

Abstract
Biologic factors limiting responsiveness to matched targeted therapies include genomic heterogeneity and complexity. Advanced tumors with unique molecular profiles can be studied by comprehensive genomic profiling (CGP) and enhance patient outcomes using principles of precision medicine. The clinical utility of CGP across all cancer types and different therapeutic interventions using overall survival (OS) and progression-free survival (PFS) data was studied in this systematic literature review. Randomized controlled, nonrandomized, and observational studies conducted in adult patients with advanced cancer, dated up to September 2022, were searched from PubMed and EMBASE databases following PRISMA guidelines. Of 14 CGP studies, 7 (50%) and 9 (64%) reported OS and PFS as an outcome, respectively. Improved OS and PFS were reported when CGP guided treatment decisions, but its clinical utility varied among cancer types. Treatments were assigned based on matching scores and with the involvement of molecular tumor board (MTB) enhanced OS and PFS. Patients following MTB recommendations had superior treatment outcomes compared with those on physician’s choice regimens. CGP clinically benefited patients with genomically matched therapies and yielded better clinical outcomes regardless of cancer type. Further, uniform clinical value-based ranking of actionable mutations can encourage oncologists to use CGP tests for patients.
Keywords: advanced cancer, comprehensive genomic profiling, precision medicine, overall survival, progression-free survival, molecular tumor board
BACKGROUND
The past decade has seen a significant transformation in the approach, treatment, and management of patients with cancer. This is further characterized by a paradigm shift from a universal treatment strategy to a growing emphasis on personalized medicine based on genomic variants.[1] Precision medicine in cancer emphasizes personalized care using the patient’s tumor-specific information for cancer diagnosis, treatment, and management. It uses identification tests like single-gene testing, multigene hotspot panels, and comprehensive genome profiling (CGP) assays for disease diagnosis and treatment prognosis.[2,3] The molecular characteristics of individual tumors play a crucial role in assisting oncologists in enhancing the traditional approach to tissue localization and tumor histology.[4] While single gene-based assays are routinely used in clinical settings,[4] there is a gradual shift toward the adoption of next-generation sequencing (NGS) as a more preferred approach in cancer diagnostics and treatment.[4] Of all the NGS-based assays, CGP uses a single genomic panel to assess hundreds of genes, including relevant cancer biomarkers, and, thus, provides broad molecular coverage of the genome. It offers multiple benefits compared with the analysis of individual genes, facilitating the concurrent identification of all categories of genomic alterations known to promote malignant proliferation.[4]
While conventional molecular tests assess the limited alteration in the specific set of genes, CGP aims to identify all the classes of genomic aberrations at a nucleotide level and genetic signatures, including microsatellite instability and tumor mutational burden.[3,5] It also provides insights into the genomic alterations and biomarkers that direct the targeted therapy with the efficient use of tissue samples.[3] Despite these benefits of CGP, the utility remains limited, primarily due to the limited access to targeted therapy.
Several individual studies have demonstrated the low actionability of CGP results. In one study by Cobain et al.,[6] 80.5% of the profiled patients had actionable genetic variations, but only 16.2% received targeted therapy. Further, of the patients who received targeted therapy, only 37.1% (4.82% of total patients profiled, n = 1138) reported clinical benefits. Hilal et al.[3] observed that the CGP assay identified actionable genomic variants in 92% of patients, but only 12% received targeted therapy, and only 2% derived clinical benefits. In another study, actionable genomic alterations were found in 40–94% of the patients; however, only a few of them (10–25%) received genomically matched therapy.[7] Additionally, studies like the SHIVA trial reported that using molecularly targeted agents did not improve progression-free survival (PFS) in patients.[8] Significant differences are also seen in the overall survival (OS) after molecularly targeted therapy.[9]
To address this issue of perceived low actionability, consolidating data from clinical trials evaluating the clinical utility of CGP can be beneficial.[10] This systematic literature review aims to investigate the clinical utility of CGP across various cancer types and therapeutic interventions, providing insights into its practical applications and potential benefits.
METHODS
The systematic literature review search, selection, and data extraction were conducted and reported using PRISMA guidelines.[11] Studies published in English up to September 2022 were searched using PubMed and EMBASE databases. Appropriate keyword searches and MESH terms including: advanced cancer, comprehensive genomic profiling, next-generation sequencing, precision medicine, companion diagnostics, personalized healthcare, overall survival (OS), progression-free survival (PFS), molecular tumor board (MTB), and quality of life were employed, along with suitable Boolean operators for the search.
In assessing the eligibility of studies that passed screening, we first excluded the following: (1) studies published in a language other than English, (2) systematic literature reviews, meta-analyses, reviews, comments, letters, editorials, case series, case reports, and thesis, and (3) studies that use models, animals, or in vitro methods. We subsequently included: (1) randomized controlled trials (RCTs), non-RCTs, and observational studies; (2) studies of adult human patients with advanced cancer; (3) molecular screening studies by CGP or hotspot NGS; (4) patients treated with targeted therapy based on CGP results; and (5) studies that evaluated survival endpoints as either OS or PFS. The inclusion and exclusion criteria details are given in Supplementary Figure 1 (available online). After eliminating the duplicates, titles and abstracts were assessed for eligibility by two independent reviewers; publications that met the inclusion criteria were further evaluated as full text.
The clinical utility of CGP in treatment selection was studied using the data for OS and PFS. All data were analyzed descriptively.
RESULTS
Overall, 1090 studies were obtained from PubMed and EMBASE. After removing the duplicates and title screening (n = 904), 186 studies were screened for abstracts. Of these, 16 studies were removed as they did not fit the study design criteria, and the full texts of 170 studies were evaluated further. Based on the inclusion and exclusion criteria defined above, only 14 studies were included after two rounds of screening with 35,975 patients across 10 tumor types (Fig. 1). Details of the baseline characteristics of these studies are given in Table 1.
Figure 1.
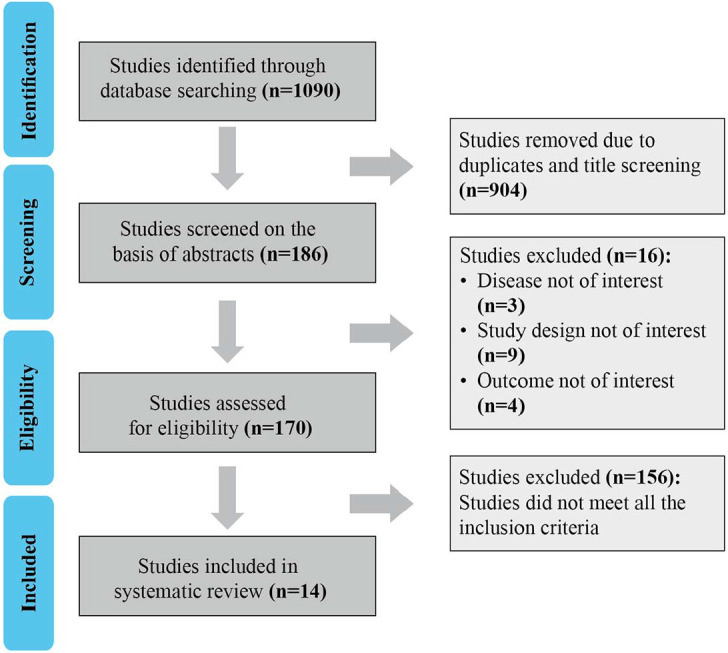
Flowchart of study selection according to PRISMA guidelines.
Table 1.
Basic characteristics of the studies included
| Study Characteristics | n (%) |
|---|---|
| Total studies included | 14 (100) |
| Tissue Type | |
| Liquid biopsy | 8 (57) |
| Tumor tissue | 14 (100) |
| Type of Cancer | |
| Breast | 3 (21) |
| Lung | 1 (7) |
| Colorectal | 1 (7) |
| Gastric | 2 (14) |
| Ovarian | 1 (7) |
| Multiple cancers | 6 (43) |
| Involvement of MTB | |
| Yes | 7 (50) |
| Treatment | |
| Targeted immune checkpoint inhibitor + hormone therapy | 1 (7) |
| Targeted immunotherapy | 9 (64) |
| Targeted immunotherapy + chemotherapy | 2 (14) |
| Targeted immunotherapy + hormone therapy | 2 (14) |
MTB: molecular tumor board.
Overall Survival (OS) After CGP Testing
In total, 7 of 14 CGP studies (50%) reported OS as an outcome. All seven studies reported improved OS after targeted therapy (alone or in combination with conventional chemotherapy or hormonal therapy) over the prior or comparator therapy.[12–18] One study[15] was conducted in advanced non–small cell lung cancer, while two studies[12,18] focused on gastric cancers and one study on colorectal cancers[16]; the rest of the studies involved multiple cancer types.[13,14,17] Three studies used circulating-tumor DNA (ctDNA) as a testing modality and its utility for successful targeted therapy.[12,13,18]
Of note is that of the Lee et al. study,[18] which used CGP for a biomarker-driven disease diagnosis and showed significantly improved outcomes in patients after biomarker-driven treatment than those following conventional therapy. Additionally, the correlation between MET-targeted therapy and increased levels of ctDNA MET amplification was noted.[18]
No two molecular profiles are identical, and hence, different patients respond differently to the same treatment regimens. Molecular analysis by CGP testing for a large panel of cancer-related genes can help investigators determine the matching scores by calculating the total number of molecular alterations matched to the drugs administered and dividing that number by the total number of characterized genomic aberrations. A study by Schwaederle et al.[14] demonstrated that OS was significantly improved in patients with higher matching scores (> 0.2) than those with lower matching scores (< 0.2) (matching score was calculated by dividing the number derived from the direct matched [defined as when ≥ 1 drug directly impacted the gene product of the molecular alteration or a differentially expressed protein] and indirect matched [a drug that affects a target removed from the molecular aberration] for each patient by the number of aberrations. For instance, if a patient harboring six genomic aberrations received two drugs, the matching score would be 2/6 or 0.33).[14] The clinical utility of CGP is increasingly evident in the study conducted by Catenacci et al.[12] in patients with newly diagnosed advanced gastroesophageal adenocarcinoma, wherein a novel study design was implemented to test an individualized treatment strategy using monoclonal antibodies matched to tumor molecular profiles in combination with chemotherapy for up to three lines of sequential treatment versus historical control. The study demonstrated that excellent outcomes can be achieved by individually optimizing chemotherapy, biomarker profiling, and matching targeted therapies at baseline and over time (OS: 15.7 vs. 9.0 months [historical controls], p = 0.05).[12]
The study conducted by Steuten et al.[17] highlighted that multigene panel testing detected more actionable mutations than single-gene mutation testing (30% vs. 23%). This, in turn, increased the probability of patients being treated with better treatment options and, hence, better treatment outcomes (21% and 16% of patients were treated with targeted therapies and immunotherapies after multigene panel testing, respectively, in contrast to 19% and 7% of patients being treated with targeted therapies and immunotherapies after single-gene mutation testing, respectively). Subsequently, the study showed how appropriate treatment decisions improved the clinical outcomes of these patients.[17] This study is one of the few that emphasized that although the cost-effectiveness of CGP versus single-gene mutation testing was moderate ($147,000 per life year gained) compared with commonly cited threshold values in the United States ($50,000–$200,000 per life year gained), as the number of available targeted therapies increase over time, and, if the incremental benefit afforded by those therapies also improves compared with current treatments, CGP will likely become more cost-effective in the future.[17]
Progression-Free Survival (PFS) After CGP Testing
Overall, 9 of 14 studies (64%) involving CGP reported PFS as an outcome. The majority of these studies (n = 8, 89%) reported improved PFS over the prior therapy or placebo. Three studies focused on advanced breast cancers,[19–21] while one each was reported in ovarian cancer[22] and metastatic gastric cancer[18]; the rest reported multiple cancers.[13,14,23–25] Molecular profiling-guided treatments yielded 30–56% more benefit in terms of PFS than the PFS achieved in prior therapies.[26]
In the ARIEL 3 study on recurrent ovarian carcinoma, it was seen that improvements in PFS in patients receiving rucaparib (versus placebo) were not just solely driven by patients with BRCA-mutant carcinoma. However, the PFS benefit was more pronounced in patients with a BRCA wild type and high loss of heterogeneity than those with a BRCA wild type and low loss of heterogeneity. This implied how CGP testing could help in using genes associated with homologous recombination deficiency as a predictive biomarker for testing treatment sensitivity.[22]
It is also seen that treatments assigned based on matching scores have better PFS outcomes.[13,24] In the study by Sicklick et al.[24] higher matching score (matching score > 50%) was an independent predictor of longer PFS (median PFS, high matching score versus low matching score: 6.5 vs. 3.1 months, p = 0.001). Furthermore, generally, PFS becomes shorter with each line of therapy administered. When PFS on the study (PFS2) was compared with the immediate prior line of unmatched therapy (PFS1), using the patient as their own control, it was seen that a high matching score was the only parameter significantly impacting the PFS ratio (PFS2/PFS1) 1.3 or greater in both the univariable (p = 0.026) and multivariable analyses (p = 0.015).[26] Consistent with this, Kato et al.[13] also demonstrated a significantly prolonged PFS in patients with high matching scores compared with patients with the lowest matching score (matching scores > 50% vs. matching scores < 25%: hazard ratio [HR], 0.47; 95% CI, 0.32–0.69; p < 0.001). Another study conducted in patients with metastatic cancers demonstrated significant improvements (p = 0.001) in PFS after treatment in patients with high matching scores than those with low matching scores, thereby suggesting how molecular screening strategy can lead to a matched targeted treatment for successfully screened patients and can help to obtain a PFS ratio above the predefined threshold in these patients.[23]
PFS Benefit and Detection of Less Common Aberrations or Alternative Pathways
CGP testing helps in detection of less common aberrations of FGFRs, RET, or targets of antibody-drug conjugate (ADC) along with well-established actionable events, such as mutations in PIK3CA, ESR1, and BRCA1/2, or amplifications and overexpression of the ErbB family of receptor tyrosine kinases in metastatic breast cancers, suggesting for the pharmacologic targeting of pathways that are not routinely targeted in the clinical management of metastatic breast cancers and can be realized only within clinical trials or with an off-label use of drugs already approved in a different setting.[19] In line with this, Hortobagyi et al.[25] indicated that while PIK3CA mutations had minimum effect on the efficacy of everolimus versus placebo in the patients with advanced breast cancers when patients were assigned to subgroups based on mutations in PIK3CA exon 20 (kinase domain) or exon 9 (helical domain), PFS benefit from everolimus was greater in patients with exon-9 mutations (HR, 0.26; 95% CI, 0.12–0.54) than in those with exon-20 mutations (HR, 0.56; 95% CI, 0.31–1.00). Thus, the study suggested that tumors dependent on a particular signal transduction pathway might yield greater treatment benefits in terms of PFS.[25] CGP is a robust tool to identify Her2 copy number gain and Her2-activating mutations in different tumor types (breast, gastric, and gastroesophageal junction cancers) that could benefit from treatment with trastuzumab deruxtecan (TDXD), an ADC-targeting Her2 TDXD therapy. It is theoretically plausible to identify potential targets for other ADCs, such as TDM1 and mirvetuximab soravtansine, through CGP analysis.[27,28]
Role of MTB in CGP Testing
Overall, seven studies had MTB involvement (Tables 2 and 3).[13,18–20,23–26] All studies confirmed that patients who received MTB-recommended regimens had better clinical outcomes and were better matched to therapy.[13,18–20,23–26] In the study conducted by Kato et al.,[13] although physicians were permitted to choose which therapy they considered best for their patients regardless of MTB discussion, it was seen that patients whose physicians adhered to MTB recommendations were more likely to receive matched targeted therapies covering a larger fraction of their tumor’s molecular alterations, and thus had a high matching score. When treated with a matching therapy, such patients (with ≥ 50% matching score) had significantly improved clinical outcomes (55.8% vs. 4.3%; p < 0.0001) than patients who received a physician’s choice regimen.
Table 2.
Clinical utility of comprehensive genomic profiling for improving overall survival in patients with advanced cancers
| Study | Indication | Specimen | Treatment | Treatment Arms |
mOS | HR, 95% CI | p-value | |
|---|---|---|---|---|---|---|---|---|
| Schwaederle (2016)[14] | Multiple cancers | Tissue | Targeted immunotderapy | Matching score > 0.2 | Matching score < 0.2 | 15.7 (matching score > 0.2) vs 10.6 (matching score 0.2) mos | 95% CI, 13.1–18.3 | 0.040 |
| Lee (2019)[18] | Metastatic gastric cancer | Tissue and ctDNA | Targeted immunotherapy | Biomarker-driven treatment group | Conventional 2L therapy group | 9.8 (biomarker driven) vs 6.9 (conventional) mos | HR, 0.58; 95% CI, 0.45–0.76 | < 0.0001 |
| Steuten (2019)[17] | Multiple cancers | FFPE or fresh tissue | Targeted immunotherapy | Targeted treatment group | Nontargeted treatment group | 2.31 (targeted) vs 1.73 (nontargeted) y | 95% CI, 0.31–4.12 vs 0.28–3.59 y | - |
| Singal (2019)[15] | NSCLC | FFPE tissue | Targeted immunotherapy | Targeted treatment group (n = 575) | Nontargeted treatment group (n = 560) | 18.6 (targeted) vs 11.4 (nontargeted) mos | 95% CI, 15.2–21.7 vs 9.7–12.5 | < 0.001 |
| Kato S (2020)[13] | Multiple cancer types | Tissue and cell-free ctDNA | Targeted immunotherapy | Group with MTB recommendations | Physician- chosen regimen | - | HR, 0.69; 95% CI, 0.49–0.98 | 0.036 |
| Stahler (2020)[16] | Metastatic colorectal cancer | Tissue | Targeted immunotherapy | SMAD4 wild-type tumors | SMAD4-mutated tumors | HR, 0.59; 95% CI, 0.34–1.01 | > 0.05 | |
| Catenacci (2021)[12] | Gastroesophageal adenocarcinoma | Tissue or ctDNA | Targeted immunotherapy+ chemotherapy | ITT group | Historical controls | 15.7 (ITT) vs 9 (control) mos | 95% CI, 13.4–17.7 vs 4.6–20.3 | 0.05 |
Dash (-) indicates data not available; 2L: second line; ctDNA: circulating-tumor deoxyribonucleic acid; FFPE: formalin-fixed paraffin-embedded; HR: hazard ratio; ITT: intention-to-treat; mOS: median overall survival; MTB: molecular tumor board; NSCLC: non–small cell lung cancer; SMAD4: mothers against decapentaplegic homolog 4.
Table 3.
Clinical utility of comprehensive genomic profiling for improving progression-free survival in patients with advanced cancers
| Study | Cancer Type | Specimen | Treatment | Treatment Arms |
mPFS | HR, 95% CI | p-value | |
|---|---|---|---|---|---|---|---|---|
| Hortobagyi (2016)[25] | Advanced breast cancer | FFPE tissue | Targeted immunotderapy | Everolimus (n = 209) | Placebo (n = 93) | 7.0 (Everolimus) vs 4.0 (Placebo) mos | 95% CI, 6.7–8.5 vs 2.6–4.2 | - |
| Schwaederle, (2016)[14] | Multiple cancers | Tissue | Targeted immunotherapy | Matched group | Unmatched group | 4.0 (matched) vs 3.0 (unmatched) | - | 0.039 |
| Massard (2017)[23] | Multiple cancers | Fresh-frozen tumor tissue, whole blood | Targeted immunotherapy | Matched therapy (PFS2) | Prior therapy (PFS1) | PFS2/PFS1 ratio was > 1.3 | 95% CI, 26–39% | - |
| Coleman (2017)[22] | Ovarian cancer | Tissue | Targeted immunotherapy | BRCA-mutant carcinoma group | Placebo group | 16.6 (BRCA) vs 5.4 (placebo) mos | 95% CI, 13.4–22.9 vs 3.4–6.7 | < 0.0001 |
| Lee (2019)[18] | Metastatic gastric cancer | Tissue and ctDNA | Targeted immunotherapy | Matched therapy | Conventional chemotherapy | 5.7 (matched) vs 3.8 (conventional) mos | - | < 0.0001 |
| Sicklick (2019)[24] | Multiple cancers | Tissue and ctDNA | Targeted immune checkpoint inhibitor + hormone therapy | High-matching score group | Low-matching score group | 6.5 (high match) vs 3.1 (low match) mos | HR, 0.42; 95% CI, 0.18–0.95 vs HR, 0.34; 95% CI, 0.19–0.62 | 0.001 |
| Tuxen (2019)[20] | Multiple cancers | Tissue and whole blood | Targeted immunotherapy+ chemotherapy | Molecular profiling–guided treatment (PFS2) | Most recent treatment (PFS1) | PFS2/PFS1 > 1.3; 32% | 95% CI, 23–42% | - |
| Kato (2020)[13] | Multiple cancers | Tissue and cell-free ctDNA | Targeted immunotherapy | Group with MTB recommendations | Physician’s choice regimens | - | HR, 0.63; 95% CI, 0.50–0.80 | < 0.001 |
| Sultova (2021)[26] | Breast cancer | FFPE and liquid biopsies | Targeted immunotherapy + hormone therapy | Group with treatment recommendation (PFS2) | Last therapy (PFS1) | PFS2/PFS1 ≥ 1.3 in 9/16 patients (56%, 9% of all patients) | - | - |
| Hlevnjak (2021)[19] | Breast cancer | Tissue and blood-derived nontumor DNA | Targeted immunotherapy + hormone therapy | Group with treatment recommendation (PFS2) | Last therapy (PFS1) | PFS2/PFS1 ≥ 1.3 in 30% of all patients | - | - |
Dash (-) indicates data not available; BRCA: breast cancer gene; ctDNA: circulating-tumor deoxyribonucleic acid; FFPE: formalin-fixed paraffin-embedded; HR: hazard ratio; MTB: molecular tumor board; PFS: progression-free survival, PFS1: PFS under immediate previous treatment line; PFS2: PFS under MTB-recommended treatment.
Another study[20] conducted on patients with advanced breast cancer demonstrated that individual treatment recommendations based on molecular profiling using CGP could improve PFS. Of 73 tumor samples analyzed, 53% had at least one actionable mutation, as classified by the MTB, and 51% had more than one molecular alteration. The most common molecular alterations across the sequenced samples were found in the PIK3CA gene (19%), followed by the TP53 gene (17%) and FGFR1 gene (15%). In total, 49% of patients received at least one treatment recommendation from the MTB, of which 18% obtained more than one treatment recommendation as their samples contained more than one actionable alteration. The most common therapy recommendation (21%) was everolimus, and 19% of patients carrying a now actionable mutation (PIK3CA) received no therapy recommendation, as the drug targeting this mutation (alpelisib) was not approved at the time of MTB presentation. Among those patients with implemented treatment recommendations, more than half had a PFS2/PFS1 ratio at NGS application was more than 1.3 (it should be noted here that NGS was done at a late line setting). Overall, this study helps to understand the potential relevance of involving targeted NGS-guided therapies in metastatic breast cancer settings and the important role of MTB in guiding the right therapies (Table 3).
DISCUSSION
Improved clinical benefits associated with targeted and immunotherapy treatments based on results of molecular diagnosis have increased demand for CGP assays. With the developments in precision medicine, complemented by an improved understanding of molecular pathways in oncology and the availability of newer molecular profiling methods, many clinicians are investigating the clinical utility of CGP in cancer genomic medicine. The Cancer Genome Atlas (TCGA) embarked on creating a comprehensive catalog of cancer genomic profiles by exploring major cancer-causing genomic alterations, analyzing large cohorts of over 30 human tumors using genome sequencing and multidimensional analyses. Studies have shown how TCGA information can be incorporated with CGP test results for treatment decisions.[29–31] In one study, CGP allowed the molecular classification of endometrial cancer into four TCGA categories (POLE ultramutated, microsatellite instable, copy-number low, and copy-number high) and allowed the identification of potential biomarkers for matched therapy in a clinical setting.[30] Another study used TCGA and CGP testing that helped in subtyping thyroid carcinoma for better treatment decisions.[31] With this increased understanding of molecular mechanisms, the clinical utility of the CGP assay for recommending targeted therapy was firmly established.[32]
Our review indicates that CGP, irrespective of the cancer type, clinically benefitted patients who received genomically matched therapy had better clinical outcomes than those who did not. This was evident in both outcomes studied (OS and PFS).
Currently, NGS technologies are applied as a screening strategy to find candidates for genomic-based clinical trials and are accepted by the oncology community.[33] This was evident in our review as well. Most studies that used NGS or CGP were either clinical studies testing novel targeted therapies or suggested in patients with failed prior therapies. Furthermore, while most studies incorporated CGP testing during the study, the actual treatment based on CGP testing was limited to subgroup analysis only. Most results of these subgroup analyses had to be interpreted with caution because of the low sample size and statistical insignificance. Hlevanjak et al.[19] demonstrate how CGP and RNA sequencing are useful in creating a deep molecular snapshot of biopsied metastatic breast lesions to guide clinical decision-making regarding disease progression. The study demonstrated how the combined strategy enabled maximal detection of genomic alterations along with concurrent analyses of both tumor and blood samples to discriminate germline from true somatic variants and identify candidates required for human genetic counseling. Furthermore, the study also demonstrated that measuring expression levels helped distinguish passengers from drivers in copy number variant analysis and provided information on whether mutations are expressed or not.[19]
One of the indicators of the clinical utility of the CGP test is whether patients received the treatment recommended by the CGP report. Several studies have indicated that while CGP in patients with advanced or metastatic solid tumors has reported a wide range of actionable genomic alterations per patient, ranging from 40–94%, only 10–25% of patients received genomically matched therapy.[7]
Consistent with the earlier findings, an important observation noted in the current review is that the clinical utility of genomic profiling is not uniform across all cancers. The proportion of patients receiving genomically matched therapy was higher among those with common cancers than noncommon cancers, thereby suggesting that the clinical utility of CGP tests may differ between patients with common and noncommon or rare cancers. It is also seen that many of these CGP platforms do not prioritize the various targetable mutations discovered, and it is often left to the oncologist to select clinically efficient, evidence-based targeted therapy.[21] Thus, there is a requirement to have a clinical value-based ranking of actionable mutations similar to the European Society of Medical Oncology scale for clinical actionability of molecular targets that would enable oncologists to select the actionable mutations with proven clinical benefits. There is also an unmet need for more discrete global and regional guidelines and recommendations for CGP testing for different cancer types. CGP has the potential to identify various genomic alterations in a single test that could be targeted for the therapy. However, oncologists have to consider multiple factors, such as existing comorbidities and toxicity-related adverse effects, before selecting the targeted therapy. Furthermore, although the scope of precision therapies continues to be immense, the patient needs to be made aware of the clinical reality that only a small fraction of patients who undergo CGP receive sequencing-directed therapies, and a smaller fraction benefit from it. Awareness of these data would help in patient counseling so that the expectations match the real-world scenario.
Our review further confirms the significance of MTB as patient outcomes improve after MTB involvement. Consistent with our findings, one recent study by Aoyagi et al.,[34] highlights how a patient whose diagnosis was changed in the MTB discussion because of the CGP results and who was treated based on the new diagnosis. In our analysis, only 45% of overall studies had MTB involvement. MTB plays a very important role in guiding the right diagnostic testing for appropriate clinical decision-making and can enhance the utility of precision medicine from laboratory to clinic. It is imperative not just to set up more MTBs but, more importantly, to standardize the MTB panel. Furthermore, developing appropriate digital platforms to integrate the genomic results and clinical findings in one place is essential for a complete MTB review. Rothwell et al.[35] developed an in-house digital solution for efficient and effective integration of clinical and genomic data and making it available for MTB review. The digital platform integrated a single overview of patients’ clinical and genomic characteristics with a web application for data visualization; thus, it enabled the MTB to review summary patient data via a single portal (and remotely if required), capture meeting outcomes in real-time, and upload information to electronic patient records.[35]
Over the years, using ctDNA and liquid biopsy has not just enhanced patient experience but also provided comparable results to tissue biopsy.[36] Results from this review further encourage routine implementation of ctDNA testing and restrict tumor analysis only in cases with lower tumor burden or low ctDNA yields where blood analysis may be unsuccessful, thereby reducing invasive procedures for patients and the associated healthcare system costs.[35] Further studies involving ctDNA are warranted to ascertain its use across all cancer types.
One of the most debatable topics today is related to the financial burden of precision medicine and CGP testing, which makes it difficult for clinicians to prescribe these tests for their patients. Stueten et al.[17] demonstrated that although the financial burden of CGP testing is moderate today, as the number of available targeted therapies increases over time, and with improved incremental benefit afforded by those therapies than conventional treatments, CGP will likely become more cost-effective in the future. This is consistent with the earlier published studies, which suggested that financial burden can be reduced considerably when CGP tests are recommended based on MTB advice.[34,37]
There was heterogeneity in the matching scores for outcome interpretation, which is a limitation of this study. Each study employed different scoring methods to predict the outcome (e.g., Sicklick et al.[24] used a matching score of > 50%, whereas Kato et al.[36] used 2 scores, namely > 50% and > 25% to predict the treatment outcomes), resulting in a lack of unified interpretation. However, despite the heterogeneity of matching scores in studies, the outcomes were favorable for the clinical utility of CGP. Lastly, this review includes publications up to September 2022; thus, publications after this date have not been considered.
In a real-world setting, more case discussions are required to evaluate the feasibility and utility of CGP tests in front-line settings in patients with advanced or metastatic solid tumors to ascertain the clinicians’ confidence in prescribing these tests. Results from ongoing trials (ClinicalTrials.gov IDs: NCT03061305, NCT02806388), especially the IMPACT trial, may provide more insights into the utility of CGP testing in patients with different solid tumors.
CONCLUSION
To summarize, CGP testing has several advantages. Treatment outcomes after CGP testing are favorable; however, these are usually done in a small subset of patients, and hence, larger trials focusing on treatment outcomes after CGP versus single-gene mutation testing are warranted for more clarity on the usefulness of CGP in clinical decisions. The use of ctDNA for CGP testing is promising, and more data on this will further encourage its utility in clinical settings.
Supplementary Material
{kind=link}
Acknowledgements
The authors thank Turacoz Healthcare Solutions Pvt. Ltd. for publication support.
References
- 1.Malone ER, Oliva M, Sabatini PJ, et al. Molecular profiling for precision cancer therapies. Genome Med. 2020;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pennell NA, Mutebi A, Zhou Z-Y, et al. Economic impact of next-generation sequencing versus single-gene testing to detect genomic alterations in metastatic non–small-cell lung cancer using a decision analytic model. JCO Precis Oncol. 2019;3:1–9. [DOI] [PubMed] [Google Scholar]
- 3.Hilal T, Nakazawa M, Hodskins J, et al. Comprehensive genomic profiling in routine clinical practice leads to a low rate of benefit from genotype-directed therapy. BMC Cancer. 2017;17:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Svoboda M, Lohajova Behulova R, Slamka T, et al. Comprehensive genomic profiling in predictive testing of cancer. Physiol Res. 2023;72(S3):S267–S75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reitsma M, Fox J, Borre PV, et al. Effect of a collaboration between a health plan, oncology practice, and comprehensive genomic profiling company from the payer perspective. J Manag Care Spec Pharm. 2019;25:601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cobain EF, Wu YM, Vats P, et al. Assessment of clinical benefit of integrative genomic profiling in advanced solid tumors. JAMA Oncol. 2021;7:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ida H, Koyama T, Mizuno T, et al. Clinical utility of comprehensive genomic profiling tests for advanced or metastatic solid tumor in clinical practice. Cancer Sci. 2022;113:4300–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16:1324–1334. [DOI] [PubMed] [Google Scholar]
- 9.Wheler JJ, Janku F, Naing A, et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 2016;76:3690–3701. [DOI] [PubMed] [Google Scholar]
- 10.Hall MJ, D’Avanzo P, Chertock Y, et al. Oncologists’ perceptions of tumor genomic profiling and the communication of test results and risks. Public Health Genom. 2021;24:304–309. [DOI] [PubMed] [Google Scholar]
- 11.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catenacci DVT, Moya S, Lomnicki S, et al. Personalized antibodies for gastroesophageal adenocarcinoma (PANGEA): a phase II study evaluating an individualized treatment strategy for metastatic disease. Cancer Discov. 2021;11:308–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato S, Kim KH, Lim HJ, et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun. 2020;11:4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwaederle M, Parker BA, Schwab RB, et al. Precision oncology: the UC San Diego moores cancer center PREDICT experience. Mol Cancer Ther. 2016;15:743–752. [DOI] [PubMed] [Google Scholar]
- 15.Singal G, Miller PG, Agarwala V, et al. Association of patient characteristics and tumor genomics with clinical outcomes among patients with non-small cell lung cancer using a clinicogenomic database. JAMA. 2019;321:1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stahler A, Stintzing S, von Einem JC, et al. Single-nucleotide variants, tumour mutational burden and microsatellite instability in patients with metastatic colorectal cancer: next-generation sequencing results of the FIRE-3 trial. Eur J Cancer. 2020;137:250–259. [DOI] [PubMed] [Google Scholar]
- 17.Steuten L, Goulart B, Meropol NJ, et al. Cost effectiveness of multigene panel sequencing for patients with advanced non-small-cell lung cancer. JCO Clin Cancer Inform. 2019;3:1–10. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Kim ST, Kim K, et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: the VIKTORY umbrella trial. Cancer Discov. 2019;9:1388–1405. [DOI] [PubMed] [Google Scholar]
- 19.Hlevnjak M, Schulze M, Elgaafary S, et al. CATCH: A prospective precision oncology trial in metastatic breast cancer. JCO Precis Oncol. 2021;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuxen IV, Rohrberg KS, Oestrup O, et al. Copenhagen prospective personalized oncology (CoPPO)-clinical utility of using molecular profiling to select patients to phase I trials. Clin Cancer Res. 2019;25:1239–1247. [DOI] [PubMed] [Google Scholar]
- 21.Jiang YZ, Liu Y, Xiao Y, et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: the FUTURE trial. Cell Res. 2021;31:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massard C, Michiels S, Ferte C, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7:586–595. [DOI] [PubMed] [Google Scholar]
- 24.Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25:744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA,, 3rd,, Pritchard KI, et al. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2016;34:419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sultova E, Westphalen CB, Jung A, et al. Implementation of precision oncology for patients with metastatic breast cancer in an interdisciplinary MTB setting. Diagnostics (Basel). 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meric-Bernstam F, Makker V, Oaknin A, et al. Efficacy and safety of trastuzumab deruxtecan in patients with HER2-expressing solid tumors: primary results from the DESTINY-PanTumor02 phase II trial. J Clin Oncol. 2024;42:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu Z, Li S, Han S, et al. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022;7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19:A68–A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prendergast EN, Holman LL, Liu AY, et al. Comprehensive genomic profiling of recurrent endometrial cancer: Implications for selection of systemic therapy. Gynecol Oncol. 2019;154:461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Jia L, Zhou H-R, et al. Development and validation of potential molecular subtypes and signatures of thyroid carcinoma based on aging-related gene analysis. Cancer Genomics Proteomics. 2024;21:102–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrot-Zhang J, Chambwe N, Damrauer JS, et al. Comprehensive analysis of genetic ancestry and its molecular correlates in cancer. Cancer Cell. 2020;37:639–654 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathew A, Joseph S, Boby J, et al. Clinical benefit of comprehensive genomic profiling for advanced cancers in India. JCO Glob Oncol. 2022;8:e2100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aoyagi Y, Kano Y, Tohyama K, et al. Clinical utility of comprehensive genomic profiling in Japan: Result of PROFILE-F study. PLoS One. 2022;17:e0266112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E, et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med. 2019;25:738–743. [DOI] [PubMed] [Google Scholar]
- 36.Kato S, Weipert C, Gumas S, et al. Therapeutic actionability of circulating cell-free DNA alterations in carcinoma of unknown primary. JCO Precis Oncol. 2021;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The cost of sequencing a human genome. National Human Genome Research Institute. Accessed May 14, 2024. www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.