

Summary
Hearing impairment (HI) is the most common neurosensory disorder globally and is reported to be more prevalent in low-income countries. In high-income countries, up to 50% of congenital childhood HI is of genetic origin. However, there are limited genetic data on HI from sub-Saharan African populations. In this study, we investigated the genetic causes of HI in the Malian populations, using whole-exome sequencing. Furthermore, cDNA was transfected into HEK293T cells for localization and expression analysis in a candidate gene. Twenty-four multiplex families were enrolled, 50% (12/24) of which are consanguineous. Clustering methods showed patterns of admixture from non-African sources in some Malian populations. Variants were found in six known nonsyndromic HI (NSHI) genes, four genes that can underlie either syndromic HI (SHI) or NSHI, one SHI gene, and one novel candidate HI gene. Overall, 75% of families (18/24) were solved, and 94.4% (17/18) had variants in known HI genes including MYO15A, CDH23, MYO7A, GJB2, SLC26A4, PJVK, OTOGL, TMC1, CIB2, GAS2, PDCH15, and EYA1. A digenic inheritance (CDH23 and PDCH15) was found in one family. Most variants (59.1%, 13/22) in known HI genes were not previously reported or associated with HI. The UBFD1 candidate HI gene, which was identified in one consanguineous family, is expressed in human inner ear organoids. Cell-based experiments in HEK293T showed that mutants UBFD1 had a lower expression, compared to wild type. We report the profile of known genes and the UBFD1 candidate gene for HI in Mali and emphasize the potential of gene discovery in African populations.
Keywords: hearing impairment, UBFD1 candidate gene, admixture mapping, Mali, Africa
Graphical abstract
Hearing impairment (HI) is the most common neurosensory disorder globally, with limited understanding of its genetic causes in Africa. This study employed exome sequencing to identify known and novel genetic variants linked to HI in Mali, highlighting the potential for gene discovery (e.g., UBFD1) in highly genetically diverse African populations.
Introduction
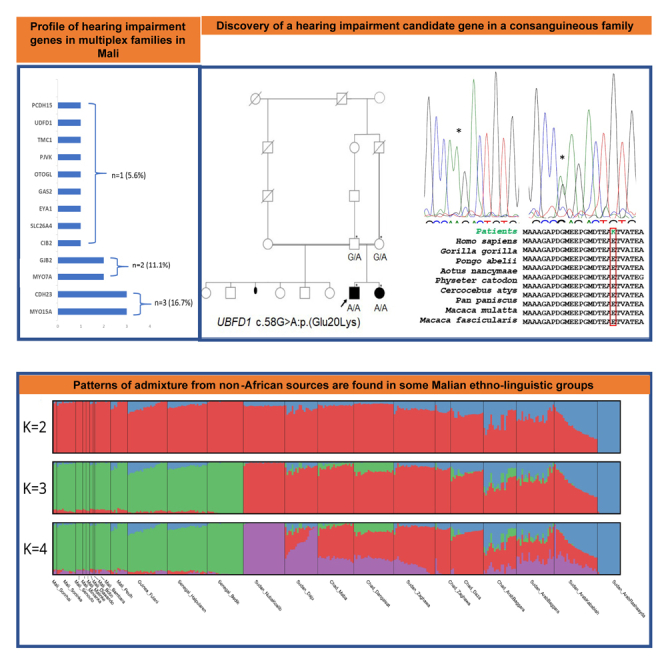
Congenital hearing impairment (HI) is the most common neurosensorial disorder, affecting 1–2/1,000 living newborns in high-income countries and up to 6/1,000 in Africa.1 Up to 30%–50% of congenital HI is associated with genetic etiology, with a higher percentage in high-income countries compared to sub-Saharan Africa (SSA), where environmental factors such as meningitis infections remain significant, and few genetics studies are performed.2,3,4,5 Inherited congenital HI is a genetically heterogeneous condition, with causative variants identified in over 120 genes to date.6 Clinically, HI can be broadly divided into nonsyndromic HI (NSHI), representing about 70%, whereas 30% are syndromic HI (SHI).7 Among populations of European ancestry, ∼50% of autosomal recessive (AR) NSHI are caused by variants in the gap junction protein beta-2 (GJB2, OMIM: 220290).8,9,10,11,12 However, systematic investigations of the GJB2 gene in numerous clinical settings among most of SSA has shown these genes to be rarely associated with HI,5,8,13,14,15 with exceptions found in Ghana and Senegal, where variants c.427C>T:p.(Arg143Trp) and c.94C>T:p.(Arg32Cys) in the GJB2 gene have been reported to have a high frequency.5,14,16 These data stress the need for extending genetic studies of HI to many African populations,3,4,15 specifically using whole-exome sequencing (WES) or whole-genome sequencing (WGS).17 In a recent study in schools for the deaf in Mali, we found that 29% of prelingual HI were assumed to be caused by genetic factors in a particular context of high consanguinity rate.2 In this study, we used ES to investigate multiplex HI families from Mali and found variants in known HI genes in MYO15A, CDH23, PCDH15, SLC26A4, OTOGL, TMC1, MYO7A, PJVK, CIB2, GJB2, GAS2, and EYA1, and identified a candidate gene, UBFD1.
Subjects, material, and methods
Ethics declaration
This study was conducted in full compliance with the Declaration of Helsinki. Institutional ethical committees/institutional review board (IRB) approvals were obtained from the Faculty of Medicine and Dentistry of the University of Sciences, Techniques, and Technologies of Bamako (Mali; no. 2020/129/CE/FMOS/FAPH), the University of Cape Town (Cape Town, South Africa; HREC ref. 691/2020), and Columbia University (New York, New York; IRB-AAAS2343). Informed consent and assent (for minor participants) were obtained before their enrollment in this study.
Participants and clinical assessments
Participants in this study were recruited mainly from the schools of the deaf in three different regions: the capital, Bamako, and Sikasso and Ségou. Only families that had at least two members affected with HI were included. All patients were carefully evaluated by medical geneticists, neurologists, and ear, nose, and throat specialists. Pure tone audiometry for air and bone conduction was performed. HI was classified according to the International Bureau for Audiophonology classification number 2/1 (BIAP; www.biap.org).
WES
DNA was extracted from peripheral blood using QIAGEN Blood DNA Kits, following the manufacturer’s instructions (QIAGEN, Germantown, MD). ES was performed following the previously published protocol.18,19 Briefly, DNA samples of probands and selected relatives of the families were sent for WES. Library and hybridization were done with an Illumina Nextera Rapid Capture Exome Kit (Illumina, San Diego, CA). Sequencing was performed on an Illumina HiSeq 2500 sequencer using the pair-end 150-bp run format. Sequencing data were processed using the Illumina DRAGEN Germline Pipeline version 3.2.8. High-quality reads were aligned to the human reference genome GRCh37/hg19 using DRAGEN software version 05.021.408.3.4.12, and after sorting and duplicate marking, variants were called, and individual genomic variant call format (GVCF) files were generated. Joint SNV and insertion or deletion (indel) variant calling were performed using Genome Analysis Toolkit (GATK) version 4.0.6.0.20 We used single sample variant calling with the GATK HaplotypeCaller module, which generated GVCFs per sample. The CombineGVCFs module was then used to merge the GVCF files into a multi-sample GVCF file. Lastly, the GenotypeGVCF module was used to jointly call variants.
Annotation and filtering strategy
Variants were annotated and filtered using ANNOVAR and custom scripts following a previously reported strategy.19 Briefly, variants were first prioritized based on the inheritance pattern following pedigree analysis, autosomal dominant (AD), and AR. Subsequently, rare variants with minor allele frequencies (MAF) <0.005 (for AR) and <0.0005 (for AD) in each population of the genome aggregation database (gnomAD, version 4.1.0) were retained. Known pathogenic (P) or likely pathogenic (LP) HI variants listed in ClinVar were also retained, regardless of their frequencies. dbNSFP version 3.0 was used to annotate, with several bioinformatic tools predicting the deleterious effects of the identified variants.21 In addition, the American College of Medical Genetics (ACMG) criteria were used to further classify variants.22 Lastly, for candidate variants, we considered whether (1) they occurred in known HI genes, (2) they had a predicted effect on protein function or pre-mRNA splicing (nonsense, missense, start-loss, frameshift, splicing, start-loss), and (3) they co-segregated with the HI phenotype within the family as previously described.19
Population’s structure
Principal-component analysis
Biallelic SNVs that passed all filters were extracted from the multi-sample VCF file into single-chromosome VCF files. Per sample, chromosome VCF files were then merged with the 1000 Genomes Project samples23 to produce a merged superset that was converted into PLINK binary format using PLINK2.24 The following filters were applied in the conversion: SNVs with MAF <0.05, missing >0.05 of their genotypes, or failed the Hardy-Weinberg equilibrium p < 0.001 were excluded. In addition, samples missing >0.10 of their genotypes were excluded. Next, SNVs obtained from the Mende population from Sierra Leone (MSL) that has been shown to harbor a large proportion of basal West African ancestry was used.25 This was achieved through the --keep command of PLINK2 while pruning to obtain only SNVs that were not in strong linkage disequilibrium using the following pruning parameters: r2 < 0.1, window size of 50 bp, and step size of 10 bp. The remaining 53,972 SNPs were then extracted from the merged superset, and 5 principal components (PCs) were computed using PLINK2. First- and second-degree relatives were excluded from PC calculations using the --king-cutoff 0.1875 parameter in PLINK2. PC plots were generated using a custom R script.
Additionally, in comparison to the present study, we examined common pathogenic variants in the top 10 recessive hearing impairment (HI) genes and the most frequently identified recessive genes in this cohort. We then profiled the allele frequencies (AF) across different genetic ancestry groups using gnomAD version 4.1.0.
Genotype array data and admixture mapping
We collected DNA samples from 50 participants who belonged to nine ethnic groups in Mali (Bambara, Bobo, Diawando, Malinke, Minianka, Peulh, Senoufo, Soninke, and Songhai). The DNA samples were genotyped on the Illumina Infinium H3Africa Consortium array (using BeadChip type H3Africa_2019_20037295_B1), designed for genotyping 2,271,503 genetic variants. The detailed protocol for the quality control, dataset information, and the genetic diversity and the admixture analysis is provided in the supplemental information.
Sanger sequencing
Sanger sequencing for available family members was performed for the segregation analysis of variants in selected families. Primer pairs covering the variants of interest were designed on the free access Integrated DNA Technologies (Coralville, IA) platform in our laboratory. PCR-amplified DNA products were Sanger sequenced using a BigDye Terminator version 3.1 Cycle Sequencing Kit and an ABI 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA) in the Division of Human Genetics, University of Cape Town.
Evolution of amino acid conservation
We aligned the sequences spanning the variants of candidate genes found here with non-human similar proteins to further support evidence of the amino acid residue conservation. A PSI-BLAST search against the non-redundant protein database was performed. Multiple sequence alignment was performed using MEGA X software.
In silico predictions of synonymous and splicing variants
The splicing variants in TMC1: c.2003 + 2T>C, OTOGL: c.209-9C>G, and MYO15A: c.5361-1G>A and the synonymous variant in SLC26A4: c.471C>T:p.(Pro157Pro) found in this study were assessed using different in silico predictors Human Splicing Finder (HSF)26 and SpliceAI.27
Secondary structural changes of the novel candidate gene
The isoform and protein sequences of UBFD1 (NM_019116.3; NP_061989.2) were obtained from the NCBI gene database in FASTA format, and secondary structures were predicted using the PSIPRED workbench.28 Three-dimensional (3D) structures of the proteins were modeled on the Swiss model using AlphaFold structures as the template with sequences retrieved from NCBI.29,30 Newly predicted protein structures were refined on the GalaxyWeb server,31 and Pymol software was used for visualization and structural analysis.32
In vitro functional HEK293T assay of the candidate genes
Site-directed mutagenesis
Mammalian expression plasmids expressing GFP-tagged UBFD1 (no. CW310112) were obtained from OriGene (Rockville, MD) and was used as template for site-directed mutagenesis (SDM) using custom primers acquired from Integrated DNA Technologies (Table S1). SDM was performed in our laboratory at the Department of Genetic Medicine, Johns Hopkins University (Baltimore, MD), using a protocol available upon request. Long-read sequencing of the plasmids was performed at Plasmidsaurus (Eugene, OR).
Cell culture, transfections, and visualization using confocal microscopy
HEK293T cells were cultured in complete DMEM (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific). Cells were cultured in a humidified incubator at 37°C with 5% CO2. The HEK293T cells were plated in 4-chamber dishes (density 4 × 105 cells/mL) 18 h before transfection. Cells were transiently transfected using TurboFect transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions, with 250 ng UBFD1 plasmid (empty, GFP-only, GFP-tagged wild type [WT], and mutant). Cells were stained with Hoechst dye for co-visualization of nuclear material (dilution 1:1,000). Live viewing was performed 48 h after transfection using an Olympus FV3000RS Confocal Microscope (Olympus, Center Valley, PA). Images were visualized and processed using the FV31S-W viewer software.
Western blots
HEK293T cells were plated (density 6 × 105 cells/mL) in 6-well plates 18 h before transfection. Cells were transfected using TurboFect transfection reagent according to the manufacturer’s instructions, with 2,000 ng of the WT- or mutant (MT)-UBFD1 plasmid containing the GFP tag and non-transfected cells with no plasmid DNA. Total protein lysates were collected at 48 h using radioimmunoprecipitation assay buffer and stored at −20°C. Proteins were briefly sonicated and concentrations were checked using a Qubit5 fluorometer. Then, 30 μg total protein was denatured (70°C, over 10 min) and loaded onto Novex WedgeWell 4%–20% Tris-glycine gel. The gel electrophoresis was run at 100 V over 90 min. Proteins were transferred onto iBlot 2 regular stacks, nitrocellulose, regular size (Invitrogen, Thermo Fisher, Scientific). Membranes were stained with Revert 700 Total Protein Stain (LI-COR Biosciences, Lincoln, NE).
The membranes were incubated with primary rabbit anti-UBFD1 antibody (Abcam, catalog no. ab240696, Waltham, MA) diluted at 3:10,000. Secondary antibody anti-rabbit (Thermo Fisher, catalog no. A11374) was diluted at 1:1,000 for UBFD1.
Results
Patients’ sociodemographic and phenotypic data
A total of 24 families with 63 affected individuals were enrolled (Figure 1A). The sex ratio favored males, representing 52.3% (n = 33), while females represented 47.7% (n = 30) of the participants. The onset of the disease was congenital or prelingual in 87.5% (n = 21) and postlingual in 12.5% (n = 3) of the families. The median age at molecular diagnosis was 15.9 years (range: 2–71 years). Parental consanguinity was reported in 50% (n = 12/24) of the families. HI was classified as being from severe to profound, and the sensorineural was the common mechanism involved (n = 17/24; 71%). In two families (9 and 18), HI was mixed moderate to severe. Following pedigree analysis and history of the disease, the inheritance pattern was consistent with AR in 87.5% cases (n = 21/24) and AD in 12.5% of cases (n = 3/24) (Figure S1). The sociodemographic and clinical characteristics are provided in Table S2.
Figure 1.

Recruitment of participants, population structure, and variants information
(A) Flow diagram of the study.
(B) Genes identified in this study.
(C) Proportion of novel variants.
(D) Type of variants identified in this study.
(E and F) Principal-component analysis (PCA) showing that samples cluster within African genomic data when compared to other non-African populations and populations from Gambia (GWD) and Sierra Leone (MSL) as geographically expected (red circle).
WES analysis
The average target region coverage was about 200×, with 96.30% of targeted regions covered at 10× or more. After applying various filtering criteria described in the subjects, material, and methods section, P and LP variants were found in 13 genes (Figures 1B–1D).
Population structure and admixture pattern
PCs constructed using genotype data obtained from families in this study were projected against data extracted from participants of continental ancestries in the 1000 Genomes reference panel (phase 3 version 5) and showed, as expected, that our samples clustered with other African populations (Figure 1E). When projected only against African populations, our samples clustered with populations from Gambia (GWD) and MSL as geographically expected (Figure 1F).
Furthermore, PC analysis (PCA) from the SNP-array data found a pattern of genetic diversity consistent with geographical differentiation on PC2 coordinates (Figure 2A), showing a west-east gradient among the studied populations with western and eastern African populations. On PC1 coordinates, there is a genetic differentiation between recent migrants in Sudan with non-African ancestry (negative values on PC1) and other African populations (positive values on PCA). Also, Fulani populations from Mali, Guinea, and Senegal (gray markers) have a cline of genetic diversity between western African populations and the recent migrants in Sudan and have gene flow from non-SSA sources. Between PC7 and PC8 coordinates (Figure 2B), Fulani individuals from different countries have their own clump due to their genetic affinities, while other studied populations from Mali have their own clump at the other side of the PCA space with Bono, Bambara, and Songhai individuals at the edges of this group. Furthermore, clustering methods showed patterns of admixture from non-SSA sources among the Fulani populations (Figure 2C).
Figure 2.

Population structure and admixture pattern
(A) PCA showing a pattern of genetic diversity consistent with geographical differentiation on PC2 coordinates.
(B) PCA7 and PCA8 showing that Fulani individuals from different countries have their own clump due to their genetic affinities, while other studied populations from Mali have their own clump at the other side of the PCA space with Bono, Bambara, and Songhai individuals at the edges of this group.
(C) Clustering methods showing patterns of admixture from non-sub-Saharan African sources (blue component) among the studied Fulani populations.
Variants in known genes associated with HI
Among the 24 families, 75% (18/24) were successfully solved or potentially solved (Figures 1A and 1B). Variants were identified in known HI genes in 94.4% of families (17/18; Figures 1C and 1D). In addition, variants in a novel candidate gene accounted for 5.6% of the solved families (1/18).
Finally, we categorized families into solved cases: P or LP variants in known HI genes, and potentially solved: variant of unknown significance (VUS) in known HI genes (Tables S3 and S4).
P and LP variants
PLP variants in known deafness genes included MYO15A (OMIM: 602666) observed in three families, CDH23 (OMIM: 605516) in two families, and GJB2 (OMIM: 220290) in two families. In addition, variants in TCM1 (OMIM: 606706), CIB2 (OMIM: 605564), SLC26A4 (OMIM: 605646), GAS2 (OMIM: 602835), MYO7A (OMIM: 276903), EYA1 (OMIM: 601653), and PJVK (OMIM: 610219) genes were each identified in a single family. Furthermore, a missense variant in the PCDH15 (OMIM: 605514) gene was found in one family with a CDH23 gene variant, suggesting a possible digenic associated HI (Figure 1B; Table 1). Among the known genes, 39.1% (n = 9) were previously reported to be associated with HI (Figure 1C).
Table 1.
Known and candidate genes associated with HI in Mali
| Gene | Family ID | Variants | Protein change | RS no. | RefSeq | Inheritance | Genotype | ACMG classification | gnomAD allele frequencya | Additional variants |
|---|---|---|---|---|---|---|---|---|---|---|
| Known SHI or NSHI genes | ||||||||||
| CDH23 | 6 | c.646C>G | p.(Leu216Val) | novel | NM_001171930 | AR | Hom | P | 0 | – |
| CDH23 | 13 | c.4445dupC c.4783G>A |
p.(Arg1483Glnfsa19) p.(Glu1595Lys) |
novel (rs778204574) | NM_022124 | AR | Comp Het | P LP |
0 0 |
– |
|
CDH23 PCDH15 |
17 | c.1353G>T c.4377C>A | p.(Leu451Phe) p.(Asp1459Glu) |
novel novel |
NM_001171933 NM_001142767 | AR | Hom | VUS LP |
0 0 |
– |
| CIB2 | 10 | c.409C>T | p.(Arg137Trp) | novel | NM_001271889 | AR | Hom | LP | 0 | |
| MYO7A | 4 | c.3978C>A | p.(Cys1326a) | known | NM_000260 | AR | Hom | P | 0.000001239 | – |
| 12 | c.6042C>G | p.(His2014Gln) | novel | NM_000260.4 | AR | Hom | VUS | 0 | CPLX2 c.37A>T:p.(Thr13Ser) | |
| SLC26A4 | 15 | c.2170G>A c.471C>T |
p.(Asp724Asn) p.(Pro157Pro) |
known known |
NM_000441.2 | AR | Comp Het | P LP |
0.000004350 0.00001673 |
CPA1 c.79C>T:p.(Arg27a) |
| Known SHI gene | ||||||||||
| EYA1 | 18b | c.1286A>G | p.(Asp429Gly) | known | NM_001370335.1 | AD | Het | LP | 0 | – |
| Known NSHI genes | ||||||||||
| GAS2 | 9 | c.533C>T | p.(Thr178Ile) | novel | NM_005256.4 | AD | Het | LP | 0 |
TXNDC11 c.697delC:p.(Leu233Cysfsa21) RPS6KA3 c.1246A>G:p.(Iso416Val) ABCC1 c.3505G>A:p.(Glu1169Lys) |
| GJB2 | 14 | c.427C>T | p.(Arg143Trp) | known (rs80338948) | NM_004004.6 | AR | Hom | P | 0.0001035 | – |
| 16 | c.427C>T | p.(Arg143Trp) | known (rs80338948) | NM_004004.6 | AR | Hom | P | 0.0001035 | – | |
| MYO15A | 2 | c.6331A>T | p.(Asn2111Tyr) | known | NM_016239 | AR | Hom | P | 0 | – |
| 3 | c.8158G>A | p.(Asp2720Asn) | known | NM_016239 | AR | Hom | P | 0.000005578 | – | |
| 11 | c.4347G>C c.5361-1G>A |
p.(Lys1449Asn) – |
known novel |
NM_016239 | AR | Comp Het | P P |
0.0000006196 | – | |
| OTOGL | 5 | c.209-9C>G c.5685C>A | -p.(Asp1895Glu) | known (rs768749743) | NM_173591 | AR | Comp Het | VUS VUS |
0.0000006637 |
PCDH15 c.3005A>G:p.(Glu1002Gly) CDH23 c.7793T>C:p.(Leu2598Pro) |
| PJVK | 7 | c.156T>G | p.(Val119Glu) | novel | NM_001042702.5 | AR | Hom | LP | 0 | – |
| TCM1 | 8 | c.2003 + 2T>C | – | novel | NM_138691 | AR | Hom | P | 0 | – |
| Candidate gene in NSHI | ||||||||||
| UBFD1 | 1 | c.58G>A | p.(Glu20Lys) | (rs567254290) novel | NM_019116 | AR | Hom | – | 0.00002844 | SLC19A2 c.47_48insGGC:p.(Ala16_Thr17insA) |
In family 9, both CDH23 and PCDH23 were found, suggesting a digenic condition, as previously reported. ACMG, American College of Medical Genetics; AD, autosomal dominant; AR, autosomal recessive; Comp Het, compound heterozygote; Het, heterozygote; Hom, homozygote; NSHI, nonsyndromic hearing impairment; rsID, reference SNP cluster ID; SHI, syndromic hearing impairment; VUS, variant of unknown significance.
Global allele frequency in gnomAD version 4.1.0 was accessed.
Clinical and genetic description of family 18 was reported previously.
Regarding the types of variants, missense variants were the most often detected (n = 17; 74%) followed by splice site variants (n = 3; 13%; Figure 1D), nonsense variants (n = 1; 4.3%; Table 1), frameshift variants (n = 1; 4.3%), and synonymous (n = 1; 4.3%). Homozygous variants were the most commonly identified (n = 12; 66.7%), which is consistent with the relatively high rate of consanguinity, followed by compound heterozygote and heterozygote (n = 4, 22.2%; n = 2, 11.1%), each. Several in silico predictions showed that these variants were deleterious (Table S3). According to ACMG criteria, 50% (n = 11/22) of variants in known HI genes were classified as P and 31.8% (n = 7/22) as LP (Table S4). Other variants in known HI genes that did not segregate with the phenotype within the families were found in five families (Table 1).
Commonly known P variants in gnomAD associated with recessive HI were not found in our cohort (Figure 3A). Similarly, variants identified in the most common recessive HI genes in this study had largely not been reported in the gnomAD database (Figure 3B).
Figure 3.

Comparative allele frequencies of common HI variants across global ancestries
(A) Allele frequency of the common variants in the top 10 recessive HI genes, showing variability across populations and not found in our cohort.
(B) Allele frequency of variants in the most common recessive genes identified in this study across different genetic ethnic groups, showing that these variants were mostly not reported in gnomAD.
VUS
Families with VUS in known HI genes were retained as potentially solved cases. In fact, VUS were identified in CDH23, MYO7A, and OTOGL (OMIM: 614925), representing 18.2% (n = 4/22; Table 1) in three different families.
Synonymous and splicing variant predictions
HSF predictions showed that the variant c.2003 + 2T>C in TMC1 affects the WT donor site. For variant c.5361-1G>A in MYO15A, two signals were identified, a broken WT acceptor site and a new acceptor splice site, both predicted to affect splicing. In addition, SpliceAI predictions showed that splice site variants in TMC1 affect the donor site, with a delta score of 0.13, and in MYO15A, the acceptor loss has a score of 0.98 and new acceptor gain has a score of 0.55. Furthermore, SpliceAI predicts two splicing site events for the synonymous variant in SLC26A4, including an acceptor gain with a delta score of 0.59 and an acceptor loss with a delta score of 0.29. However, HSF and SpliceAI predictions of the splicing variant in OTOGL (c.209-9C>G) do not reveal significant impacts on the splicing sites. The details are summarized in Tables S5–S7.
Candidate gene for HI
A novel candidate gene was identified in one family, UBFD1 (Figures 4A–4C; Table 1). A homozygous missense variant, c.58G>A:p.(Glu20Lys), was identified in the UBFD1 in family 1. This gene has not previously been associated with HI. UBFD1 is highly expressed in the structure and substructure of the inner ear in embryonic and adult mice and in human inner ear organoids (Figure 4D).
Figure 4.

Pedigree, genetic and expression data, and three-dimensional (3D) structure of candidate genes
(A) The pedigree of family 1 demonstrates an autosomal recessive inheritance pattern with consanguinity, indicated by double horizontal lines between the parents. The affected male is represented by a filled black square, and the affected female is represented by a filled black circle. Unaffected males and females are shown as unfilled squares and circles, respectively. An episode of miscarriage is represented with black dot symbols, asterisks indicate individuals genotyped in this study, and black arrows indicate the probands and capital letters are genotype information.
(B) Electropherograms showing the nucleotide change in family 1, indicated by asterisks.
(C) Portion of amino acids sequence showing the conservation of the amino acids of interest (red box) across a wide range of species.
(D) Uniform manifold approximation and projection 1 plots of UBFD1 gene expression profile in human inner ear organoids.
(E) The 3D structures of wild-type (WT) UBFD1 in green and mutant in light blue, showing major structural changes in mutant, including loss of helix and β sheets (red arrow).
Sanger sequencing and in silico predictions of variants in candidate genes
Variants identified in ES data were confirmed to segregate with HI using Sanger sequencing in family members with available DNA samples (Figures 4B and S1).
Evolutionary conservation of amino acids of novel candidate gene UBFD1
The NCBI PSI-BLAST search of the candidate gene UBFD1 against the non-redundant protein database found all residues to be highly conserved across a wide range of species retrieved from BLAST hits (Figures 4C and S1). As expected, there was substantial conservation across a wide range of species, consistent with the GERP and PhyloP scores for conservation, indicating a strong evolutionary and functional constraint on the region of the protein.
Secondary structures and 3D prediction of the novel candidate gene
Several major structural changes involving the alpha helices located at 18EAETVAT24, 66AAQ68, and beta sheets 161VLAV164, 173DAKAEE178, and 273MAFQ276 were observed in the mutant protein UBFD1. In addition, other changes were observed, including gain of new helical structures in the mutant protein located at 170AQ171 (Figures 4E and S3). These perturbations are expected to disrupt the normal function of UBFD1.
Expression of candidate genes in human inner ear organoids and mouse cochlear tissue
To investigate the expression pattern of the candidate gene (Ubfd1) identified in this study, we accessed human inner ear organoids single-cell RNA sequencing (RNA-seq) and small nuclear RNA-seq, and single-cell mouse RNA-seq data accessible from gEAR (http://umgear.org) at different ages (embryonic day 16, postnatal days [P] 1, P7, P15, and P30). The candidate gene had moderate expression in human inner ear organoids hair cells (HCs) and otic epithelial cells (Figure 4D).
Cochlear tissue and age-specific expression showed that Ubfd1 has strong expression profiles in different cell types of the cochlea and ages from embryonic age to P30. Interestingly, these Ubfd1s are similarly or more expressed in HCs when compared to the specific gene markers (Myo7a, Fgf8, and Slc26a5 for HCs, inner HCs, and outer HCs, respectively) for different cell types of the inner ear. Moreover, these genes were shown to be highly expressed in supporting and lateral wall cells (Figures S2A and S2B).
In vitro functional HEK293T assay of the candidate gene UBFD1
Live co-localization analysis with confocal microscopy showed that WT UBFD1 and MT UBFD1 reach normal subcellular cytoplasmic and nuclear localization. However, the expression was lower in MT compared to WT (Figures 5A–5D), 48 h after transfection. This was consistent with the western blot findings, with the specific band at the expected size of ∼60 kDa (Figure 5E).
Figure 5.

Confocal microscopy images and western blot data
(A–D) Co-visualization at 10× of the nuclear material (blue) with the GFP (green) signal in WT UBFD1 (A) and mutant (B), and at 40× of WT (C) and mutant (D) showing cytoplasmic localization, with decreased expression in mutant.
(E) Western blot analysis showing that both WT and mutant UBFD1 were at the expected size, with lower expression in the mutant.
Unsolved families through WES
In six families (25%), ES did not identify putative variants underlying HI (Figures 1A and S1). In family 22, compound heterozygote variants in ATP6V0A4 c.2155G>A:p.(Val719Ile) and c.1316A>T:p.(Asn439Ile) were identified, but they were not segregating with HI in the family. In family 24, two compound heterozygote variants were identified in known HI genes, one in USH2A c.1409_1419del:p.(Phe470Serf∗24) and c.1387_1405del:p.(Tyr463Thrfs∗12) and another in MYO15A c.5168T>G:p.(Val1723Gly) and c.386_387del:p.(Cys131Profs∗23). In Family 23, a segregating heterozygous pathogenic variant (c.2T>C:p.(Met1?)) was identified in the BLM gene. Although BLM has not been previously associated with HI, it is well-known for its association with Bloom syndrome (OMIM: 210900). This suggests that the finding is likely incidental. Lastly, no candidate gene variants were identified in three families, 19, 20, and 21.
Discussion
Exome sequencing (ES) has been widely used both in clinical and research settings in the past few decades to identify variants underlying human diseases.33 With about 1% of all human genes involved in the process of hearing and hundreds of HI patients with the Mendelian pattern, there are still many HI genes to discover.7,34 Despite the context of high consanguinity rate and interethnic marriages, genetic studies of HI have not been extensively performed in Mali.35,36,37 Sensorineural HI was the common mechanism of the disease found in this study, supporting previous findings.38 In addition, we took advantage of the WES data to provide a comprehensive admixture pattern of some ethnic groups from Mali, likely the first from that country. Interestingly, clustering methods showed patterns of admixture from non-SSA sources (blue component) among the studied Fulani populations as anticipated and surprisingly with other non-Fulani populations probably due to local admixture (Figure 2C). In Mali, the Fulani population, known as Peulh, have individuals with different degrees of admixture and without gene flow in contract with other non-Fulani populations. Their genetic diversity is similar to that of other western African populations (green component at K = 3 and K = 4), which is different than the components inferred for central and eastern Sahelian populations.
We successfully identified several variants associated with HI (Figure 1). Interestingly, we identified a novel candidate gene in this study, UBFD1. UBFD1 encodes for Ubiquitin Family Domain Containing 1 protein and is located at 16p12.2, an intracellular protein.39 UBFD1 is a ubiquitous gene that is expressed in mouse cochlear-specific tissue and human inner ear organoids. Molecularly, UBFD1 plays a role in RNA and cadherin binding function40,41 and was reported as having a direct interaction with the TMEM248 protein, a structural cell transmembrane protein that apparently plays a critical role in cell membrane integrity in humans.42 Using HEK293T experiments, we showed that the variant p.(Glu20Lys) decreases the expression of UBFD1 protein (Figure 5). These findings could suggest that in humans, the low expression of UBFD1 could lead to decreased TMEM248 expression, thus disrupting the cell membrane integrity. These cell experiments provided data to support the pathogenicity of the variant.
Of the known deafness genes, P or LP variants were found in MYO15A and CDH23, representing one-third of the cohort (Table 1), similar to the recent study from Ghana, when excluding families with GJB2-associated genes.15,17 In fact, several cohort studies reported these commonly HI-associated genes.43,44 CDH23 is associated with NSHI and Usher syndrome.45 In our cohort, CDH23 was associated with NSHI in three different families. However, considering the possible late onset of the retinal symptoms in Usher syndrome, these families with CDH23 variants could be either syndromic or nonsyndromic. In addition, in one family, a variant in PCDH15 was found in addition to CDH23, suggesting a digenic condition, as in previous studies.46 Moreover, variants in other known HI genes, including MYO7A, SLC26A4, PJVK, TMC1, CIB2, GAS2, and GJB2, were identified in our study. Importantly, most variants found in the most common HI genes in this study had not been previously reported in the gnomAD database, except for the GJB2 variant (p.Arg143Trp) among African and African American populations, which will contribute greatly to improving curation of global HI-gene pairs.
Notably, the compound heterozygote variants identified in SLC26A4 in family 11 included a synonymous variant c.471C>T:p.(Pro157Pro), which was reported in patients with NSHI.47 Furthermore, in a previous in vitro assay, this variant was shown to lead to the partial deletion of exon 5 and produce a truncated protein p.(Gly139Alafs∗6), which is classified as P, according to ACMG criteria.47 A segregating nonsense variant c.79C>T:p.(Arg27∗) in the CPA1 gene (not known to cause HI),48 was also identified in family 11, which was harboring the compound heterozygote variants in SLC26A4.48 The LP start loss variant c.2T>C:p.(Met1?) in BLM identified in family 23, has not been reported to be associated with HI. However, this variant was recently reported in ClinVar as LP for Bloom syndrome and hereditary cancer predisposition syndrome. It is more likely an incidental finding in this family, further supported by the fact that the proband has a medical history of surgery for an unspecified brain tumor. Therefore, more family members will need to be screened for this variant and be provided surveillance and anticipatory guidance for possible tumor development.
GJB2 variants have not been commonly reported to be associated with HI in most SSA populations, except for those in Ghana and Senegal.5,8,14,16 The Ghanaian founder variant p.(Arg143Trp), observed in families 14 and16, was recently reported in the Senegalese population, although these two countries do not share borders.16 The authors argued that this variant may have not been introduced in Senegal through regional migration, but rather the forced movement of people during the transatlantic slave trade.16 Further studies will provide more insights into the origin and migration pattern of this variant.
Although there is no approved treatment for HI to date, a recent study showed that a single dose of gene therapy has resulted in a robust recovery among Chinese and US children with recessive severe to profound HI caused by variants in OTOF gene.49,50,51 With gene therapy technology becoming increasingly promising, identifying the underlying genetic causes of HI is an imperative to better anticipate the therapeutic perspectives.
Conclusion
We report here the most comprehensive genetic study of HI in Mali to date, using WES, which displays a relatively high solved rate, of genetic causes among multiplex families. The study emphasizes the high level of genetic and allelic heterogeneity for HI genes, while revealing a high rate of homozygous variants in families, due to the high consanguinity rate in Mali. Most families had unique variants in known HI genes, with MYO15A and CDH23 being the most common, suggesting that future clinical diagnostic approaches should ideally use next-generation sequencing techniques, including the HI gene panel, exome sequencing, or genome sequencing. The UBFD1 gene had not previously been reported to be associated with HI. Further studies using a mouse model could reveal the mechanism by which UBFD1 leads to HI. The data emphasize the potential to discover genes in understudied African populations, which harbors the highest human genetic diversity. The data will improve the disease-gene pairs curation and advance the understanding of HI pathobiology globally.
Data and code availability
All the data supporting the present study are available from the corresponding author upon reasonable request. Additionally, variants found in this study were submitted to ClinVar (SCV005420291 and SCV005420311).
Acknowledgments
The authors are deeply grateful to all participants and their families; the Association Malienne de Sourds; the schools for the deaf in Bamako, Sikasso, and Ségou, Mali; and the President of the Association of Parents with Deaf Children. We thank the Center Hospitalier Universitaire du Point G, Bamako, Mali. This study was funded by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke grant no. U01HG007044 to G.L.; the Wellcome Trust, grant no. 107755Z/15/Z to A.W.; NIH/National Institute of Mental Health grant no. U01MH127692 and NIH/National Human Genome Research Institute grant no. U01-HG-009716 to A.W.; the African Academy of Science/Wellcome Trust grant no. H3A/18/001 to A.W.; and NIH/National Institute on Deafness and Other Communication Disorders grant nos. R01 DC01165, R01 DC003594, and R01 DC016593 to S.M.L.
Author contributions
Conceptualization: G.L., A.W., and S.M.L. Data curation: A.Y., I.S., A.W., S.M.L., and G.L. Formal analysis: A.Y., I.S., A.A., T.B., K.E., S.B., C.A.F.-L., and R.L. Funding acquisition: G.L., S.M.L., and A.W. Investigation: A.Y., G.L., O.T., K.E., A.d.B.M., C.A.K.C., M.A.S., C.d.K., E.T.A., and R.L. Project administration: C.d.K., M.J., and S.B. Resources: C.O.G., G.L., A.W., and S.M.L. Supervision: G.L., A.W., and S.M.L. Validation: G.L., S.M.L., and A.W. Writing – original draft: A.Y. Writing – review & editing: A.Y., A.W., I.S., S.M.L., R.L., and G.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xhgg.2024.100391.
Web resources
World Health Organization: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss
Hearing Loss homepage: https://hereditaryhearingloss.org/
Genome Aggregation Database: https://gnomad.broadinstitute.org/
Supplemental information
References
- 1.WHO Deafness and hearing loss. 2022. https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss
- 2.Yalcouyé A., Traoré O., Taméga A., Maïga A.B., Kané F., Oluwole O.G., Guinto C.O., Kéita M., Timbo S.K., DeKock C., et al. Etiologies of Childhood Hearing Impairment in Schools for the Deaf in Mali. Front. Pediatr. 2021;9 doi: 10.3389/fped.2021.726776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wonkam Tingang E., Noubiap J.J., Fokouo J.V., Oluwole O.G., Nguefack S., Chimusa E.R., Wonkam A. Hearing Impairment Overview in Africa: the Case of Cameroon. Genes. 2020;11:233. doi: 10.3390/genes11020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wonkam A., Noubiap J.J.N., Djomou F., Fieggen K., Njock R., Toure G.B. Aetiology of childhood hearing loss in Cameroon (sub-Saharan Africa) Eur. J. Med. Genet. 2013;56:20–25. doi: 10.1016/j.ejmg.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Adadey S.M., Wonkam-Tingang E., Aboagye E.T., Quaye O., Awandare G.A., Wonkam A. Hearing loss in Africa: current genetic profile. Hum. Genet. 2022;141:505–517. doi: 10.1007/s00439-021-02376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Camp G.S.R. Hereditary Hearing Loss Homepage. https://hereditaryhearingloss.org
- 7.Helga V.T.S., Smith D. Oxford University Press; 2013. Hereditary Hearing Loss and its Syndromes; p. 749. [Google Scholar]
- 8.Adadey S.M., Wonkam-Tingang E., Twumasi Aboagye E., Nayo-Gyan D.W., Boatemaa Ansong M., Quaye O., Awandare G.A., Wonkam A. Connexin Genes Variants Associated with Non-Syndromic Hearing Impairment: A Systematic Review of the Global Burden. Life. 2020;10 doi: 10.3390/life10110258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beheshtian M., Babanejad M., Azaiez H., Bazazzadegan N., Kolbe D., Sloan-Heggen C., Arzhangi S., Booth K., Mohseni M., Frees K., et al. Heterogeneity of Hereditary Hearing Loss in Iran: a Comprehensive Review. Arch. Iran. Med. 2016;19:720–728. [PMC free article] [PubMed] [Google Scholar]
- 10.Sloan-Heggen C.M., Bierer A.O., Shearer A.E., Kolbe D.L., Nishimura C.J., Frees K.L., Ephraim S.S., Shibata S.B., Booth K.T., Campbell C.A., et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016;135:441–450. doi: 10.1007/s00439-016-1648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang Y.B., Tang S.H., Li H.Z., Xu C.Y., Chen C., Xu Y.Z., Ding L.R., Xu X.Q. Mutation analysis of common deafness-causing genes among 506 patients with nonsyndromic hearing loss from Wenzhou city, China. Int. J. Pediatr. Otorhinolaryngol. 2019;122:185–190. doi: 10.1016/j.ijporl.2019.04.024. [DOI] [PubMed] [Google Scholar]
- 12.Plevova P., Tvrda P., Paprskarova M., Turska P., Kantorova B., Mrazkova E., Zapletalova J. Genetic Aetiology of Nonsyndromic Hearing Loss in Moravia-Silesia. Medicina (Kaunas) 2018;54 doi: 10.3390/medicina54020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wonkam A., Bosch J., Noubiap J.J.N., Lebeko K., Makubalo N., Dandara C. No evidence for clinical utility in investigating the connexin genes GJB2, GJB6 and GJA1 in non-syndromic hearing loss in black Africans. S. Afr. Med. J. 2015;105:23–26. doi: 10.7196/samj.8814. [DOI] [PubMed] [Google Scholar]
- 14.Adadey S.M., Manyisa N., Mnika K., de Kock C., Nembaware V., Quaye O., Amedofu G.K., Awandare G.A., Wonkam A. GJB2 and GJB6 Mutations in Non-Syndromic Childhood Hearing Impairment in Ghana. Article. Front. Genet. 2019;10 doi: 10.3389/fgene.2019.00841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wonkam A., Manyisa N., Bope C.D., Dandara C., Chimusa E.R. Whole exome sequencing reveals pathogenic variants in MYO3A, MYO15A and COL9A3 and differential frequencies in ancestral alleles in hearing impairment genes among individuals from Cameroon. Hum. Mol. Genet. 2021;29:3729–3743. doi: 10.1093/hmg/ddaa225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dia Y., Adadey S.M., Diop J.P.D., Aboagye E.T., Ba S.A., De Kock C., Ly C.A.T., Oluwale O.G., Sène A.R.G., Sarr P.D., et al. GJB2 Is a Major Cause of Non-Syndromic Hearing Impairment in Senegal. Biology. 2022;11 doi: 10.3390/biology11050795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wonkam A., Adadey S.M., Schrauwen I., Aboagye E.T., Wonkam-Tingang E., Esoh K., Popel K., Manyisa N., Jonas M., deKock C., et al. Exome sequencing of families from Ghana reveals known and candidate hearing impairment genes. Commun. Biol. 2022;5:369. doi: 10.1038/s42003-022-03326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yalcouyé A., Traoré O., Diarra S., Schrauwen I., Esoh K., Kadlubowska M.K., Bharadwaj T., Adadey S.M., Kéita M., Guinto C.O., et al. A monoallelic variant in EYA1 is associated with Branchio-Otic syndrome in a Malian family. Mol. Genet. Genom. Med. 2022;10 doi: 10.1002/mgg3.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wonkam-Tingang E., Schrauwen I., Esoh K.K., Bharadwaj T., Nouel-Saied L.M., Acharya A., Nasir A., Adadey S.M., Mowla S., Leal S.M., Wonkam A. Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment. Genes. 2020;11 doi: 10.3390/genes11111249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X., Wu C., Li C., Boerwinkle E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skoglund P., Thompson J.C., Prendergast M.E., Mittnik A., Sirak K., Hajdinjak M., Salie T., Rohland N., Mallick S., Peltzer A., et al. Reconstructing Prehistoric African Population Structure. Cell. 2017;171:59–71.e21. doi: 10.1016/j.cell.2017.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B., et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 28.McGuffin L.J., Bryson K., Jones D.T. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 29.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko J., Park H., Heo L., Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40:W294–W297. doi: 10.1093/nar/gks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chimera M., PyMOL L. PyMOL (Version 2.3). Schrödinger. 2019. http://www.pymol.org
- 33.Cabanillas R., Diñeiro M., Cifuentes G.A., Castillo D., Pruneda P.C., Álvarez R., Sánchez-Durán N., Capín R., Plasencia A., Viejo-Díaz M., et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med. Genom. 2018;11:58. doi: 10.1186/s12920-018-0375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Camp G.S.R. Hereditary Hearing Loss Homepage. https://hereditaryhearingloss.org
- 35.Landouré G., Dembélé K., Diarra S., Cissé L., Samassékou O., Bocoum A., Yalcouyé A., Traoré M., Fischbeck K.H., Guinto C.O., H3Africa Consortium A novel variant in the spatacsin gene causing SPG11 in a Malian family. J. Neurol. Sci. 2020;411 doi: 10.1016/j.jns.2020.116675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landouré G., Mochel F., Meilleur K., Ly M., Sangaré M., Bocoum N., Bagayoko K., Coulibaly T., Sarr A.M., Bâ H.O., et al. Novel mutation in the ATM gene in a Malian family with ataxia telangiectasia. J. Neurol. 2013;260:324–326. doi: 10.1007/s00415-012-6738-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yalcouyé A., Diallo S.H., Coulibaly T., Cissé L., Diallo S., Samassékou O., Diarra S., Coulibaly D., Keita 1 M., Guinto C.O., et al. A novel mutation in the GARS gene in a Malian family with Charcot-Marie-Tooth disease. Article. Mol. Genet. Genom. Med. 2019;7 doi: 10.1002/mgg3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lebeko K., Bosch J., Noubiap J.J.N., Dandara C., Wonkam A. Genetics of hearing loss in Africans: use of next generation sequencing is the best way forward. Pan Afr. Med. J. 2015;20:383. doi: 10.11604/pamj.2015.20.383.5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fenner B.J., Scannell M., Prehn J.H.M. Identification of polyubiquitin binding proteins involved in NF-kappaB signaling using protein arrays. Biochim. Biophys. Acta. 2009;1794:1010–1016. doi: 10.1016/j.bbapap.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M., et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 41.Guo Z., Neilson L.J., Zhong H., Murray P.S., Zanivan S., Zaidel-Bar R. E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci. Signal. 2014;7 doi: 10.1126/scisignal.2005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N., et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Budde B.S., Aly M.A., Mohamed M.R., Breß A., Altmüller J., Motameny S., Kawalia A., Thiele H., Konrad K., Becker C., et al. Comprehensive molecular analysis of 61 Egyptian families with hereditary nonsyndromic hearing loss. Clin. Genet. 2020;98:32–42. doi: 10.1111/cge.13754. [DOI] [PubMed] [Google Scholar]
- 44.Yan D., Tekin D., Bademci G., Foster J., 2nd, Cengiz F.B., Kannan-Sundhari A., Guo S., Mittal R., Zou B., Grati M., et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Article. Hum. Genet. 2016;135:953–961. doi: 10.1007/s00439-016-1697-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bork J.M., Peters L.M., Riazuddin S., Bernstein S.L., Ahmed Z.M., Ness S.L., Polomeno R., Ramesh A., Schloss M., Srisailpathy C.R., et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Q.Y., Yan D., Ouyang X.M., Du L.L., Yu H., Chang B., Johnson K.R., Liu X.Z. Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum. Mol. Genet. 2005;14:103–111. doi: 10.1093/hmg/ddi010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smits J.J., de Bruijn S.E., Lanting C.P., Oostrik J., O'Gorman L., Mantere T., DOOFNL Consortium, Cremers F.P.M., Roosing S., Yntema H.G., et al. Exploring the missing heritability in subjects with hearing loss, enlarged vestibular aqueducts, and a single or no pathogenic SLC26A4 variant. Hum. Genet. 2022;141:465–484. doi: 10.1007/s00439-021-02336-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amberger J.S., Hamosh A. Searching Online Mendelian Inheritance in Man (OMIM): A Knowledgebase of Human Genes and Genetic Phenotypes. Curr. Protoc. Bioinformatics. 2017;58:1.2.1–1.2.12. doi: 10.1002/cpbi.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lv J., Wang H., Cheng X., Chen Y., Wang D., Zhang L., Cao Q., Tang H., Hu S., Gao K., et al. AAV1-hOTOF gene therapy for autosomal recessive deafness 9: a single-arm trial. Lancet. 2024;403:2317–2325. doi: 10.1016/S0140-6736(23)02874-X. [DOI] [PubMed] [Google Scholar]
- 50.Smith C., Zafeer M.F., Tekin M. Gene therapy for hereditary deafness. Nat. Med. 2024;30:1828–1829. doi: 10.1038/s41591-024-03004-8. [DOI] [PubMed] [Google Scholar]
- 51.Rold C.D. Gene therapy allows a deaf child to hear for the first time. 2024. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data supporting the present study are available from the corresponding author upon reasonable request. Additionally, variants found in this study were submitted to ClinVar (SCV005420291 and SCV005420311).