycanopathies: an update. Neuromuscul. Disord. 2021;31(10):1021–1027. doi: 10.1016/j.nmd.2021.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Rosales X.Q., Al-Dahhak R., Tsao C.Y. Childhood-onset of limb-girdle muscular dystrophy. Pediatr. Neurol. 2012;46(1):13–23. doi: 10.1016/j.pediatrneurol.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 3.El Kerch F., Ratbi I., Sbiti A., Laarabi F.Z., Barkat A., Sefiani A. Carrier frequency of the c.525delT mutation in the SGCG gene and estimated prevalence of limb girdle muscular dystrophy type 2C among the Moroccan population. Genet. Test. Mol. Biomarkers. 2014;18(4):253–256. doi: 10.1089/gtmb.2013.0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernández-Eulate Gorka, Leturcq France, Laforêt Pascal, Richard Isabelle, Stojkovic Tanya. Les sarcoglycanopathies: État des lieux et perspectives thérapeutiques. Médecine/sciences. 2020;36:22–27. doi: 10.1051/medsci/2020243. [DOI] [PubMed] [Google Scholar]
- 5.Ferreira A.F., Carvalho M.S., Resende M.B., Wakamatsu A., Reed U.C., Marie S.K. Phenotypic and immunohistochemical characterization of sarcoglycanopathies. Clinics (Sao Paulo) 2011;66(10):1713–1719. doi: 10.1590/s1807-59322011001000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathew G., Agha R.A., Sohrabi C., Franchi T., Nicola M., Kerwan A., Agha R for the PROCESS Group Preferred reporting of case series in surgery (PROCESS) 2023 guidelines. Int. J. Surg. 2023 doi: 10.1097/JS9.0000000000000940. (article in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuisset J.M., Maurage C.A., Carpentier A., Briand G., Thévenon A., Rouaix N., Vallée L. Intérêt de la biopsie musculaire chez l’enfant en 2012 (Muscle biopsy in children: usefulness in 2012) Rev. Neurol. (Paris) 2013;169(8–9):632–639. doi: 10.1016/j.neurol.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 8.Walters J., Baborie A. Muscle biopsy: what and why and when? Pract. Neurol. 2020;20(5):385–395. doi: 10.1136/practneurol-2019-002465. [DOI] [PubMed] [Google Scholar]
- 9.Filosto M., Tonin P., Vattemi G., Bertolasi L., Simonati A., Rizzuto N., Tomelleri G. The role of muscle biopsy in investigating isolated muscle pain. Neurology. 2007;68(3):181–186. doi: 10.1212/01.wnl.0000252252.29532.cc. Jan 16. [DOI] [PubMed] [Google Scholar]
- 10.Mornet Dominique. De l’importance de la biopsie musculaire chez les patients atteints de dystrophinopathie. Cah Myol. 2018;17:30. doi: 10.1051/myolog/201817008. [DOI] [Google Scholar]
- 11.Nigro V., Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 2014;33:1–12. [PMC free article] [PubMed] [Google Scholar]
- 12.Dınız G., Hazan F., Yildirim H.T., Unalp A., Polat M., Serdaroğlu G., Güzel O., Bağ O., Seçıl Y., Fdoi Ozgönül, Türe S., Akhan G., Tükün A. Histopathological and genetic features of patients with limb-girdle muscular dystrophy type 2C. Turk Patoloji Derg. 2014;30(2):111–117. doi: 10.5146/tjpath.2014.01239. [DOI] [PubMed] [Google Scholar]
- 13.Vermeer S., Verrips A., Willemsen M.A., ter Laak H.J., Ginjaar I.B., Hamel B.C. Novel mutations in three patients with LGMD2C with phenotypic differences. Pediatr. Neurol. 2004;30(4):291–294. doi: 10.1016/j.pediatrneurol.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Alonso-Pérez J., González-Quereda L., Bello L., Guglieri M., Straub V., Gallano P., et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain. 2020;143(9):2696–2708. doi: 10.1093/brain/awaa228. [DOI] [PubMed] [Google Scholar]
- 15.Scano Martina, Benetollo Alberto, Barba Francesco Dalla, Sandonà Dorianna. Advanced therapeutic approaches in sarcoglycanopathies. Curr. Opin. Pharmacol. 2024;76 doi: 10.1016/j.coph.2024.102459. [DOI] [PubMed] [Google Scholar]
- 16.Vainzof Mariz, Souza Lucas S., Gurgel-Giannetti Juliana, Zatz Mayana. Sarcoglycanopathies: an update. Neuromuscul. Disord. 2021 Oct;31(10):1021–1027. doi: 10.1016/j.nmd.2021.07.014. [DOI] [PubMed] [Google Scholar]
- 17.Klinge L., Dekomien G., Aboumousa A., Charlton R., Epplen J.T., Barresi R., Bushby K., Straub V. Sarcoglycanopathies: can muscle immunoanalysis predict the genotype? Neuromuscul. Disord. 2008 Dec;18(12):934–941. doi: 10.1016/j.nmd.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Chang N.C., Chevalier F.P., Rudnicki M.A. Satellite cells in muscular dystrophy - lost in polarity. Trends Mol. Med. 2016;22(6):479–496. doi: 10.1016/j.molmed.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Souza G.T., Zanette Rde S., do Amaral D.L., da Guia F.C., Maranduba C.P., de Souza C.M., Guimarães Eda S., Rettore J.V., Rabelo N.C., do Carmo A.M., Silva Fde S., Maranduba C.M. Satellite cells: regenerative mechanisms and applicability in muscular dystrophy. Stem Cells Int. 2015;2015 doi: 10.1155/2015/487467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.González-Jamett A., Vásquez W., Cifuentes-Riveros G., Martínez-Pando R., Sáez J.C., Cárdenas A.M. Oxidative stress, inflammation and connexin hemichannels in muscular dystrophies. Biomedicines. 2022;10(2):507. doi: 10.3390/biomedicines10020507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan K.M., Morgan K.G. The importance of dystrophin and the dystrophin-associated proteins in vascular smooth muscle. Front. Physiol. 2022;13:1059021. doi: 10.3389/fphys.2022.1059021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coral-Vazquez R., Cohn R.D., Moore S.A., Hill J.A., Weiss R.M., Davisson R.L., Straub V., Barresi R., Bansal D., Hrstka R.F., Williamson R., Campbell K.P. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98(4):465–474. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- 23.Ohlendieck K., Swandulla D. Complexity of skeletal muscle degeneration: multi-systems pathophysiology and organ crosstalk in dystrophinopathy. Pflugers Arch. 2021;473(12):1813–1839. doi: 10.1007/s00424-021-02623-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this research.