

Abstract
The CDKN2A gene, responsible for encoding the tumor suppressors p16(INK4A) and p14(ARF), is frequently inactivated in non-small cell lung cancer (NSCLC). Herein, an uncharacterized long non-coding RNA (lncRNA) (ENSG00000267053) on chromosome 19p13.12 was found to be overexpressed in NSCLC cells with an active, wild-type CDKN2A gene. This lncRNA, named cyclin-dependent kinase inhibitor 2A-regulated lncRNA (CyKILR), also correlated with an active WT STK11 gene, which encodes the tumor suppressor, liver kinase B1. CyKILR displayed two splice variants, CyKILRa (exon 3 included) and CyKILRb (exon 3 excluded), which are cooperatively regulated by CDKN2A and STK11 as knockdown of both tumor suppressor genes was required to induce a significant loss of exon 3 inclusion in mature CyKILR RNA. CyKILRa localized to the nucleus, and its downregulation using antisense RNA oligonucleotides enhanced cellular proliferation, migration, clonogenic survival, and tumor incidence. In contrast, CyKILRb localized to the cytoplasm, and its downregulation using small interfering RNA reduced cell proliferation, migration, clonogenic survival, and tumor incidence. Transcriptomics analyses revealed the enhancement of apoptotic pathways with concomitant suppression of key cell-cycle pathways by CyKILRa demonstrating its tumor-suppressive role. CyKILRb inhibited tumor suppressor miRNAs indicating an oncogenic nature. These findings elucidate the intricate roles of lncRNAs in cell signaling and tumorigenesis.
Keywords: MT: Non-coding RNAs, long non-coding RNAs, RNA splicing, non-small cell lung cancers, antisense oligonucleotides, CDKN2A, LKB1, STK11
Graphical abstract
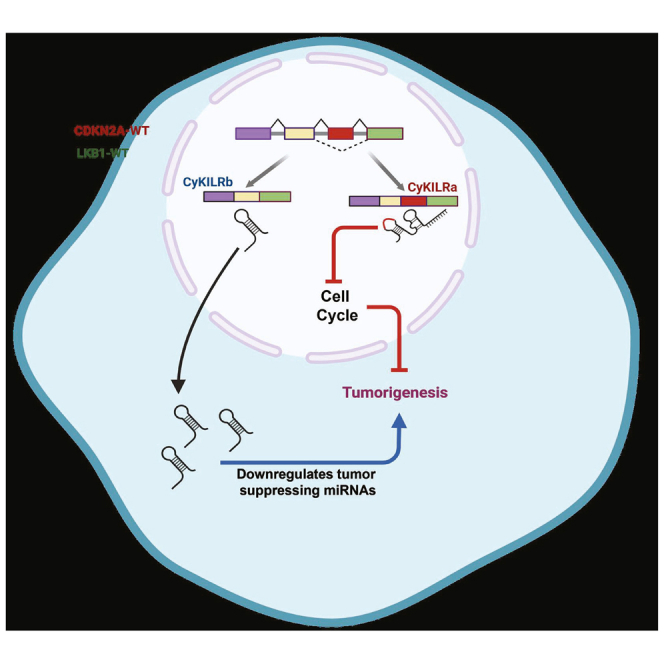
Chalfant and colleagues identified a novel lncRNA, CyKILR, regulated by CDKN2A and STK11, that exhibits two splice variants in NSCLC. CyKILRa suppresses cell-cycle pathways, acting as a tumor suppressor, while CyKILRb inhibits tumor suppressor microRNAs (miRNAs), linked to its oncogenic role. These findings highlight the intricate involvement of lncRNAs in NSCLC tumorigenesis.
Introduction
Non-small cell lung cancers (NSCLCs) encompass a diverse group of lung malignancies, accounting for approximately 85% of all lung cancer cases.1,2,3,4 Despite significant progress in diagnosis and treatment, NSCLCs remain a leading cause of cancer-related deaths worldwide.5 The intricate molecular landscape of NSCLCs, characterized by complex tumor microenvironments and diverse genetic alterations, presents considerable challenges in understanding their pathogenesis and developing effective therapeutic strategies. In regard to genetic alterations, the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene encodes the tumor suppressors, p16(INK4a) and p14(ARF), and is found inactivated or deleted in NSCLC at approximately 30%.6,7 p14ARF is an alternate reading frame protein product of the CDKN2A locus, and p14ARF is induced in response to elevated mitogenic stimulation, such as aberrant growth signaling from the MYC and RAS genes. It accumulates mainly in the nucleolus, where it forms stable complexes with NPM or Mdm2. p16INK4a (also known as CDKN2A, multiple tumor suppressor 1, and numerous other synonyms) is a protein that suppresses cell division by slowing the progression of the cell cycle from the G1 phase to the S phase, thereby acting as a tumor suppressor. Loss of either p14ARF or p16INK4a has dramatic effects on the landscape of gene expression and RNA transcription, increasing the susceptibility of cells to undergo oncogenic transformation.
Effects on gene expression can also include long non-coding RNAs (lncRNAs), which were for many years dismissed as transcriptional noise. In recent years, lncRNAs have gained significant attention due to their potential roles in various diseases including cancer.8,9,10,11 LncRNAs are RNA transcripts with lengths exceeding 200 nucleotides and differ from protein-coding genes in not encoding for functional proteins. Instead, they exert their biological effects through diverse molecular mechanisms. With unique secondary and tertiary structures, lncRNAs can regulate gene expression through processes like chromatin modification, RNA splicing, and mRNA transcriptional and post-transcriptional regulation via RNA-DNA, RNA-miRNA, or RNA-protein interactions.12,13,14,15,16 In cancer biology, dysregulation and aberrant expression of lncRNAs have been linked to tumor initiation, progression, and therapeutic responses.17 Distinct lncRNA expression patterns have been observed in various cancer types, including lung, breast, prostate, and colorectal cancers.18,19,20,21,22 Furthermore, accumulating evidence suggests that lncRNAs can act as oncogenes or tumor suppressors exerting both pro-tumorigenic or anti-tumorigenic effects.23,24
Like protein-coding genes, lncRNA genes consist of introns and exons that undergo alternative splicing. Splicing of lncRNA plays a pivotal role in generating transcriptomic diversity and expanding the functional repertoire of lncRNAs. Alternative splicing events give rise to distinct isoforms of lncRNA, which can differ in their sequences, structures, and subcellular localization. These splice variants possess the remarkable capacity to differentially modulate gene expression and cellular function in a context-dependent manner.25,26,27 Although lncRNAs and their splicing variants have emerged as crucial players in the intricate landscape of cellular gene regulation,28 identifying lncRNAs and their various splice variants remains challenging despite advancements in high-throughput sequencing technologies and bioinformatics approaches. Notably, one identified lncRNA, Neat1, undergoes alternative splicing regulated by RBM10. The resulting two splice variants, Neat1-1 and Neat1-2, play distinct roles in the invasion and metastasis of NSCLCs.29 Another reported lncRNA has multiple splice variants, SOX2OT (4 and 7), which have oncogenic roles in NSCLCs.30 Thus, the alternative splicing of lncRNAs is becoming appreciated for their distinct and novel roles in NSCLC and cancer, but are significantly understudied.
In this study, we have made the exciting discovery of a novel lncRNA (ENSG00000267053) located on the forward strand of chromosome 19p13.12 that exhibits multiple splice variants. This lncRNA, termed CDKN2A-regulated lncRNA (CyKILR), is almost exclusively present in NSCLC cells with both an active and wild-type (WT) CDKN2A and STK11 gene. Based on the presence of exon 3, the CyKILR splice variants are classified into two distinct groups, CyKILRa (exon 3 included) and CyKILRb (exon 3 excluded). Mechanistic studies show that CyKILRa predominantly localizes in the nucleus and demonstrates a tumor-suppressive function. In contrast, CyKILRb localizes to the cytoplasm and exhibits pro-survival functions. The balance between the two groups of CyKILR RNA splicing variants is dynamically regulated by gene products of both the CDKN2A and STK11 genes, thereby finely tuning cellular homeostasis in these NSCLC cell lines. Our findings shed light on the potential diagnostic, prognostic, and therapeutic implications of targeting lncRNAs and their specific splicing variants in NSCLCs, opening new avenues for precision medicine approaches. Indeed, this study also unravels the intricate regulatory networks orchestrated by lncRNAs and their splice variants, offering an exciting opportunity to decipher their complex roles in cancer biology.
Results
The association of the expression of a novel lncRNA and CDKN2A/LKB1 status in NSCLCs
The CDKN2A gene is found inactivated or deleted in NSCLC at a high percentage, which also means that a number of NSCLC tumors and cell lines have a WT, active, and functional CDKN2A gene. To examine if there are specific gene expression patterns between WT CDKN2A versus mutant/inactive CDKN2A NSCLC cell lines, 12 NSCLC cell line with a differing CDKN2A status were compared (6 with WT CDKN2A [H838, H522, H2009, H1299, H1792, and H1734] and 6 with deleted/mutant CDKN2A [H2030, H810, H460, A549, H838, and H1648]) using unbiased transcriptomics. A top hit for a differentially expressed lncRNA associated with a WT CDKN2A gene was a novel, uncharacterized lncRNA (ENSG00000267053) (Table S1) located on chromosome 19p13.12 (Figure S1). Validation studies using qRT-PCR corroborated a strong link between this CyKILR and the WT CDKN2A gene, but a perfect correlation was not observed (Figures 1A–1C). Indeed, the DESeq2 and plot3 packages in R studio were also used to generate both 3D principal component analysis (PCA) and correlation plots of CDKN2A mRNA expression with CyKILR expression, but the PCA showed that the 12 NSCLC lines did not exhibit strong clustering based solely on CDKN2A status/expression (Figure 1D). Examining additional oncogenotypes to explain the inconsistency showed that the five NSCLC cell lines with high CyKILR expression (H358, H522, H2009, H1299, and H1792) also correlated with the expression of the tumor suppressor, liver kinase B1 (LKB1), a product of the STK11 gene (Figures 1E and 1F). The protein levels of the CDKN2A gene products, p14 ARF and p16 INK4A, as well as LKB1 were also examined to validate our transcriptomic and qRT-PCR findings (Figures 1G and 1H). These results were consistent with the mRNA expression findings across the 12 cell lines. Importantly, our analyses revealed high expression of CyKILR in only H358, H522, H2009, H1299, and H1792, which were cell lines positive for both WT CDKN2A and STK11 genes and demonstrated expression of their gene products at the protein level (Figures 1G and 1H). These results demonstrate that, in NSCLC cell lines, CyKILR is almost exclusively expressed when the cells possess both active and WT CDKN2A and STK11 genes.
Figure 1.

The association of CyKILR expression with an active, WT STK11, and CDKN2A genes in NSCLCs
Total RNA was collected from the indicated NSCLC cell lines and reverse transcribed (RT). qRT-PCR and deep RNA-seq were conducted. The R program was used for analysis of deep RNA-seq data. Western blotting was conducted on 12 NSCLC lines to assess the expression of CDKN2A and STK11 gene products with CyKILR expression. (A) The mRNA level of CDKN2A across the 12 NSCLC lines normalized to GAPDH mRNA levels. (B) Correlation plot of deep RNA-seq data displaying the relationship between CDKN2A gene status and the expression of the lncRNA, CyKILR. (C) CyKILR RNA levels by qRT-PCR across the 12 NSCLC lines normalized to GAPDH mRNA levels. Data in (A) and (C) are presented as mean (SD) (n ≥ 3 replicates). (D) Three-dimensional PCA plot representing the variation of CDKN2A and CyKILR expression for the 12 NSCLC lines. (E) The mRNA level of the STK11 gene product, LKB1, across the 12 NSCLC lines normalized to GAPDH mRNA levels. Data in (E) are presented as mean ± SD (n ≥ 3 replicates). (F) Correlation plot of deep RNA-seq data to an active, WT STK11 gene displaying the relationship between STK11 mRNA expression and CyKILR expression. (G) Relative protein levels of the indicated factors across a panel of 12 cell lines with β-actin used for normalization. Data are representative of n = 3. (H) The protein levels of the CDKN2A gene products, p14 and p16, the STK11 gene product, LKB1, across the 12 NSCLC lines with β-actin used for normalization. Data in (H) are presented as mean ± SD (n ≥ 3 replicates). Data in (B) and (F) were analyzed by unpaired t test with Welch’s correction. Reverse transcription (RT) “minus” controls (−RT) were included in all RT-PCR experiments and replicates with no signal observed.
CyKILR classifies into two distinct isoform groups based on the alternative RNA splicing of exon 3
RNA sequencing (RNA-seq) data revealed the presence of multiple variants of CyKILR, as depicted in Figures 2A and 2B. To categorize these variants, we used a classification based on the status of exon 3. Those variants containing exon 3 were designated as CyKILRa, while those lacking exon 3 were denoted as CyKILRb. To evaluate their relative expression patterns, we conducted RT-PCR across a panel of 12 NSCLC lines. As shown in Figure 2C, expression of the CyKILRa group was easily detected in H358, H522, H2009, H1299, H1792, and H1648 cell lines. Moreover, a discernable level of expression was also observed in H2030, H1734, and H810 cell lines. In contrast, H460, A549, and H838 showed minimal to negligible expression of the CyKILRa group, the cell lines completely devoid of both CDKN2A and STK11 gene products as assessed by western blotting (Figure 1G). Conversely, the cohort of CyKILRb displayed expression exclusively within the H358, H522, H2009, H1299, and H1792 cell lines (Figure 2C), NSCLC cell lines with discernable expression of both CDKN2A and STK11 gene products by western blotting (Figure 1G). To quantify the expression level of CyKILRa and CyKILRb across the 12 NSCLC lines, we used customized quantitative primers and probes (Figure 2D). To target exon 3 inclusion variants (CyKILRa), the primers and probes were designed to be complementary to exon 3 (Figure 2D). In contrast, for the identification of exon-exclusive variants (CyKILRb), the forward primer corresponded with exon 1, while the reverse primer aligned with exon 5, complemented by a probe spanning the junction of exon 1 and exon 5 (Figure 2D). Our results revealed that both CyKILRa and CyKILRb were predominantly expressed in H358, H522, H2009, H1299, and H1792 cell lines (Figure 2D), which are characterized by the presence of both active and WT CDKN2A and STK11 genes (expression of gene products derived from WT CDKN2A and WT STK11 NSCLC lines verified by western blotting) (Figure 1G) vs. other NSCLC cell lines (p = 0.000729 for n = 4/cell line using one-way ANOVA). These data also demonstrate that CyKILRb has more distinct expression linked to these tumor suppressing genes.
Figure 2.

CyKILR is characterized by two distinct isoform groups based on exon 3 inclusion/exclusion
Total RNA was collected from the indicated NSCLC cell lines. Short-read deep RNA-seq was conducted on these RNA samples. The R program was used for analysis of deep RNA-seq data. (A) The predicted pre-mRNA structure of CyKILR based on data from Ensembl. The CyKILR RNA splice variants were categorized into two distinct groups based on the inclusion or exclusion of exon 3 into the mature RNA transcript. Predicted exon and full-length CyKILR splice variants are annotated. Of note, the designated exon 6 is predicted to have multiple 5′ splice sites for several CyKILRa variants. (B) The expression level of CyKILR isoforms across the 12 cell lines from the deep RNA-seq conducted on RNA samples from these cell lines. The R program was used for analysis of deep RNA-seq data. (C) Relative RNA levels of CyKILRa (including exon 3) and CyKILRb (excluding exon 3) variants across a panel of 12 NSCLC cell lines using RT-PCR. (D) The RNA expression levels of CyKILRa and CyKILRb are depicted by using a customized quantitative primer and probe strategy for the detection of CyKILRa and CyKILRb variants by qRT-PCR and normalized to GAPDH mRNA levels. The data in (D) are presented as mean (SD) (n ≥ 3 replicates). −RT controls were included in all experiments and replicates with no signal observed.
CyKILRa and CyKILRb RNA splicing variants display differential cellular localization
Unlike mRNA, lncRNA is frequently detected in both the cytoplasm and nucleus. Research in the area suggests that an AGCCC motif plays a crucial role in the nuclear localization of numerous lncRNAs.31 Following a sequence analysis, the specific presence of an AGCCC motif in exon 3 of CyKILR was identified (Figure 3A). This discovery prompted us to investigate the localization patterns of the two distinct CyKILR groups by isolating total RNA from both the nucleus and cytoplasm. Subsequent multiplex and qRT-PCR analyses highlighted a prevalent nuclear distribution for the variants containing exon 3 (CyKILRa), in stark contrast with the predominant cytoplasmic localization observed for the variants lacking exon 3 (CyKILRb) (Figures 3B and 3C). Indeed, qRT-PCR analyses showed that more than 80% of the CyKILRa variants clearly situated within the nucleus, while a corresponding percentage of more than 80% of the CyKILRb variants consistently resided in the cytoplasm (Figure 3C). To validate our fractionation methodology, lncRNAs with reported cellular localizations were examined. CyKILRa localization correlated with the known nuclear-localized lncRNA, NEAT1, while CyKILRb localization correlated with the known cytoplasmic-localized lncRNA, TUG1 (Figure 3D).32,33 To determine whether the AGCCC motif of CyKILR exon 3 was regulating the nuclear localization, this sequence was removed by deletion mutagenesis. Ectopic expression of WT CyKILRa in NSCLC cells showed a high nuclear CyKILRa/cytoplasmic CyKILRa ratio, showing the expected predominant nuclear localization (Figure 3E). In contrast, the ectopic expression of a CyKILRa mutant lacking the AGCCC motif (Del-Mut) demonstrated a dramatic and significant reduction in the nuclear CyKILRa/cytoplasmic CyKILRa ratio (Figure 3E). These data demonstrate that the presence or absence of exon 3 in the mature CyKILR transcript regulates the cellular localization of this lncRNA via the AGCCC motif.
Figure 3.

CyKILRa and CyKILRb demonstrate differential cellular localization regulated by an exon 3 AGCCC motif
(A) Sequence analysis of exon 3 of CyKILR reveals a nuclear localization motif, AGCCC. (B–D) NSCLC cells from the depicted cell lines were fractionated for nuclei and cytoplasm. Total RNA was extracted from these fractions, reverse transcribed (RT), and subjected to multiplex PCR (B), qPCR analysis for CYKILRa and CYKILRb (C), or qPCR for NEAT1 and TUG1 (D). The data are presented as boxplots (n = 3 replicates). (E) WT or an AGCCC deletion mutant of CyKILRa (Del-Mut) were ectopically expressed in two non-CyKILR-expressing cell lines (A549 and H833). Cells were fractionated for nuclei and cytoplasm. Total RNA was extracted from these fractions, reverse transcribed (RT), and subjected to qPCR analysis for CYKILRa. The ratio of nuclear CyKILRa/cytoplasmic CyKILRa for WT and Del-Mut are depicted. Data in (B)–(E) are presented as boxplots (n = 3 replicates) and were analyzed by an unpaired t test with Welch’s correction. −RT controls were included in all experiments and replicates with no signal observed.
CDKN2A and LKB1 regulate CyKILR exon 3 exclusion
Our data showing a strong correlation between WT, active CDKN2A, and STK11 genes with CyKILR expression suggested that the gene products of these two tumor suppressors cooperated to drive CyKILR expression. Surprisingly, the downregulation of mRNAs stemming from the CDKN2A and STK11 genes, individually or concomitantly, by small interfering RNA (siRNA) (Figure 4A) did not yield consistent or conclusive effects on the total CyKILR RNA levels in NSCLC lines, although a significant reduction in CyKILR expression was observed in non-cancerous, primary human bronchial epithelial cells (HBECs) (Figures 4A and 4B). In additional mechanistic studies, the RNA levels of the two splice variant groups of CyKILR (CyKILRa and CyKILRb) were examined individually, and whereas the individual downregulation of either CDKN2A or STK11 mRNAs by siRNA did not yield significant changes in the ratio between the two splice variant groups, the co-downregulation of the mRNA products of these two tumor suppressors led to a dramatic increase in the ratio of CyKILRb/CyKILRa variants compared with control siRNA (Figure 4C). These data demonstrate that LKB1 and CDKN2A gene products act to induce the inclusion of exon 3 into CyKILR RNA, which will enhance the expression of nuclear CyKILRa.
Figure 4.

CDKN2A and LKB1 concomitantly regulate the inclusion of exon 3 into CyKILR RNA
The depicted cell lines were transfected with scrambled control (SCR), CDKN2A, LKB1, or CDKN2A/LKB siRNAs. After 48 h, total protein and RNA were extracted. (A) Protein levels of CDKN2A (p14 and p16) and LKB1 in samples subjected to single or double gene siRNA downregulation compared with scrambled siRNA controls (SCR). (B) The level of total CyKILR RNA was determined by qRT-PCR normalized to GAPDH in samples subjected to single or double gene siRNA downregulation compared with scrambled siRNA controls. (C) The ratio of CyKILRb to CyKILRa RNA levels determined by qRT-PCR in samples subjected to single or double gene siRNA downregulation compared with scrambled siRNA controls. (D) The fold change of proliferation analysis of CyKILRb silencing on double gene downregulation by siRNA (CDKN2A and LKB1) compared with scrambled siRNA controls (SCR) after 48 h silencing (day 0). Data in (B)–(D) were analyzed by one-way ANOVA with Tukey’s multiple comparisons test and presented as boxplots (n ≥ 3 replicates). Data in (A) are representative of n = 3. −RT controls were included in all experiments and replicates with no signal observed.
The above findings implied that the induction of exon 3 inclusion into the mature CyKILR RNA transcript was biologically pertinent in relation to functional CDKN2A and STK11 genes. To investigate whether the suppression of CyKILRb expression affected biological phenotypes suppressed by the CDKN2A and STK11 gene products, proliferation was assessed when these tumor suppressors were again co-downregulated by siRNA. The downregulation of both CDKN2A and STK11-derived mRNAs induced a significant increase in cell proliferation (Figure 4D). The simultaneous downregulation of CyKILRb attenuated the increased proliferation observed after CDKN2A and LKB1 co-silencing as compared with control siRNA cells (Figure 4D). These data show that the suppression of CyKILRb expression via inclusion of exon 3 is an important mechanism by which LKB1 and CDKN2A subdue cell proliferation. To further interrogate these biological findings, all CyKILR transcripts were downregulated in multiple NSCLC cell lines with WT STK11 and CDKN2A genes, which required the use of DNA/RNA chimeric antisense oligonucleotides (ASOs) targeting intron 1 of the pre-mRNA to silence nuclear CyKILR; siRNA has been shown ineffective for these nuclear forms of RNA.34 The downregulation of CyKILR pre-mRNA induced significant increases in cellular proliferation (Figure S2), and, thus, CyKILR, in general, is a suppressor of cell growth and proliferation, while the CyKILRb splice variant possesses an opposing function.
Nuclear CyKILRa and cytoplasmic CyKILRb have opposing functions in vitro and in vivo
Our data suggested that CyKILR has biological functions linked to cancer cell phenotypes (e.g., cell proliferation), which may also be opposing depending on alternative RNA splicing and cellular localization. Thus, the role of the individual CyKILR isoform groups was examined for effects on cell proliferation, cell clonogenic survival, and cell migration. First, the functional role of nuclear-localized CyKILR variants (CyKILRa) was assessed, which again required the use of DNA/RNA chimeric ASOs to silence nuclear CyKILRa, as siRNA has been shown ineffective for these nuclear forms of RNA.34 The silencing efficacy of our ASOs exceeded 50% across all four tested cell lines (Figure 5A). The downregulation of CyKILRa resulted in significant enhancement of cell proliferation (Figure 5B), and a greater than 2-fold acceleration in cell migration (Figure 5C). Furthermore, cell clonogenic survival was significantly increased in these cell lines by downregulation of CyKILRa (Figure 5D). In addition, downregulation of CyKILRa had a significant and profound effect on subcutaneous (sc) tumor formation in mice for both the H2009 and H1792 cell lines. Specifically, the downregulation of CyKILRa with ASOs induced significant tumor growth and formation for both cell lines injected sc in the flanks of mice after 4 weeks, a time point when tumors were not appreciably formed in the control cell lines (Figure 5E). These cellular effects could not be attributed to artifactual or regulatory effects on the expression of genes located in close proximity to the CyKILR transcription locus (Figure S1), as the downregulation of CyKILRa had no effect on the expression of these genes (Figure S3). In stark contrast with CyKILRa, the downregulation of CyKILRb by siRNA (Figure 6A) resulted in a significant reduction in cell proliferation (Figure 6B). Additionally, a significant decrease in both cell migration and cell clonogenic survival was observed (Figures 6C and 6D). The downregulation of CyKILRb by siRNA in H2009 and H1792 cells also induced a significant reduction in sc tumor growth in mice when control tumors were allowed to achieve at least 200 mm3 in volume (Figure 6E). In all replicates, the downregulation of CyKILRa and CyKILRb produced profound effects on tumor growth over the experimental time course (Figure S4). These data demonstrate that nuclear CyKILRa has specific cellular functions and acts to suppress key pro-survival phenotypes observed in NSCLC cells. Furthermore, cytosolic CyKILRb has an opposing function and acts as a pro-survival, pro-tumorigenic, and pro-growth factor.
Figure 5.

Nuclear CyKILRa acts as a tumor suppressing factor
The depicted cell lines were transfected with SCR ASO or CyKILRa ASO (ASO). After 48 h, total RNA was extracted or in concomitant experiments, the cells were assessed for the indicated biological mechanisms with the 48 h after transfection acting as the 0 time point (A–D). In addition, 48 h post-transfected H2009 and H1792 cells were subsequently mixed with Matrigel in a 1:1 ratio and injected sc into the flanks of 5-week-old female NOD-SCID mice (1 × 106 cells per mouse per flank). The left flank received control-treated cells, while the right flank received knockdown cells (E). (A) RNA levels of CyKILRa in the depicted cell lines after ASO treatment normalized to the mRNA levels of GAPDH. −RT controls were included in all experiments and replicates with no signal observed. (B) Proliferation of CyKILRa ASO-treated cells compared with SCR ASO-treated cells at the 48-h time point. (C) Migration of CyKILRa ASO-treated samples compared with SCR ASO-treated cells at the 24-h time point. (D) Plate clonogenic survival of CyKILRa ASO-treated cells compared with SCR ASO-treated cells. (E) Tumor volume of ASO-Con or CyKILRa ASO treated H2009 or H1792 in mice. The data are presented as boxplots (n ≥ 3 replicates), and statistical analysis was performed using the unpaired t test with Welch’s correction.
Figure 6.

Cytoplasmic CyKILRb is a pro-survival factor
The depicted cell lines were transfected with scrambled control (SCR) or CyKILRb siRNA (siCyKILRb). After 48 h, total RNA was extracted or the cells were assessed for the indicated biological mechanisms with the 48 h after transfection acting as the 0 time point (A–D). In addition, 48-h post-transfected H2009 and H1792 cells were subsequently mixed with Matrigel in a 1:1 ratio and injected sc into the flanks of 5-week-old female NOD-SCID mice (1 × 106 cells per mouse per flank). The left flank received Scramble-treated cells, while the right flank received knockdown cells (E). (A) RNA levels of CyKILRb siRNA-treated samples compared with scrambled siRNA control (SCR) samples normalized to GAPDH mRNA. −RT controls were included in all experiments and replicates with no signal observed. (B) Proliferation of CyKILRb siRNA-treated cells compared with scrambled siRNA control cells at the 48-h time point. (C) Migration of CyKILRb siRNA-treated cells compared with scrambled siRNA control cells at the 24-h time point. (D) Plate clonogenic survival of CyKILRb siRNA-treated cells compared with scrambled siRNA control cells. (E) Tumor volume of siRNA-scramble control or CyKILRb siRNA treated H2009 or H1792. The data are presented as boxplots (n ≥ 3 replicates), and statistical analysis was performed using the unpaired t test with Welch’s correction.
CyKILRa and CyKILRb have opposing functions on cell signaling pathways
Since the modulation of CyKILR splice variants showed strong effects on tumor biology and cancer cell phenotypes, the mechanistic underpinnings were explored. In this regard, the downregulation of CyKILRa and CyKILRb followed by deep RNA-seq and pathway analysis was undertaken. The downregulation of CyKILRa by ASOs showed decreased expression of factors central in apoptosis induction (e.g., Bak1, CHOP, and PUMA), as well as significantly increased expression of factors known to drive the cell cycle, cell proliferation, and cell growth (e.g., CDK1, CDK2, and CHEK2) (Figures 7A–7C, S5, S6, and S7A–S7D). The downregulation of CyKILRb, in contrast, led to the induction of pro-survival pathways (Figure S7E) and tumor suppressing miRNAs, hsa-miR-424–3p and has-miR-503-p (Figures 7D–7F). Treatment of H2009 and H1299 cells with mimics of these two miRNAs partially but significantly suppressed key cancer cell proliferation (Figure 7G). Highlighting the complexity of CyKILR in the regulation of cell signaling and gene expression, CyKILRb also serves as a suppressor of CyKILRa expression, as the downregulation of CyKILRb led to a significant increase in CyKILRa expression (Figure S8). This is in stark contrast with CyKILRa, as ASO downregulation of this isoform did not significantly affect CyKILRb expression (Figure S8). These data show that CyKILR splice variants have complex roles in the modulation of key pathways linked to both tumor suppression and tumorigenesis.
Figure 7.

CyKILRa and CyKILRb apposingly regulate cell growth signaling pathways
H2009 or H1792 cells were treated with either SCR ASO (SCR) or CyKILRa ASO (ASO), and H1299 and H2009 cells were treated with either siRNA-Con (SCR) or CyKILRb siRNA (siCyKILRb). Total RNA from cell subjected to these treatments underwent deep RNA-seq. Following data filtering and clean read alignment, gene expression levels and differential expression gene analysis were conducted using RSEM (v1.2.28) and DESeq2. Kyoto Encyclopedia of Genes and Genomes enrichment analysis of annotated differentially expressed genes was performed using R Phyper. The CyKILRa ASO-treated groups exhibited regulation of cell apoptosis and cell-cycle pathways compared with the control-treated groups (A–C). While the CyKILRb siRNA-treated groups exhibited the regulation of miRNAs (D–G). (A) RNA levels of key apoptosis markers were down-regulated by CyKILRa ASO in all cell lines as assessed by qRT-PCR. −RT controls were included in all experiments and replicates with no signal observed. (B) Protein levels of key apoptosis markers were down-regulated by CyKILRa ASO in all cell lines as assessed by western blotting. (C) Protein levels of key cell-cycle markers were up-regulated by CyKILRa ASO in all cell lines. (D) Up-regulated miRNAs by CyKILRb knockdown in both cell lines. (E) The Venn diagram displays the upregulated miRNAs resulting from CyKILRb knockdown in both H1299 and H2009 cell lines. (F) RNA levels of miR-424–3P and miR-503-3P in H1299 and H2009 cells treated with si-CyKILRb vs. scramble control (SCR). (G) Cell proliferation assay comparing miR-424-3P and miR-503-3P mimic-treated H1299 and H2009 cells vs. scramble-treated (SCR) cells 48 h after transfection. The data in (A), (F), and (G) are presented as boxplots (n ≥ 3 replicates), and statistical analysis was performed using the unpaired t test with Welch’s correction. Data in (B) and (C) are representative of n = 3.
Discussion
In this study, we have discovered a novel lncRNA, which we termed CyKILR due to its expression being mainly confined to NSCLC lines harboring a WT and active CDKN2A gene. Additional studies showed that an oncogenotype consisting of both a WT CDKN2A and STK11 gene correlated more strongly with the expression of CyKILR, and downregulation of the mRNA of either gene led to significant loss of CyKILR expression in primary HBECs. The loss of CyKILR expression observed in NSCLC cell lines lacking either a WT STK11 or CDKN2A gene correlated and corroborated this finding, which strongly suggested that CyKILR has a possible tumor suppressor role linked to the cell signaling pathways activated by the LKB1 and CDKN2A gene products. In support of this conclusion, downregulation of the CyKILRa isoforms (exon 3 included) significantly enhanced cell clonogenic survival, migration, and proliferation, as well as tumor growth. In contrast, the downregulation of CDKN2A and STK11 mRNAs in NSCLC cell lines with both WT STK11 and CDKN2A genes surprisingly demonstrated minimal or no significant effect on total CyKILR expression. Thus, depending on the cellular context and oncogenic landscape, the maintenance of CyKILR expression becomes independent of these genes. Shedding light on a plausible adaptive mechanism was the additional characterization of CyKILR, which identified multiple RNA splice variants, including an exon 3 excluded isoform designated CyKILRb. In contrast with CyKILRa, this splice variant class was pro-survival, and notably, the ratio of CyKILRb/a, was very low in non-cancerous, HBECs, when compared with the NSCLC cell lines that expressed CyKILR. We posit that shifting from the CyKILRa isoform to the CyKILRb isoform through the modulation of exon 3 inclusion or exclusion by alternative RNA splicing is required for transformed NSCLC cells to maintain cell homeostasis in the presence of these key tumor suppressors. Indeed, the downregulation of CyKILRb had dramatic effects in reducing the clonogenic survival and tumor growth of NSCLC cells with WT STK11 and CDKN2A genes. Specific downregulation of CyKILRb also blocked any enhancement of cellular proliferation observed when both of these genes were inactivated. Therefore, the CyKILRb splice variants of this lncRNA are plausible proto-oncogenes, or, more specifically, these data suggest that the exclusion of exon 3 from CyKILR RNA removes a key nuclear lncRNA required for LKB1 and CDKN2A gene products to act as efficient tumor suppressors.
The observation that the STK11 and CDKN2A gene products co-regulated the alternative RNA splicing of CyKILR was an unexpected finding. Indeed, a tremendous 30-fold increase in the CyKILRb/a RNA ratio in HBECs was observed when CDKN2A and STK11 mRNAs are downregulated. This is a 10-fold higher response than the NSCLC cells examined in this study. These data suggest that transformed cells with an active and WT CDKN2A and STK11 gene also have active oncogenic pathways with opposing functions on downstream RNA splicing, which act to suppress exon 3 inclusion. How the products of these tumor suppressor genes regulate the inclusion or exclusion of CyKILR exon 3 is unknown, opening avenues of unanswered questions in relation to their tumor-suppressing functions and the regulation of alternative RNA splicing. Dysregulated alternative RNA splicing is well established in cancer with RNA trans-factors acting as proto-oncogenes,35 but there is essentially nothing reported in the literature as to alternative RNA splicing regulation downstream of the signaling pathways regulated by CDKN2A and STK11 gene products. There are reported links to the CDKN2A gene product, p16, in regulating the association of the RNA-binding protein, AUF1 to CDKN1A mRNA with regard to RNA stability.36 The expression of hnRNP B1, an RNA splicing factor, was reported as inversely regulated in relation to p16 expression in lung cancer tissue.37 CDKN2A gene products, as well as LKB1, will repress activation of the AKT pathway, and thus, when CDKN2A and LKB1 are downregulated, AKT activation will occur, which is known to have distinct effects on RNA splicing.38,39,40,41 This signaling paradigm is logical as loss of both pathways may cooperate to enhance the expression of specific AKT isoforms, modulate specific post-translational modifications related to AKT activation, and alter cellular topology via PIP3 production in specific organelles such as the nuclear membrane or nucleus itself. As to the latter, it has been previously surmised and shown that AKT can directly phosphorylate RNA trans-factors in the nucleus to modulate specific RNA splicing events.38,39,40 Thus, the coordinated modulation or suppression of nuclear AKT signaling may be one plausible route for the loss of these tumor suppressors to cooperate in regulating specific alternative splicing events. Additionally, our study has not examined the alternative RNA splicing in the NSCLC cells lacking appreciable CyKILR expression. Thus, the downregulation and suppression of total CyKILR expression may occur via the induction of CyKILR RNA splice variants removed by nonsense mediate RNA decay,42 which is an efficient mechanism to remove aberrant mRNA transcripts that may arise from alternative RNA splicing. Future studies are required to determine how loss of the STK11 and CDKN2A tumor suppressors leads to the suppression of CyKILR expression in NSCLC.
The characterization on CyKILR RNA splice variants in this study led to the observation of distinct subcellular distributions for CyKILRa and CyKILRb, which corresponded with their divergent functions. Indeed, lncRNAs serve as operational units or scaffolds, and their subcellular localization plays a pivotal role in governing their functions.43 For example, a substantial number of lncRNAs primarily reside within the nucleus, where they perform diverse regulatory functions through a multitude of combinatorial mechanisms optimized for nuclear enrichment.44,45,46,47 In our study, we found that CyKILRa has a distinct nuclear localization in contrast to the cytoplasmic predominant localization of CyKILRb. Through in silico analysis, a potential nuclear retention motif, AGCCC, was identified with −8 (T or A) and −3 (G or C) relative to the first nucleotide of the pentamer in exon 3 of CyKILR.31 Thus, CyKILRa predominance in localizing to the nucleus, in contrast with CyKILRb, is logical as this sequence only resides in exon 3, and our empirical experimental results validated the requirement of this motif in regulating CyKILRa nuclear localization. As to how CyKILRa functions as a tumor suppressor, its nuclear localization may be key to exerting regulatory influence over cellular processes, possibly through interactions with epigenetic factors involved in histone modification, complex scaffolding, or recruitment, as previously reported for other lncRNAs.48 Additionally, nuclear lncRNAs often assume additional roles, including the modulation of pre-mRNA processing and the involvement in alternative splicing events.49 Uncovering the specific regulatory mechanisms governing CyKILRa within the nucleus holds great promise for precision-targeted tumor therapy; our transcriptomics analyses show that AKT levels were significantly increased in two separate NSCLC cells lines when CyKILRa was downregulated by ASO, and this pathway is potent in both survival signaling, oncogenesis, and modulating alternative RNA splicing. Indeed, systems-level analyses of the transcriptome showed that the apoptosis genes were downregulated and other pathways linked to enhancing the cell cycle were increased, which our laboratory confirmed by qRT-PCR and western blotting. These data support the implication that CyKILRa acts as a nuclear scaffold for regulating gene expression, specifically the suppression of cell-cycle pathways while simultaneously maintaining the levels of factors allowing for apoptosis to occur upon oncogenic stress.
In contrast with CyKILRa, CyKILRb functions as a pro-tumorigenic factor; like CyKILRa, it is currently unknown as to how CyKILRb functions in this role. Emerging research also suggests that the population of cytoplasmic lncRNAs may be more abundant than previously recognized.50,51,52 Cytoplasmic lncRNAs have been implicated as a miRNA sponge, sequestering miRNAs or regulating protein stability.53 When lncRNAs function as miRNA sponges, these non-coding RNAs act as competing endogenous RNAs,54 which are reported to play key roles in cancer disease states.55 In this regard, systems-level analyses of CyKILRb downregulation in two NSCLC cell lines with WT STK11 and CDKN2A genes found a consistent increase in two reported, tumor suppressing miRNAs, hsa-miR-424-3p and has-miR-503-p.56,57 Interestingly, there is no complementary sequence for these miRNAs in CyKILRb, suggesting that the regulation of these miRNAs by CyKILRb is not through a sponge mechanism, but possibly via the modulation of cell signaling pathways affecting the expression of these miRNAs. Indeed, CyKILRb may act as a sponge for other miRNAs that then regulate cell signaling pathways modulating the expression of these tumor suppressing miRNAs analogous to other cytoplasmic lncRNAs like TUG1.33 Regardless, examining this novel link between CyKILRb and the expression of hsa-miR-424-3p and has-miR-503-p is a future mechanistic avenue to explore.
In conclusion, our study characterizes a novel lncRNA with functional roles in NSCLC with a specific oncogenotype. The association with CDKN2A and LKB1 status, the differential subcellular localization, and the impact on cellular behavior and in vivo tumorigenicity suggest that CyKILR splice variants are important players in the pathogenesis of NSCLC. Further research is needed to elucidate the precise molecular mechanisms underlying the functions of the individual CyKILR variants and their expression. Indeed, this intricate interplay between the two subcellular groups of CyKILR underscores the critical importance of their relative ratios offering a precise means to finely tune cellular progression. Additionally, exploration as to its potential as a therapeutic target in NSCLC and potentially other cancer types is warranted as RNA-based therapies are now plausible.58 Overall, this study provides a foundation for future investigations into the diagnostic and therapeutic implications of targeting specific lncRNA splicing variants in NSCLCs, and the future exploration of lncRNAs like CyKILR offers a promising avenue for precision medicine approaches in cancer therapy.
Materials and methods
Cell lines and materials
NCI-358, NCI-H522, NCI-H2009, NCI-H810, NCI-H460, A549, NCI-H838, NCI-H1734, NCI-H1648, NCI-H2030, NCI-H1299, NCI-H1792, and primary HBECs were purchased from ATCC. RNA Subcellular Isolation Kit was from Active Motif (25501). The SuperScript IV VILO Master Mix with ezDNase Enzyme (11766050), TaqMan Universal PCR Master Mix (4324018), Opti-MEM I Reduced Serum Medium (31985062), Lipofectamine RNAiMAX Transfection Reagent (13778150), and Pierce Fast Western Blot Kit (35050) were from Thermo Fisher Scientific. The Cell Titer-Glo 2.0 Assay Kit was purchased from Promega (G9242). The mycoplasma contamination detection kit was purchased from InvivoGen (rep-mys-50).
Cell culture
All cells were cultured following the procedures described previously and were used within 6 months or 20 passages, whichever came first, from the time of receipt and thawing.59 Throughout the entire study, cell lines were subjected to mycoplasma testing every 2 months using a mycoplasma contamination detection kit. Parental cell lines were authenticated by short tandem repeat profiling.
Transcriptomics and splicomic analyses
Short-Read
RNA-seq was conducted using 150 bp paired-end reads, aiming for a depth of approximately 90 million reads for each RNA sample. Sequencing reads were aligned to the GRCh38.p14 reference genome using STAR software (v2.7.10b), and post-alignment, BAM files were filtered and indexed with Samtools (v1.16.1) to remove low-quality reads.60,61 For differential gene expression analysis, the R package DESeq2 was used on previously described RNA-seq data processed with Salmon 1.10.0.62,63 Additional R packages used included tximport for importing data, biomaRt for genomic annotation, tidyverse for statistical analyses, RColorBrewer for color palettes, and org.Hs.eg.db for gene annotations.64,65,66 Isoform detection and quantification were performed using the previously described Salmon processed RNA-seq data. Transcript IDs for genes of interest were identified from the GENCODE v45 comprehensive annotation.gtf file. These IDs facilitated parsing of Salmon.sf files to obtain transcripts per million (TPM) values for each transcript, which informed our transcript abundance calculations. Finally, TPM values for selected transcripts were visualized using the tidyverse package in R version 4.3.3.64 Differential alternative splicing analysis was performed on these filtered BAM files using rMATS turbo (v4.1.2).67 Subsequently, R programming language was used to filter and generate a list of splicing targets from the data produced by rMATS.
RNA extraction and qRT-PCR
Total RNA and subcellular RNA were isolated from cultured cells using the RNA Subcellular Isolation Kit from Active Motif (25501) following the manufacturer’s instructions with DNase treatment to remove genomic DNA contamination. RNA quantification was performed using a NanoDrop NP-1000 spectrometer, and RNA (2 μg) was used for the first-strand cDNA synthesis using SuperScript IV VILO Master Mix with ezDNase Enzyme. Subsequently, qRT-PCR was conducted as done on a Quant Studio 5 instrument (Applied Biosystems).38,39,40,48,49,53,68 Superscript minus (−RT) controls were included in all experiments to confirm a lack of target amplification caused by genomic DNA contamination. The housekeeping gene GAPDH was used for normalization using the ΔΔCt method. The specific primers and probes used in the study are listed in Table S2. A customized quantitative primer and probe strategy for the detection of CyKILRa and CyKILRb variants was also used. For CyKILRa exon 3 inclusion variants, we designed primers and probes specifically targeting exon 3, as all CyKILRa variants in this group contain exon 3. In contrast, for CyKILRb exon 3 exclusion variants, which lack exons 2–4, the forward primer aligns with exon 1, the reverse primer is complementary to exon 5, and a probe spans the exon 1 to exon 5 junction.
Multiplex RT-PCR
A reaction mixture was prepared by combining cDNA (25 ng) with both sets of primers at a final concentration of 0.1 μM. To achieve a total volume of 50 μL, the cDNA-primer mixture was mixed with 2X master mix and water. The PCR was performed using the hot-start method, with an annealing temperature of 58°C for 26 cycles. The specific primers used in the study are listed in Table S2.
siRNA and ASOs transfection
Cells were plated in a full medium without antibiotics the day before transfection, ensuring they reached 50% confluency at the time of transfection. To dilute siRNA or ASOs, Opti-MEM I Reduced Serum Medium was used. For siRNA transfection, Lipofectamine RNAiMAX was used as done previously,54,55,69,70,71 while Lipofectamine 3000 was used for ASOs transfection. The final concentration of both siRNA and ASOs in the cell culture medium was 20 nM. The sequences of siRNAs and ASOs are listed in Table S2. ASO was customized 2′-O-methyl-locked DNA chimera with phosphorothioate bonds backbone. siRNA was customized 19–22 basepair duplex RNA with 3′ UU overhangs.
Plasmid of WT or deletion mutant of CyKILRa transfection
The full-length CyKILRa (ENST00000586345.6) and the AGCCC deletion mutant were synthesized and inserted into the plasmid pcDNA3.1 between the Not I and Xho I sites. A total of A549 or H838 cells (1 × 106) were seeded in a 10-cm plate and allowed to incubate overnight before transfection. Plasmids (5 μg) were transfected using Lipofectamine 3000 (12.5 μL) according to the manufacturer’s instructions. Subcellular RNA extraction was performed 48 h after transfection.
Proliferation analysis
To investigate cell proliferation, the Cell Titer-Glo Luminescent Cell Viability kit was used following the manufacturer’s instructions. Cells were initially seeded in 96-well plates at a density of 2,000 cells per well in RPMI1640 supplemented with 10% fetal bovine serum (FBS). After treatment with ASO or siRNA for 48 h, the plates were carefully decanted and refilled with 50 μL of Cell Titer-Glo reagent and 50 μL of PBS per well. The plates were then incubated for 10 min at room temperature in the dark. Subsequently, the luminescence readings were taken using the Gene5 Multi-Mode Microplate Reader (Molecular Devices) with a 250-ms integration.
Cell clonogenic survival assays
After ASO or siRNA treatment, cells were harvested and re-suspended in complete growth media. Subsequently, the cells (2–4 × 102) were seeded onto six-well plates and allowed to grow until visible colonies formed as done elsewhere,38,39,40,48,68,72,73,74 which typically occurred within 7–21 days depending on the cell line. Once the colonies became visible, they were fixed with cold methanol, stained with 0.25% crystal violet in 25% methanol, washed, and then left to air dry. Any colony with greater than 40 cells/colony were counted.
Cell migration
The scratch migration assay was conducted following the procedure previously described.75,76,77,78 Briefly, ASOs- or siRNA-treated cells were cultured in six-well plates with RPMI 1640 medium containing 10% FBS until they reached 90% confluency. An approximately 1-mm cell-free gap was created using a sterile pipette tip. After washing away dislodged cells with PBS, the plates were refilled with fresh medium and incubated for 24 h before capturing images. Cell migration was quantified using ImageJ software, analyzing the change in the cell-free area at 0 and 24 h.
Western blotting
Cells were lysed in RIPA buffer containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78440). For protein concentration determination, the Pierce BCA Protein Assay kit (Thermo Fisher Scientific, 23227) was used.49,79,80,81 Whole cell lysates were mixed with Laemmli loading buffer, boiled, separated by 4%–20% SDS-PAGE, and transferred to a polyvinylidene fluoride membrane as done previously.79,81,82,83,84,85,86 Subsequently, immunoblot analyses were performed using specific antibodies (see source companies, catalog numbers, and dilution ratios listed in Table S3). The specific protein bands were visualized by using a Western Blotting Kit (Pierce, 35050).
Mouse tumor xenograft models
Five-week-old female NOD-SCID mice were injected sc with H1792 or H2009 cells (1 × 106) mixed in Matrigel solution (50%) into the hind flanks as previously done by us.39,40,68,72,73 Tumor size was measured two to three times per week using a vernier caliper. When the tumor size reached 1,000 mm3, the mice were euthanized, and the tumors were excised for weight measurement and histological analysis. Throughout the study, all animals were maintained according to the University of Virginia (protocol#4372-11-21) and Richmond VAMC (IRBnet ID#1654460-4) approved IACUC protocols.
Transcriptomics analysis
The sequencing depth was 90 million reads per RNA sample from the 2 included cell lines with 3 replicates per cell line. The sequencing data was filtered with SOAPnuke (v1.5.6) (removing reads containing sequencing adapter and low-quality base ratio), then the clean reads were stored in FASTQ format.87 Differential expression gene analysis was performed using the DESeq2 with a Q value of 0.05 or less.63 Kyoto Encyclopedia of Genes and Genomes/Gene Ontology enrichment analysis of annotated different expression gene was performed by R Phyper (v120.0) based on a hypergeometric test. The significant levels of terms and pathways were corrected by Q value with a rigorous threshold (Q value of ≤0.05).
Statistical analysis
Graphing and statistics were performed using Prism GraphPad (Prism Software). The following models were used for statistical analysis as described, unpaired t test with Welch’s correction were used when only two experimental groups were being compared. Repeated measures ANOVA were used when analyzing comparison groups that contained a dependent variable that was measured several times over the course of the experiment. One-way ANOVA with Tukey’s multiple comparisons test was used when comparing three or more independent groups. Two-way ANOVA with Šídák’s multiple comparisons were used when an examination of how the combination of two independent variables affected a dependent variable. All data reported as boxplot or mean (SD) where applicable; a p value of less than 0.05 was considered statistically significant. All bioinformatic analyses were conducted using the R program.
Data and code availability
All RNA-seq data are in the process of submission to NCBI Sequence Read Archive (SRA) database for public access (Submission number: sub14341995). The GEO accession number is pending a database instrument update.
Acknowledgments
The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the United States Government. This work was mainly supported by the Veteran’s Administration (VA Merit Review, BX001792 [to C.E.C.], VA Merit Review award, BX 006063 [to C.E.C.], and a Senior Research Career Scientist Award, IK6BX004603 [to C.E.C.]). This work was peripherally supported by way of research cores, methodology and technology development, and software development by the National Institutes of Health by way of P01 CA171983 (Project 1 to C.E.C.), NIH/NCI Cancer Center Support Grant P30 CA044579, and the R01s AI139072 (to C.E.C.), and DK126444 (to C.E.C.). This work was also supported by funds from the Department of Medicine in the University of Virginia-School of Medicine in regard to software licensing and equipment purchases.
Author contributions
X.X.: Designing research studies, conducting experiments, acquiring data, analyzing data, and writing the manuscript. H.P.M.: Developing and analyzing short and long read deep RNA-seq data, manuscript method writing, and figure composition. A.L.L.: Conducting experiments, acquiring data, and analyzing data. C.E.C.: Overall supervision, designing research studies, analyzing data, providing reagents and funding, editing manuscript, and writing the manuscript.
Declaration of interests
The authors declare that they have no conflicts of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102412.
Supplemental information
References
- 1.Chen Z., Fillmore C.M., Hammerman P.S., Kim C.F., Wong K.K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao R., Choi B.Y., Lee M.H., Bode A.M., Dong Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine. 2016;8:30–39. doi: 10.1016/j.ebiom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger J.H., Bardeesy N. Modeling INK4/ARF tumor suppression in the mouse. Curr. Mol. Med. 2007;7:63–75. doi: 10.2174/156652407779940477. [DOI] [PubMed] [Google Scholar]
- 4.Smith-Sorensen B., Hovig E. CDKN2A (p16INK4A) somatic and germline mutations. Hum. Mutat. 1996;7:294–303. doi: 10.1002/(SICI)1098-1004(1996)7:4<294::AID-HUMU2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 5.Fitzmaurice C., Abate D., Abbasi N., Abbastabar H., Abd-Allah F., Abdel-Rahman O., Abdelalim A., Abdoli A., Abdollahpour I., Abdulle A.S.M., et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019;5:1749–1768. doi: 10.1001/jamaoncol.2019.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan M.R., Avino M., Wellinger R.J., Laurent B. Distinct regulatory functions and biological roles of lncRNA splice variants. Mol. Ther. Nucleic Acids. 2023;32:127–143. doi: 10.1016/j.omtn.2023.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olivero C.E., Martinez-Terroba E., Zimmer J., Liao C., Tesfaye E., Hooshdaran N., Schofield J.A., Bendor J., Fang D., Simon M.D., et al. p53 Activates the Long Noncoding RNA Pvt1b to Inhibit Myc and Suppress Tumorigenesis. Mol Cell. 2020;77:761–774. doi: 10.1016/j.molcel.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan J.H., Liu X.N., Wang T.T., Pan W., Tao Q.F., Zhou W.P., Wang F., Sun S.H. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat. Cell Biol. 2017;19:820–832. doi: 10.1038/ncb3538. [DOI] [PubMed] [Google Scholar]
- 11.Jiang M.C., Ni J.J., Cui W.Y., Wang B.Y., Zhuo W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019;9:1354–1366. [PMC free article] [PubMed] [Google Scholar]
- 12.Novikova I.V., Hennelly S.P., Sanbonmatsu K.Y. Tackling structures of long noncoding RNAs. Int. J. Mol. Sci. 2013;14:23672–23684. doi: 10.3390/ijms141223672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitz S.U., Grote P., Herrmann B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016;73:2491–2509. doi: 10.1007/s00018-016-2174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kazemzadeh M., Safaralizadeh R., Orang A.V. LncRNAs: emerging players in gene regulation and disease pathogenesis. J. Genet. 2015;94:771–784. doi: 10.1007/s12041-015-0561-6. [DOI] [PubMed] [Google Scholar]
- 15.Rinn J.L., Kertesz M., Wang J.K., Squazzo S.L., Xu X., Brugmann S.A., Goodnough L.H., Helms J.A., Farnham P.J., Segal E., Chang H.Y. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mondal T., Subhash S., Vaid R., Enroth S., Uday S., Reinius B., Mitra S., Mohammed A., James A.R., Hoberg E., et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015;6:7743. doi: 10.1038/ncomms8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibb E.A., Brown C.J., Lam W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang F., Li J., Ge Q., Zhang Y., Zhang M., Zhou J., Wang H., Du J., Gao S., Liang C., Meng J. Non-coding RNAs: Emerging roles in the characterization of immune microenvironment and immunotherapy of prostate cancer. Biochem. Pharmacol. 2023;214 doi: 10.1016/j.bcp.2023.115669. [DOI] [PubMed] [Google Scholar]
- 19.Ge W.J., Huang H., Wang T., Zeng W.H., Guo M., Ren C.R., Fan T.Y., Liu F., Zeng X. Long non-coding RNAs in hepatocellular carcinoma. Pathol. Res. Pract. 2023;248 doi: 10.1016/j.prp.2023.154604. [DOI] [PubMed] [Google Scholar]
- 20.Mohammadpour S., Naderi Noukabadi F., Torshizi Esfahani A., Kazemi F., Esmaeili S., Zafarjafarzadeh N., Sarpash S., Nazemalhosseini-Mojarad E. Non-coding RNAs in Precursor Lesions of Colorectal Cancer: Their Role in Cancer Initiation and Formation. Curr. Mol. Med. 2023;24:565–575. doi: 10.2174/1566524023666230523155719. [DOI] [PubMed] [Google Scholar]
- 21.Yin Q., Ma H., Bamunuarachchi G., Zheng X., Ma Y. Long Non-Coding RNAs, Cell Cycle, and Human Breast Cancer. Hum. Gene Ther. 2023;34:481–494. doi: 10.1089/hum.2023.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baba S.K., Baba S.K., Mir R., Elfaki I., Algehainy N., Ullah M.F., Barnawi J., Altemani F.H., Alanazi M., Mustafa S.K., et al. Long non-coding RNAs modulate tumor microenvironment to promote metastasis: novel avenue for therapeutic intervention. Front. Cell Dev. Biol. 2023;11 doi: 10.3389/fcell.2023.1164301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chao C., Tang R., Zhao J., Di D., Qian Y., Wang B. Oncogenic roles and related mechanisms of the long non-coding RNA MINCR in human cancers. Front. Cell Dev. Biol. 2023;11 doi: 10.3389/fcell.2023.1087337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guzel E., Okyay T.M., Yalcinkaya B., Karacaoglu S., Gocmen M., Akcakuyu M.H. Tumor suppressor and oncogenic role of long non-coding RNAs in cancer. North. Clin. Istanb. 2020;7:81–86. doi: 10.14744/nci.2019.46873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan M.R., Wellinger R.J., Laurent B. Exploring the Alternative Splicing of Long Noncoding RNAs. Trends Genet. 2021;37:695–698. doi: 10.1016/j.tig.2021.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Geisler S., Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013;14:699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ulitsky I., Bartel D.P. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batista P.J., Chang H.Y. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cong S., Di X., Li R., Cao Y., Jin X., Tian C., Zhao M., Wang K. RBM10 regulates alternative splicing of lncRNA Neat1 to inhibit the invasion and metastasis of NSCLC. Cancer Cell Int. 2022;22:338. doi: 10.1186/s12935-022-02758-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saghaeian Jazi M., Samaei N.M., Ghanei M., Shadmehr M.B., Mowla S.J. Overexpression of the non-coding SOX2OT variants 4 and 7 in lung tumors suggests an oncogenic role in lung cancer. Tumour Biol. 2016;37:10329–10338. doi: 10.1007/s13277-016-4901-9. [DOI] [PubMed] [Google Scholar]
- 31.Zhang B., Gunawardane L., Niazi F., Jahanbani F., Chen X., Valadkhan S. A novel RNA motif mediates the strict nuclear localization of a long noncoding RNA. Mol. Cell Biol. 2014;34:2318–2329. doi: 10.1128/MCB.01673-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hutchinson J.N., Ensminger A.W., Clemson C.M., Lynch C.R., Lawrence J.B., Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007;8:39. doi: 10.1186/1471-2164-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du Z., Sun T., Hacisuleyman E., Fei T., Wang X., Brown M., Rinn J.L., Lee M.G.S., Chen Y., Kantoff P.W., Liu X.S. Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nat. Commun. 2016;7 doi: 10.1038/ncomms10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng Y., Cullen B.R. RNA interference in human cells is restricted to the cytoplasm. RNA. 2002;8:855–860. doi: 10.1017/s1355838202020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faustino N.A., Cooper T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 36.Al-Khalaf H.H., Aboussekhra A. p16(INK4A) positively regulates p21(WAF1) expression by suppressing AUF1-dependent mRNA decay. PLoS One. 2013;8 doi: 10.1371/journal.pone.0070133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tominaga M., Sueoka N., Irie K., Iwanaga K., Tokunaga O., Hayashi S.i., Nakachi K., Sueoka E. Detection and discrimination of preneoplastic and early stages of lung adenocarcinoma using hnRNP B1 combined with the cell cycle-related markers p16, cyclin D1, and Ki-67. Lung Cancer. 2003;40:45–53. doi: 10.1016/s0169-5002(02)00529-9. [DOI] [PubMed] [Google Scholar]
- 38.Vu N.T., Park M.A., Shultz J.C., Goehe R.W., Hoeferlin L.A., Shultz M.D., Smith S.A., Lynch K.W., Chalfant C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 2013;288:8575–8584. doi: 10.1074/jbc.M112.443333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shultz J.C., Goehe R.W., Wijesinghe D.S., Murudkar C., Hawkins A.J., Shay J.W., Minna J.D., Chalfant C.E. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 2010;70:9185–9196. doi: 10.1158/0008-5472.CAN-10-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goehe R.W., Shultz J.C., Murudkar C., Usanovic S., Lamour N.F., Massey D.H., Zhang L., Camidge D.R., Shay J.W., Minna J.D., Chalfant C.E. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Invest. 2010;120:3923–3939. doi: 10.1172/JCI43552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shultz J.C., Chalfant C.E. Caspase 9b: a new target for therapy in non-small-cell lung cancer. Expert Rev. Anticancer Ther. 2011;11:499–502. doi: 10.1586/era.11.23. [DOI] [PubMed] [Google Scholar]
- 42.Lykke-Andersen S., Jensen T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- 43.Bridges M.C., Daulagala A.C., Kourtidis A. LNCcation: lncRNA localization and function. J. Cell Biol. 2021;220 doi: 10.1083/jcb.202009045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo C.J., Xu G., Chen L.L. Mechanisms of Long Noncoding RNA Nuclear Retention. Trends Biochem. Sci. 2020;45:947–960. doi: 10.1016/j.tibs.2020.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Ariel F., Lucero L., Christ A., Mammarella M.F., Jegu T., Veluchamy A., Mariappan K., Latrasse D., Blein T., Liu C., et al. R-Loop Mediated trans Action of the APOLO Long Noncoding RNA. Mol. Cell. 2020;77:1055–1065.e4. doi: 10.1016/j.molcel.2019.12.015. [DOI] [PubMed] [Google Scholar]
- 46.Mishra K., Kanduri C. Understanding Long Noncoding RNA and Chromatin Interactions: What We Know So Far. Noncoding. RNA. 2019;5 doi: 10.3390/ncrna5040054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato M., Carninci P. Genome-Wide Technologies to Study RNA-Chromatin Interactions. Noncoding. RNA. 2020;6 doi: 10.3390/ncrna6020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shultz J.C., Goehe R.W., Murudkar C.S., Wijesinghe D.S., Mayton E.K., Massiello A., Hawkins A.J., Mukerjee P., Pinkerman R.L., Park M.A., Chalfant C.E. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol. Cancer Res. 2011;9:889–900. doi: 10.1158/1541-7786.MCR-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chalfant C.E., Watson J.E., Bisnauth L.D., Kang J.B., Patel N., Obeid L.M., Eichler D.C., Cooper D.R. Insulin regulates protein kinase CbetaII expression through enhanced exon inclusion in L6 skeletal muscle cells. A novel mechanism of insulin- and insulin-like growth factor-i-induced 5' splice site selection. J. Biol. Chem. 1998;273:910–916. doi: 10.1074/jbc.273.2.910. [DOI] [PubMed] [Google Scholar]
- 50.Benoit Bouvrette L.P., Cody N.A.L., Bergalet J., Lefebvre F.A., Diot C., Wang X., Blanchette M., Lécuyer E. CeFra-seq reveals broad asymmetric mRNA and noncoding RNA distribution profiles in Drosophila and human cells. RNA. 2018;24:98–113. doi: 10.1261/rna.063172.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carlevaro-Fita J., Rahim A., Guigó R., Vardy L.A., Johnson R. Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA. 2016;22:867–882. doi: 10.1261/rna.053561.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Heesch S., van Iterson M., Jacobi J., Boymans S., Essers P.B., de Bruijn E., Hao W., MacInnes A.W., Cuppen E., Simonis M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014;15 doi: 10.1186/gb-2014-15-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mietla J.A., Wijesinghe D.S., Hoeferlin L.A., Shultz M.D., Natarajan R., Fowler A.A., Chalfant C.E. 3rd, Chalfant CE: Characterization of eicosanoid synthesis in a genetic ablation model of ceramide kinase. J. Lipid Res. 2013;54:1834–1847. doi: 10.1194/jlr.M035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettus B.J., Bielawska A., Subramanian P., Wijesinghe D.S., Maceyka M., Leslie C.C., Evans J.H., Freiberg J., Roddy P., Hannun Y.A., Chalfant C.E. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 2004;279:11320–11326. doi: 10.1074/jbc.M309262200. [DOI] [PubMed] [Google Scholar]
- 55.Lamour N.F., Stahelin R.V., Wijesinghe D.S., Maceyka M., Wang E., Allegood J.C., Merrill A.H., Jr., Cho W., Chalfant C.E. Ceramide kinase uses ceramide provided by ceramide transport protein: localization to organelles of eicosanoid synthesis. J. Lipid Res. 2007;48:1293–1304. doi: 10.1194/jlr.M700083-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Zhang M., Zeng J., Zhao Z., Liu Z. Loss of MiR-424-3p, not miR-424-5p, confers chemoresistance through targeting YAP1 in non-small cell lung cancer. Mol. Carcinog. 2017;56:821–832. doi: 10.1002/mc.22536. [DOI] [PubMed] [Google Scholar]
- 57.Sun Y., Li L., Xing S., Pan Y., Shi Y., Zhang L., Shen Q. miR-503-3p induces apoptosis of lung cancer cells by regulating p21 and CDK4 expression. Cancer Biomark. 2017;20:597–608. doi: 10.3233/CBM-170585. [DOI] [PubMed] [Google Scholar]
- 58.Zhu Y., Zhu L., Wang X., Jin H. RNA-based therapeutics: an overview and prospectus. Cell Death Dis. 2022;13:644. doi: 10.1038/s41419-022-05075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chalfant C.E., Rathman K., Pinkerman R.L., Wood R.E., Obeid L.M., Ogretmen B., Hannun Y.A. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J. Biol. Chem. 2002;277:12587–12595. doi: 10.1074/jbc.M112010200. [DOI] [PubMed] [Google Scholar]
- 60.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wickham H., Advanced R. 2019. Second Edition. CRC Press/Taylor & Francis Group; p. 1. [Google Scholar]
- 65.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Durinck S., Spellman P.T., Birney E., Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen S., Park J.W., Huang J., Dittmar K.A., Lu Z.X., Zhou Q., Carstens R.P., Xing Y. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vu N.T., Park M.A., Shultz M.D., Bulut G.B., Ladd A.C., Chalfant C.E. Caspase-9b Interacts Directly with cIAP1 to Drive Agonist-Independent Activation of NF-kappaB and Lung Tumorigenesis. Cancer Res. 2016;76:2977–2989. doi: 10.1158/0008-5472.CAN-15-2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simanshu D.K., Kamlekar R.K., Wijesinghe D.S., Zou X., Zhai X., Mishra S.K., Molotkovsky J.G., Malinina L., Hinchcliffe E.H., Chalfant C.E., et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature. 2013;500:463–467. doi: 10.1038/nature12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lamour N.F., Subramanian P., Wijesinghe D.S., Stahelin R.V., Bonventre J.V., Chalfant C.E. Ceramide 1-phosphate is required for the translocation of group IVA cytosolic phospholipase A2 and prostaglandin synthesis. J. Biol. Chem. 2009;284:26897–26907. doi: 10.1074/jbc.M109.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mishra S.K., Gao Y.G., Deng Y., Chalfant C.E., Hinchcliffe E.H., Brown R.E. CPTP: A sphingolipid transfer protein that regulates autophagy and inflammasome activation. Autophagy. 2018;14:862–879. doi: 10.1080/15548627.2017.1393129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vu N.T., Kim M., Stephenson D.J., MacKnight H.P., Chalfant C.E. Ceramide Kinase Inhibition Drives Ferroptosis and Sensitivity to Cisplatin in Mutant KRAS Lung Cancer by Dysregulating VDAC-Mediated Mitochondria Function. Mol. Cancer Res. 2022;20:1429–1442. doi: 10.1158/1541-7786.MCR-22-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim M., Vu N.T., Wang X., Bulut G.B., Wang M.H., Uram-Tuculescu C., Pillappa R., Kim S., Chalfant C.E. Caspase 9b Drives Cellular Transformation, Lung Inflammation, and Lung Tumorigenesis. Mol. Cancer Res. 2022;20:1284–1294. doi: 10.1158/1541-7786.MCR-21-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Massiello A., Chalfant C.E. SRp30a (ASF/SF2) regulates the alternative splicing of caspase-9 pre-mRNA and is required for ceramide-responsiveness. J. Lipid Res. 2006;47:892–897. doi: 10.1194/jlr.C600003-JLR200. [DOI] [PubMed] [Google Scholar]
- 75.Xie X., Piao L., Cavey G.S., Old M., Teknos T.N., Mapp A.K., Pan Q. Phosphorylation of Nanog is essential to regulate Bmi1 and promote tumorigenesis. Oncogene. 2014;33:2040–2052. doi: 10.1038/onc.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maus K.D., Stephenson D.J., Ali A.N., MacKnight H.P., Huang H.J., Serrats J., Kim M., Diegelmann R.F., Chalfant C.E. Ceramide kinase regulates acute wound healing by suppressing 5-oxo-ETE biosynthesis and signaling via its receptor OXER1. J. Lipid Res. 2022;63 doi: 10.1016/j.jlr.2022.100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maus K.D., Stephenson D.J., Macknight H.P., Vu N.T., Hoeferlin L.A., Kim M., Diegelmann R.F., Xie X., Chalfant C.E. Skewing cPLA(2)alpha activity toward oxoeicosanoid production promotes neutrophil N2 polarization, wound healing, and the response to sepsis. Sci. Signal. 2023;16 doi: 10.1126/scisignal.add6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.MacKnight H.P., Stephenson D.J., Hoeferlin L.A., Benusa S.D., DeLigio J.T., Maus K.D., Ali A.N., Wayne J.S., Park M.A., Hinchcliffe E.H., et al. The interaction of ceramide 1-phosphate with group IVA cytosolic phospholipase A(2) coordinates acute wound healing and repair. Sci. Signal. 2019;12 doi: 10.1126/scisignal.aav5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamamoto M., Acevedo-Duncan M., Chalfant C.E., Patel N.A., Watson J.E., Cooper D.R. Acute glucose-induced downregulation of PKC-betaII accelerates cultured VSMC proliferation. Am. J. Physiol. Cell Physiol. 2000;279:C587–C595. doi: 10.1152/ajpcell.2000.279.3.C587. [DOI] [PubMed] [Google Scholar]
- 80.Patel N.A., Chalfant C.E., Yamamoto M., Watson J.E., Eichler D.C., Cooper D.R. Acute hyperglycemia regulates transcription and posttranscriptional stability of PKCbetaII mRNA in vascular smooth muscle cells. FASEB J. 1999;13:103–113. doi: 10.1096/fasebj.13.1.103. [DOI] [PubMed] [Google Scholar]
- 81.Patel N.A., Apostolatos H.S., Mebert K., Chalfant C.E., Watson J.E., Pillay T.S., Sparks J., Cooper D.R. Insulin regulates protein kinase CbetaII alternative splicing in multiple target tissues: development of a hormonally responsive heterologous minigene. Mol. Endocrinol. 2004;18:899–911. doi: 10.1210/me.2003-0391. [DOI] [PubMed] [Google Scholar]
- 82.Wijesinghe D.S., Subramanian P., Lamour N.F., Gentile L.B., Granado M.H., Bielawska A., Szulc Z., Gomez-Munoz A., Chalfant C.E. Chain length specificity for activation of cPLA2alpha by C1P: use of the dodecane delivery system to determine lipid-specific effects. J. Lipid Res. 2009;50:1986–1995. doi: 10.1194/jlr.M800367-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Subramanian P., Vora M., Gentile L.B., Stahelin R.V., Chalfant C.E. Anionic lipids activate group IVA cytosolic phospholipase A2 via distinct and separate mechanisms. J. Lipid Res. 2007;48:2701–2708. doi: 10.1194/jlr.M700356-JLR200. [DOI] [PubMed] [Google Scholar]
- 84.Caslin H.L., Abebayehu D., Abdul Qayum A., Haque T.T., Taruselli M.T., Paez P.A., Pondicherry N., Barnstein B.O., Hoeferlin L.A., Chalfant C.E., Ryan J.J. Lactic Acid Inhibits Lipopolysaccharide-Induced Mast Cell Function by Limiting Glycolysis and ATP Availability. J. Immunol. 2019;203:453–464. doi: 10.4049/jimmunol.1801005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ghosh N., Patel N., Jiang K., Watson J.E., Cheng J., Chalfant C.E., Cooper D.R. Ceramide-activated protein phosphatase involvement in insulin resistance via Akt, serine/arginine-rich protein 40, and ribonucleic acid splicing in L6 skeletal muscle cells. Endocrinology. 2007;148:1359–1366. doi: 10.1210/en.2006-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Norton S.K., Wijesinghe D.S., Dellinger A., Sturgill J., Zhou Z., Barbour S., Chalfant C., Conrad D.H., Kepley C.L. Epoxyeicosatrienoic acids are involved in the C(70) fullerene derivative-induced control of allergic asthma. J. Allergy Clin. Immunol. 2012;130:761–769.e2. doi: 10.1016/j.jaci.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li R., Li Y., Kristiansen K., Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq data are in the process of submission to NCBI Sequence Read Archive (SRA) database for public access (Submission number: sub14341995). The GEO accession number is pending a database instrument update.