

Abstract
The second and third amino acid residues of the N-terminal arm of most Hox protein homeodomains are basic (lysine or arginine), whereas they are asparagine and alanine, respectively, in the Hoxa1 homeodomain. Previous reports pinpointed these residues as specificity determinants in the function of Hoxa1 when it is acting as a monomer. However, in vitro data supported that these residues do not influence the target specificity of Hoxa1 in Pbx1a–Hoxa1 heterodimers. Here, we have analysed the transcriptional activity of a Hoxa1(NA-KR) mutant for which the asparagine and alanine residues of the homeodomain have been replaced by lysine and arginine, respectively. Comparison between the wild-type and mutant Hoxa1 reveals that they show distinct activity on the TSEII enhancer of the somatostatin gene, but that they are equally active in the presence of Pbx and Prep cofactors. This therefore corroborates the biochemical evidence having shown that the second and third residues of the homeodomain do not contribute to the DNA binding of Hoxa1–Pbx dimers. However, on the hoxb1 autoregulatory enhancer, Hoxa1 and Hoxa1(NA-KR) may display distinct activity despite the presence of Pbx, in a cell-type dependent manner. Therefore, our data suggest that, depending on the enhancer, these residues may contribute to the functional specificity of Hoxa1 and that this contribution may not be abrogated by the interaction with Pbx.
INTRODUCTION
Proteins of the Hox family are transcription factors contributing to the regulation of important developmental pathways (1,2). They belong to the huge class of proteins which share the evolutionary conserved homeodomain involved in the recognition of, and the binding to, target DNA sites (3,4). Hox proteins bind in vitro to similar sites with similar affinities (5,6). However, in vivo studies provided data proving their high specificity of action. It is therefore possible that slight changes in DNA-binding specificity and/or affinity in vitro can lead to more important functional differences in vivo. Moreover, the high functional specificity of Hox proteins is ensured at least in part by interacting with cofactors. Some of these cofactors like Pbx, Meis, Prep, Sox or Oct proteins, may act by modulating the DNA-binding specificity or the transcriptional activity of Hox proteins through the formation of multimers (7–10). Consistently, the cell-type dependent activity that Hox proteins may display on a given target enhancer can be explained by the availability of such modulatory proteins (7,11–13).
Hox–Pbx dimerisation relies on a conserved motif obeying a YPWM consensus in the Hox sequence (hereafter referred to as the hexapeptide) (4) and causes a change in the DNA recognition specificity (7). This specificity shift involves the second nucleotide of the Hox bound TNAT core sequence which is contacted by residues of the N-terminal arm of the homeodomain. Strikingly, it has been reported that, in vitro, the specificity of DNA recognition by Hox proteins as monomers or in dimer with Pbx relies on distinct N-terminal arm residues (9).
Residues 2 and 3 of the homeodomain N-terminal arm are basic residues in most of the Hox proteins. However, they are asparagine (N) and alanine (A), respectively, in Hoxa1. Previous studies reported that changing the NA residues into KR modified the DNA-binding specificity of Hoxa1 as a monomer (14). However, these amino acid substitutions did not affect either the DNA-binding specificity or the binding affinity for cognate sites by Hoxa1–Pbx dimers (9). Therefore, these biochemical data resulted in the conclusion that residues 2 and 3 of the homeodomain do not contribute to the binding of DNA by Hoxa1 when it acts in combination with Pbx.
Here, we have analysed the transcriptional activity of Hoxa1 and Hoxa1 mutant proteins in different cell lines on the well characterised hoxb1 autoregulatory enhancer (ARE) (15) as well as on the TSEII enhancer involved in the pancreatic control of the Somatostatin coding gene (16). Our data show that changing residues 2 and 3 of the Hoxa1 homeodomain into the basic residues K and R may induce a loss of activity on the ARE as well as a gain of activity on TSEII. The difference between the wild-type Hoxa1 and the Hoxa1(NA-KR) mutant activity on TSEII is only observed in the absence of Pbx which is consistent with the model drawn from biochemical data (9,14). However, on the hoxb1 ARE, the wild-type and mutant proteins may show distinct activity even in the presence of Pbx, but in a cell-type dependent fashion.
MATERIALS AND METHODS
Plasmid construction
The pAdMLARE plasmid (15,17) contains the TATA box and transcriptional start site from the Adenovirus-2 Major Late promoter (AdML) (18), downstream of the hoxb1 ARE enhancer. The TSEII-luc reporter plasmid contains two copies of the TSEII enhancer inserted upstream of the minimal promoter of the growth hormone gene (16). pCMVlacZ plasmid was constructed by inserting the Escherichia coli lacZ coding region into the multiple cloning site (MCS) of pCMX-PL1 (19).
The different expression vectors are derived from the pCMX-PL1 plasmid. pGIH327 contains the intronless hoxa1 sequence subcloned from pPGK-Hoxa1(HD+), kindly provided by M.S. Featherstone and described elsewhere (9,20). Similarly, the pGIH328 and pGIH329 plasmids are pCMX-PL1-based vectors coding for the Hoxa1(WM-AA) and Hoxa1(NA-KR) mutant proteins, and were derived from pPGKHoxa1(HD+)(WM-AA) and pPGKHoxa1(HD+)(NA-KR), respectively (9,14). The Hoxa1(QN-AA) mutant protein is encoded by pGIH512. The two amino acid substitution in the homeodomain (QN to AA) was created by site-directed mutagenesis using a PCR approach (mutagenic primer: 5′-GTGAAGATCT GGTTCGCGGC CCGCCGCATG AAGCAG-3′). The plasmids coding for the fusion between the VP16 activation domain and, Hoxa1 (pGIH807), Hoxa1(WM-AA) (pGIH421) and Hoxa1(NA-KR) (pGIH422), were obtained by swapping the 3′ end of the Hoxa1 coding sequence fused to the VP16 coding sequence, from pPGKHoxa1VP16 (9), into pGIH327, pGIH328 and pGIH329, respectively. The fusion protein Hoxa1(QN-AA)–VP16 is encoded by a plasmid (pGIH811) obtained from pGIH807, by swapping a SnaBI–AccI fragment for the corresponding fragment containing the mutations which cause the QN to AA substitutions.
The Pbx1a coding sequence was isolated from the pBSK-Pbx1a plasmid (20) as a XbaI–EcoRV fragment and was cloned into the pCMX-PL1 MCS (pGIH346). The expression vector for Prep1, pCS2-Prep, has been described by Goudet et al. (21) and is based on pCS2+, a mammalian expression vector bearing the CMV promoter.
Cell culture and transient transfection
EC P19, COS7 and HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 7.5% (EC P19) or 10% (COS7 and HEK293) fetal calf serum (PAA Laboratories, GmbH), 100 IU/ml penicillin and 100 µg/ml streptomycin (Sigma), at 37°C in a humidified, 5% CO2 atmosphere. Before transfections, exponentially proliferating cells were trypsinised and 105 cells were plated per 35 mm culture dish. Cells were transfected by a standard calcium phosphate precipitation procedure (22), 24 h after plating.
Cotransfections were carried out with a total amount of 10 µg of DNA (EC P19 or COS7 cells), containing, when appropriate: 3 or 4 µg of reporter plasmid; 1.5 or 2 µg of Hox expression vector; 3 or 4 µg of Pbx expression vector; 3 or 4 µg of Prep expression vector; and 0.2 µg of internal standard plasmid (pCMVlacZ). In transfections involving HEK293 cells, the same proportions of reporter and expression vectors were used for a total of 1.7 µg of DNA.
Cells were harvested 48 h after transfection for enzymatic assays. Lysis and enzymatic activity dosages were performed with the β-gal Reporter Gene Assay (Chemiluminescent) kit (Roche) and the Luciferase Reporter Gene Assay (High Sensitivity) kit (Roche).
Statistical analysis of the cotransfection data was performed by the use of t-tests (23).
RT and PCR reactions
Forty-eight hours after transfection, total RNA was isolated from cells with the Trizol Reagent (Gibco BRL, Life Technologies), 1.5 µg of RNA was then treated with DNase I and subjected to reverse transcription (RT). cDNA synthesis occurred for 2 h at 42°C using Avian Myeloblastosis Virus Reverse Transcriptase (U.S. Biochemicals), after priming with random hexamers at a final concentration of 76 µg/ml [pd(N)6, Pharmacia Biotech]. PCR reactions were performed in a final volume of 25 µl with 2 µl of cDNA, each primer at 100 nM, deoxyribonucleoside triphosphates at 250 nM, 0.02 U Taq DNA polymerase (Takara), and the buffer supplied with the enzyme. As a control for cDNA synthesis, the β-actin sequence was amplified (23–25 amplification cycles with primers 5′-GGCATCGTGATGGACTCCG-3′ and 5′-GCTGGAAGGTGGACAGCGA-3′). The efficiency of transfection for the different expression vectors was evaluated (30–35 amplification cycles with primers 5′-CAGAGACGTCTTCTCCAGCG-3′ and 5′-TGCGCCATCGCCACGTCCTC-3′). And finally, variations in the level of hoxb1 endogenous expression were determined (30–37 amplification cycles with primers 5′-TTATTCTGTGGGTCAGTCGG-3′ and 5′-CGTTTCTTCTGCTTCATGCG-3′).
To quantify the activation of the genomic hoxb1 by the Hoxa1–VP16 variants (NA-KR, WM-AA and QN-AA substitutions), 32P-dCTP was incorporated with the amplification reactions, the output of the RT–PCR was normalised to that for the β-actin mRNA and the relative amount of hoxb1 amplification product was reported to that obtained for the hoxa1–VP16 variants. Therefore, this ratio rules out experimental variations imputable to transfection efficiency, mRNA extraction and cDNA synthesis.
RESULTS
Substituting the second (N) and third (A) residues in the N-terminal arm of the Hoxa1 homeodomain by K and R results in increased activation through the Somatostatin gene enhancer TSEII
Amino acid residues 2 and 3 are atypically asparagine and alanine, respectively, in the Hoxa1 homeodomain sequence. To evaluate the effect of the N-terminal arm NA to KR substitutions on the activity of the murine Hoxa1, a Hoxa1(NA-KR) mutant expression vector was cotransfected with luciferase reporter plasmids harbouring the hoxb1 ARE (pAdMLARE) (15,17) or the somatostatin gene enhancer TSEII (TSEII-luc) (16). The hoxb1 ARE contains three binding sites for Pbx–Hoxb1 heterodimers (15,17) and a Meis/Prep binding site that allow a synergistic association of Meis/Prep together with Pbx and Hoxb1 (24,25). Transcription of the somatostatin gene is controlled by several cis-acting sequences, among which is the TSEII element, which is recognised by the pancreatic specific homeodomain factor Pdx1, which possesses a homeodomain related to those of Hox proteins (16).
Cotransfection experiments performed in COS7 cells revealed that the wild-type Hoxa1 protein provided a modest 2–3-fold luciferase activation via the hoxb1 ARE enhancer (Fig. 1A), and did not activate TSEII-mediated reporter expression (Fig. 1B). Although significant, the Hoxa1(NA-KR) activity on ARE was also very low and, thus, comparable to that of the wild-type protein. In contrast, Hoxa1(NA-KR) induced a 5-fold luciferase activation via TSEII (Fig. 1B). The difference between Hoxa1 and Hoxa1(NA-KR) activity on TSEII was reproducibly observed in several independent experiments involving amounts of expression vectors decreased by a factor of up to 40, thus precluding that the observed difference was due to transfection artifacts like the squelching of endogenous transcription factors (Table 1) (26).
Figure 1.

Comparison between Hoxa1 and Hoxa1(NA-KR) activity in COS7 cells. (A) A reporter construct (pAdMLARE) consisting of the luciferase gene placed under the control of the hoxb1 ARE enhancer was transfected in COS7 cells, alone (Control) or in combination with expression vectors for Hoxa1 (Hoxa1 WT), and mutant Hoxa1 (Hoxa1 NA-KR, Hoxa1 WM-AA, Hoxa1 QN-AA) proteins. (B) A reporter construct (TSEII-luc) in which the TSEII enhancer controls the expression of the luciferase reporter was transfected in COS7 cells, alone (Control) or in combination with expression vectors for Hoxa1 (Hoxa1 WT), and mutant Hoxa1 (Hoxa1 NA-KR, Hoxa1 WM-AA, Hoxa1 QN-AA) proteins. Open boxes correspond to transfections performed in the absence of both Pbx1a and Prep1 expression vectors. Dark boxes correspond to transfections for which both Pbx1a and Prep1 expression vectors were added. Values are expressed as fold activation over transfection of the reporter plasmid alone. Bars indicate the standard deviation of six independent experiments. The difference between Hoxa1 and Hoxa1(NA-KR) activity on TSEII in the absence of Pbx1a was significant according to t statistics with t > t0.001.
Table 1. Gene dosage and Hoxa1(NA-KR) versus Hoxa1 relative activity in cotransfection experiments.
| Amount of expression vector per 105 cells | Hoxa1(NA-KR)/Hoxa1 relative activity (%) | t statistics (n = 6) |
|---|---|---|
| On the TSEII enhancer in COS7 cells | ||
| 50 ng | 214 | t > t0.001 |
| 500 ng | 299 | t > t0.02 |
| 2.0 µg | 302 | t > t0.001 |
| On the ARE enhancer in EC P19 cells | ||
| 50 ng | 72 | t > t0.01 |
| 500 ng | 66 | t > t0.02 |
| 2.0 µg | 40 | t > t0.001 |
As controls, Hoxa1 did not activate enhancer-less reporter constructs (data not shown). Similarly, the activity of the reporter was not modified when the pAdMLARE or TSEII-luc constructs were cotransfected with the expression vector for Hoxa1(QN-AA) in which the homeodomain residues glutamine 50 and asparagine 51 were changed into alanines, and which is impaired in its DNA binding ability (3,4,10). Finally, a Hoxa1 variant mutated in its hexapeptide sequence (WM residues changed to AA) was also shown to be inactive on both ARE and TSEII (Fig. 1). Similar WM to AA substitutions have been shown to abolish the interaction between Hox and Pbx proteins (9,10,20). The WM-AA mutation abolished the modest activation provided via the ARE, suggesting that the low activity of Hoxa1 on this enhancer relies on the endogenous Pbx proteins available in the nuclei of COS7 cells.
To rule out the possibility that differences between transcriptional activation by Hoxa1 and Hoxa1(NA-KR) resulted from different protein stability and/or abundance, the amount of Hoxa1 protein was estimated by western blotting of extracts obtained from transfected cells. Quantitative analysis revealed that the difference in activity observed for the different Hoxa1 variants cannot be correlated to differences in abundance of the proteins in transfected cells (data not shown). Altogether, therefore, this shows that the NA to KR substitution in the Hoxa1 homeodomain sequence allowed the protein to be active on the somatostatin TSEII enhancer.
Hoxa1 and Hoxa1(NA-KR) display similar activity in the presence of Pbx1A and Prep1 cofactors in COS7 cells
Biochemical data reported previously by Phelan and Featherstone (9) showed that the second and third residues of the Hoxa1 homeodomain do not contribute to the DNA binding of Hoxa1–Pbx heterodimers. Cotransfections involving Pbx1a and Prep1 expressing vectors were performed in order to evaluate the relative activities of Hoxa1 and Hoxa1(NA-KR) in the presence of these cofactors. Prep1 may enhance the Hox–Pbx mediated transcriptional activation on ARE (7,24,25) and TSEII (B. Peers, personal communication), but it is also involved in the nuclear accumulation of Pbx proteins (11). Providing Pbx1a and Prep1 in COS7 cells, allowed an equal 15–16-fold activation of the ARE driven reporter by both Hoxa1 and Hoxa1(NA-KR) (Fig. 1A). Similarly, addition of Pbx1a and Prep1 induced a synergistic activation of TSEII-luc with either Hoxa1 or Hoxa1(NA-KR), whereas only Hoxa1(NA-KR) was active on this enhancer when provided alone (Fig. 1B). Therefore, Hoxa1 and Hoxa1(NA-KR) may display differential activity in COS7 cells, but the effect of the NA to KR mutation on Hoxa1 activity is abolished in the presence of Pbx1a and Prep1.
The effect of the NA to KR substitution on Hoxa1 activity is not suppressed by Pbx1a in EC P19 and HEK 293 cells
Wild-type Hoxa1 induced a 6–7-fold activation of the ARE-controlled reporter in murine teratocarcinoma EC P19 cells (Fig. 2A). In contrast, the Hoxa1(NA-KR) mutant showed a 2–3-fold reduction of activity on the ARE, as compared with Hoxa1. Such a reduction was reproducibly observed in several independent experiments involving amounts of expression vectors that were decreased by a factor of up to 40 (Table 1).
Figure 2.

Comparison between Hoxa1 and Hoxa1(NA-KR) activity in ECP19 and HEK293 cells. (A) The pAdMLARE reporter construct was transfected in EC P19 cells, alone (Control) or in combination with expression vectors for Hoxa1 (Hoxa1 WT), and mutant Hoxa1 (Hoxa1 NA-KR, Hoxa1 WM-AA, Hoxa1 QN-AA) proteins. (B) Similar experiments were performed in HEK293 cells. (C) The TSEII-luc construct was transfected in EC P19 cells, alone (Control) or in combination with expression vectors for Hoxa1 (Hoxa1 WT), and mutant Hoxa1 (Hoxa1 NA-KR, Hoxa1 WM-AA, Hoxa1 QN-AA) proteins. These experiments were performed in the absence (open boxes) or in the presence (grey boxes) of a Pbx1a coding plasmid; or in the presence of both the Pbx1a and Prep1 expression plasmids (dark boxes). Bars indicate the standard deviation of six (EC P19 cells) or two (HEK293 cells) independent experiments. The difference between Hoxa1 and Hoxa1(NA-KR) activity on ARE in EC P19 and HEK293 cells was significant according to t statistics with t > t0.001 and t > t0.05, respectively.
In these experiments, the activation of the luciferase reporter controlled by the hoxb1 ARE was observed in the absence of a Pbx expression vector. Moreover, addition of Pbx1a and/or Prep1 expression vectors to the different cotransfection series did not change Hoxa1 and Hoxa1(NA-KR) activity on the ARE (Fig. 2A), and did not rescue the partial loss of function displayed by the Hoxa1(NA-KR) mutant. This is in sharp contrast with previously reported data supporting that Pbx proteins are required for the transcriptional activation of the pAdMLARE luciferase gene by HOX proteins (17). However, the WM-AA mutation abolished the capacity of Hoxa1 to activate transcription via the ARE (Fig. 2A). This supports that, on this enhancer, Hoxa1 requires its interaction with a Pbx partner to be active and, thereby, that Pbx—or a substituting cofactor—is available in the EC P19 cells.
Very similar observations were made with cotransfections in HEK293 cells (Fig. 2B). First, Hoxa1 and Hoxa1(NA-KR) were active on ARE in the absence of Pbx1a vector, whereas Hoxa1(WM-AA) was inactive, suggesting also that endogenous Pbx proteins were available in HEK293 cell nuclei. Secondly, the Hoxa1(NA-KR) protein was severely impaired in its ability to activate the reporter through ARE, keeping only 20–25% of the wild-type activity (Fig. 2B). Addition of Pbx1a did not rescue this loss of activity.
Both Hoxa1 and Hoxa1(NA-KR) proteins failed to activate the TSEII-luc in EC P19 cells (Fig. 2C). This lack of activation was also observed upon cotransfection with Pbx1a expression vector. However, upon concomitant addition of both Pbx1a and Prep1, Hoxa1 and Hoxa1(NA-KR) provided an equal 7-fold activation of the reporter. Under similar cotransfection conditions, Hoxa1(WM-AA) remained inactive on TSEII.
Altogether, these cotransfection experiments show that even when combined with Pbx, the Hoxa1(NA-KR) protein behaves like a partial loss-of-function mutant of Hoxa1 on the ARE, in both EC P19 and HEK293 cells.
Hoxa1–VP16 and Hoxa1(NA-KR)–VP16 fusion proteins differentially activate the genomic hoxb1 gene
To check whether the NA to KR substitution in the homeodomain of Hoxa1 affects its ability to activate the hoxb1 gene in its genomic context, quantitative RT–PCR amplifications were run on the hoxb1 mRNA. After 35 amplification cycles, a very faint hoxb1 specific band is detected with RNA samples from non-transfected EC P19 cells (Fig. 3). However, quantitative RT–PCR did not reveal an increase in hoxb1 expression upon transfection of the Hoxa1 and Pbx1a coding plasmids (data not shown).
Figure 3.
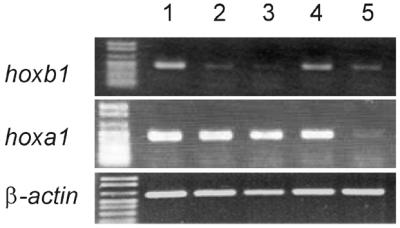
A Hoxa1–VP16 fusion protein activates the transcription of the genomic hoxb1 gene. Top, RT–PCR amplifications (35 cycles) of the hoxb1 mRNA were performed on RNA extracted from EC P19 cells transfected with the Pbx1a expression vector together with those for Hoxa1–VP16 (lane 1), Hoxa1(QN-AA)–VP16 (lane 2), Hoxa1(WM-AA)–VP16 (lane 3) or Hoxa1(NA-KR)–VP16 (lane 4). The result of RT–PCR amplification arising from EC P19 transfected with the Pbx1a vector alone is shown on lane 5. Centre, RT–PCR run on the hoxa1 sequence shared by the different Hoxa1–VP16 expression vectors (33 amplification cycles). Lanes 1–5 correspond to those shown in the top panel. Bottom, RT–PCR amplification of the β-actin mRNA were performed as controls (25 amplification cycles). Lanes 1–5 correspond to those shown in the top panel.
Transfection of a vector coding for a Hoxa1–VP16 fusion protein resulted in a 3.6-fold increase of the hoxb1 RT–PCR product (Fig. 3, lane 1; see Materials and Methods for the quantitation procedure). As controls, the mutated Hoxa1(QN-AA)–VP16 and Hoxa1(WM-AA)–VP16 fusion proteins did not induce a detectable increase in hoxb1 mRNA (Fig. 3, lanes 2 and 3). Therefore, these data show that the Hoxa1–VP16 protein is active on hoxb1 control elements, presumably the ARE, and support that Hoxa1–VP16 activity requires Pbx. Finally, the Hoxa1(NA-KR)–VP16 fusion protein activated the genomic hoxb1 gene (Fig. 3, lane 4). However, the relative amount of the amplified product reached only 69% of that obtained upon activation by Hoxa1–VP16.
Therefore, as in experiments involving a transfected reporter target, Hoxa1 and Hoxa1(NA-KR) display differential activity on a genomic target, showing that homeodomain residues 2 and 3 contribute to the functional specificity of these proteins.
DISCUSSION
The N-terminal residues 2 and 3 of the homeodomain of most Hox proteins are basic residues, but in Hoxa1, these are asparagine and alanine, respectively. When the N and A residues of the N-terminal arm of the Hoxa1 homeodomain are converted to K and R, corresponding to the Hoxd4 homeodomain residues 2 and 3, this mutant protein binds to, and is transcriptionally active on, the enhancer mediating hoxd4 autoregulation, while the wild-type Hoxa1 does not (14). However, upon its cooperative binding with Pbx1a on Hox–Pbx recognition sites in vitro, the mutant Hoxa1(NA-KR) behaves like Hoxa1, showing that homeodomain residues 2 and 3 of Hoxa1 do not contribute to the binding specificity of Hoxa1–Pbx dimers (9).
Here we have shown that the wild-type Hoxa1 and the NA to KR mutant display similar activity on ARE in COS7 cells and on TSEII in EC P19 cells, but they show differential activity on TSEII in COS7 cells and on ARE in HEK293 and EC P19 cells. On the TSEII enhancer, Hoxa1 and Hoxa1(NA-KR) have similar activity in EC P19 cells: neither the wild-type nor the mutant protein allowed TSEII-mediated reporter activation as long as Pbx and Prep expression vectors were not cotransfected; but they both activated the reporter at a similar level upon addition of Pbx and Prep vectors. Inversely, in COS7 cells, the Hoxa1(NA-KR) mutant caused a 5-fold activation of the TSEII reporter in the absence of Pbx and Prep, whereas Hoxa1 was inactive. Again, both proteins showed an equivalent activity in the presence of Pbx1a and Prep1 proteins. Therefore, the results obtained with the TSEII enhancer are equivalent to those reported with the hoxd4 autoregulatory element: the wild-type and mutant show distinct activities when they act alone, i.e. in the absence of Pbx cofactor (9,14). Thus, these data are in agreement with the conclusions drawn from the biochemical data reported by Phelan and Featherstone (9): the modulatory action of Pbx confers equivalent DNA-binding specificities and affinities to Hoxa1 and Hoxa1(NA-KR).
Conversely, the situation is somewhat different with the ARE enhancer. Although both Hoxa1 and Hoxa1(NA-KR) showed similar activities in COS7 cells, Hoxa1(NA-KR) was significantly less active than Hoxa1 on the ARE in EC P19 and HEK293 cells. This loss of activity was observed despite the availability of Pbx proteins in these cells, and was still observed when additional Pbx and Prep proteins were provided upon transfection. Similarly, Hoxa1–VP16 and Hoxa1(NA-KR)–VP16 fusion proteins provided a differential activation of the genomic hoxb1 target in EC P19 cells, as monitored by RT–PCR. Finally, this NA to KR substitution effect was also observed when changing the hoxa1 and hoxa1(NA-KR) gene dosage by a factor of 40 upon transfection. Therefore, since Hoxa1 and Hoxa1(NA-KR) proteins display distinct activity on the ARE in EC P19 cells, but are equally active on this enhancer in COS7 cells, the difference in activity Hoxa1 and Hoxa1(NA-KR) may show cannot simply be due to different intrinsic DNA binding. We propose that Hoxa1 and Hoxa1(NA-KR) undergo differently the modulatory activity of a cofactor available in EC P19 cells and absent from COS7 cells. The difference in endogenous abundance of such a modulator may also explain why Hoxa1(NA-KR) alone is active on TSEII in COS7 cells but not in EC P19 cells. In support of this hypothesis, it has been shown that the N-terminal arm of the homeodomain of certain proteins is involved in protein–protein contacts (27–29).
Several recent reports presented evidence that the availability of such modulators may vary from one cell type to another and in particular may explain the differences in activity of paralogy group-1 Hox proteins on enhancers like the ARE (12,13). HOXB1 and HOXA1 bind the ARE with the same affinity in vitro, but they differ in their ability to functionally interact with ARE, as a consequence of their differential dependence on Sox/Oct proteins to activate transcription. It is worth noting that the availability of Sox/Oct was pinpointed as responsible for the differential activity HOXB1 and HOXA1 show in EC P19 versus COS7 cells (13).
Acknowledgments
ACKNOWLEDGEMENTS
We thank B. Peers, F. Lemaigre, P. Bogaert, B. Hallet and the members of the Unit of Developmental Genetics (UCL) for discussions and reading the manuscript. We are grateful to M. S. Featherstone for providing the pPGKHoxa1(HD+), pPGKHoxa1(HD+)(WM-AA), pPGKHoxa1(HD+)(NA-KR), pPGKHoxa1VP16, pBSK-Pbx1a and pAdMLARE plasmids; to B. Peers for providing the pCS2-Prep and TSEII-luc plasmids; and to Th. Perlmann for the pCMX-PL1 expression vector. This work was supported by grant CT98-0227 from the BIOTECH Programme of the European Commission, and by the Belgian National Foundation for Scientific Research. S.R., C.M. and X.L. hold a FRIA fellowship from the Belgian National Foundation for Scientific Research.
REFERENCES
- 1.Maconochie M., Nonchèv,S., Morrison,A. and Krumlauf,R. (1996) Paralogous Hox genes: function and regulation. Annu. Rev. Genet., 30, 529–556. [DOI] [PubMed] [Google Scholar]
- 2.Favier B. and Dollé,P. (1997) Developmental functions of mammalian Hox genes. Mol. Hum. Reprod., 3, 115–131. [DOI] [PubMed] [Google Scholar]
- 3.Gehring W.J., Qian,Y.Q., Billeter,M., Furukubo-Tokunaga,K., Schier,A.F., Resendez-Perez,D., Affolter,M., Otting,G. and Wüthrich,K. (1994) Homeodomain-DNA recognition. Cell, 78, 211–223. [DOI] [PubMed] [Google Scholar]
- 4.Piper D.E., Batchelor,A.H., Chang,C.-P., Cleary,M.L. and Wolberger,C. (1999) Structure of a HoxB1–Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell, 96, 587–597. [DOI] [PubMed] [Google Scholar]
- 5.Pellerin I., Schnabel,C., Catron,K.M. and Abate,C. (1994) Hox proteins have different affinities for a consensus DNA site that correlate with the positions of their genes on the hox cluster. Mol. Cell. Biol., 14, 4532–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catron K.M., Iler,N. and Abate,C. (1993) Nucleotides flanking a conserved TAAT core dictate the DNA binding specificity of three murine homeodomain proteins. Mol. Cell. Biol., 13, 2354–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mann R.S. and Affolter,M. (1998) Hox proteins meet more partners. Curr. Opin. Genet. Dev., 8, 423–429. [DOI] [PubMed] [Google Scholar]
- 8.Li X., Murre,C. and McGinnins,W. (1999) Activity regulation of a Hox protein and a role for the homeodomain in inhibiting transcriptional activation. EMBO J., 18, 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phelan M.L. and Featherstone,M.S. (1997) Distinct HOX N-terminal arm residues are responsible for specificity of DNA recognition by HOX monomers and HOX.PBX heterodimers. J. Biol. Chem., 272, 8635–8643. [DOI] [PubMed] [Google Scholar]
- 10.Shanmugam K., Green,N.C., Rambaldi,I., Saragovi,H.U. and Featherstone,M.S. (1999) PBX and MEIS as non-DNA-binding partners in trimeric complexes with HOX proteins. Mol. Cell. Biol., 19, 7577–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Affolter M., Marty,T. and Vigano,M.A. (1999) Balancing import and export in development. Genes Dev., 13, 913–915. [DOI] [PubMed] [Google Scholar]
- 12.Saleh M., Rambaldi,I., Yang,X.-J. and Featherstone,M.S. (2000) Cell signaling switches HOX–PBX complexes from repressors to activators of transcription mediated by histone deacetylases and histone acetyltransferases. Mol. Cell. Biol., 20, 8623–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiRocco G., Gavalas,A., Pöpperl,H., Krumlauf,R., Mavilio,F. and Zappavigna,V. (2001) The recruitment of SOX/OCT complexes and the differential activity of HOXA1 and HOXB1 modulate the Hoxb1 auto-regulatory enhancer function. J. Biol. Chem., 276, 20506–20515. [DOI] [PubMed] [Google Scholar]
- 14.Phelan M.L., Sadoul,R. and Featherstone,M.S. (1994) Functional differences between HOX proteins conferred by two residues in the homeodomain N-terminal arm. Mol. Cell. Biol., 14, 5066–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pöpperl H., Bienz,M., Studer,M., Chan,S.-K., Aparicio,S., Brenner,S., Mann,R.S. and Krumlauf,R. (1995) Segmental expression of Hoxb-1 is controlled by a highly conserved autoregulatory loop dependent upon exd/pbx. Cell, 81, 1031–1042. [DOI] [PubMed] [Google Scholar]
- 16.Peers B., Sharma,S., Johnson,T., Kamps,M. and Montminy,M. (1995) The pancreatic islet factor STF-1 binds cooperatively with Pbx to a regulatory element in the Somatostatin promoter: importance of the FPWMK motif and of the homeodomain. Mol. Cell. Biol., 15, 7091–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Rocco G., Mavilio,F. and Zappavigna,V. (1997) Functional dissection of a transcriptionally active, target-specific Hox–Pbx complex. EMBO J., 16, 3644–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponglikitmongkol M., White,J.H. and Chambon,P. (1990) Synergistic activation of transcription by the human estrogen receptor bound to tandem responsive elements. EMBO J., 9, 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Umesono K., Murakami,K.K., Thompson,C.C. and Evans,R.M. (1991) Direct repeats as selective response elements for the thyroid hormone, retinoic acid and vitamin D3 receptors. Cell, 65, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phelan M.L., Rambaldi,I. and Featherstone,M.S. (1995) Cooperative interactions between HOX and PBX proteins mediated by a conserved peptide motif. Mol. Cell. Biol., 15, 3989–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goudet G., Delhalle,S., Biemar,F., Martial,J.A. and Peers,B. (1999) Functional and cooperative interactions between the homeodomain PDX1, Pbx and Prep1 factors on the somatostatin promoter. J. Biol. Chem., 274, 4067–4073. [DOI] [PubMed] [Google Scholar]
- 22.Graham F.L. and van der Erb,A.J. (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- 23.Zar J.H. (1996) Biostatistical Analysis, 3rd Edn. Prentice Hall, Upper Saddle River, New Jersey, USA.
- 24.Ferretti E., Marshall,H., Pöpperl,H., Maconochie,M., Krumlauf,R. and Blasi,F. (2000) Segmental expression of Hoxb2 in r4 requires two separate sites that integrate cooperative interactions between Prep1, Pbx and Hox proteins. Development, 127, 155–166. [DOI] [PubMed] [Google Scholar]
- 25.Jacobs Y., Schnabel,C.A. and Cleary,M.L. (1999) Trimeric association of Hox and TALE homeodomain proteins mediates Hoxb2 hindbrain enhancer activity. Mol. Cell. Biol., 19, 5134–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matis C., Chomez,P., Picard,J. and Rezsöhazy,R. (2001) Differential and opposed transcriptional effects of protein fusions containing the VP16 activation domain. FEBS Lett., 499, 92–96. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H., Hu,G., Wang,H., Sciavolino,P., Iler,N., Shen,M.M. and Abate-Shen,C. (1997) Heterodimerization of Msx and Dlx homeoproteins results in functional antagonism. Mol. Cell. Biol., 17, 2920–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zappavigna V., Sartori,D. and Mavilio,F. (1994) Specificity of HOX protein function depends on DNA-protein and protein-protein interactions, both mediated by the homeo domain. Genes Dev., 8, 732–744. [DOI] [PubMed] [Google Scholar]
- 29.Zappavigna V., Falciola,L., Helmer Citterich,M., Mavilio,F. and Bianchi,M.E. (1996) HMG1 interacts with HOX proteins and enhances their DNA binding and transcriptional activation. EMBO J., 15, 4981–4991. [PMC free article] [PubMed] [Google Scholar]