


Abstract
Altered expression of c-myc is implicated in pathogenesis and progression of many human cancers. Triple helix-forming oligonucleotides (TFOs) directed to a polypurine/polypyrimidine sequence in a critical regulatory region near the c-myc P2 promoter have been shown to inhibit c-myc transcription in vitro and in cells. However, these guanine-rich TFOs had moderate binding affinity and required high concentrations for activity. The 23 bp myc P2 sequence is split equally into AT- and GC-rich tracts. Gel mobility analysis of a series of short TFOs directed in parallel and anti-parallel orientation to the purine strand of each tract showed that only parallel CT and anti-parallel GT TFOs formed stable triplex on the AT- and GC-rich tracts, respectively. A novel full-length GTC TFO was designed to bind simultaneously in parallel and anti-parallel orientation to the polypurine strand. Gel-shift and footprinting assays showed that the new TFO formed a triple helix in physiological conditions with significantly higher affinity than an anti-parallel TFO. Protein-binding assays showed that 1 µM GTC TFO inhibited binding of nuclear transcription factors to the P2 promoter sequence. The novel TFO can be developed into a potent antigene agent, and its design strategy applied to similar genomic sequences, thus expanding the TFO repertoire.
INTRODUCTION
Triple helix formation offers a direct means of selectively manipulating gene expression in cells. Synthetic triple helix-forming oligonucleotides (TFOs) bind with high affinity and specificity to the purine strand in the major groove of homopurine–homopyrimidine sequences in double-stranded DNA. TFOs have proved effective in various gene-targeting strategies in living cells and, recently, in animals (reviewed in 1). Binding characteristics of TFOs depend on their nucleotide composition. Thus, oligonucleotides composed of pyrimidine bases (C and T) bind through Hoogsteen hydrogen bonds and are oriented parallel to the purine-rich strand of the target duplex, forming C+·G–C and T·A–T triplets. TFOs composed of purine bases (G and A), or of mixed purine/pyrimidine (G and T) bind in anti-parallel orientation through reverse Hoogsteen bonds, forming G·G–C and A·A–T or T·A–T triplets (2). In certain circumstances GT TFOs can bind in parallel orientation (3). Protonation of cytosine is required to form two Hoogsteen bonds in C+·G–C triplets, so that binding of CT TFOs is favored by low pH. Triplex formation by GA and GT TFOs is pH independent. Binding in each of these motifs can occur in physiological conditions, although cytosines must be modified to overcome pH dependence of CT TFOs.
Purine-rich tracts are frequently found in gene promoter regions and TFOs directed to these regulatory sites have been shown to selectively reduce transcription of the targeted genes, likely by blocking binding of transcriptional activators and/or formation of initiation complexes (reviewed in 4). Triplex-mediated modulation of transcription has potential application in therapy since it can be used, for example, to reduce levels of proteins thought to be important in disease processes. TFOs can also be useful molecular tools for studying gene expression and function. We are investigating the triplex approach as a means of down-regulating expression of the c-myc oncogene in cancer cells. c-myc is an attractive target for antigene agents in cancer cells because its expression drives cell proliferation. The importance of c-myc in cancer cell growth is emphasized by the findings that it is frequently amplified or involved in chromosomal rearrangements and that its expression is deregulated and augmented through various mechanisms in many cancers (5,6). Functions of the c-Myc protein, in tandem with its obligatory binding partner Max, include transcriptional activation and repression. Both activities appear to promote cell proliferation (7,8). Recent studies demonstrated that reduced expression of inducible c-myc was sufficient to cause regression of hematologic tumors in mice, suggesting that that this approach may have therapeutic potential (9).
Several sequences suitable for triplex formation are present in the c-myc gene. Of particular interest is a highly conserved polypurine/polypyrimidine tract in the c-myc P2 promoter region. P2 is the major c-myc promoter and gives rise to 75–90% of transcripts in almost all cells, with P1 contributing most of the remainder. The polypurine sequence lies from –39 to –61 relative to the P2 start site, and includes or overlaps binding sites for a number of transcription factors including Sp1 and Sp3 (10), ZF87/MAZ (11), ets (12), E2F (13,14) and Stat 3 (15). The site is required for transcription from P2 in the murine gene (11,16). Mutating the sequence in an episomal vector carrying the human gene severely disrupted transcription from both the P1 and P2 sites (17). A survey of TFOs directed to four different sequences in c-myc, including one in the P1 promoter region, showed that a P2-targeted TFO had highest antigene activity (18). Other studies showed that TFOs directed to this sequence had transcriptional inhibitory activity in vitro and in cells (19–21). However, all P2-targeted TFOs tested so far required high micromolar concentrations for triplex formation in vitro. This implied that high concentrations were required for antigene activity in cells. High oligonucleotide concentrations might be harmful in vivo, and cause non-specific effects that could confuse studies of cellular responses to reduced c-myc expression.
In the study reported here, we identified target sequence elements contributing to moderate binding affinity of P2-targeted TFOs. Based on our findings we designed a novel TFO incorporating parallel and anti-parallel binding motifs. Gel-shift and footprinting assays showed that the new TFO had significantly higher binding affinity than an anti-parallel TFO directed to the P2 sequence. In assays using nuclear extracts in vitro, the TFO at <1 µM inhibited binding of transcription factors to the targeted region, whereas similar concentrations of the anti-parallel TFO had little effect. The presence of double 5′ ends conferred resistance to digestion in vitro by nucleases in fetal bovine serum, but did not prevent rapid degradation of the TFO in cells. Intracellular instability was probably responsible for the TFO’s modest inhibitory activity in reporter gene assays. Our findings encourage progress towards synthesizing a nuclease-resistant TFO for testing in cells. Also, the strategy used to optimize triplex formation on the c-myc target can be applied to similar sequences in the genome, expanding the repertoire of sequences available for oligonucleotide-directed high-affinity triplex formation.
MATERIALS AND METHODS
Oligodeoxyribonucleotides
Unmodified oligonucleotides were purchased from Sigma-Genosys (The Woodlands, TX). Gel-purified oligonucleotides with 3′–3′ central linkages, double 5′ ends and 5-methyl cytosines were purchased from Oligos Etc. (Wilsonville, OR). All oligonucleotides were dissolved in water, and concentrations were determined spectrophotometrically using extinction coefficients for each base as follows: A, 15 400; C, 7300; G, 11 700; T, 8800.
Cell culture
MCF-7 and MDA-MB-231 breast cancer cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and passaged twice weekly.
Electrophoretic mobility shift assays (EMSA) to detect triplex formation
Either of two protocols was used as previously described (18). Briefly, either the pyrimidine-rich strand of the 23 bp target duplex, or TFOs were 5′-end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. Increasing concentrations of unlabeled TFO or duplex were added to labeled duplex or TFO, respectively, and reactions were incubated overnight at 37°C, except short CT TFOs, which were incubated at 4°C. Binding buffers contained either 90 mM Tris, 90 mM borate (TBM) pH 8; 50 mM 2-[N-morpholino] ethane sulfonic acid (Mes) pH 5.6; or 50 mM N-2-hydroxyethylpiperazine-N′-2-ethane sulfonic acid (HEPES) pH 7–7.4. All buffers contained 10 mM MgCl2. Electrophoresis at 10 or 20°C in native gels was done using the same buffer as in binding reactions. Films were scanned and bands quantified by densitometric analysis using Gel Pro software. Apparent dissociation constant values (Kd) were defined as concentration of TFO required to shift 50% of duplex to triplex DNA.
DMS footprinting
A 339 bp fragment containing the myc P2 promoter region was prepared and samples processed as described (21), except that TFO-binding reactions were incubated overnight at 37°C in HEPES–MgCl2, pH 7.2–7.4. Densitometric analysis of scanned X-ray films was done by determining total optical density units in the TFO target region, and correcting for unequal loading using values obtained from an unprotected region of identical size.
EMSA to examine protein binding
MCF-7 and MDA-MB-231 breast cancer nuclear extracts were prepared using the method of Dignam (22), with minor modifications as described (23). Double-stranded 32P-labeled probes with myc P1 and P2 promoter sequences were prepared by annealing complementary, gel-purified single-stranded oligonucleotides then filling in 5′ overhangs using Klenow fragment and [32P]dCTP (3000 Ci/mmol; Amersham Pharmacia, Piscataway, NJ). Labeled probes were further purified on native gels and eluted as described (24). The sequence of the polypurine strand of each probe was: Myc P1, 5′-GCGCTTATGGGGAGGGTGGGGAGGGTGGGGAAGGTGGGGA-GGAGACTCAGCC-3′; Myc P2, 5′-GAGGCTTGGCGGGAAAAAGAACGGAGGGAGGGATCGCGCT-3′. Double-stranded Sp1/Sp3 and control probes used as competitors were purchased from Promega (Madison, WI) or Geneka Biotechnology Inc. (Montreal, Quebec). Antibodies used in supershift assays were from Geneka. To detect nuclear protein binding to labeled probes, nuclear extracts (∼5 µg) were pre-incubated with 1 µg poly(dI–dC) (Sigma) for 10 min on ice, in 10 mM HEPES pH 7.4, 50 mM NaCl, 4% glycerol, 1 mM MgCl2, 1 mM EDTA, 0.5 mM DTT. Extracts were then added to ∼20 fmol probe (final concentration 1 nM) and incubated for 20 min at 10°C. Competitor oligonucleotides were added to extracts immediately before labeled probes. Antibodies were added to extracts pre-incubated with poly(dI–dC) as above, then incubation was extended for a further 20 min at 10°C before probes were added. To pre-form triplex, probes were pre-incubated with TFOs for 2–3 h at 37°C in HEPES pH 7.2–7.4, 10 mM MgCl2 before nuclear extract was added. Samples were resolved on 4% polyacrylamide gels (45 mM Tris, 45 mM borate, 0.5 mM EDTA) run at 200 V for ∼2 h at 10°C. Gels were dried and exposed at –70°C to X-ray films with intensifying screens.
Assay of oligonucleotide degradation in cell culture medium and in breast cancer cells
Oligonucleotides (200 pmol) were 5′-end-labeled and incubated at 37°C in 20 µl DMEM containing 10% fetal bovine serum which had been heat inactivated at 55°C for 30 min. Reactions were stopped at successive time points by adding 20 µl buffer containing 98% formamide and 10% EDTA, then heating at 90°C for 10 min. Samples were electrophoresed on a 15% polyacrylamide–7 M urea gel, and visualized by exposing the dried gel to X-ray film overnight. To test oligonucleotide stability in cells, MDA-MB-231 cells were plated in 24-well plates and grown for 24 h. Oligonucleotide 5′-end-labeled with 32P was mixed with unlabeled oligo to give a final concentration of 100 nm (∼6 × 106 c.p.m./ml). Cells were transfected for 6 h with labeled oligos using DOTAP reagent (Roche). Samples of transfection mix were retained for subsequent analysis. Transfection medium was removed, cells were washed three times with DMEM, and harvested by scraping into formamide/EDTA buffer, and heating at 90°C for 10 min, or grown for a further 24 or 48 h. Similar washes preceded each harvest. Radioactivity in each sample was determined, and equal numbers of counts loaded on a denaturing gel. The amount of intact TFO in each sample was determined by autoradiography and densitometric analysis.
Luciferase assays
MDA-MB-231 cells were transfected with a plasmid containing the luciferase gene driven by the c-myc P2 minimal promoter (pMyc262), and with the Renilla luciferase-expressing plasmid pRLTK, as control for transfection efficiency. TFO and control oligonucleotides were co-transfected with plasmids. Luciferase assays were done according to the manufacturer’s protocol (Dual Luciferase System, Promega).
RESULTS
Survey of short TFOs directed to different domains of the c-myc P2 target sequence
In an attempt to improve binding affinity of a TFO targeted to a critical regulatory sequence near the c-myc P2 transcriptional start site, we first identified the optimal binding motif for TFOs directed to two discrete domains present in the sequence. The 23 bp tract has an almost equal number of AT and GC base pairs. Distribution is uneven, however, with A grouped at the 5′, and G at the 3′ end of the purine strand. Anti-parallel motif TFOs were found previously to bind the P2 sequence with moderate affinity (19–21). We investigated whether the unbalanced composition of the target might be responsible for triplex instability. Using EMSA, we determined binding of a series of short 11mer PO TFOs targeted to either the A-rich 5′ or the G-rich 3′ segment of the 23 bp target duplex. In these initial studies, we tested GT TFOs in the parallel and anti-parallel motifs; GA TFOs in the anti-parallel motif, CT TFOs and a parallel GT TFO containing one cytosine (Table 1). This approach allowed us to identify the preferred binding motif for each segment of the target. We 32P-labeled the TFOs and incubated them with increasing concentrations of unlabeled duplex to detect a shift from single-stranded to triple-stranded DNA. The use of labeled TFOs also allowed us to determine whether the TFOs formed alternative structures with themselves or other single-stranded DNA. In comparing TFOs targeted to the G-rich 3′ sequence, we determined that an 11mer GT TFO (1-GT) binding in the anti-parallel motif was able to form triple helix on the 23-bp target (Table 1). Triplex was detected at 50 nM added duplex and increased to a maximum level at 0.5–1 µM. Two prominent gel-shifted bands were seen with 1-GT, possibly indicating that binding of this short TFO was partially unstable under gel-running conditions. The shifted bands represented complexes of TFO with duplex DNA since under identical conditions, 1 µM of unlabeled TFO, or of each single strand of the duplex caused no mobility shift (data not shown). This also indicated that the GT TFO had little tendency to associate with itself and other G- or C-rich single-stranded oligonucleotides. The formation of triplex was further confirmed in experiments where the 23-bp target was labeled and incubated with increasing concentrations of 1-GT (data not shown).
Table 1. Triplex formation by TFOs directed to c-myc P2 sequence.

aShort TFOs 1- and 2-targeted to regions 1 and 2 of the myc P2 sequence, respectively.
bTFO orientation to the purine-rich strand of the target sequence.
cTriple helix formation detected by EMSA as described in Materials and Methods.
dAssayed at pH 5.6.
eSynthesized with 5-methyl cytosine (C) and central 3′–3′ linkage (–).
fSequence non-complementary to the target sequence.
An anti-parallel 11mer GA TFO (1-GA) also formed triplex but predominant multiple bands of higher mobility than the double-stranded target were detected at all concentrations of duplex (data not shown). These non-triple-helical structures may represent TFO detached from the target during electrophoresis, or complexes of TFO with unlabeled purine or pyrimidine strands detached from the duplex. We did not characterize this further, since the GA TFO appeared to form triplex less efficiently than its GT counterpart. No other short TFO formed detectable triplex with the G-rich region (Table 1). We then tested a series of TFOs directed to the A-rich segment of the target. A CT TFO (2-CT), binding in parallel orientation and at acidic pH formed triplex that was essentially complete in the presence of 1 µM added duplex (Table 1). The triplex formed by 2-CT was also unstable and could be detected only when samples were incubated and gels run at low temperatures (4 and 10°C, respectively). No other short TFO formed detectable triplex on this sequence under any conditions, indicating that weak binding to this region probably contributed to the moderate affinity of full-length anti-parallel TFOs.
Design and binding affinity of parallel/anti-parallel GTC TFO
These results suggested that the preferred TFO-binding motif varied within the 23-bp myc P2 target sequence. The G-rich 3′ segment was most efficiently bound by an anti-parallel GT TFO, while only a parallel CT TFO was able to form triplex on the A-rich 5′ segment. The 11mer TFOs, however, could not be proposed as antigene agents since they did not form stable triplexes at physiological temperatures. To initiate design of a TFO with high binding affinity and specificity, we investigated whether the GT and CT short TFOs could be linked to create a molecule capable of binding simultaneously in parallel and anti-parallel orientations to the target purine strand.
A possible pitfall was that intermolecular base-pairing between groups of guanines and cytosines in the TFO could interfere with triplex formation. To investigate this, we incubated 2-CT at 100-fold molar excess with 32P-labeled 1-GT, and used EMSA to detect possible complex formation. No gel-shifted complexes were observed, and 2-CT had no adverse effect on triplex formation by 1-GT, suggesting that the TFOs were not associating in stable base-paired complexes. To test whether the two unlinked short oligos might act cooperatively to form a stable triplex, we used EMSA in which both TFOs together at 10 µM were incubated with labeled duplex at low pH to allow binding by 2-CT as well as 1-GT. No difference in binding affinity was observed between the short TFOs alone or paired, and a full-length CT TFO bound with ∼2-fold higher affinity, suggesting that short TFOs did not form a triple helix in a cooperative manner (data not shown). This is consistent with results of a recent study of short TFOs targeting neighboring sequences (25).
These findings prompted design of a novel TFO in which 1-GT and 2-CT were synthesized contiguously to form equal domains with opposite polarity (Table 1). The novel TFO had two 5′ ends with a central 3′–3′ phosphodiester linkage. Cytosines were methylated at the C5 position to promote binding of the CT portion at physiological pH (26). The central cytosine present in the purine-rich target strand was matched with a thymine in the parallel portion of the TFO. Thymine, which can form a single Hoogsteen bond with cytosine, is recognized as the best natural base to target this pyrimidine inversion (27).
EMSA using labeled target duplex incubated with increasing concentrations of unlabeled TFO confirmed that binding by the GTC TFO at pH 5.6 and 7.2 was similar, with 50% binding observed at concentrations between 100 and 250 nM in both conditions (Fig. 1). This represented a 50–125-fold molar excess of TFO over target. Triplex formation was complete at a TFO molar excess of 250–500-fold. A full-length CT TFO without modified cytosines bound with comparable affinity to the GTC TFO at pH 5.6. Stability of the full-length TFO must have been conferred by binding to the A-rich region, since no binding to the G-rich portion of the target was detected with the short CT TFO at pH 5.6. The full-length TFO did not form detectable triplex at pH 7.2.
Figure 1.

Triplex formation by 23mer TFOs on 23 bp c-myc P2 sequence. The pyrimidine-rich strand of the target duplex was labeled with 32P and annealed to the complementary purine-rich strand. Indicated concentrations of TFOs Myc CT or Myc GTC were incubated with duplex (2 nM) overnight at 37°C and pH 5.6 or 7.2, except Myc CT at pH 7.2, where incubation was at 4°C. Complexes were resolved on non-denaturing gels at the same pH and at 20°C. T, triplex DNA; D, duplex DNA.
We then used DMS footprinting to examine binding affinity and specificity of the parallel/anti-parallel TFO. This assay also allowed direct comparison between the GTC TFO and a full-length anti-parallel GT TFO Myc-GT. We previously drew attention to the difficulty of using EMSA to examine binding affinity of the GT TFO due to the small mobility shift observed (21). Binding reactions for footprinting were carried out at pH 7.2–7.4. Figure 2A shows that the GTC TFO formed triple helix with equally high specificity and significantly improved affinity compared with an anti-parallel GT TFO. Densitometric analysis showed that the GTC TFO at 0.01, 0.1 and 1 µM reduced cleavage in the target sequence to ∼72, ∼25 and ∼17%, respectively, of a control sample without oligonucleotide, whereas 10 µM GT TFO caused ∼55% reduction. The effect of Myc-GT was consistent with previous DMS footprinting experiments (21). Footprinting also showed that the single guanine at the 3′ end of the A-rich sequence was protected by 1 µM GTC but not by 20 µM anti-parallel TFO, indicating that only the former was able to bind this region of the target (Fig. 2B). The three guanines at the 5′ end of the target were not protected, which may reflect unstable binding by the adjacent methyl cytosines in the GTC TFO. The pH-dependent CT TFO caused no footprint at pH 7.2, confirming that triplex formation was required to protect from cleavage.
Figure 2.

DMS footprinting assays showing specificity and extent of triplex formation by parallel/anti-parallel TFO. A 339 bp fragment the of c-myc gene was labeled with 32P on the strand containing the 23 bp polypurine target sequence. The fragment was incubated alone, or with TFOs at concentrations indicated above each lane, overnight at 37°C in HEPES–MgCl2 at pH 7.2–7.4. Samples were treated with 0.5% DMS for 3 min, reactions were stopped, then DNA was recovered and treated with piperidine at 95°C for 30 min to cleave at methylated guanines. DNA was subjected to three rounds of lyophilization, then resuspended in formamide loading buffer and run on sequencing gels. The position and sequence of the target site, confirmed by sequencing, is shown on the right of each panel. The guanine protected by Myc GTC, but not by Myc GT, is indicated with arrowheads in each panel. (A) View of entire gel. (B) Target region of a separate gel isolated and magnified.
Inhibition of nuclear protein binding by parallel/anti-parallel TFO
Since the GTC TFO displayed improved affinity without loss of target specificity, we investigated its ability to block binding of nuclear proteins to the c-myc promoter sequence. A 40 bp probe including 5′ and 3′ c-myc sequences flanking the TFO target site was used in gel-shift assays with breast cancer cell nuclear extracts. To ensure that proteins known to bind the target sequence were present in MCF-7 nuclear extracts we first identified major bands observed when extracts were incubated with the 32P-labeled 40 bp probe containing the target sequence. All bands except one (labeled as non-specific) were competed away by excess unlabeled myc probe but not by unrelated DNA probes, confirming sequence-specificity of protein binding (data not shown). A probe containing a high-affinity binding site for Sp1 and other Sp family transcription factors competed away three major bands (data not shown). To specifically identify Sp family proteins binding to the P2 probe, we pre-incubated nuclear extracts with antibodies against Sp1 and Sp3. Figure 3A shows that one major band was specifically abrogated by an antibody against Sp1, and the Sp3 antibody removed two secondary bands. This indicated that antibodies either blocked Sp1/Sp3 DNA-binding sites, or formed large protein complexes that could not enter the gel. Control immunoglobulin had no effect on protein binding. These findings confirmed previous reports that Sp1 and Sp3 were among the transcription factors binding the P2 polypurine tract of the c-myc promoter (10).
Figure 3.

Identification of proteins recognizing myc P2 double-stranded, but not single-stranded DNA. A double-stranded probe containing the TFO target and flanking sequence was labeled using [32P]dCTP and the Klenow fragment of DNA polymerase then incubated with nuclear extracts from MCF-7 breast cancer cells. Protein–DNA complexes were resolved on 4% polyacrylamide non-denaturing gels at 10°C in 0.5× TBE. (A) Nuclear extracts were pre-incubated with rabbit polyclonal antibodies against Sp1 or Sp3, or with normal rabbit IgG, before adding Myc P2 DNA probe, incubating again then resolving complexes by gel electrophoresis. Arrowheads and arrows indicate Sp1 and Sp3 protein complexes, respectively. (B) Single-stranded oligos were added to labeled P2 probe simultaneously with nuclear extracts, incubated, then resolved on a gel. U, unbound DNA probe; ns, band representing protein(s) binding non-specifically to P2 probe.
We next investigated whether Sp1 and Sp3 could bind to either of the single strands making up the Myc2 probe, or to the GTC TFO and control oligo. If this occurred, inhibition by the TFO of protein binding to target DNA could be due to competition for single-stranded DNA binding (decoy effect) rather than to triplex formation. Conditions of this experiment were identical to those used to examine effects of double-stranded DNA competitors. Oligonucleotides (1 µM) were added to the probe simultaneously with nuclear extracts, to minimize any possible triplex formation by the TFO. In these conditions, protein binding was not reduced by either of the single strands making up the c-myc probe, the GTC TFO or a GTC oligo with scrambled sequence that did not form triplex (GTC-C, Table 1). This indicated that proteins detected in the assay recognized only the double-stranded myc P2 sequence, and that the TFO had no decoy effect (Fig. 3B).
Figure 4 shows that when the probe was pre-incubated at pH 7.2 for 2 h with 0.25, 0.5 and 1 µM GTC TFO before addition of nuclear extracts, the Sp1 and Sp3 complexes decreased in a concentration-dependent fashion. Pre-incubation with pH-dependent CT and non-triplex-forming GTC oligos had no inhibitory effect. Densitometric analysis of the Sp1 complex showed that 1 µM GTC TFO (∼1000-fold excess of TFO over probe) decreased the intensity of the band by ∼50% compared with the 1 µM GTC control oligo. The GT TFO at 1 µM reduced Sp1 binding by ∼10%. Other sequence-specific bands were similarly decreased by the TFOs, but not by control oligos. These results showed that triplex formation was required to block protein binding and that the GTC TFO blocked more efficiently than the GT TFO.
Figure 4.
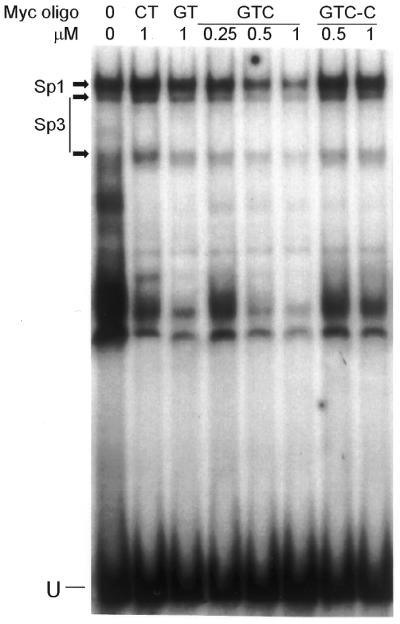
TFO-mediated inhibition of nuclear protein binding to myc P2 sequence. Myc P2 probe was pre-incubated alone (0) or with indicated concentrations of TFOs and control oligos in HEPES–MgCl2 for 2 h at 37°C. MCF-7 nuclear extracts were added, mixtures incubated, and complexes resolved on a polyacrylamide gel as described in Figure 3. Sp1 and Sp3 complexes are indicated. U, unbound DNA probe.
We then tested target specificity of the GTC TFO by examining its effect on a polypurine/polypyrimidine sequence in the c-myc P1 promoter region, which is also recognized by Sp1/Sp3 (28). Pre-incubation with an antibody against Sp1, done as described for the P2 probe, specifically removed two major complexes, confirming Sp1 binding (Fig. 5A). The Sp3 antibody also removed smaller bands and, in this case, caused a supershift (data not shown). Pre-incubation of the P1 probe with 1 µM GTC TFO did not affect binding of any proteins, whereas the Sp1 complex on the P2 probe was again reduced by ∼50% compared with the control chimeric oligo (Fig. 5B). This series of protein binding experiments clearly showed that the GTC TFO selectively blocked transcription factor binding to the P2 promoter sequence with increased efficiency compared with a conventional TFO. Similar results were obtained in experiments where nuclear extracts from MDA-MB-231 cells were used, showing that the TFO was effective in a different cell background.
Figure 5.

Specificity of GTC TFO inhibition of Sp1 and Sp3 binding to myc P2 probe. Gel-shift assays used double-stranded probes containing either the TFO target and flanking sequence (Myc P2) or a polypurine/polypyrimidine tract and flanking sequence from the c-myc P1 promoter region (Myc P1). (A) Antibody pre-incubation with the P1 probe was done as described in Figure 3. (B) Pre-incubation of P1 (left) or P2 (right) probes with 1 µM Myc GTC or GTC-C and subsequent protein-binding assays and electrophoresis were done as described in Figure 6. Arrowheads and arrows indicate Sp1 and Sp3 protein complexes, respectively. U, unbound probe.
Assay of nuclease resistance and activity in cells of parallel/anti-parallel TFO
Unmodified phosphodiester oligos are rapidly degraded by exo- and endonucleases. Since the GTC TFO lacks a 3′ end, we investigated whether double 5′ ends would increase resistance to 3′ exonuclease activity. We compared in vitro stability of the GTC and the GT TFOs in cell culture medium that contained 10% fetal bovine serum. Nuclease resistance of modified and unmodified oligos has previously been assessed by this method (29,30). We found that the GTC TFO was stable in serum-containing medium for at least 24 h while the unmodified GT TFO was significantly degraded within 2 h (Fig. 6A). We next tested GTC TFO resistance to intracellular nucleases by transfecting MDA-MB-231 breast cancer cells with 5′-radiolabeled TFO. Slight degradation of the TFO was already detectable at 6 h, and at 24 h only ∼35% of TFO present in the transfection mix remained intact (Fig. 6B). The half-life in cells of unmodified phosphodiester oligos has been estimated at 15–30 min, with both exonuclease and endonuclease activity causing degradation (31). Our results indicated that the presence of double 5′ ends prolonged partially the intracellular half-life of the GTC TFO, but did not ensure long-term stability in cells.
Figure 6.

Myc GTC resistance to degradation by nuclease activity in fetal bovine serum, and partial resistance in cells. Unmodified phosphodiester TFOs Myc GTC and Myc GT were labeled using [32P]ATP and T4 kinase. (A) Labeled oligos were incubated for the indicated times at 37°C in cell culture medium alone (–) or with 10% fetal bovine serum (+). Formamide loading buffer was added and samples resolved on a 15% polyacryamide–7 M urea denaturing gel. (B) Labeled oligo was left unprocessed (lane 1), or mixed with transfection reagent and cell culture medium (lanes 2–4). Cells were transfected for 6 h, then washed and lysed in formamide sample buffer (lane 3) or grown for a further 24 h (lane 4) before lysis. Equal amounts of radioactivity were loaded in each lane of a 7 M urea denaturing gel. Dried gels were exposed to X-ray films.
Lack of long-term stability was reflected in limited activity of Myc-GTC in reporter gene assays in breast cancer cells. We assayed activity of the TFO in cells by transfecting MDA-MB-231 cells with Myc-GTC or Myc-GTC-C along with a plasmid containing a Myc promoter-driven luciferase reporter gene. The TFO had only limited inhibitory effect (∼20% inhibition of luciferase activity at concentrations up to 1 µM in the transfection medium, data not shown). Since in vitro studies strongly suggest that the GTC TFO is the best approach to targeting the c-myc major promoter, future studies will focus on selecting optimal, practicable modifications to enhance nuclease resistance, allowing further examination of antigene effects in cells.
DISCUSSION
We addressed the problem of moderate binding affinity demonstrated by anti-parallel design TFOs targeted to a critical regulatory site close to the c-myc P2 promoter. We found that two discrete 11 bp domains in the target, one G-rich and the other A-rich, were bound most efficiently by 11mer anti-parallel GT (1-GT) and parallel CT TFOs (1-CT), respectively. Besides the GT and CT TFOs mentioned above, only one other TFO of the series examined was able to bind to either element of the target sequence. This was an anti-parallel GA TFO (1-GA), which formed a triplex on the G-rich sequence, but with low efficiency. Some GA TFOs can bind duplex DNA with very high affinity, while others tend to self-associate in hairpin, homoduplex and quadruplex structures, which can affect their ability to form triplex (32–34). TFOs directed to the c-myc P2 site appear to fall into the latter category, perhaps due to the long A-tract in both the target and the TFO. We previously reported that a full-length GA TFO directed to the myc P2 site had decreased ability to form triplex compared to its GT counterpart, and formed complexes that migrated more slowly than triplex in gel-shift assays (18). A phosphorothioate TFO with identical sequence was nevertheless able to reduce expression in HeLa cells of a luciferase gene driven by the c-myc promoter, but this required incubation of the plasmid with 10 000-fold excess of GA TFO before transfection, again suggesting low efficiency binding (20). Debin et al. (35) reported that optimal stability of triplex formation by 13mer GA TFOs depended on a high guanine content (∼85%) in the target sequence. A sub-optimal level of guanine in the P2 sequence (72% in the G-rich domain and 52% in the 23 bp tract) may also have contributed to low efficiency binding by short and full-length GA TFOs.
Although the 11mer GT TFO bound to the G-rich target, the triplex was unstable in native gels at temperatures ≥25°C (data not shown). We reported a similar result with the full-length P2-directed GT TFO, suggesting that this TFO may have formed only a partial triplex, likely on the G-rich but not the A-rich domain (21). The failure of the 11mer oligo (2-GT) forming the 3′ portion of the long TFO to form triplex, and the partial DMS footprint observed with the 23mer TFO, support this conclusion.
In contrast to 2-CT, the short TFO targeted to the A-rich part of the target sequence, 1-CT was not able to bind the G-rich portion even at low pH and temperature. The latter TFO has a high number of adjacent cytosines, which may have affected triplex formation by causing the TFO to self-associate through interaction of cytosines in the i-motif (36). In addition, adjacent C+·G–C triplets may be destabilized by electrostatic repulsion between the protonated cytosines (37). The full-length CT TFO formed a triplex quite efficiently at 37°C in low pH, indicating that the T·A–T triplets had a general stabilizing effect. The stability of these triplets was emphasized by our finding that only the short CT TFO was able to form a detectable triplex on the A-rich part of the target. A parallel-binding 11mer GT TFO (2-GTC) with a single cytosine opposite the isolated guanine in the target did not bind to the A-rich sequence. Interestingly, a similar TFO was found to be optimal for binding a 16 bp target sequence with an extremely unbalanced distribution of GC and AT pairs (38). In this case, six guanines, clustered at the 3′ end of the purine strand, were matched with guanine in the third strand. One guanine present in a 5′ stretch of eight adenines was matched with a third strand cytosine. Clearly, the c-myc sequence, although somewhat similar, did not favor this unusual third strand composition.
Detailed examination of the myc P2 sequence resulted in design of a novel parallel/anti-parallel TFO with significantly improved binding affinity. To the best of our knowledge, this approach to targeting an unbalanced sequence has not been reported before. Use of TFOs with central 3′–3′ linkages has been described previously in the alternate strand binding strategy, where purine tracts adjacent but on opposite strands can be targeted by TFOs that cross over from one strand to the other. These TFOs consist of two domains binding in the same triplex motif (either parallel or anti-parallel), usually with a linker structure at the central interface (39,40). Continuously synthesized TFOs without linkers, that bind to alternate strands in opposite triplex motifs, have also been developed (41). We reasoned that weak binding of thymine to cytosine at the pyrimidine inversion would offer flexibility at the 3′–3′ junction. However, we did not test any alternative linker structures. It is possible that non-nucleotide linkers may improve binding affinity of parallel/anti-parallel TFOs such as Myc-GTC.
The GTC TFO described here, binding with opposite polarity to adjacent sequences on the same strand, showed higher affinity than any previously described TFO targeted to the same critical sequence in c-myc. Further improvements in design of this TFO might enhance binding affinity even more, and reduce concentrations required to compete with transcriptional activators in cells. For example, in the CT portion of the TFO, replacement of methyl C with 8-oxo-2′ deoxyadenosine would likely improve binding at neutral pH (42). Alternatively, replacing DNA in the pyrimidine portion with 2′-alkyl-modified RNA could improve binding and increase nuclease resistance (43,44). Other base and backbone modifications have been found to improve binding and nuclease resistance of CT TFOs, and may be suitable for the c-myc sequence (reviewed in 45,46).
Since we found that double 5′-ended structure was only partially protective against degradation of the TFO by nucleases, modifications to enhance nuclease resistance will be required for the TFO to have extended life in cells. Phosphorothioate (PS) modification, in which sulfur replaces non-bridging oxygens in internucleotide linkages, has been widely used for antisense oligonucleotides, and has little, if any, inhibitory effect on binding of anti-parallel TFOs. Unfortunately, PS linkages severely reduce binding by parallel motif TFOs and cannot be used in this context. Conversely, other backbone modifications, like 2′-alkylated ribose and N3′-P5′ phosphoramidate linkages, are advantageous for parallel-binding TFOs but detrimental to anti-parallel binding (46). These limitations, as well as possibilities for improving binding affinity along with nuclease resistance, will help guide the choice of strategy for developing a c-myc TFO for use in cells.
Our approach to designing an improved TFO for the unbalanced sequence in c-myc may be useful for other genes. Database searches revealed other sequences in the human genome similar to the one in c-myc, some associated with known genes. As the genome becomes more fully characterized, and genes important in disease are identified, extending the repertoire of sites available for high-affinity triplex formation will be of increasing interest.
Acknowledgments
ACKNOWLEDGEMENTS
MCF-7 and MDA-MB-231 cells were kind gifts from Steven Rosenszweig and Michael Mitas, respectively, Medical University of South Carolina. This work was supported by NIH grant CA70735 to C.V.C. E.M.M. is supported by post-doctoral award DAMD17-00-1-0339 from US Army Breast Cancer Research Program.
REFERENCES
- 1.Casey B.P. and Glazer,P.M. (2001) Gene targeting via triple-helix formation. Prog. Nucleic Acid Res. Mol. Biol., 67, 163–192. [DOI] [PubMed] [Google Scholar]
- 2.Praseuth D., Guieysse,A.L. and Helene,C. (1999) Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta, 1489, 181–206. [DOI] [PubMed] [Google Scholar]
- 3.de Bizemont T., Duval-Valentin,G., Sun,J.S., Bisagni,E., Garestier,T. and Helene,C. (1996) Alternate strand recognition of double-helical DNA by (T,G)-containing oligonucleotides in the presence of a triple helix-specific ligand. Nucleic Acids Res., 24, 1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winters T.A. (2000) Gene targeted agents: new opportunities for rational drug development. Curr. Opin. Mol. Ther., 2, 670–681. [PubMed] [Google Scholar]
- 5.Spencer C.A. and Groudine,M. (1991) Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res., 56, 1–48. [DOI] [PubMed] [Google Scholar]
- 6.Grandori C., Cowley,S.M., James,L.P. and Eisenman,R.N. (2000) The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell. Dev. Biol., 16, 653–699. [DOI] [PubMed] [Google Scholar]
- 7.Dang C.V. (1999) c-Myc target genes involved in cell growth, apoptosis and metabolism. Mol. Cell. Biol., 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coller H.A., Grandori,C., Tamayo,P., Colbert,T., Lander,E.S., Eisenman,R.N. and Golub,T.R. (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling and adhesion. Proc. Natl Acad. Sci. USA, 97, 3260–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felsher D.W. and Bishop,J.M. (1999) Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell, 4, 199–207. [DOI] [PubMed] [Google Scholar]
- 10.Majello B., De Luca,P., Suske,G. and Lania,L. (1995) Differential transcriptional regulation of c-myc promoter through the same DNA binding sites targeted by Sp1-like proteins. Oncogene, 10, 1841–1848. [PubMed] [Google Scholar]
- 11.Bossone S.A., Asselin,C., Patel,A.J. and Marcu,K.B. (1992) MAZ, a zinc finger protein, binds to c-MYC and C2 gene sequences regulating transcriptional initiation and termination. Proc. Natl Acad. Sci. USA, 89, 7452–7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roussel M.F., Davis,J.N., Cleveland,J.L., Ghysdael,J. and Hiebert,S.W. (1994) Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F-1. Oncogene, 9, 405–415. [PubMed] [Google Scholar]
- 13.Hiebert S.W., Lipp,M. and Nevins,J.R. (1989) E1A-dependent trans-activation of the human MYC promoter is mediated by the E2F factor. Proc. Natl Acad. Sci. USA, 86, 3594–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thalmeier K., Synovzik,H., Mertz,R., Winnacker,E.L. and Lipp,M. (1989) Nuclear factor E2F mediates basic transcription and trans-activation by E1a of the human MYC promoter. Genes Dev., 3, 527–536. [DOI] [PubMed] [Google Scholar]
- 15.Kiuchi N., Nakajima,K., Ichiba,M., Fukada,T., Narimatsu,M., Mizuno,K., Hibi,M. and Hirano,T. (1999) STAT3 is required for the gp130-mediated full activation of the c-myc gene. J. Exp. Med., 189, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asselin C., Nepveu,A. and Marcu,K.B. (1989) Molecular requirements for transcriptional initiation of the murine c-myc gene. Oncogene, 4, 549–558. [PubMed] [Google Scholar]
- 17.Albert T., Wells,J., Funk,J.O., Pullner,A., Raschke,E.E., Stelzer,G., Meisterernst,M., Farnham,P.J. and Eick,D. (2001) The chromatin structure of the dual c-myc promoter P1/P2 is regulated by separate elements. J. Biol. Chem., 276, 20482–20490. [DOI] [PubMed] [Google Scholar]
- 18.Catapano C.V., McGuffie,E.M., Pacheco,D. and Carbone,G.M. (2000) Inhibition of gene expression and cell proliferation by triple helix-forming oligonucleotides directed to the c-myc gene. Biochemistry, 39, 5126–5138. [DOI] [PubMed] [Google Scholar]
- 19.Kim H.-G. and Miller,D.M. (1995) Inhibition of in vitro transcription by a triplex-forming oligonucleotide targeted to human c-myc P2 promoter. Biochemistry, 34, 8165–8171. [DOI] [PubMed] [Google Scholar]
- 20.Kim H.G., Reddoch,J.F., Mayfield,C., Ebbinghaus,S., Vigneswaran,N., Thomas,S., Jones,D.E.,Jr and Miller,D.M. (1998) Inhibition of transcription of the human c-myc protooncogene by intermolecular triplex. Biochemistry, 37, 2299–2304. [DOI] [PubMed] [Google Scholar]
- 21.McGuffie E.M., Pacheco,D., Carbone,G.M. and Catapano,C.V. (2000) Antigene and antiproliferative effects of a c-myc-targeting phosphorothioate triple helix-forming oligonucleotide in human leukemia cells. Cancer Res., 60, 3790–3799. [PubMed] [Google Scholar]
- 22.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller T.L., Jin,Y., Sun,J.M., Coutts,A.S., Murphy,L.C. and Davie,J.R. (1996) Analysis of human breast cancer nuclear proteins binding to the promoter elements of the c-myc gene. J. Cell. Biochem., 60, 560–571. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Xodo L.E., Rathinavelan,T., Quadrifoglio,F., Manzini,G. and Yathindra,N. (2001) Targeting neighbouring poly(purine.pyrimidine) sequences located in the human bcr promoter by triplex-forming oligonucleotides. Eur. J. Biochem., 268, 656–664. [DOI] [PubMed] [Google Scholar]
- 26.Lee J.S., Woodsworth,M.L., Latimer,L.J. and Morgan,A.R. (1984) Poly(pyrimidine).poly(purine) synthetic DNAs containing 5-methylcytosine form stable triplexes at neutral pH. Nucleic Acids Res., 12, 6603–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gowers D.M. and Fox,K.R. (1999) Towards mixed sequence recognition by triple helix formation. Nucleic Acids Res., 27, 1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DesJardins E. and Hay,N. (1993) Repeated CT elements bound by zinc finger proteins control the absolute and relative activities of the two principal human c-myc promoters. Mol. Cell. Biol., 13, 5710–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peyman A. and Uhlmann,E. (1996) Minimally modified oligonucleotides - combination of end-capping and pyrimidine-protection. Biol. Chem. Hoppe-Seyler, 377, 67–70. [DOI] [PubMed] [Google Scholar]
- 30.Lacoste J., Francois,J.C. and Helene,C. (1997) Triple helix formation with purine-rich phosphorothioate-containing oligonucleotides covalently linked to an acridine derivative. Nucleic Acids Res., 25, 1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisher T.L., Terhorst,T., Cao,X. and Wagner,R.W. (1993) Intracellular disposition and metabolism of fluorescently-labeled unmodified and modified oligonucleotides microinjected into mammalian cells. Nucleic Acids Res., 21, 3857–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noonberg S.B., Francois,J.C., Garestier,T. and Helene,C. (1995) Effect of competing self-structure on triplex formation with purine-rich oligodeoxynucleotides containing GA repeats. Nucleic Acids Res., 23, 1956–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arimondo P.B., Barcelo,F., Sun,J.S., Maurizot,J.C., Garestier,T. and Helene,C. (1998) Triple helix formation by (G,A)-containing oligonucleotides: asymmetric sequence effect. Biochemistry, 37, 16627–16635. [DOI] [PubMed] [Google Scholar]
- 34.Arimondo P.B., Garestier,T., Helene,C. and Sun,J.S. (2001) Detection of competing DNA structures by thermal gradient gel electrophoresis: from self-association to triple helix formation by (G,A)-containing oligonucleotides. Nucleic Acids Res., 29, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Debin A., Laboulais,C., Ouali,M., Malvy,C., Le Bret,M. and Svinarchuk,F. (1999) Stability of G,A triple helices. Nucleic Acids Res., 27, 2699–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lacroix L., Mergny,J.L., Leroy,J.L. and Helene,C. (1996) Inability of RNA to form the i-motif: implications for triplex formation. Biochemistry, 35, 8715–8722. [DOI] [PubMed] [Google Scholar]
- 37.Kiessling L.L., Griffin,L.C. and Dervan,P.B. (1992) Flanking sequence effects within the pyrimidine triple-helix motif characterized by affinity cleaving. Biochemistry, 31, 2829–2834. [DOI] [PubMed] [Google Scholar]
- 38.Giovannangeli C., Rougee,M., Garestier,T., Thuong,N.T. and Helene,C. (1992) Triple-helix formation by oligonucleotides containing the three bases thymine, cytosine, and guanine. Proc. Natl Acad. Sci. USA, 89, 8631–8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horne D.A. and Dervan,P.B. (1990) Recognition of mixed sequence duplex DNA by alternate-strand triple-helix formation. J. Am. Chem. Soc., 112, 2435–2437. [Google Scholar]
- 40.Froehler B.C., Terhorst,T., Shaw,J.-P. and McCurdy,S.N. (1992) Triple-helix formation and cooperative binding by oligodeoxynucleotides with a 3′–3′ internucleotide junction. Biochemistry, 31, 1603–1609. [DOI] [PubMed] [Google Scholar]
- 41.Brodin P., Sun,J.S., Mouscadet,J.F. and Auclair,C. (1999) Optimization of alternate-strand triple helix formation at the 5′-TpA-3′ and 5′-ApT-3′ junctions. Nucleic Acids Res., 27, 3029–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krawczyk S.H., Milligan,J.F., Wadwani,S., Moulds,C., Froehler,B.C. and Matteucci,M.D. (1992) Oligonucleotide-mediated triple helix formation using an N3-protonated deoxycytidine analog exhibiting pH-independent binding within the physiological range. Proc. Natl Acad. Sci. USA, 89, 3761–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blommers M.J.J., Natt,F., Jahnke,W. and Cuenoud,B. (1998) Dual recognition of double stranded DNA by 2′-aminoethoxy-modified oligonucleotides: the solution structure of an intramolecular triplex obtained by NMR spectroscopy. Biochemistry, 37, 17714–17725. [DOI] [PubMed] [Google Scholar]
- 44.Stutz A.M., Hoeck,J., Natt,F., Cuenoud,B. and Woisetschlager,M. (2001) Inhibition of interleukin-4- and CD40-induced IgE germline gene promoter activity by 2′-aminoethoxy-modified triplex-forming oligonucleotides. J. Biol. Chem., 276, 11759–11765. [DOI] [PubMed] [Google Scholar]
- 45.Lacroix L. and Mergny,J.L. (2000) Chemical modification of pyrimidine TFOs: effect on i-motif and triple helix formation. Arch. Biochem. Biophys., 381, 153–163. [DOI] [PubMed] [Google Scholar]
- 46.Francois J.C., Lacoste,J., Lacroix,L. and Mergny,J.L. (2000) Design of antisense and triplex-forming oligonucleotides. Methods Enzymol., 313, 74–95. [DOI] [PubMed] [Google Scholar]